
Development of a Molecular Snail Xenomonitoring Assay to Detect Schistosoma haematobium and Schistosoma bovis Infections in their Bulinus Snail Hosts

 , ,
, ,
Abstract
:1. Introduction
2. Results
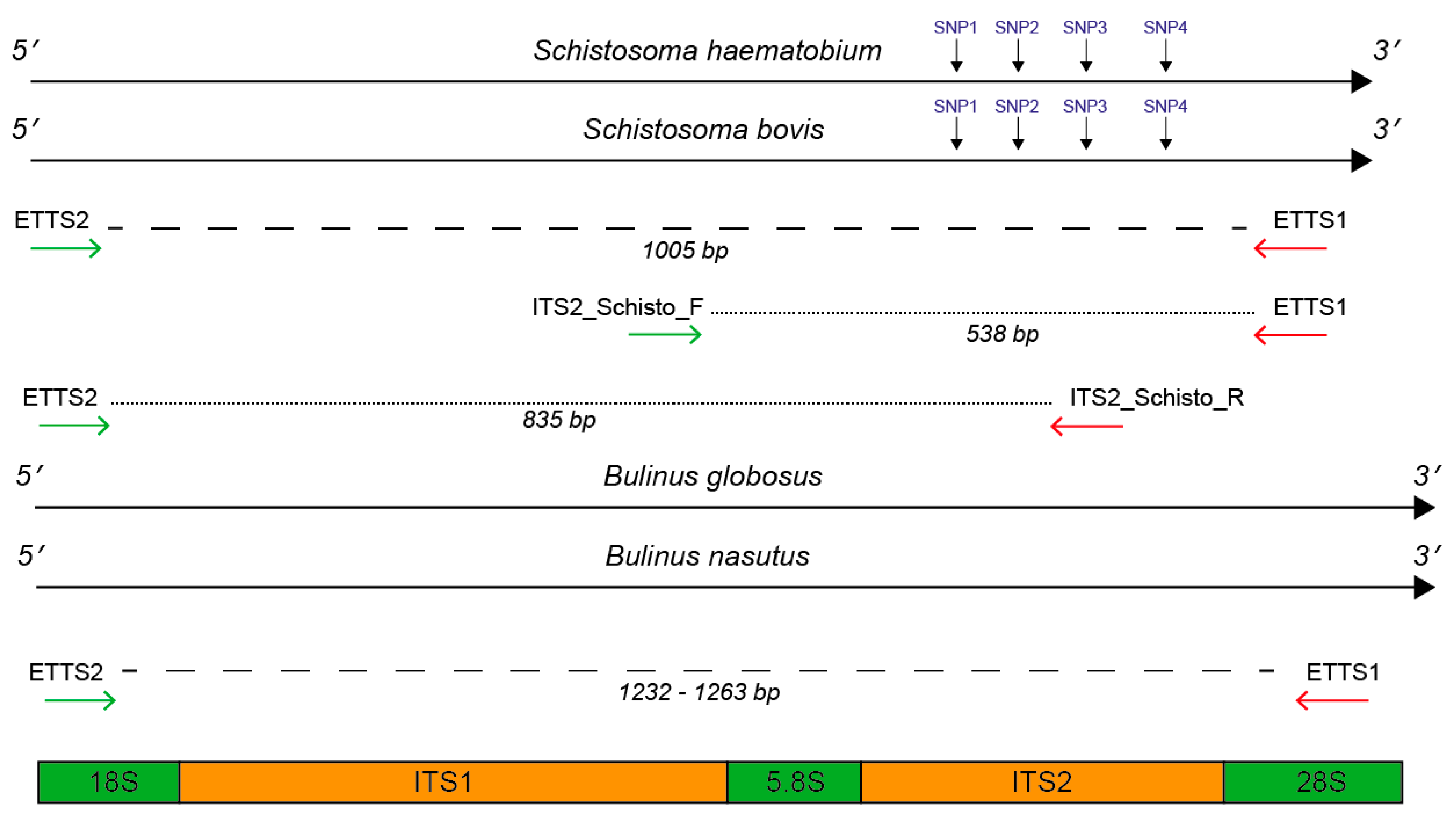
2.1. In Silico and In Vitro Primer Evaluation
2.2. Analytical Sensitivity
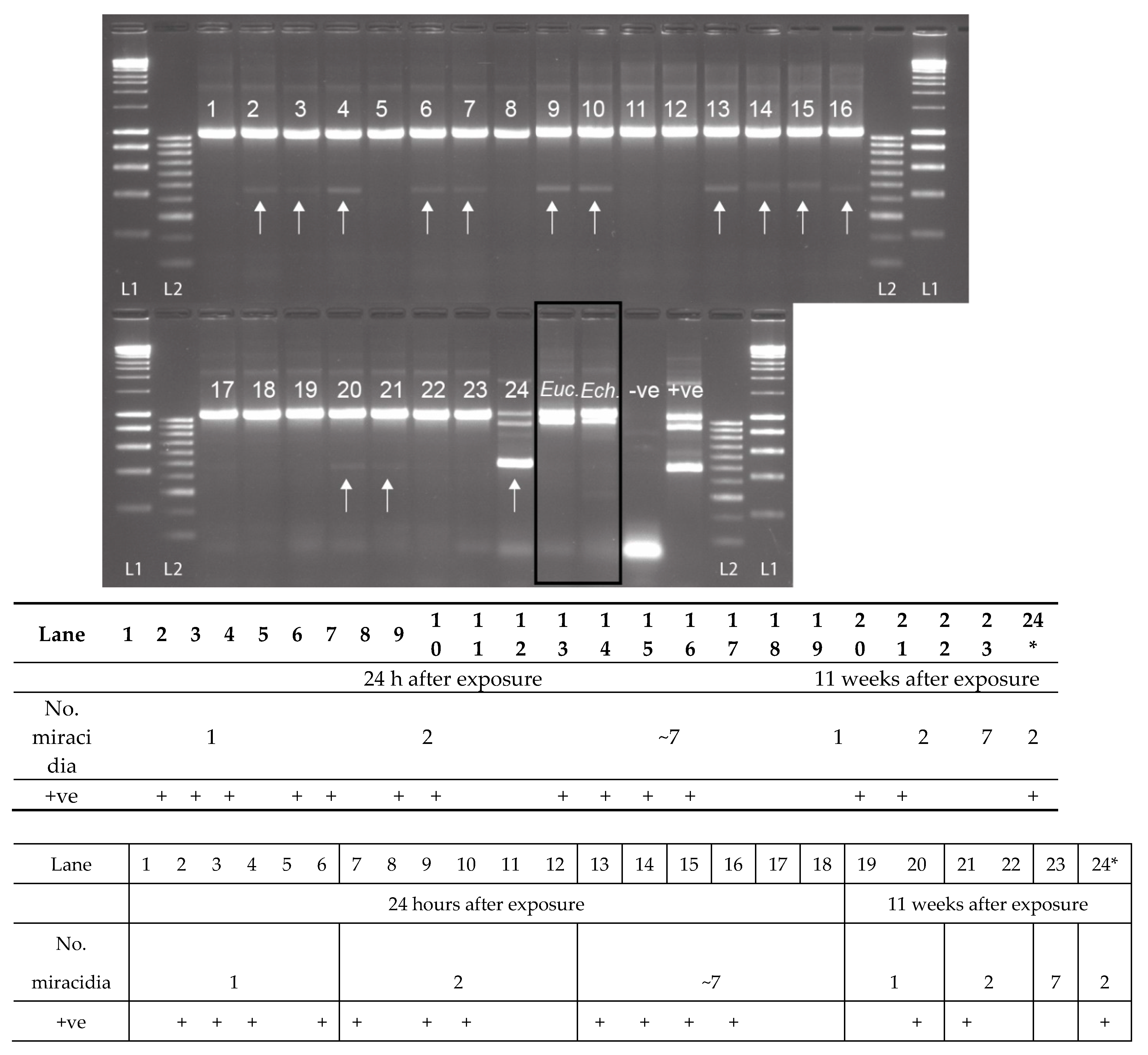
2.3. Experimental Snail Infections
2.4. Specificity Testing
2.5. Testing on Field Samples
2.6. Schistosoma spp. cox1 RD-PCR
3. Discussion
3.1. The Sensitivity of MIX PCR Assay
3.2. Benefits of an Updated Molecular Xenomonitoring Protocol for Schistosomiasis Surveillance
3.3. Limitations of Molecular Xenomonitoring Approaches for Schistosomiasis Surveillance
4. Materials and Methods
4.1. Primer Selection and In Silico Evaluation
4.2. Bulinus and Schistosoma Genomic DNA Extractions
4.3. PCR Conditions, Amplicon Visualization, and Sequencing
4.4. In Vitro Primer Testing and Assay Validation
4.5. Sensitivity Testing
4.6. Specificity Testing and Validation on Field Samples
