
High-Resolution Mass Spectrometry-Based Approaches for the Detection and Quantification of Peptidase Activity in Plasma

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
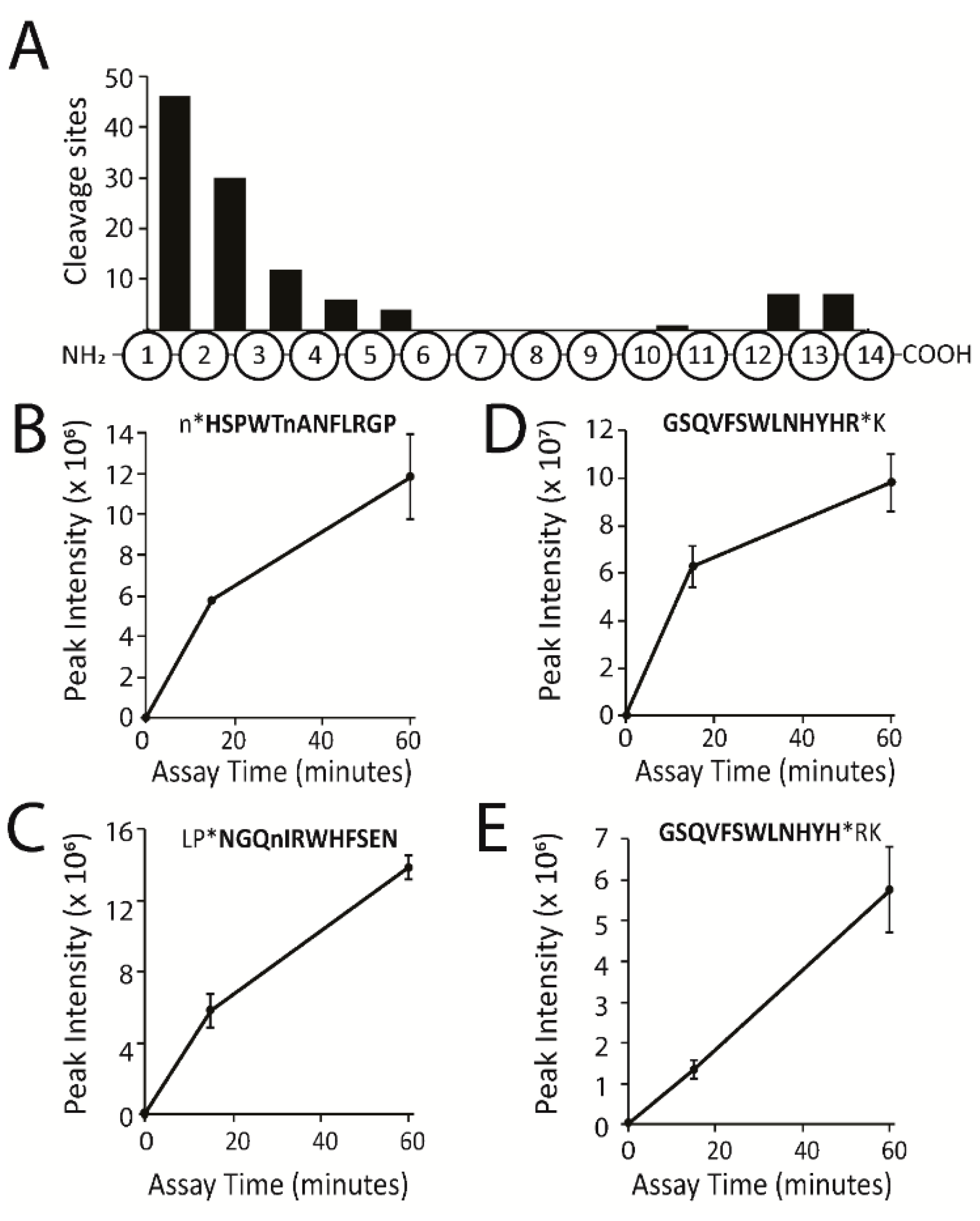
2.1. Global Protease Activity Profiling of Pig Plasma by MSP-MS
2.2. Peptidomics of Pig Plasma
3. Discussion
4. Materials and Methods
4.1. Plasma Sample Preparation
4.2. Fluorogenic Reporter Assays
4.3. Peptide Cleavage Site Identification by Multiplex Substrate Profiling (MSP) Mass Spectrometry
4.4. Nano-LC-ESI-MS/MS Mass Spectrometry-Based Shotgun Peptidomics
4.5. Data Presentation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Aletti, F.; Maffioli, E.; Negri, A.; Santamaria, M.H.; DeLano, F.A.; Kistler, E.B.; Schmid-Schönbein, G.W.; Tedeschi, G. Peptidomic Analysis of Rat Plasma: Proteolysis in Hemorrhagic Shock. Shock 2016, 45, 540–554. [Google Scholar] [CrossRef] [Green Version]
- Aletti, F.; Conti, C.; Ferrario, M.; Ribas, V.; Pinto Bollen, B.; Herpain, A.; Post, E.; Medina, E.R.; Barlassina, C.; de Oliveira, E.; et al. ShockOmics: Multiscale approach to the identification of molecular biomarkers in acute heart failure induced by shock. Scand. J. Trauma Resusc. Emerg. Med. 2016, 24, 9. [Google Scholar] [CrossRef] [Green Version]
- Bauzá-Martinez, J.; Aletti, F.; Pinto, B.B.; Ribas, V.; Odena, M.A.; Díaz, R.; Romay, E.; Ferrer, R.; Kistler, E.B.; Tedeschi, G.; et al. Proteolysis in septic shock patients: Plasma peptidomic patterns are associated with mortality. Br. J. Anaesth. 2018, 121, 1065–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid-Schönbein, G.W.; Chang, M. The autodigestion hypothesis for shock and multi-organ failure. Ann. Biomed. Eng. 2014, 42, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Carrara, M.; Babini, G.; Baselli, G.; Ristagno, G.; Pastorelli, R.; Brunelli, L.; Ferrario, M. Blood pressure variability, heart functionality, and left ventricular tissue alterations in a protocol of severe hemorrhagic shock and resuscitation. J. Appl. Physiol. (1985) 2018, 125, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, M.; Brunelli, L.; Su, F.; Herpain, A.; Pastorelli, R. The Systemic Alterations of Lipids, Alanine-Glucose Cycle and Inter-Organ Amino Acid Metabolism in Swine Model Confirms the Role of Liver in Early Phase of Septic Shock. Front. Physiol. 2019, 10, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwenk, J.M.; Omenn, G.S.; Sun, Z.; Campbell, D.S.; Baker, M.S.; Overall, C.M.; Aebersold, R.; Moritz, R.L.; Deutsch, E.W. The Human Plasma Proteome Draft of 2017: Building on the Human Plasma PeptideAtlas from Mass Spectrometry and Complementary Assays. J. Proteome Res. 2017, 16, 4299–4310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [Green Version]
