2.2. Disaccharides
Sucrose is a non-reducing disaccharide constituted of
d-glucose and
d-fructose units which are glycosidically bonded through their anomeric carbon atoms. Sucrose (or saccharose) is broadly present in plants and is the main disaccharide feedstock across the globe. It is an important carbohydrate reserve compound and one of the main energy sources required in the human’s diet. It is produced from sugar cane and sugar beet (130 × 10
6 t/year) [
16] and is present in honey, maple syrup, fruits, and vegetables. Sucrose itself is widely used in the food industry for its sweetening ability. Sucrose can be functionalized at various positions and therefore can be converted to various interesting additives. Monocarboxylic acids of sucrose are valuable intermediates in the synthesis of monoesters and monoamides of sucrose, which are used as for their tensioactive capacities. However, examples of chemical and electrochemical transformations of sucrose are scarce due to the poor selectivity of the reactions.
One of the most noticeable examples of electrosynthesis employing disaccharides as starting material was reported by Lamy and co-authors in 1993 [
17]. The authors documented a thorough study of the electrochemical behaviour of sucrose in various reaction conditions, as well as the capacity of a range of electrodes to perform the desired oxidation reaction. Due to the known capacity of saccharose to hydrolyse under acidic condition an alkaline media was therefore used to conduct the experiments. A vast number of electrode materials were tested and evaluated by using voltammetry analysis. Ir, Fe, and Co showed no activity toward the oxidation of sucrose; Cu, Ag, Ni, Rh, and Pd displayed some activity but too weak to be explored further. Lastly, Au and Pt showed good reactivity for the oxidation of sucrose. Gold produced a much higher current density than Pt, but required a much higher working potential for the oxidation to occur. The authors then turned their attention to parameters such as temperature and initial sucrose concentration. They used a potential step program, designed to clean the electrode of species which could remain adsorbed to the electrode surface. With the optimal reaction conditions at hand, a long term electrolysis was performed on a large Au plate electrode with sucrose (10 mM) in NaOH solution (0.1 M) for 12.5 h, aiming for the specific oxidation of primary alcohol groups, without cleaving the C-O-C bond. After 12.5 h of electrolysis, 67% of sucrose was converted to a range of products (
Table 1).
In a follow up work, the same group was able to scale up their reaction by adapting it to a flow cell reactor [
18]. The working electrode used in this novel system was a lead-modified platinum electrode. A similar potential step program as previously reported was employed in order to diminish to a maximum the electrode poisoning during the reaction, and therefore maintaining the high activity and selectivity of the electrocatalyst. After 8.5 h, the electrolysis was stopped and the products of the reaction analysed by different characterisation methods. The overall sucrose conversion was estimated to 60%, with a selectivity of 80% towards 6-monoacid of sucrose (
10) and 10% toward the 1′-monoacid of sucrose (
11). In order to confirm the structure of the reaction products, compounds
10 and
11 were hydrolysed and the resulting monosaccharide derivatives were analysed (
Scheme 6).
Hence, the authors were able to assign with certainty the structure and the selectivity of the electrochemical oxidative process.
A mediated electro-oxidation of sucrose was also demonstrated by employing TEMPO as the homogenous mediator [
10]. Within the structure of sucrose, the three primary alcohols groups are the most likely to be oxidised by TEMPO, due to its high selectivity toward primary alcohol oxidation. When the electrolysis was stopped after the consumption of 6970 C (4.8 F/mol), 83% sucrose was converted and the three possible sucrose monocarboxylic acid species (such as
10 and
11) could be observed in various amounts (low to moderate yields). However, when the electrolysis was stopped after 28,450 C (19.7 F/mol) were consumed, only the tricarboxylic acid species could (
16) be observed and isolated in 39% yield (
Scheme 7).
Other disaccharides were also submitted to electrochemical transformations. Trehalose is a non-reducing sugar constituted of two
d-glucose units, linked by a α(1→1) glycosidic bond. Both direct and indirect electrochemical oxidation of trehalose were studied. Schnatbaum et al. applied a similar reaction procedure than for the electrochemical oxidation of sucrose, and were able to demonstrate the ability of TEMPO to act as a selective mediator for the oxidation of α,α-trehalose (
17) toward α,α-trehalose dicarboxylic acid (
18) [
10]. The desired product was isolated in 61% yield after consumption of 2010 C (8.8 F/mol). In a later study, Kokoh and co-workers were able to further improve the TEMPO-mediated electrochemical oxidation of trehalose by fine tuning the electrolysis potential They were also able to improve the analytical process by transforming
18 into its trimethylsilylated equivalent for a better detection by GC-MS [
19]. Using this processes, the authors were able to report a full conversion of trehalose (
17) to the dicarboxylic product (
18).
Parpot et al. studied a direct electrochemical oxidation of trehalose using a gold electrode or a gold-nickel (70/30) alloy, in alkaline media [
20]. It was postulated that combining gold and nickel in an alloy would produce a great selectivity towards the oxidation of primary alcohols (due to gold), and high current density because of the nickel atoms. Indeed, saccharide oxidation at nickel electrode is reported to occur at a high anodic potential, in NiOOH region, but generates low molecular weight carboxylic acid species. The authors first noticed that trehalose was not showing significant oxidation signals in carbonate buffer. They attributed this lack of reactivity to the absence of aldehyde/hemiacetal group, which are found in reducing sugars. Considering this aspect, the authors report changing the electrolyte from carbonate to NaOH (0.1 M) was significantly beneficial to the reactivity of trehalose with Au and Au/Ni electrodes. In order to maximise the efficiency of the electrodes, the authors used a pulse potential program for long-term electrolysis. The aim of this potential program was to perform the oxidation at potential 0.25 V vs. RHE for 60 s, followed by a cleaning pulse at 1.7 V vs. RHE for 1 s. This pulse was used to remove any species which might have remained adsorbed on the surface of the electrode after the oxidation reaction was done. The end products of the reactions were characterised by GC-MS and LC-MS. Independently of the electrode used, the main product was the trehalose monocarboxylic acid (
19). Alongside this product, significant amount of trehalose dicarboxylic acid (
17) and glucose were also detected, as well as other acids of low molecular masses (
Table 2).
This study showed no significant difference between a pure gold electrode and a gold/nickel alloy electrode toward the selectivity or the efficiency of the reaction (
Table 2) [
21,
22].
Reducing sugars are carbohydrates with an unprotected aldehyde or hemiacetal group. The reactivity order of the various groups present in reducing sugars toward an oxidative process are as follow: equatorial secondary hydroxy < axial secondary hydroxy < primary hydroxy < hemiacetal hydroxy. Therefore, the unprotected hemiacetal group present in disaccharides such as cellobiose, d-maltose, and d-lactose is the most likely to react first, which could bring further selectivity issues than with non-reducing sugars.
Cellobiose (
19) is constituted by two glucose units, linked by a β(1→4) glycosidic bond. It can be obtained from materials such as cotton, jute, or paper after hydrolysis of the cellulose.
d-maltose (
23) is a stereoisomer of cellobiose, formed of two glucose units which are is this case linked by an α(1→4) bond. Due to their similar structure, cellobiose and maltose were often studied in parallel. Both disaccharides were submitted to similar electromediated oxidation processes by Liaigre et al. [
23]. Both
19 and
23 were oxidised electrochemically using TEMPO as a homogeneous mediator, and in alkaline media (1.0 M carbonated buffer). However, the selectivity of these transformations toward the corresponding triacid products (
20 and
24 respectively) were very low when the reactions were performed at room temperature, and lead to compounds of low molecular masses such as tartaric acid and oxalic acid. Cooling the reaction mixture to 2 °C improved significantly the selectivity of the reaction and the desired tricarboxylic acid products
20 and
24 were reported in moderate to good yields (
Scheme 8).