4.7. Testing the Schistosoma cox1 Rapid-Diagnostic PCR (RD-PCR) for Secondary Species Identification
4.8. Ethical Statement
Author Contributions
Funding
Conflicts of Interest
References
- Colley, D.G.; Secor, W.E. Immunology of human schistosomiasis. Parasite Immunol. 2014, 36, 347–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.S. Freshwater Snails of Africa and Their Medical Importance, 1st ed.; Taylor & Francis Ltd.: London, UK, 1994. [Google Scholar]
- WHO Schistosomiasis Fact Sheets. Available online: https://www.who.int/news-room/fact-sheets/detail/schistosomiasis (accessed on 27 April 2020).
- De Bont, J.; Vercruysse, J. Schistosomiasis in Cattle. Adv. Parasitol. 1998, 41, 285–364. [Google Scholar] [CrossRef] [PubMed]
- De Bont, J.; Vercruysse, J. The epidemiology and control of cattle schistosomiasis. Parasitol. Today 1997, 13, 255–262. [Google Scholar] [CrossRef]
- Pennance, T.; Ame, S.M.; Amour, A.K.; Suleiman, K.R.; Allan, F.; Rollinson, D.; Webster, B.L. Occurrence of Schistosoma bovis on Pemba Island, Zanzibar: Implications for urogenital schistosomiasis transmission monitoring. Parasitology 2018, 145, 1727–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennance, T.; Allan, F.; Emery, A.; Rabone, M.; Cable, J.; Garba, A.D.; Hamidou, A.A.; Webster, J.P.; Rollinson, D.; Webster, B.L. Interactions between Schistosoma haematobium group species and their Bulinus spp. intermediate hosts along the Niger River Valley. Parasites Vectors 2020, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Guidelines for the Evaluation of Soil-Transmitted Helminths and Schistosomiasis at Community Level. Available online: https://apps.who.int/iris/handle/10665/63821 (accessed on 23 July 2020).
- Ending the Neglect to Attain the Sustainable Development Goals: A Road Map for Neglected Tropical Diseases 2021–2030. Available online: https://www.who.int/neglected_diseases/Ending-the-neglect-to-attain-the-SDGs--NTD-Roadmap.pdf?ua=1 (accessed on 23 July 2020).