- O’Donoghue, A.J.; Alegra Eroy-Reveles, A.; Knudsen, G.M.; Ingram, J.; Zhou, M.; Statnekov, J.B.; Greninger, A.L.; Hostetter, D.R.; Qu, G.; Maltby, D.A.; et al. Global identification of peptidase specificity by multiplex substrate profiling. Nat. Methods 2012, 9, 1095–1100. [Google Scholar] [CrossRef] [Green Version]
- Lapek, J.D., Jr.; Jiang, Z.; Wozniak, J.M.; Arutyunova, E.; Wang, S.C.; Lemieux, M.J.; Gonzalez, D.J.; O’Donoghue, A.J. Quantitative Multiplex Substrate Profiling of Peptidases by Mass Spectrometry. Mol. Cell Proteom. 2019, 18, 968–981. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, A.J.; Ye, J.; Knudsen, G.M.; Perera, N.C.; Jenne, D.E.; Murphy, J.E.; Craik, C.S.; Hermiston, T.W. Global Substrate Profiling of Proteases in Human Neutrophil Extracellular Traps Reveals Consensus Motif Predominantly Contributed by Elastase. PLoS ONE 2013, 8, e75141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivry, S.L.; Sharib, J.M.; Dominguez, D.A.; Roy, N.; Stacy, E.; Hatcher, S.E.; Yip-Schneider, M.T.; Schmidt, C.M.; Randall, E.; Brand, R.E.; et al. Global Protease Activity Profiling Provides Differential Diagnosis of Pancreatic Cysts. Clin. Cancer Res. 2017, 23, 4865–4874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravemann, U.; Kusch, M.; Koenig, H.; Mohr, H.; Mueller, T.H. Thrombin Generation Capacity of Methylene Blue-Treated Plasma Prepared by the Theraflex MB Plasma System. Transfus. Med. Hemother. 2009, 36, 122–127. [Google Scholar] [CrossRef]
- Biroc, S.L.; Gay, S.; Hummel, K.; Magill, C.; Palmer, J.T.; Spencer, D.R.; Sa, S.; Klaus, J.L.; Michel, B.A.; Rasnick, D.; et al. Cysteine protease activity is up-regulated in inflamed ankle joints of rats with adjuvant-induced arthritis and decreases with in vivo administration of a vinyl sulfone cysteine protease inhibitor. Arthritis Rheum. 2001, 44, 703–711. [Google Scholar] [CrossRef]
- Shimizu, C.; Yoshida, S.; Shibata, M.; Kato, K.; Momota, Y.; Matsumoto, K.; Shiosaka, T.; Midorikawa, R.; Kamachi, T.; Kawabe, A.; et al. Characterization of recombinant and brain neuropsin, a plasticity-related serine protease. J. Biol. Chem. 1998, 273, 11189–11196. [Google Scholar] [CrossRef] [Green Version]
- Thai, T.; Alastair, G.S. Protease-activated receptor (PAR)-independent growth and pro-inflammatory actions of thrombin on human cultured airway smooth muscle. Br. J. Pharmacol. 2003, 138, 865–875. [Google Scholar] [CrossRef]
- Iacovos, P.M.; Sotiropoulou, G.; Pampalakis, G.; Magklara, A.; Ghosh, M.; Wasney, G.; Diamandis, E.P. Biochemical and enzymatic characterization of human kallikrein 5 (hK5), a novel serine protease potentially involved in cancer progression. J. Biol. Chem. 2005, 280, 14628–14635. [Google Scholar] [CrossRef] [Green Version]
- Massey, A.P.; Harley, W.R.; Pasupuleti, N.; Gorin, F.A.; Nantz, M.H. 2-Amidino analogs of glycine-amiloride conjugates: Inhibitors of urokinase-type plasminogen activator. Bioorganic Med. Chem. Lett. 2012, 22, 2635–2639. [Google Scholar] [CrossRef] [Green Version]