In a subsequent study Schämann et al. reported the scaling up of the electrochemical oxidation process of
d-maltose (up to 67.5 mmol), and the further transformation of the tricarboxylic acid in the corresponding trimethyl ester in 50% yield [
15,
24]. In a comparative study between reductive and non-reductive sugars, Parpot et al. were able to demonstrate that the selectivity of TEMPO for the electro-oxidation of
23 was lower than the one observed for trehalose [
19]. They attributed this to the presence of the free aldehyde group. During their investigation the authors were able to detect by GC-MS the low molecular weight compounds, but also mono-, di-, and tricarboxylic acid products, in significant amounts.
d-Lactose (
25) is also a reducing sugar, it is formed of a galactose and a
d-glucose subunit, linked by a β(1→4) glycosidic bond. Lactose is naturally present in milk, in 2 to 8% by weight and is thus available in large quantities (about 300,000 t/year). Its TEMPO-electromediated oxidation was also studied by Shafer and co-workers [
15].
d-Lactose was electro-oxidised in alkaline media (NaHCO
3/Na
2CO
3) at constant potential (E = 0.77 V vs. RHE). Under these reaction conditions,
d-lactose was consumed quantitatively, however, the selectivity of the reaction was poor. Indeed, five different products could be identified by ESI-MS but were not isolated (
Scheme 9). The expected mono, di, and tricarboxylic acid products
26-28 were present, as well as the diketo product
29. The monosaccharide galactaric acid (
30) was also identified, and arises from the cleavage of the β(1→4) glycosidic bond. This phenomenon was also observed during the TEMPO electromediated oxidation of
d-cellobiose (
19).
The electrochemical behaviour of disaccharides has also been studied in direct oxidation processes on rare metal electrodes. As previously mentioned, a direct electrochemical transformation gives more insights on the reaction itself, and in theory, allows a better control on the reaction by being able to tune the potential at which the transformation is occurring. Whilst a wide range of metal electrodes were tested for the electrochemical oxidation of disaccharides (Au, Pt, Ni, Cu, Co, Ru, Cd, Ir), most reports only documented voltammetric studies, evaluating the electrical response of disaccharides on the metal electrode. These types of studies were looking for application in sensors, and did not report the products of the reaction. Only few reports are giving information on the reaction products formed after a long term electrolysis.
Druliolle et al. studied the oxidation of
d-Lactose (
25) on platinum and on modified platinum electrodes in alkaline medium toward the formation of lactobionic acid (
26) [
25,
26]. The authors first studied the electrocatalytic ability of a smooth platinum electrode to oxidise lactose in a sodium hydroxide solution (0.1 M), at room temperature using cyclic voltammetry (CV). However, from this preliminary tests, lactose did not demonstrate good electrochemical reactivity on platinum and no lactose was oxidised during a long term electrolysis. Then, they evaluated the effect of different underpotential deposited (upd) metal adatoms. Indeed, the ability of upd adatom to enhance the catalytic activity of noble metals for electro-transformation of organic compounds is well documented [
27,
28,
29,
30]. The authors studied the catalytic effect of Tl, Bi, and Pb adatoms on a platinum electrode in 10 mM of lactose and 0.1 M NaOH. The CV measurements showed some significant differences in terms of optimal oxidation potential and some drastic increase of current density.
Long term electrolysis of lactose was carried out with bismuth perchlorate salt in NaOH solution (0.1 M). A potential step program was used to maximise the rate of the oxidation reaction, while depositing the Bi metal adatoms onto the electrode, and minimising the poisoning of the electrode. After two hours of electrolysis, the recorded quantity of electricity passed was Qexp = 20.7 C. After analysing the crude material of the reaction by HPLC, the authors reported that lactose was converted in 70% exclusively to lactobionic acid (26), and that no other products could be detected by their analytical method, therefore awarding a nearly 100% selectivity toward 26, with a Faradaic yield of 75%. The lower Faradaic yield compared to the overall selectivity of the reaction can be explained by the slow decomposition of 26 under the reaction conditions.
Long term electrolysis of
d-Lactose on lead adatoms modified platinum electrode in carbonated buffer was also studied [
26]. A similar potential step program was used, with the adatom deposition plateau, the oxidative plateau, and the final step to remove the poisoning species from the surface of the electrode. After three hours of electrolysis, the recorded quantity of electricity passed was Q
exp = 15.1 C. After analysing the resulting mixture, the authors report that 78% of the starting lactose was converted to the desired lactobionic acid with nearly 100% selectivity. If the reaction was continued to reach four hours, 95% of lactose was converted, but the selectivity toward
26 decreased to 76%, which can be explained by the slow decomposition of lactobionic acid under the reaction conditions.
Druliolle et al. then turned their attention to gold electrode for the oxidation of
d-Lactose (
25) [
31]. The ability of gold based electrocatalyst to oxidise selectively alcohol groups in alkaline medium was previously reported by Larew and co-workers [
32]. Furthermore, gold is known to be less subject to poisoning than palladium, which explain the increase of attention which was directed toward this noble-metal for electrochemical transformations. To determine the optimal reaction conditions for the electro-oxidation of lactose on a gold electrode, the authors used voltammetric measurements. They report that a potential step program was used to maximise the oxidation of
25, while minimising the poisoning of the gold electrode, under alkaline medium (carbonated buffer, 0.1 M). Then, the authors evaluated the impact of the starting concentration of
d-Lactose, as well as the effect of small variation on the oxidative potential plateau. With the optimal reaction conditions ([lactose] = 10 mM) in hand, 95% of
25 was converted to lactobionic acid, with 98% selectivity after five hours of electrolysis.
By using two complementary IR analytical methods (SPAIRS and SNIFTIRS), the authors were able to identify the possible steps of the reaction, and therefore to propose a possible mechanism for the electro-oxidation of
d-Lactose at a gold electrode (
Scheme 10). During this investigation, the authors noticed that two different signals, depending on the oxidative potential applied. When the applied potential of the reaction is below 1.1 V vs. RHE, a characteristic signal of vibration mode was observed, which can be attributed to an asymmetric O-C-O bond stretching (
Scheme 9, left hand side cycle). At higher potential, a signal characteristic of a symmetric O-C-O stretching (
Scheme 9, right hand side cycle) bond could be identified. Gold being known for its oxygen adsorption properties, it was assumed that at low potential, a small quantity of hydroxyl species can be adsorbed on the gold electrode, and at higher potential, a much larger quantity of gold hydroxide species could be generated. Despite the differences observed by infra-red spectroscopy, only lactobionic acid was observed after the reaction, which indicate that at different potential, different mechanisms are taking places, yet delivering the same product. The authors postulated that once
d-Lactose was adsorbed on the gold surface, either a hydroxide group could attack on the glycosidic carbon, therefore opening the ring (
Scheme 9, left hand side cycle), or if the concentration of gold hydroxide species was sufficient, another Au-OH group could open the ring (
Scheme 9, right hand side cycle). Both intermediate could then deliver the desired product
26.
In a following study, Kokoh et al. documented the electrocatalytic oxidation of
d-Lactose on nanoparticles of Au-colloids (5 nm), immobilised on a carbon felt electrode, in a flow reactor [
33]. The optimal reaction conditions were first determined using CV, which demonstrated that the gold nanoparticles were able to generate a higher current density than when a plain gold electrode of three times the size was used. With the information gathered during the preliminary investigation, the authors determined that a two potential plateau program had to be employed to limit the poisoning of the electrode, as well as to maximise the oxidation reaction. The electro-oxidation was then carried out in carbonated buffer (0.1 M, pH = 10.2), and delivered lactobionic acid (
26) in 91% yield.
Recently, Parpot et al. described the electrochemical oxidation of cellobiose (
19) and
d-maltose (
23) on a gold electrode in alkaline media [
20]. For each disaccharide, the author conducted preliminary investigations using CV. From these tests, it was possible to determine the optimal reaction conditions. As previously mentioned, gold electrodes are subject to poisoning. Therefore, in order to diminish this undesired phenomenon, a multi-potential plateau program was used for the long term electrolysis of both cellobiose and
d-maltose.
With the optimal reaction conditions at hand, 95% of cellobiose (
19) could be converted almost exclusively to its monocarboxylic acid equivalent, cellobionic acid (
27), resulting from the oxidation of the aldehyde at carbon C1. The dicarboxylic acid product could also be detected in very small amount (3%) as well as trace amounts of the products resulting from C-C bond cleavage (
Scheme 11A).