- Mutsaka-Makuvaza, M.J.; Zingoni, Z.M.; Tshuma, C.; Ray, S.; Zhou, X.-N.; Webster, B.L.; Midzi, N. Reinfection of urogenital schistosomiasis in pre-school children in a highly endemic district in Northern Zimbabwe: A 12 months compliance study. Infect. Dis. Poverty 2018, 7, 102. [Google Scholar] [CrossRef] [Green Version]
- Tchuente, L.-A.T.; Rollinson, D.; Stothard, J.R.; Molyneux, D.H. Moving from control to elimination of schistosomiasis in sub-Saharan Africa: Time to change and adapt strategies. Infect. Dis. Poverty 2017, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- King, C.H.; Sturrock, R.F.; Kariuki, H.C.; Hamburger, J. Transmission control for schistosomiasis—Why it matters now. Trends Parasitol. 2006, 22, 575–582. [Google Scholar] [CrossRef]
- Rollinson, D.; Knopp, S.; Levitz, S.; Stothard, J.R.; Tchuenté, L.-A.T.; Garba, A.; Mohammed, K.A.; Schur, N.; Person, B.; Colley, D.G.; et al. Time to set the agenda for schistosomiasis elimination. Acta Trop. 2013, 128, 423–440. [Google Scholar] [CrossRef]
- Secor, W.E.; Colley, D.G. When Should the Emphasis on Schistosomiasis Control Move to Elimination? Trop. Med. Infect. Dis. 2018, 3, 85. [Google Scholar] [CrossRef] [Green Version]
- Pennance, T.; Person, B.; Muhsin, M.A.; Khamis, A.N.; Muhsin, J.; Khamis, I.S.; Mohammed, K.A.; Kabole, F.; Rollinson, D.; Knopp, S. Urogenital schistosomiasis transmission on Unguja Island, Zanzibar: Characterisation of persistent hot-spots. Parasites Vectors 2016, 9, 646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayssade-Dufour, C. Chétotaxies cercariennes comparées de dix espèces de Schistosomes. Ann. Parasitol. Hmm. Comp. 1982, 57, 467–485. [Google Scholar] [CrossRef]
- Bayssade-Dufour, C.; Cabaret, J.; Ngendahayo, L.; Albaret, J.-L.; Carrat, C.; Chabaud, A. Identification of schistosoma haematobium, S. bovis and S. curassoni by multivariate analysis of cercarial papillae indices. Int. J. Parasitol. 1989, 19, 839–846. [Google Scholar] [CrossRef]
- Cunningham, L.J.; Lingley, J.K.; Haines, L.R.; Ndung’U, J.M.; Torr, S.J.; Adams, E.R. Illuminating the Prevalence of Trypanosoma brucei s.l. in Glossina Using LAMP as a Tool for Xenomonitoring. PLoS Negl. Trop. Dis. 2016, 10, e0004441. [Google Scholar] [CrossRef] [PubMed]
- Garrod, G.; Adams, E.R.; Lingley, J.K.; Saldanha, I.; Torr, S.J.; Cunningham, L.J. Identification of Trypanosoma brucei gambiense and T. b. rhodesiense in vectors using multiplexed high-resolution melt analysis. bioRxiv 2020. Available online: https://www.biorxiv.org/content/10.1101/2020.04.21.052928v1 (accessed on 22 April 2020). [CrossRef] [Green Version]
- Cook, D.A.; Pilotte, N.; Minetti, C.; Williams, S.A.; Reimer, L. A superhydrophobic cone to facilitate the xenomonitoring of filarial parasites, malaria, and trypanosomes using mosquito excreta/feces. Gates Open Res. 2018, 1, 1. [Google Scholar] [CrossRef] [Green Version]
- Minetti, C.; LaCourse, E.J.; Reimer, L.; Stothard, J.R. Focusing nucleic acid-based molecular diagnostics and xenomonitoring approaches for human helminthiases amenable to preventive chemotherapy. Parasitology 2016, 2, 16. [Google Scholar] [CrossRef] [Green Version]
- Rathinasamy, V.; Hosking, C.; Tran, L.; Kelley, J.; Williamson, G.; Swan, J.; Elliott, T.; Rawlin, G.; Beddoe, T.; Spithill, T.W. Development of a multiplex quantitative PCR assay for detection and quantification of DNA from Fasciola hepatica and the intermediate snail host, Austropeplea tomentosa, in water samples. Vet. Parasitol. 2018, 259, 17–24. [Google Scholar] [CrossRef]