- Morita, T.; Kato, H.; Iwanaga, S.; Takada, K.; Kimura, T. New fluorogenic substrates for alpha-thrombin, factor Xa, kallikreins, and urokinase. J. Biochem. 1977, 82, 1495–1498. [Google Scholar] [CrossRef]
- Colaert, N.; Helsens, K.; Martens, L.; Vandekerckhove, J.; Gevaert, K. Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 2009, 6, 786–787. [Google Scholar] [CrossRef] [PubMed]
- Altshuler, A.E.; Penn, A.H.; Yang, J.A.; Kim, G.R.; Schmid-Schönbein, G.W. Protease activity increases in plasma, peritoneal fluid, and vital organs after hemorrhagic shock in rats. PLoS ONE 2012, 7, e32672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kara, E.; Manna, D.; Løset, G.A.; Schneider, E.L.; Craik, C.S.; Kanse, S. Analysis of the substrate specificity of Factor VII activating protease (FSAP) and design of specific and sensitive peptide substrates. Thromb. Haemost 2017, 117, 1750–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dashkevich, N.M.; Ovanesov, M.V.; Balandina, A.N.; Karamzin, S.S.; Shestakov, P.I.; Soshitova, N.P.; Tokarev, A.A.; Panteleev, M.A.; Ataullakhanov, F.I. Thrombin Activity Propagates in Space During Blood Coagulation as an Excitation Wave. Biophys. J. 2012, 103, 2233–2240. [Google Scholar] [CrossRef] [Green Version]
- Keane, F.M.; Yao, T.; Seelk, S.; Gall, M.G.; Chowdhury, S.; Poplawski, S.E.; Lai, J.H.; Li, Y.; Wu, W.; Farrell, P.; et al. Quantitation of fibroblast activation protein (FAP)-specific protease activity in mouse, baboon and human fluids and organs. FEBS Open Bio 2013, 4, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.; Chen, Z.; Xu, X.; Mikhailova, M.; Steffensen, B. Co-purified gelatinases alter the stability and biological activities of human plasma fibronectin preparations. J. Periodontal Res. 2010, 45, 292–295. [Google Scholar] [CrossRef] [Green Version]
- Winter, M.B.; Salcedo, E.C.; Lohse, M.B.; Hartooni, N.; Gulati, M.; Sanchez, H.; Takagi, J.; Hube, B.; Andes, D.R.; Johnson, A.D.; et al. Global Identification of Biofilm-Specific Proteolysis in Candida albicans. mBio 2016, 7, e01514-16. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Gu, Y.; Lewis, D.F.; Alexander, J.S.; Granger, D.N. Elevated plasma chymotrypsin-like protease (chymase) activity in women with preeclampsia. Hypertens. Pregnancy 2010, 29, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Wang, Y.; O’Donoghue, A.J.; Jiang, Z.; Carballar-Lejarazú, R.; Liang, G.; Hu, X.; Wang, R.; Xu, L.; Guan, X.; et al. Engineering of multiple trypsin/chymotrypsin sites in Cry3A to enhance its activity against Monochamus alternatus Hope larvae. Pest. Manag. Sci. 2020, in press. [Google Scholar] [CrossRef]