Then,
d-maltose was submitted to its corresponding optimal reaction conditions for long term electrolysis. However, when
d-maltose was submitted to electro-oxidation in NaOH solution (0.1 M), the reaction proved to be unselective, producing a variety of small carboxylic acids, such as tartaric acid, formic acid, etc., in non-negligible quantities. Furthermore only 50% of the initial
d-maltose was converted after eight hours of electrolysis. A second electrolysis of
d-maltose was then carried out in carbonated buffer (0.1 M, pH = 10), using the same potential plateau program. After twenty hours, 79% of the initial amount of
d-maltose (
20) was converted, with a selectivity up to 92% toward maltobionic acid (
28), with only 3% of the dicarboxylic acid equivalent being detected, and no degradation products (
Scheme 11B).
2.3. Monosaccharides
Significant interest has been drawn to the oxidation reaction of monosaccharides due to the variety of high-added-value chemicals which could potentially be obtained. Enzymatic and chemical processes, stoichiometric and catalytic, as well as homogeneous and heterogeneous catalytic systems have been reported for such transformations. However, these processes are beyond the scope of this work, and several reviews focusing on those aspects have been documented [
34,
35,
36]. Concerning the electrochemical transformation of monosaccharides, the amount of reports is scarce. Most of the existing literature is focusing on the development of glucose biosensors to help for diabetic treatment, or as a reagent for fuel cells for clean energy production. However, in such studies, the structure of the products generated are generally not investigated. Amongst the electrosynthesis reports of the transformation of monosaccharides,
d-glucose was the most widely investigated because it is one of the most abundant bio-based chemicals, and it can be converted to a variety of commodity chemicals [
35,
37]. In 1910, Lob described one of the first electrochemical oxidation of
d-glucose, in sulfuric acid, with a lead anode and a platinum cathode [
38]. The various products obtained were
d-arabinonic acid and
d-glucaric acid, as well as some fragmentation by products such as
d-arabinose and formaldehyde. The slow degradation of
d-glucose to lower aldoses during an electrolysis in non-aqueous media was investigated in more details by Hay et al. [
39] although, the lack of detailed information such as yield, efficiency, and selectivity are limiting the comparative analysis.
Isbell and co-workers reported series of efficient indirect electrochemical oxidation of various monosaccharides into their corresponding aldonic acid salts [
40,
41,
42,
43]. Their process is based on the electro-oxidation of sodium or calcium bromide, generating free bromine at the anode, which reacts with the saccharide to give the corresponding aldonic acid. The calcium carbonate present in the reaction mixture quenches the acid, maintaining a constant pH (6.2) during the electrolysis (
Table 3). The bromide is continuously regenerated, allowing the conversion of a large amount of sugars while using only a catalytic amount of bromide salt [
40,
41]. Most of the aldonic acid salts then crystallises and the acid form can then be recovered by reacidification and recrystallization [
42]. The acid salt products could also be converted to the corresponding δ- or γ-lactones via an acid catalysed process.
When xylose was submitted to the electro-oxidation, the current density and the conversion of the starting material were very promising. However, calcium xylonate appeared to be very soluble, and therefore could not be separated by crystallisation in an efficient manner. The authors then applied similar reaction conditions for the electro-oxidation of xylose, but in the presence of strontium or magnesium bromide and carbonate. This allowed them to isolate the corresponding xylonate salt in high yields 49% and 80% respectively (
Table 3, entry 5) [
43].
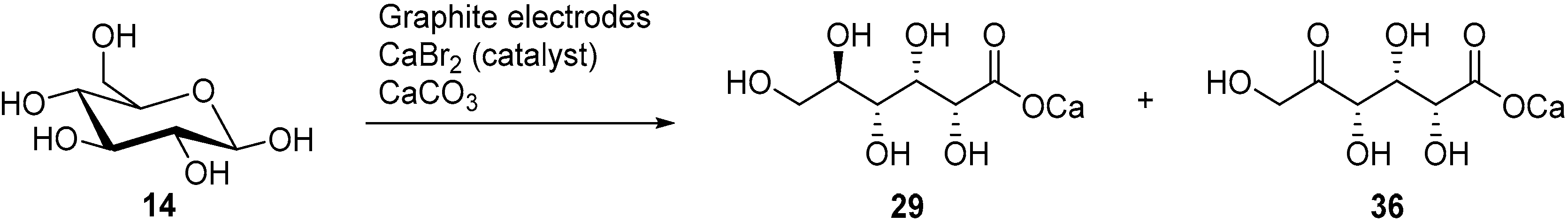
In a follow up study, the same authors, and others groups, proposed a semi-industrial procedure for the production of calcium
d-gluconate [
41,
44]. In this process, the calcium
d-gluconate was recovered after the addition of calcium hydroxide, which generate a less soluble basic salt species. Despite continuing the electrolysis longer than theoretically needed to oxidise glucose to gluconic acid, a reducing compound remained in solution. After repeating Isbell’s procedure for an electrolysis, Cook et al. were able to isolate the calcium salt of 5-keto-gluconic acid (
36) in approximately 5% yield [
45]; therefore proving that gluconic acid was not the only product of the CaBr
2 mediated electro-oxidation of glucose (
Scheme 12).
Inspired by the examples of chemical oxidation of simple alcohols and carbohydrates by ruthenium complex catalysts [
46]. Kokoh and co-workers decided to explore the possibility for a selective electro-oxidation of glucose using Ru-based complex catalysts [
47]. Azopyridine (azpy) ruthenium complexes are known to maintain the ruthenium metal at a low redox potential state Ru(IV), which could be suitable for a selective electrochemical oxidation of carbohydrates [
46,
48,
49,
50,
51]. The authors reported the synthesis of two isomers γ-RuCl
2(azpy)
2 and δ-RuCl
2(azpy)
2 and their immobilisation on carbon fibre materials prior to their electrochemical characterisation. Using CV measurements on crude RuCl
2(azpy)
2, the authors could determine that the redox couple observed in the positive scans was Ru(IV)/Ru(III). Voltammograms of the crude ruthenium complex recorded in the presence on
d-glucose in alkaline medium (0.1 M NaHCO
3/Na
2CO
3) showed no reduction peak, thus suggesting a fast reaction between the Ru(IV) and glucose. After having proved that the ruthenium complex was able to react with glucose, the authors set out to perform long term electrolysis. Using their optimal reaction conditions,
d-glucose was electro-oxidised during 45 h at fixed potential in presence of the crude RuCl
2(azpy)
2 catalyst. Analysis of the crude reaction mixture showed a 22% conversion of glucose toward gluconic acid (
29, 11% yield, 50% selectivity) and 2-keto-gluconic acid (
37, 7% yield, 33% selectivity). Trace amounts of a tricarboxylic acid could also be detected by LC-MS analysis, as well as oxalic and tartaric acid. The presence of these low molecular weights acids suggested that the tricarboxylic acid was hydrolysed which resulted in the C2-C3 bond cleavage [
52]. The authors postulated that glucose is getting oxidised to gluconic acid (
29) via the intermediate δ-gluconolactone, but that the gluconic acid can itself be further oxidised and therefore deliver 2-keto-gluconic acid (
Scheme 13).