- Hamburger, J.; Hoffman, O.; Kariuki, H.C.; Muchiri, E.M.; King, C.H.; Ouma, J.H.; Koech, D.K.; Sturrock, R.F. Large-Scale, Polymerase Chain Reaction–Based Surveillance Of Schistosoma Haematobium DNA In Snails from Transmission Sites in Coastal Kenya: A New Tool for Studying the Dynamics of Snail Infection. Am. J. Trop. Med. Hyg. 2004, 71, 765–773. [Google Scholar] [CrossRef]
- Lu, L.; Zhang, S.-M.; Mutuku, M.W.; Mkoji, G.M.; Loker, E.S. Relative compatibility of Schistosoma mansoni with Biomphalaria sudanica and B. pfeifferi from Kenya as assessed by PCR amplification of the S. mansoni ND5 gene in conjunction with traditional methods. Parasites Vectors 2016, 9, 166. [Google Scholar] [CrossRef] [Green Version]
- Allan, F.; Rollinson, D.; Smith, J.; Dunn, A.M. Host choice and penetration bySchistosoma haematobiummiracidia. J. Helminthol. 2009, 83, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Allan, F.; Dunn, A.M.; Emery, A.M.; Stothard, J.R.; Johnston, D.A.; Kane, R.A.; Khamis, A.N.; Mohammed, K.A.; Rollinson, D. Use of sentinel snails for the detection of Schistosoma haematobium transmission on Zanzibar and observations on transmission patterns. Acta Trop. 2013, 128, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Schols, R.; Carolus, H.; Hammoud, C.; Mulero, S.; Mudavanhu, A.; Huyse, T. A rapid diagnostic multiplex PCR approach for xenomonitoring of human and animal schistosomiasis in a ‘One Health’ context. Trans. R. Soc. Trop. Med. Hyg. 2019, 113, 722–729. [Google Scholar] [CrossRef]
- Casotti, M.O.; Gryschek, R.C.B.; De Paula, F.M.; Gomes-Gouvêa, M.; Pinho, J.R.R.; Tuan, R.; Dias-Neto, E.; Luna, E.J.D.A.; Espírito-Santo, M.C.C.D. Molecular detection of prepatent Schistosoma mansoni infection in Biomphalaria glabrata snail vectors. Rev. Inst. Med. Trop. Sao Paulo 2020, 62, e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiglová, A.; Changoma, M.J.S.; Špajdelová, J.; Jakubcová, D.; Bírová, K. Urinary schistosomosis in patients of rural medical health centers in Kwale county, Kenya. J. Helminthol. 2020, 57, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbasi, I.; Webster, B.L.; King, C.H.; Rollinson, D.; Hamburger, J. The substructure of three repetitive DNA regions of Schistosoma haematobium group species as a potential marker for species recognition and interbreeding detection. Parasites Vectors 2017, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, R.A.; Rollinson, D. Repetitive sequences in the ribosomal DNA internal transcribed spacer of Schistosoma haematobium, Schistosoma intercalatum and Schistosoma mattheei. Mol. Biochem. Parasitol. 1994, 63, 153–156. [Google Scholar] [CrossRef]
- Webster, B.L. Isolation and preservation of schistosome eggs and larvae in RNAlater® facilitates genetic profiling of individuals. Parasites Vectors 2009, 2, 50. [Google Scholar] [CrossRef] [Green Version]
- Webster, B.L.; Rollinson, D.; Stothard, J.R.; Huyse, T. Rapid diagnostic multiplex PCR (RD-PCR) to discriminate Schistosoma haematobium and S. bovis. J. Helminthol. 2009, 84, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Schmaedick, M.A.; Koppel, A.L.; Pilotte, N.; Torres, M.; Williams, S.A.; Dobson, S.L.; Lammie, P.J.; Won, K.Y. Molecular Xenomonitoring Using Mosquitoes to Map Lymphatic Filariasis after Mass Drug Administration in American Samoa. PLoS Neglected Trop. Dis. 2014, 8, e3087. [Google Scholar] [CrossRef]