- Waliczek, M.; Kijewska, M.; Rudowska, M.; Setner, B.; Stefanowicz, P.; Szewczuk, Z. Peptides Labeled with Pyridinium Salts for Sensitive Detection and Sequencing by Electrospray Tandem Mass Spectrometry. Sci. Rep. 2016, 6, 37720. [Google Scholar] [CrossRef]
- Wang, J.; Sun, Z.; Jiang, J.; Wu, D.; Liu, X.; Xie, Z.; Chen, E.; Zhu, D.; Ye, C.; Zhang, X.; et al. Proteomic Signature of Acute Liver Failure: From Discovery and Verification in a Pig Model to Confirmation in Humans. Mol. Cell Proteom. 2017, 16, 1188–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, C.; Yi-Lun, L.; Guiqing, P.; Fang, L. Structural basis for multifunctional roles of mammalian aminopeptidase N. Proc. Natl. Acad. Sci. USA 2012, 109, 17966–17971. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271.e8–280.e8. [Google Scholar] [CrossRef]
- Vernocchi, V.; Morselli, M.G.; Varesi, S.; Nonnis, S.; Maffioli, E.; Negri, A.; Tedeschi, G.; Luvoni, G.C. Sperm ubiquitination in epididymal feline semen. Theriogenology 2014, 82, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, G.; Albani, E.; Borroni, E.M.; Parini, V.; Brucculeri, A.M.; Maffioli, E.; Negri, A.; Nonnis, S.; Maccarrone, M.; Levi-Setti, P.E. Proteomic profile of maternal-aged blastocoel fluid suggests a novel role for ubiquitin system in blastocyst quality. J. Assist. Reprod. Genet. 2017, 34, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Vizcaíno, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef]
Sample Availability: No. |





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maffioli, E.; Jiang, Z.; Nonnis, S.; Negri, A.; Romeo, V.; Lietz, C.B.; Hook, V.; Ristagno, G.; Baselli, G.; Kistler, E.B.; et al. High-Resolution Mass Spectrometry-Based Approaches for the Detection and Quantification of Peptidase Activity in Plasma. Molecules 2020, 25, 4071. https://doi.org/10.3390/molecules25184071
Maffioli E, Jiang Z, Nonnis S, Negri A, Romeo V, Lietz CB, Hook V, Ristagno G, Baselli G, Kistler EB, et al. High-Resolution Mass Spectrometry-Based Approaches for the Detection and Quantification of Peptidase Activity in Plasma. Molecules. 2020; 25(18):4071. https://doi.org/10.3390/molecules25184071
Chicago/Turabian StyleMaffioli, Elisa, Zhenze Jiang, Simona Nonnis, Armando Negri, Valentina Romeo, Christopher B. Lietz, Vivian Hook, Giuseppe Ristagno, Giuseppe Baselli, Erik B. Kistler, and et al. 2020. "High-Resolution Mass Spectrometry-Based Approaches for the Detection and Quantification of Peptidase Activity in Plasma" Molecules 25, no. 18: 4071. https://doi.org/10.3390/molecules25184071
APA StyleMaffioli, E., Jiang, Z., Nonnis, S., Negri, A., Romeo, V., Lietz, C. B., Hook, V., Ristagno, G., Baselli, G., Kistler, E. B., Aletti, F., O’Donoghue, A. J., & Tedeschi, G. (2020). High-Resolution Mass Spectrometry-Based Approaches for the Detection and Quantification of Peptidase Activity in Plasma. Molecules, 25(18), 4071. https://doi.org/10.3390/molecules25184071