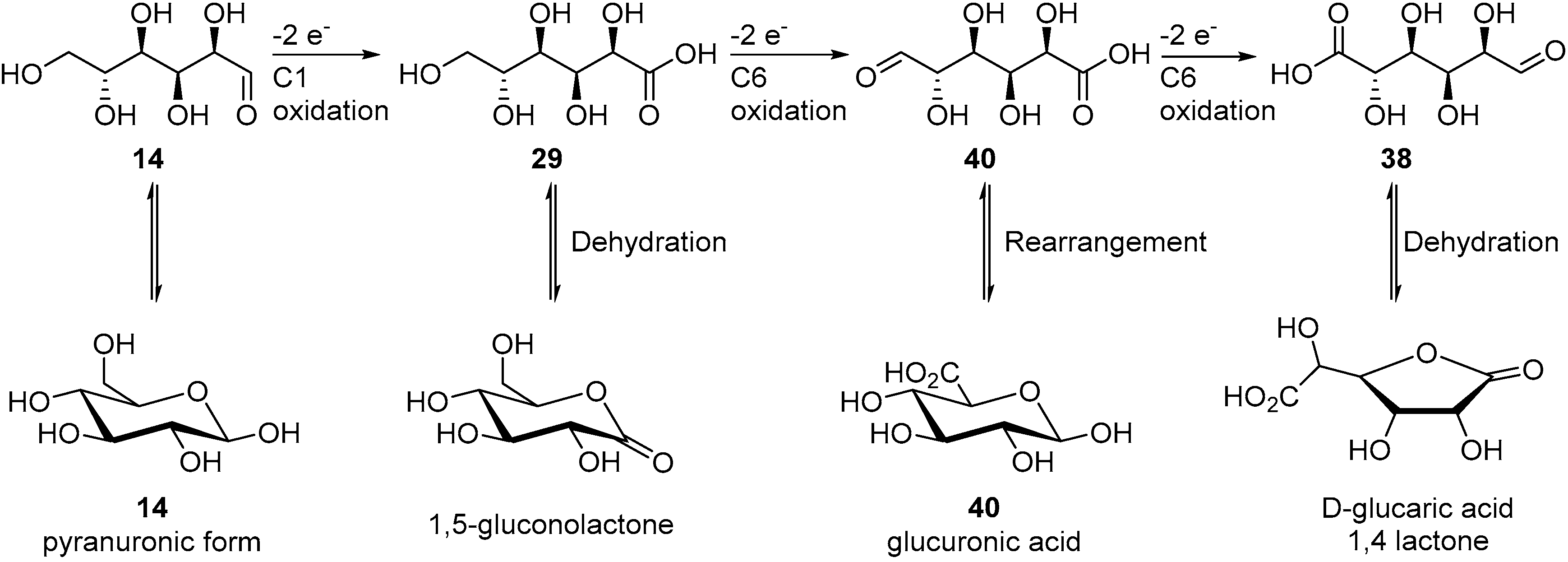
Long term electrolyses were carried out using pure γ-RuCl2(azpy)2 and δ-RuCl2(azpy)2, using the same reaction conditions as the one used for crude RuCl2(azpy)2 catalyst. When γ-RuCl2(azpy)2 was employed, the conversion of glucose increased to 50%, and delivered 29 and 37 in 30% and 18% yield, respectively. The mediated electro-oxidation of glucose by δ-RuCl2(azpy)2 gave only 10% conversion toward 29 mainly (70% selectivity), but gluconic, glucuronic, tartaric and oxalic acid could also be observed in small quantities. These results show that γ-RuCl2(azpy)2 catalyst is the most active isomer and can deliver 2-ketogluconic acid (37) in a selective manner.
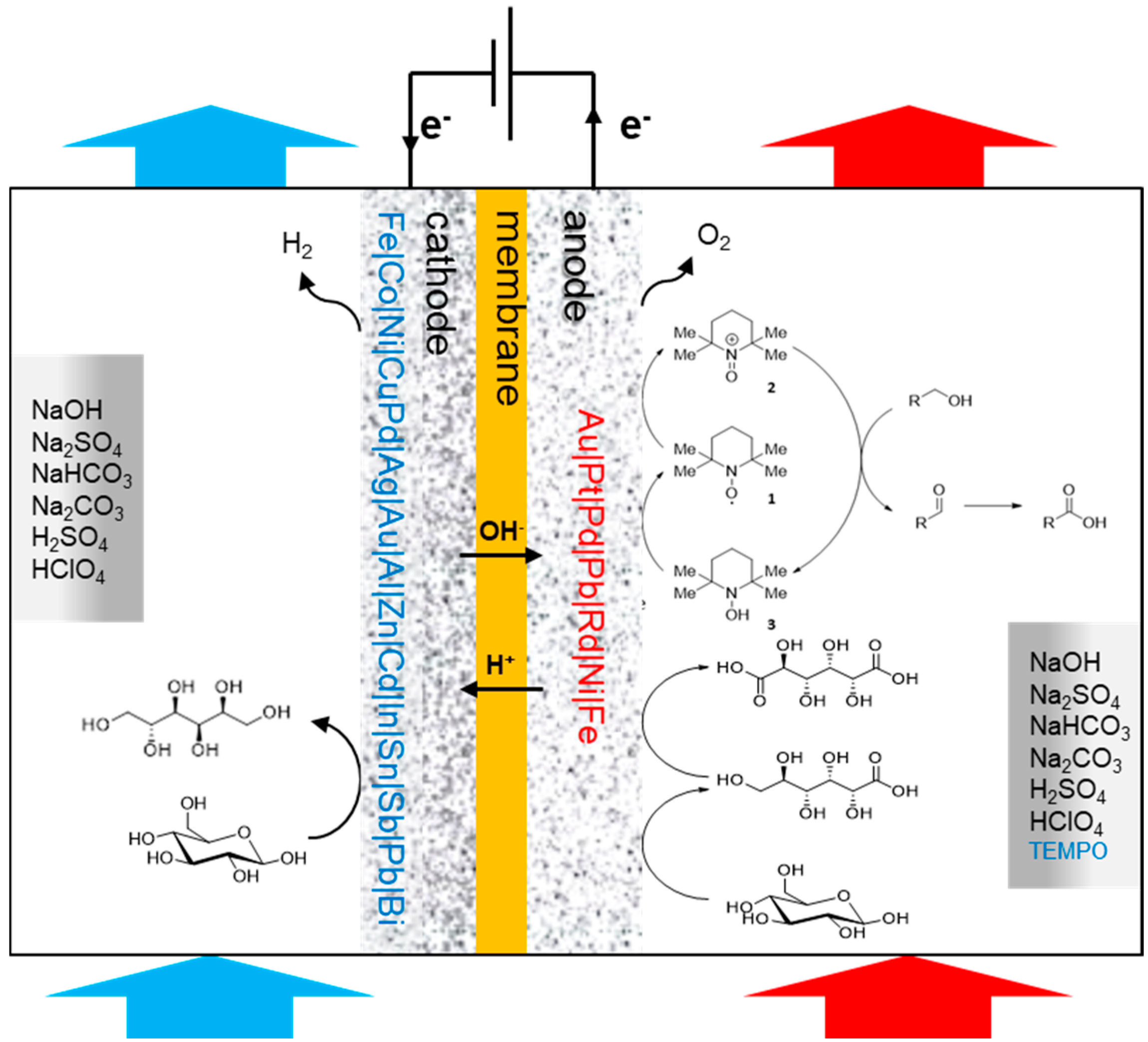
As previously mentioned, 2,2,6,6-tetramethyl-1-piperidinyl free radical (TEMPO) and its derivatives are very selective for alcohol oxidation. Such organic homogeneous mediators provide a metal free option of alcohol oxidation and usually shows great reactivity in mild conditions. Therefore, after having thoroughly studied the chemical oxidation of glucose with TEMPO and several co-oxidants [
53,
54,
55]. Ibert et al., decided to investigate the TEMPO mediated electro-oxidation of glucose in alkaline medium [
56]. The authors first evaluated the ability of glassy carbon and platinum to oxidise TEMPO efficiently. Platinum showed much lower reactivity toward TEMPO than glassy carbon, which also showed great stability after 10 CV experiments. Glassy carbon was therefore used as the working electrode. The chemical oxidation of carbohydrates is known to be sensitive to pH changes [
53], therefore, the authors turned their attention to the effect of the pH on the electrochemical oxidation of glucose by studying the current density during CV measurements. Various carbonated solutions were used with pH ranging from 7 to 12. The clear trend was that a basic pH is beneficial to the oxidation of glucose, demonstrating the important role played by the base in such transformation. With the optimal reaction conditions in hand, it was possible for the authors to attempt a preparative TEMPO mediated electrochemical oxidation of glucose, targeting glucaric acid (
38) as the main product. The first attempts in undivided cells showed significant limitation due to a lack of control over the parameters of the reaction. This pushed the authors to move to a jacketed reactor with a control module to regulate the pH and the temperature of the reaction mixture. In these conditions, the full conversion of glucose was achieved with 20% faradaic excess (i.e., the reaction was stopped after the total current collected exceeded the theoretical value for full conversion by 20%), however, a large amount of by-products were detected (low molecular weight products from degradation reaction) and the desired glucaric acid was only formed in poor yield. One of the main by-products detected was a tricarboxylic acid compound which had never been observed in chemical oxidation of glucose. By using GC analysis, combined with NMR studies, Ibert et al. were able to unambiguously determine the exact structure of the tricarboxylic acid (
39) [
57]. This information, combined with various analytical information, made it possible for the authors to propose a mechanism explaining the formation of
39 (
Scheme 14). They postulated that the triacid resulted from a benzylic rearrangement at the C-4, C-5, and C-6 centers. This rearrangement could occur via the formation of a diketone intermediate, due to the over-oxidation of the secondary alcohols of glucaric acid (
38). In order to prove their hypothesis, the authors performed several electrochemical oxidations with compounds such as 2-keto-gluconate, which they postulated to be intermediates in the formation of
39.