- Pilotte, N.; Unnasch, T.R.; Williams, S.A. The Current Status of Molecular Xenomonitoring for Lymphatic Filariasis and Onchocerciasis. Trends Parasitol. 2017, 33, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Pilotte, N.; Zaky, W.I.; Abrams, B.P.; Chadee, D.D.; Williams, S.A. A Novel Xenomonitoring Technique Using Mosquito Excreta/Feces for the Detection of Filarial Parasites and Malaria. PLoS Neglected Trop. Dis. 2016, 10, e0004641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caron, Y.; Righi, S.; Lempereur, L.; Saegerman, C.; Losson, B. An optimized DNA extraction and multiplex PCR for the detection of Fasciola sp. in lymnaeid snails. Vet. Parasitol. 2011, 178, 93–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cucher, M.A.; Carnevale, S.; Prepelitchi, L.; Labbé, J.H.; Wisnivesky-Colli, C. PCR diagnosis of Fasciola hepatica in field-collected Lymnaea columella and Lymnaea viatrix snails. Vet. Parasitol. 2006, 137, 74–82. [Google Scholar] [CrossRef]
- Kozak, M.; Wedrychowicz, H. The performance of a PCR assay for field studies on the prevalence of Fasciola hepatica infection in Galba truncatula intermediate host snails. Vet. Parasitol. 2010, 168, 25–30. [Google Scholar] [CrossRef]
- Magalhães, K.G.; Passos, L.K.J.; Carvalho, O.D.S. Detection of Lymnaea columella infection by Fasciola hepatica through Multiplex-PCR. Mem. Instit. Oswaldo Cruz 2004, 99, 421–424. [Google Scholar] [CrossRef] [Green Version]
- Mostafa, O.M.S.; A Taha, H.; Ramadan, G. Diagnosis of Fasciola gigantica in snail using the polymerase chain reaction (PCR) assay. J. Egypt. Soc. Parasitol. 2003, 33, 733–742. [Google Scholar]
- Velusamy, R.; Singh, B.; Raina, O. Detection of Fasciola gigantica infection in snails by polymerase chain reaction. Vet. Parasitol. 2004, 120, 85–90. [Google Scholar] [CrossRef]
- Kaplan, R.M.; Dame, J.; Reddy, G.; Courtney, C. A repetitive DNA probe for the sensitive detection of Fasciola hepatica infected snails. Int. J. Parasitol. 1995, 25, 601–610. [Google Scholar] [CrossRef]
- Carolus, H.; Muzarabani, K.C.; Hammoud, C.; Schols, R.; Volckaert, F.A.; Barson, M.; Huyse, T. A cascade of biological invasions and parasite spillback in man-made Lake Kariba. Sci. Total Environ. 2018, 659, 1283–1292. [Google Scholar] [CrossRef]
- Born-Torrijos, A.; Poulin, R.; Raga, J.A.; Holzer, A.S. Estimating trematode prevalence in snail hosts using a single-step duplex PCR: How badly does cercarial shedding underestimate infection rates? Parasites Vectors 2014, 7, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumagai, T.; Furushima-Shimogawara, R.; Ohmae, H.; Wang, T.-P.; Lu, S.; Chen, R.; Wen, L.; Ohta, N. Detection of Early and Single Infections of Schistosoma japonicum in the Intermediate Host Snail, Oncomelania hupensis, by PCR and Loop-Mediated Isothermal Amplification (LAMP) Assay. Am. J. Trop. Med. Hyg. 2010, 83, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Tong, Q.; Chen, R.; Zhang, Y.; Yang, G.-J.; Kumagai, T.; Furushima-Shimogawara, R.; Lou, D.; Yang, K.; Wen, L.-Y.; Lu, S.-H.; et al. A new surveillance and response tool: Risk map of infected Oncomelania hupensis detected by Loop-mediated isothermal amplification (LAMP) from pooled samples. Acta Trop. 2015, 141, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Hamburger, J.; Ruppel, A.; Jourdane, J.; Na, H.; Ramzy, R.M.; Xin, X.Y. A polymerase chain reaction assay for detecting snails infected with bilharzia parasites (Schistosoma mansoni) from very early prepatency. Am. J. Trop. Med. Hyg. 1998, 59, 872–876. [Google Scholar] [CrossRef]