The presence of 39 and degradation products (oxalic, tartaric, and malonic acid) in large amount, pushed the authors to reoptimize their reaction conditions. By lowering the temperature to 5 °C, and increasing the pH of the reaction to 12.2, most of the degradation and over-oxidation products formation could be inhibited (below 5% was generated). With these new optimal reaction conditions, an electrolysis of d-gluconic acid instead of d-glucose was performed, and the desired d-glucaric acid was delivered in 85% yield.
Due to its large availability and the added value of the potential reaction products, glucose has been the most studied monosaccharide for direct electrochemical transformation. Although the anodic oxidation of glucose has been studied with a range of metal (Ag, Hg, Ru, Rh, Cu, Ni, Ir, Co, Mg), most of the literature arguably focuses on the use of Au and Pt working electrodes. Indeed, both metals reactivity and selectivity for glucose electro-oxidation have first been thoroughly studied and compared via voltammetry experiments [
58,
59,
60,
61,
62]. These studies gave insights on various important parameters, such as the adsorption/desorption ability of the metal electrodes [
60,
61,
62], as well as the potential mechanisms for the direct-electro-oxidation of glucose at the surface of the electrodes [
58,
59]. It was found that in neutral and alkaline medium, gold electrodes displayed a greater electro-oxidation rate than any other metals, however, gold was also very prone to poisoning. To gain a deeper understanding of the electro-oxidation process and improve the performance of the electrodes, the metal surface was covered by various oxide species, or via underpotential-deposited adatoms (Pb, Tl, Bi) [
27,
30]. By partially covering the surface of the electrodes, Pt and Au were less sensitive to poisoning, therefore a higher current response could be observed when put in presence of glucose. However, these preliminary studies only used voltammetric measurements and did not perform long term electrolysis to test the stability and the selectivity of the electrodes for glucose oxidation. By basing their process on the results gathered by these preliminary studies, Belgsir et al. demonstrated that
d-glucose could efficiently be electro-oxidised by a gold anode in alkaline medium [
63]. At first, the authors employed a bare gold electrode using a three-step pulsed potential program to avoid poisoning by glucose or the reaction products. After 25 h of electrolysis, gluconic acid (
29) could be observed in 77% yield, and glucaric acid (
38) in trace amounts. Glucaric acid being a chemical with high added value, the authors decided to modify the surface of the gold electrode with Pb-adatoms, hoping to increase the reactivity of the electrode and thus the selectivity toward glucaric acid. The authors tested two different oxidation potential plateaus, and showed that the selectivity can clearly be changed by the addition of Pb adatoms and by tuning the potential used for the reaction (
Table 4).
Although the faradaic yield decreased and more degradation products could be observed, these long term electrolysis showed that it was possible to modify the selectivity and reactivity of a gold electrode for glucose electro-oxidation by using a suitable potential program and by modifying the electrode surface with foreign metal adatoms.
Kokoh and co-workers then set out to fully investigate the influence of upd adatoms (Bi, Tl, and Pb) on both Au and Pt electrodes, in acidic, neutral, and basic mediums for the electrochemical oxidation of
d-glucose [
64,
65]. The voltammograms recorded in acidic medium (HClO
4, 0.1 M) showed that regardless of the electrode metal and of the adatom used, the current densities of glucose oxidation are very low, suggesting that the reactivity of glucose in acidic conditions is limited. Indeed, when the reaction mixtures were analysed after 25 h electrolysis, only a very low amount of glucose was converted to a variety of oxidation and degradation products. When voltammograms were recorded on Pt and Au anodes in neutral medium (KH
2PO
4/NaOH, pH = 7.3), a much greater current density was observed than under acidic conditions. However, only a limited amount of glucose was converted (up to 15%) to gluconic acid (up to 12% yield) and traces of glucaric acid with Au anode or traces of glucuronic acid with Pt electrodes. The authors then moved to investigate the behaviour of Au and Pt in presence of glucose in basic medium (NaOH 0.1 M) [
64]. It is known that Au is the most efficient electrocatalyst for glucose oxidation in basic conditions, because it generates higher current densities and is less sensitive to poisoning than Pt [
32,
66,
67]. Kokoh et al. studied the influence of the concentration of glucose (from 1 mM to 50 mM) on a pure gold anode to gain insights on the rate of the reaction and on its mechanism using voltammetry. At low initial concentration of glucose, the reaction is of order one, but at high concentration, a saturation phenomenon is observed and the overall order drops to zero. The effect of upd Tl, Pb, and Bi adatoms was also studied on a gold electrode in alkaline media. Although the voltammograms remained unchanged with and without adatoms, the current density increased during the electrolysis of glucose in presence of upd adatoms. When comparing the results of long electrolysis of glucose (
14) performed on pure Au, Au-Bi, Au-Tl, and Au-Pb, gluconic acid (
29) appeared to be the main product for every system. However, the Au-Pb adatom electrocatalyst also produced a significant amount of glucaric acid (
38). This system was then studied further, by modifying the potential at which the oxidation reaction was occurring (0.5, 0.6, and 0.9 V vs. RHE). The trends observed were that the highest concentration of gluconic acid was obtained at lower potential (75% of the oxidised glucose), and that at higher potential, glucaric acid could was becoming the major product of the reaction (37% of the oxidised glucose) after 24 h of electrolysis. However, at high potential more degradation products (tartaric, oxalic, formic, and glyoxylic acid) could be observed in small quantities. The authors then looked at the glucose electro-oxidation on a Pt anode in basic conditions. After 21 h of electrolysis, 70% of glucose was converted to gluconic acid (
29) as the main product (50% yield), and other degradation products. They then moved to electro-oxidation of glucose on a Pt electrode with Pb adatoms, using the same reaction conditions as the one used for the pure Pt electrode. A significant increase in current density was observed during the 24 h electrolysis. Within 4 h, the conversion of glucose (
14) was nearly complete, and the concentration of gluconic acid (
29) reached its maximum (70% yield). After the first 4 h, the gluconic acid concentration dropped to almost zero and the concentration of by-products (oxalic acid: 60%, 2-ketogluconic acid (
37): 30%, other degradation products: traces) increased. The difference observed in the distribution of the by-products of the reaction when Pb adatoms were added to a Pt anode, lead the authors to propose possible mechanisms. For the electro-oxidation of glucose on pure Au or Pt, Kokoh and co-workers suggest that the mechanism follow similar steps (
Scheme 15A). First is the adsorption/dehydrogenation of glucose at the electrode surface. Then, the free sites of the electrodes oxidise to metal-hydroxide species. The interaction of the two chemisorbed intermediates can then react and release gluconic acid.
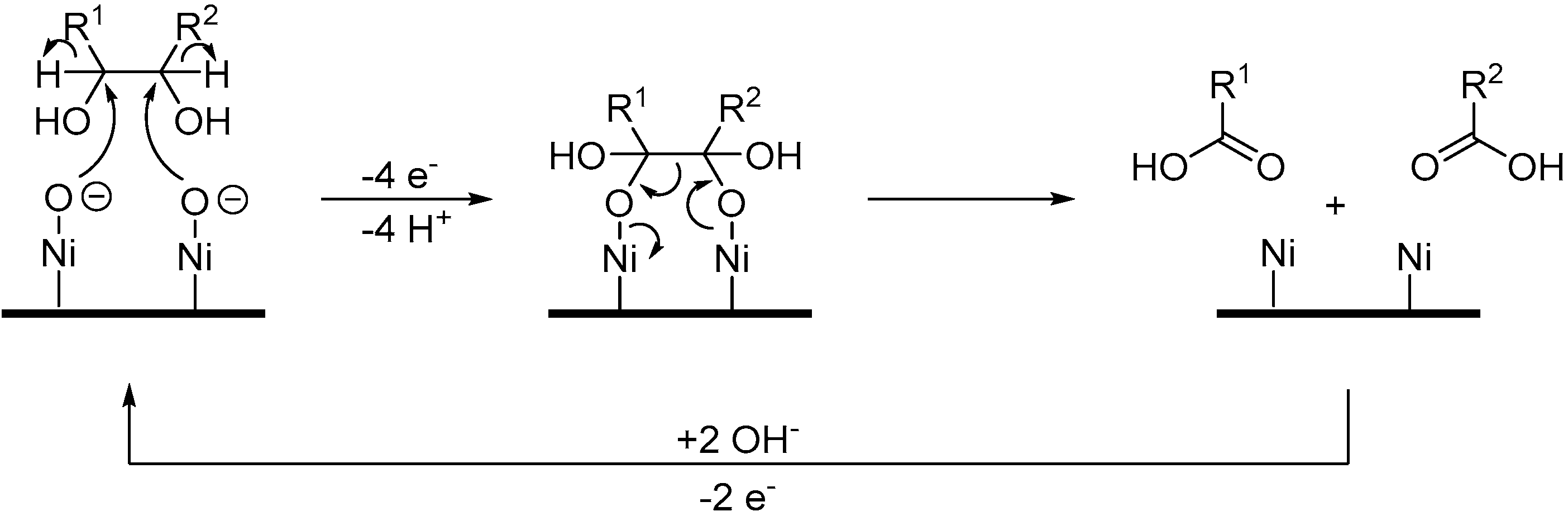
When Pb adatoms were adsorbed on the electrode surface,
d-gluconic acid (
29) was generated rapidly and then consumed, leading to the formation of by-products, and more particularly 2-keto-gluconic acid. Taking this phenomenon into account, the authors generated the following hypothesis to explain the formation of this by-product mostly in the presence of Pb adatoms (
Scheme 15B). It is postulated that the formation of gluconic acid follows the same pathway as it does with pure metal electrodes. However, once the gluconic acid is desorbed from the surface, it is possible that a new surface/chemical interaction occurs. Indeed, due to their free p-orbitals, Pb adatoms are able to coordinate with one oxygen from the carboxylic group and the oxygen from the C2 carbon. Then a free site the Pt electrode surface can dehydrogenate the C2 carbon, generating the 2-ketogluconic acid. Kokoh et al. then studied the electrochemical oxidation of
d-gluconic acid at Pt and Pt-Pb adatoms anodes under basic conditions (0.1 M NaOH) [
68]. After long term electrolysis with either pure Pt or Pt-Pb adatoms electrode, the distribution of products was significantly impacted by the presence of the Pb on the electrode (
Scheme 16). The oxidation of gluconic acid oxidation on a pure Pt electrode appeared to be very slow and yielded mainly glucuronic acid (
40). When Pb adatoms were adsorbed on the surface of the anode, much higher current density could be reached, and the main product of the reaction was 2-ketogluconic acid (
37), while glucuronic acid was not observed. These observations were confirming the hypothesis that Pb was orienting the oxidation of gluconic acid on the C2 carbon by coordinating to the carboxylic acid group and the C2 hydroxyl group as shown in
Scheme 15B.
Similar results were obtained when Tl adatoms were adsorbed on a Pt anode for glucose electro-oxidation [
69]. Glucuronic acid (
40) was detected in very small amount, gluconic acid was the major product of the reaction, and the concentration of 2-keto-gluconic acid (
37) increased with the potential of the reaction (1% yield at E = −0.15 V vs. RHE; 14% yield at E = −0.02.V vs. RHE).
To deepen the understanding of the reactivity of
d-glucose on a Pt electrode for anodic oxidation, Largeaud et al. set out to study the difference in behaviour of the two main dissolved anomeric forms of glucose (α and β) [
70]. Indeed, the free linear aldehyde form of
d-glucose is known to be the minor form in solution, and the α and β-glucopyranose forms are in an equilibrium of approximately 33% and 67% respectively. The authors evaluated the stability of α
d-glucose and β
d-glucose at low temperature in acidic, neutral, and basic conditions by polarimetric measurements. In acidic and neutral medium, the starting form of α or β-glucose used to make the solution remained the major anomer in solution after 5 h in solution. However, in alkaline medium, β
d-glucose rapidly became the main anomer in solution, independently of the crystalline form used to make the solution. The authors then performed voltammetric measurements, in acidic and basic reaction conditions at low temperature. In basic medium, the β
d-glucose being the major form in solution, it is difficult to draw conclusions on the reactivity of the α anomeric form. However, the CV measurements in acidic condition of the α and of the β
d-glucose forms showed some clear difference of reactivity. Indeed, the β
d-glucose showed much higher current density than the α
d-glucose, suggesting a much higher reactivity of the β-anomer on a Pt anode. The hypothesis proposed by the authors is that the β conformation allows the most planar approach of the glucose molecule to the electrode surface, leading to a better interaction and a more rapid reaction. By analysing the solution during and after long term electrolysis experiments in basic medium, gluconic acid seems to be the final product of the reaction, but δ-gluconolactone could be detected and was assumed to be an intermediate during the transformation.
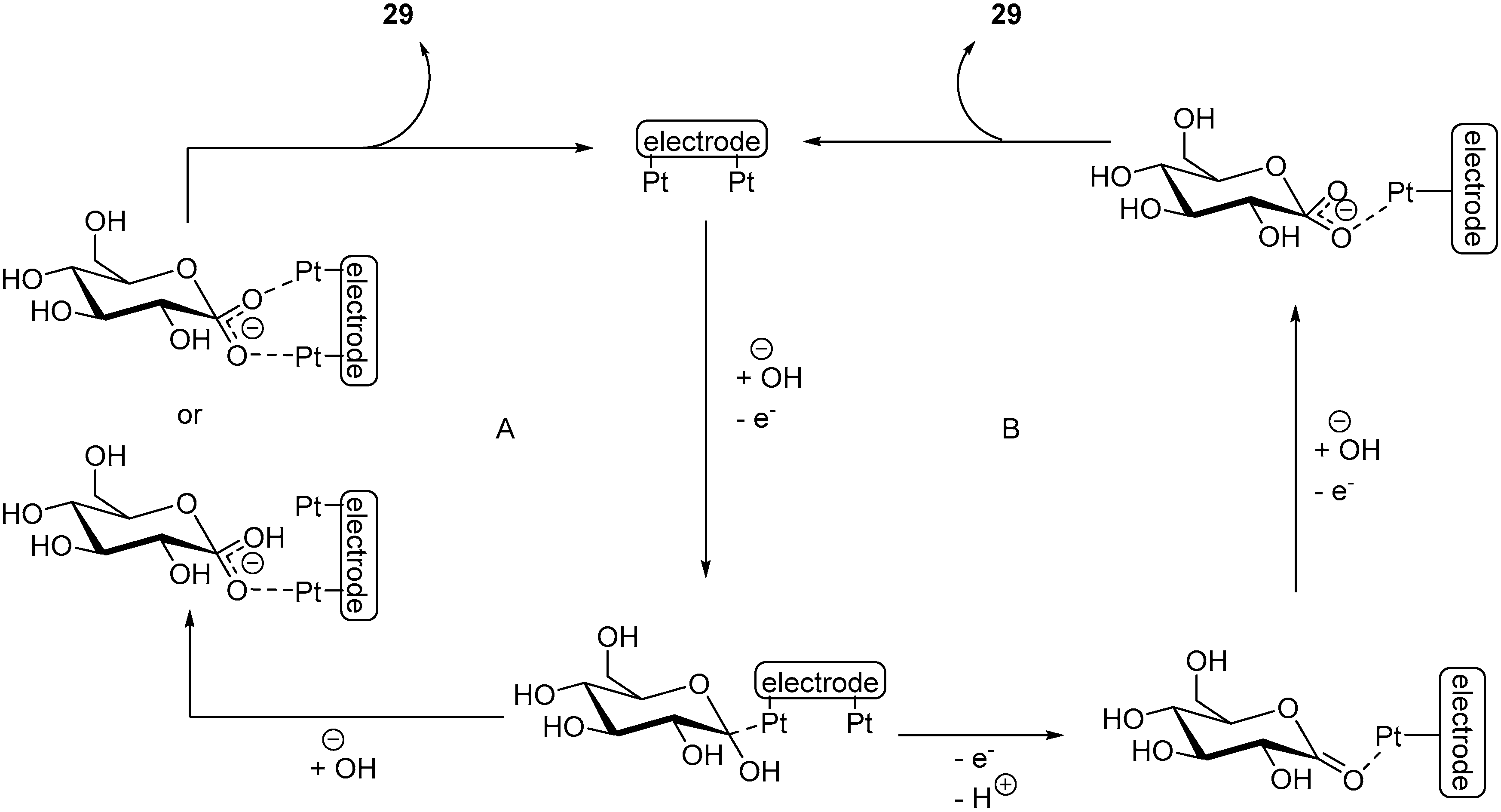
By using in situ reflectance infrared spectroscopic techniques (SPAIRS and SNIFTIRS), Beden et al. were able to give more details to the electro-oxidation of the β
d-glucose anomer on a Pt anode in alkaline media [
71]. With these investigations, the authors were able to propose a detailed mechanism for the electro-oxidation of β
d-glucose in alkaline medium on a Pt anode. The first step is the chemisorption of glucose onto the surface at a free platinum site. At a potential lower than 0.6 V vs. RHE, the adsorbed dehydrated species can then be further oxidized as a weakly adsorbed gluconate via two different possible mechanisms, which depend on the potential applied to the reaction. The gluconate can easily desorb from the surface as a gluconate species (
Scheme 17A). At potential higher than 0.6 V vs. RHE, the adsorbed dehydrated glucose species generates by oxidation a δ-gluconolactone species. This species can desorb and by hydrolysed to deliver gluconic acid or can remain weakly bonded to the surface while being oxidized, leading to a weakly bonded gluconate (
Scheme 17B).