- Melo, F.L.; Gomes, A.L.D.V.; Barbosa, C.S.; Werkhäuser, R.P.; Abath, F.G. Development of molecular approaches for the identification of transmission sites of schistosomiasis. Trans. R. Soc. Trop. Med. Hyg. 2006, 100, 1049–1055. [Google Scholar] [CrossRef]
- Fernández-Soto, P.; Arahuetes, J.G.; Hernández, A.S.; López-Abán, J.; Santiago, B.V.; Muro, A. A Loop-Mediated Isothermal Amplification (LAMP) Assay for Early Detection of Schistosoma mansoni in Stool Samples: A Diagnostic Approach in a Murine Model. PLoS Neglected Trop. Dis. 2014, 8, e3126. [Google Scholar] [CrossRef] [Green Version]
- Gandasegui, J.; Fernández-Soto, P.; Hernández-Goenaga, J.; López-Abán, J.; Vicente, B.; Muro, A. Biompha-LAMP: A New Rapid Loop-Mediated Isothermal Amplification Assay for Detecting Schistosoma mansoni in Biomphalaria glabrata Snail Host. PLoS Neglected Trop. Dis. 2016, 10, e0005225. [Google Scholar] [CrossRef]
- Abbasi, I.; King, C.H.; Muchiri, E.M.; Hamburger, J. Detection of Schistosoma mansoni and Schistosoma haematobium DNA by Loop-Mediated Isothermal Amplification: Identification of Infected Snails from Early Prepatency. Am. J. Trop. Med. Hyg. 2010, 83, 427–432. [Google Scholar] [CrossRef] [Green Version]
- Caldeira, R.L.; Jannotti-Passos, L.K.; Carvalho, O.D.S. Use of Molecular Methods for the Rapid Mass Detection of Schistosoma mansoni (Platyhelminthes: Trematoda) in Biomphalariaspp. (Gastropoda: Planorbidae). J. Trop. Med. 2017, 2017, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Kane, R.A.; Stothard, J.R.; Rollinson, D.; Leclipteux, T.; Evraerts, J.; Standley, C.J.; Allan, F.; Betson, M.; Kaba, R.; Mertens, P.; et al. Detection and quantification of schistosome DNA in freshwater snails using either fluorescent probes in real-time PCR or oligochromatographic dipstick assays targeting the ribosomal intergenic spacer. Acta Trop. 2013, 128, 241–249. [Google Scholar] [CrossRef]
- Hamburger, J.; Ruppel, A.; Ramzy, R.M.; Na, H.; Jourdane, J.; Abbasi, I. Polymerase chain reaction assay based on a highly repeated sequence of Schistosoma haematobium: A potential tool for monitoring schistosome-infested water. Am. J. Trop. Med. Hyg. 2001, 65, 907–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudko, S.P.; Reimink, R.L.; Froelich, K.; Gordy, M.A.; Blankespoor, C.L.; Hanington, P.C. Use of qPCR-Based Cercariometry to Assess Swimmer’s Itch in Recreational Lakes. EcoHealth 2018, 15, 827–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudko, S.P.; Reimink, R.L.; Peter, B.; White, J.; Hanington, P.C. Democratizing water monitoring: Implementation of a community-based qPCR monitoring program for recreational water hazards. PLoS ONE 2020, 15, e0229701. [Google Scholar] [CrossRef] [PubMed]
- Rudko, S.P.; Turnbull, A.; Reimink, R.L.; Froelich, K.; Hanington, P.C. Species-specific qPCR assays allow for high-resolution population assessment of four species avian schistosome that cause swimmer’s itch in recreational lakes. Int. J. Parasitol. Parasites Wildl. 2019, 9, 122–129. [Google Scholar] [CrossRef]
- Webster, B.L.; Culverwell, C.L.; Khamis, I.S.; Mohammed, K.A.; Rollinson, D.; Stothard, J.R. DNA barcoding of Schistosoma haematobium on Zanzibar reveals substantial genetic diversity and two major phylogenetic groups. Acta Trop. 2013, 128, 206–217. [Google Scholar] [CrossRef]