The ability to change the selectivity of the electro-oxidation of glucose by tuning the reaction potential was also exemplified on Au electrode by Moggia et al. [
72]. Indeed, the authors studied the reactivity of
d-glucose in alkaline media on a pure gold electrode, targeting
d-glucaric acid as the main product of the reaction, using voltammetric measurements. In order to assign the peaks observed during CV experiments with
d-glucose, they studied the response of the potential intermediates of the reaction, glucuronic acid (
40), gluconic acid (
29), and glucaric acid (
38).
These measurements allowed them to clearly identify the potential at which glucose was oxidised to gluconic acid (0.55 V vs. RHE), and the potential for the oxidation of gluconic to glucaric acid (1.3 V vs. RHE). Then, by applying the appropriate potential for gluconic acid, the authors report 87% selectivity toward 29 and only trace amounts of 38 detected. When the potential was then increased to favour glucaric acid formation, the selectivity reached 14% and the selectivity toward gluconic acid decreased to 66%, thus demonstrating the relationship between the potential of the reaction and the selectivity of the outcome.
Due to the scarcity and the high price of noble metals such as Pt and Au, pure metal electrodes are not easily scalable because high costs that are involved. In order to lower the amount of metal loading on the anode, whilst preserving the selectivity and the reactivity of the metal, Holade et al. reported a selective electro-oxidation of glucose using Au nanoparticles on carbon as the anode material [
73,
74]. The aim of their work was to develop a highly efficient fuel cell which harvests electrical power from the oxidation of glucose, while producing added value chemicals. At first, the authors studied the reactivity of glucose toward the Au nanoparticles via CV paired with infrared analysis. These analyses revealed that glucose was successfully adsorbed on the Au nanoparticles, and could be oxidised rapidly without generating any C-C bond cleavage, and a fast hydrolysis of gluconolactone, which proved to be an inevitable intermediate. Long term electrolysis were then performed for 7.5 h under basic conditions (0.1 M NaOH). Gluconate was detected as the major product of the reaction (65% yield) with a nearly 100% faradaic efficiency. The reaction was followed by IR spectroscopy to verify that at any stage of the reaction, no C-C bond cleavage occurred.
Bimetallic nano-objects were also investigated as electro-catalyst for glucose electro-oxidation in NaOH solution (0.1 M). Rafaideen et al. recently reported the study of carbon-supported PdAu nanomaterials for selective oxidation of monosaccharides [
75]. At first, the authors evaluated separately the activity of Pd and Au on carbon. They noticed that Au was significantly more active toward glucose oxidation. The authors then set out to test the electrocatalytic activity of the bimetallic Pd
xAu
10-x/C for monosaccharide oxidation in alkaline medium. By using voltammetric measurements, it was possible to prove that Pd rich catalysts and Au rich catalysts behaved similarly to their respective pure metal equivalents. However, the changes detected were believed to be related to the surface composition rather than to the bulk composition. It was observed that at low potential (<0.9 V vs. RHE), the activity of the catalyst increases with the increase of Au content up to 70% and then decreases, with Pd
3Au
7/C showing the highest catalytic activity. With the optimised reaction conditions,
d-glucose (14) was electrolysed at a Pd
3Au
7/C anode in basic medium. After 6 h of electrolysis, 67% of glucose was consumed, and gluconate (
29) was detected in 58% yield (selectivity towards gluconate of 87%), and very small amount of over-oxidation products could be detected. The authors then applied the same process to the electro-oxidation of
d-xylose (
34), which consumed up to 52% of xylose after 6 h of electrolysis, to deliver xylonate (
35) in 48% yield and 92% selectivity.
Although most of the research arguably focused on the electrochemical behaviour of
d-glucose, other monosaccharides could also be successfully electro-oxidised on Au or Pt electrodes. Xylose, as previously mentioned, has been electrochemically oxidised on Au and Pt electrode by Governo et al. [
76]. The authors studied the catalytic activity of both metals for anodic oxidation of
d-xylose (
34) in alkaline medium using voltammetric measurements. They evaluated the effect of the concentration of xylose and of the temperature. By studying the effect of the pH, it was possible to show that on both Au and Pt, a high concentration of NaOH could contribute to the inhibition of the reaction. To avoid the electrode poisoning, a program of potentials was designed for each electrode to perform electrolysis. Au showed significantly better reactivity than Pt for the electro-oxidation of xylose (98% and 26% conversion respectively) and selectivity toward xylonic acid (
35) (
Table 5).
The oxidation of
d-galactose (
30), a C-4 epimer of
d-glucose, has been reported under homogeneous and heterogeneous catalysis [
77,
78]. However, its electrochemical oxidation has been scarcely investigated. Parpot et al. documented the electrochemical transformation of
d-galactose (
30), and compared the selectivity and reactivity of Pt, Pt-Pb, and Au electrodes in alkaline medium [
79]. For the long term electrolysis, the appropriate potential step programs were used. These potential programs were optimised with the information gathered with preliminary voltammetric measurements with the corresponding electrode. The electro-oxidation of
d-galactose was carried out for 25 h at room temperature in 0.1 M NaOH solution. For the three electrodes,
d-galactonic acid (
31) was the main product of the reaction, however, Pt-Pb and Au electrode were significantly more selective toward
31 than the smooth Pt electrode, and Au was also more reactive, converting a greater amount of
30 than any other electrode (
Table 6).
In this study, the authors propose a mechanism which follows the same steps as the ones described for the electrochemical oxidation of
d-glucose at Pt, Pt-Pb and Au electrode (see
Scheme 15A,B). The amount of galactaric acid generated during the electro-oxidation of galactose could be significantly increased by readjusting the oxidation plateau potential used [
22]. Parpot et al. showed by employing voltammetry experiments that the enediol tautomer plays an important role in the oxidation of the primary alcohol of
d-galatose on Au electrodes. Indeed, without this enediol form, the electro-oxidation of the primary alcohol is significantly disfavoured compared to the electro-oxidation of the aldehyde/hemiacetal group. It was also possible to show that the adsorption of the enediol species occurs at higher potential than the one used for aldehyde adsorption. Furthermore this study demonstrated that the relative position of the hydroxyl groups has an influence on the current density. Indeed,
trans-diols are known to be more reactive than their
cis-isomer on at Au electrode [
80]. which seems to also apply for saccharides electro-oxidation. Therefore, by modifying the potential program to favour the enediol adsorption/oxidation, the authors were able to generate galactaric acid in 20% yield after 7 h of electrolysis under basic conditions (0.1 M NaOH). However, more degradation products, resulting from C-C bond cleavage, could also be observed.