- Webster, B.L.; Emery, A.M.; Webster, J.P.; Gouvras, A.N.; Garba, A.; Diaw, O.; Seye, M.M.; Tchuente, L.A.T.; Simoonga, C.; Mwanga, J.; et al. Genetic Diversity within Schistosoma haematobium: DNA Barcoding Reveals Two Distinct Groups. PLoS Neglected Trop. Dis. 2012, 6, e1882. [Google Scholar] [CrossRef]
- Emery, A.M.; Allan, F.; Rabone, M.; Rollinson, D. Schistosomiasis collection at NHM (SCAN). Parasites Vectors 2012, 5, 185. [Google Scholar] [CrossRef] [Green Version]
- Lewis, F.A.; Liang, Y.-S.; Raghavan, N.; Knight, M. The NIH-NIAID Schistosomiasis Resource Center. PLOS Neglected Trop. Dis. 2008, 2, e267. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples (DNA extracts of snails and parasites) are available from the authors upon appropriate request. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Schistosoma Species | ITS 2 Schistosome Species-Specific SNP Positions (bp) | |||
|---|---|---|---|---|
| SNP1 (90) | SNP2 (145) | SNP3 (195) | SNP4 (265) | |
| S. haematobium | S. h (G) | S. h (C) | S. h (G) | S. h (C) |
| S. bovis | S. b (A) | S. b (T) | S. b (A) | S. b (T) |
| Primer (Direction) | Primer Sequence (5′-3′) | Primer Position | State | Reference |
|---|---|---|---|---|
| ETTS1 (Reverse) | TGCTTAAGTTCAGCGGG | 28S 5′ end (ITS2 3′ flanking region) | U | Kane et al. (1994) [31] |
| ETTS2 (Forward) | TAACAAGGTTTCCGTAGGTGA | 18S 3′ region (ITS1 5′ flanking region) | U | Kane et al. (1994) [31] |
| ITS2_Schisto_F (Forward) | GGAAACCAATGTATGGGATTATTG | ITS1 3′ end (5.8S 5′ flanking region) | S | Schols et al. (2019) [27] |
| ITS2_Schisto_R (Reverse) | ATTAAGCCACGACTCGAGCA | ITS2 (middle) | S | Schols et al. (2019) [27] |
| Infection Group | No. of B.t. Exposed | No. of S.h. Miracidia Used | No. of B.t. Preserved at 24 h | No. of B.t. Checked for Patency at 11 wks and Preserved (no. Shedding + ve) |
|---|---|---|---|---|
| 1 | 45 | 1 | 22 | 22 1 (0) |
| 2 | 43 | 2 | 21 | 19 1 (2) |
| 3 | 45 | ~7 | 23 | 21 1 (7) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pennance, T.; Archer, J.; Lugli, E.B.; Rostron, P.; Llanwarne, F.; Ali, S.M.; Amour, A.K.; Suleiman, K.R.; Li, S.; Rollinson, D.; et al. Development of a Molecular Snail Xenomonitoring Assay to Detect Schistosoma haematobium and Schistosoma bovis Infections in their Bulinus Snail Hosts. Molecules 2020, 25, 4011. https://doi.org/10.3390/molecules25174011
Pennance T, Archer J, Lugli EB, Rostron P, Llanwarne F, Ali SM, Amour AK, Suleiman KR, Li S, Rollinson D, et al. Development of a Molecular Snail Xenomonitoring Assay to Detect Schistosoma haematobium and Schistosoma bovis Infections in their Bulinus Snail Hosts. Molecules. 2020; 25(17):4011. https://doi.org/10.3390/molecules25174011
Chicago/Turabian StylePennance, Tom, John Archer, Elena Birgitta Lugli, Penny Rostron, Felix Llanwarne, Said Mohammed Ali, Amour Khamis Amour, Khamis Rashid Suleiman, Sarah Li, David Rollinson, and et al. 2020. "Development of a Molecular Snail Xenomonitoring Assay to Detect Schistosoma haematobium and Schistosoma bovis Infections in their Bulinus Snail Hosts" Molecules 25, no. 17: 4011. https://doi.org/10.3390/molecules25174011
APA StylePennance, T., Archer, J., Lugli, E. B., Rostron, P., Llanwarne, F., Ali, S. M., Amour, A. K., Suleiman, K. R., Li, S., Rollinson, D., Cable, J., Knopp, S., Allan, F., Ame, S. M., & Webster, B. L. (2020). Development of a Molecular Snail Xenomonitoring Assay to Detect Schistosoma haematobium and Schistosoma bovis Infections in their Bulinus Snail Hosts. Molecules, 25(17), 4011. https://doi.org/10.3390/molecules25174011