The direct electrochemical oxidation of
d-mannose (
32) was also studied on Pt and Au electrodes [
81]. Parpot et al. studied the behaviour of
d-mannose to acidic, neutral, and basic conditions on each electrode. They found that, like
d-glucose,
d-mannose showed very limited reactivity toward Au or Pt in acidic media, and that the current density increased with the increase of the pH. Therefore, for the rest of the study,
d-mannose was only submitted to basic conditions (0.1 M NaOH). The authors then looked at the influence of temperature and initial concentration of
d-mannose. The smooth Pt electrode displayed low current density compared to Au, therefore upd Pb adatoms were also studied and proved to be very beneficial to the current density displayed. With the optimal reaction conditions in hand,
d-mannose was then submitted to long term electrolysis at Au and Pt-Pb adatoms electrodes. At a Au electrode, 85% of the initial
d-mannose was converted in 6 h of electrolysis and showed 49% selectivity toward
d-mannonic acid (
33, 42% yield). Beside
33, a few low molecular weight acids arising from C-C bond cleavage could be detected in small amount (
Table 7). For the direct electrolysis of
d-mannose at the Pt-Pb adatoms electrode, 80% conversion of
d-mannnose (
32) was reached in 11 h. The main product was also
d-mannonic acid (33, 50% yield), showing a slightly better selectivity than the pure Au electrode (
Table 7).
Although Au- and Pt-based electrodes have been the most widely studied, other metal-based electrodes have been reported for the selective, direct electrochemical oxidation of monosaccharides. For instance, the electrochemical oxidation of monosaccharides at various copper-oxide-modified electrodes has been investigated, mainly for detection purposes [
21,
82,
83]. However, some studies do report the products resulting from prolonged electrolysis [
84,
85,
86]. The long term electrolysis of glucose on a Cu based electrode (plain Cu plate, Cu-oxides or Cu alloys) mostly resulted in the formation of one product: formate (
Scheme 18). Indeed, each mol of initial glucose could be transformed via C-C bond cleavage to 6 moles of formic acid in basic media (0.1 M NaOH). Sorbitol and xylose appeared to follow the same process, generating 6 moles and 5 moles, respectively.
Certain kind of Rh porphyrins, adsorbed on a carbon electrode have also been reported for their high activity and selectivity toward glucose electro-oxidation [
87]. In their study, Yamazaki et al. have employed four different porphyrin ligands to generate Rh complexes. They evaluated the performance of each catalyst by CV in alkaline medium (0.1 M NaOH) and selected the most active form of the Rh porphyrin complex, [Rh
II(DPDE)]
+. With the appropriate catalyst, glucose was electro-oxidised for 3 h in 0,1 M NaOH. Gluconate was the desired product in this reaction, and its concentration was determined by enzymatic method using gluconate kinase and 6-phosphogluconic dehydrogenase. Only a very small amount of gluconate could be detected (<1% yield), however a faradaic yield up to 73% was achieved due to the very low current running through the electrochemical cell.
To control the electro-oxidation of glucose to gluconic acid (
29) and glucaric acid (
38), Bin et al. employed an electrocatalytic reactor with a tubular porous Ti anode loaded by sol-gel method with nano-sized MnO
2 [
88]. The flow rate used during the electrolysis was adjusted to ensure that the maximum amount of d-glucose could be oxidised, while maintaining a high selectivity and avoiding over-oxidation. The effect of the loading of MnO
2 on the Ti electrode was also investigated, as well as the pH. Surprisingly, the conversion of glucose was not strongly dependent on the pH. Indeed, from acidic (pH = 2) to basic (pH = 10), the conversion was always above 90%. However, the selectivity toward
29 and
38 appeared to be improved when the pH was neutral. The temperature also had an impact on the selectivity but also on the overall conversion of glucose, which significantly decreased when the reaction was performed at a temperature above 30 °C. The authors explain this observation by the fact that the adsorption process is unfavoured with high temperatures. Performing the electrolysis at the most favourable conditions, 93% of the initial
d-glucose was converted mainly to both gluconic acid (42% yield) and glucaric acid (44% yield).
Nickel-based electrodes have also been investigated for the electro-oxidation of mono-saccharides. Indeed, Parpot et al. first documented the direct electrochemical oxidation of
d-galactose (
30) on a nickel electrode in alkaline medium (0.1 M NaOH) [
79]. Two types of long term electrolysis were carried out: one with a constant potential and one with a step potential program. In both cases, the conversion of
d-galactose was low (17% and 43% respectively). Although
d-galactonic acid (
31) was detected as the major product of the reaction, only small amounts were generated (9% at constant potential and 7% with the plateaus potential program). These electrolysis also produced various low molecular weight acids (formic, glycolic, oxalic acid). The poor yield of
31 can be explained by the competition reaction between the desired oxidation, the degradation responsible for low molecular weight compounds formation, and water oxidation. The oxidation of galactose on a Ni anode occurs near the oxygen evolution potential region. This suggests that the surface is covered with NiOH and NiO species which can react with galactose (or galactonic acid) and generate the C-C bond cleavage products (
Scheme 19).
Similar results were observed for the electrochemical oxidation of
d-mannose (
32) on a Ni electrode under basic conditions (0.1 M NaOH) at constant potential [
81]. The conversion of
d-mannose was very low (35%), and a significant amount of degradation products were presents, some in larger quantities than the desired
d-mannonic acid (
33, 3% yield).
More recently, Liu et al. reported that nanostructured NiFe oxides (NiFeO
x) and NiFe nitrides (NiFeN
x) catalysts exhibit a great activity and selectivity toward the electrochemical oxidation of
d-glucose (
14) [
89]. The authors used nickel foam as a support as a Ni source for the catalyst and to ensure a 3D structure to the electrode. Then, the resulting oxides and nitride electrodes were tested in 1.0 M KOH solution (pH = 13.9), for
d-glucose oxidation. The preliminary tests showed that the electro-oxidation of glucose happened at a lower potential than the oxygen evolution reaction, which was presumed to be the main undesired side reaction. NiFeO
x appeared to be more active than NiFeN
x toward glucose oxidation. By analysing the electrode materials after electrolysis of glucose, NiOOH and FeOOH species were detected. The authors postulated that these species are the catalytic active sites of the electrodes responsible for the glucose electro-oxidation reaction. Then, they turned their attention to evaluating various parameters such as the reaction potential and the initial glucose concentration. The authors were able to carry out long term electrolysis with both catalysts. Both NiFeO
x and NiFeN
x converted glucose very efficiently (91% and 93% respectively), and with great selectivity toward glucaric acid (
38, 71% and 64% yield respectively) after 18 h. The monitoring of the reaction allowed the authors to see the initial formation of gluconic acid (
29) and then its conversion to glucaric acid (
38). The catalysts also showed great stability, when five chronoamperometric measurements were carried out the conversion of glucose slightly decreased but the faradaic efficiency remained almost unchanged. By using IR and NMR spectrometry, the authors were able to identify various intermediates and products of the reaction, and proposed the following reaction pathway for the NiFe anodic oxidation of
d-glucose (
Scheme 20).





























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
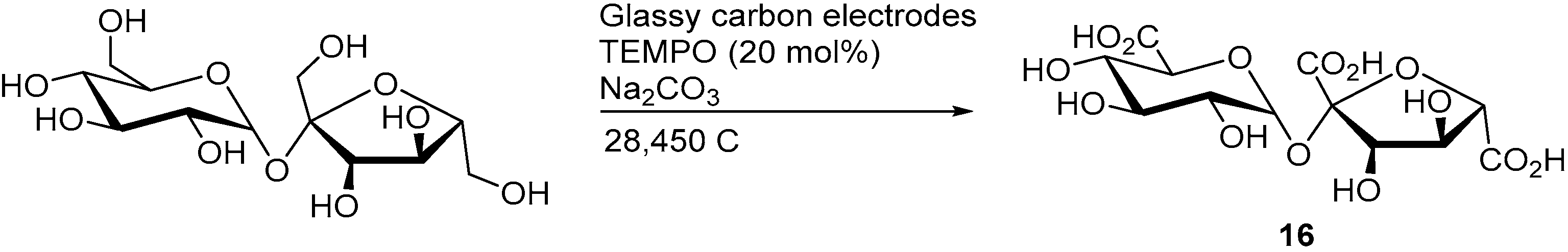{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
















