
The wPDI Redox Cycle Coupled Conformational Change of the Repetitive Domain of the HMW-GS 1Dx5—A Computational Study



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
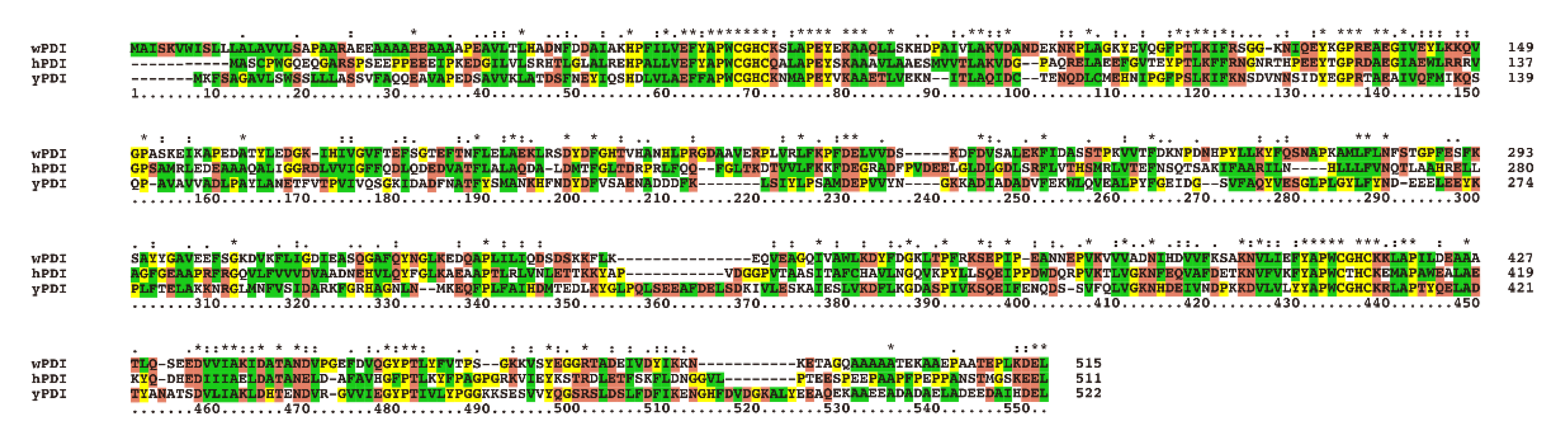
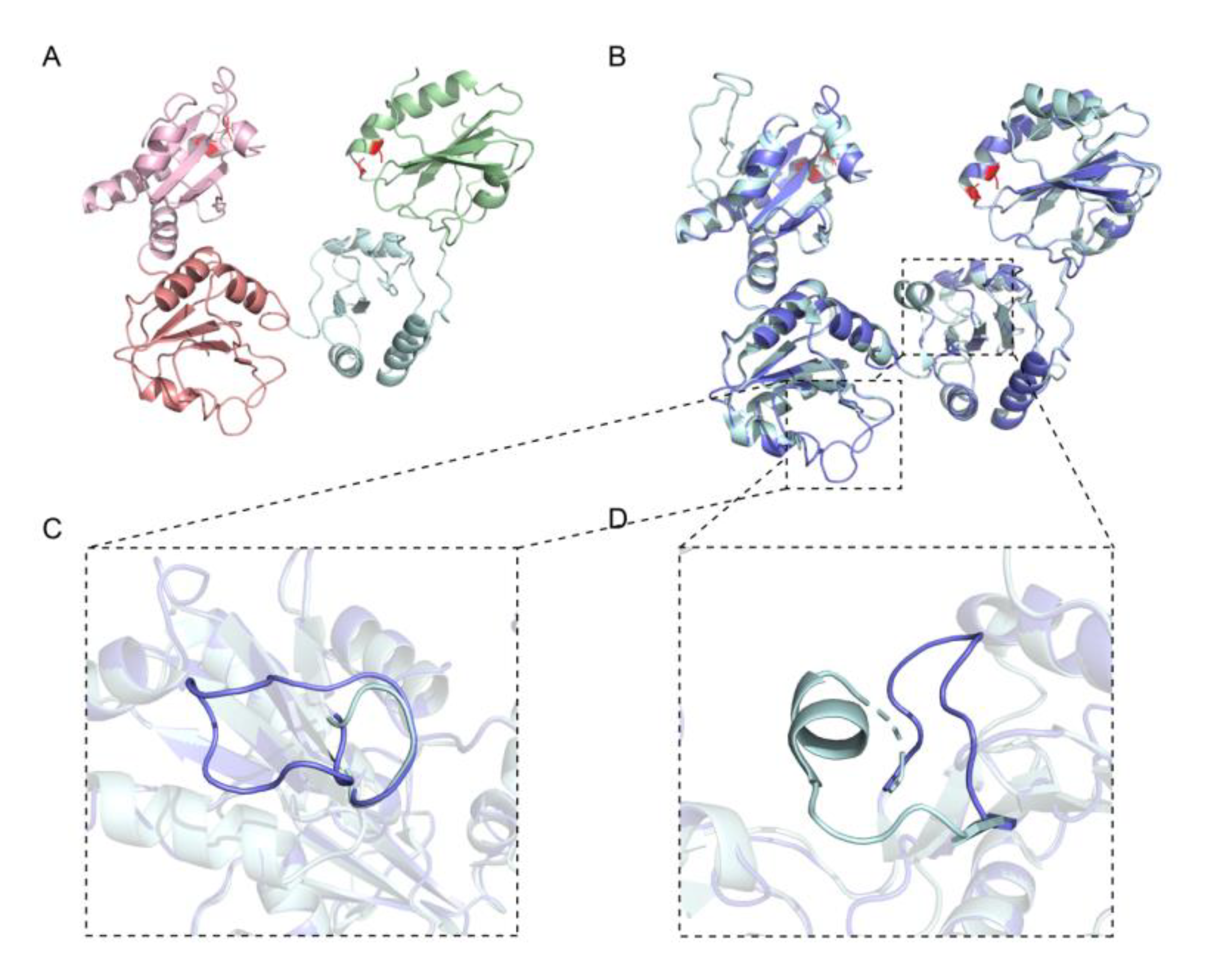
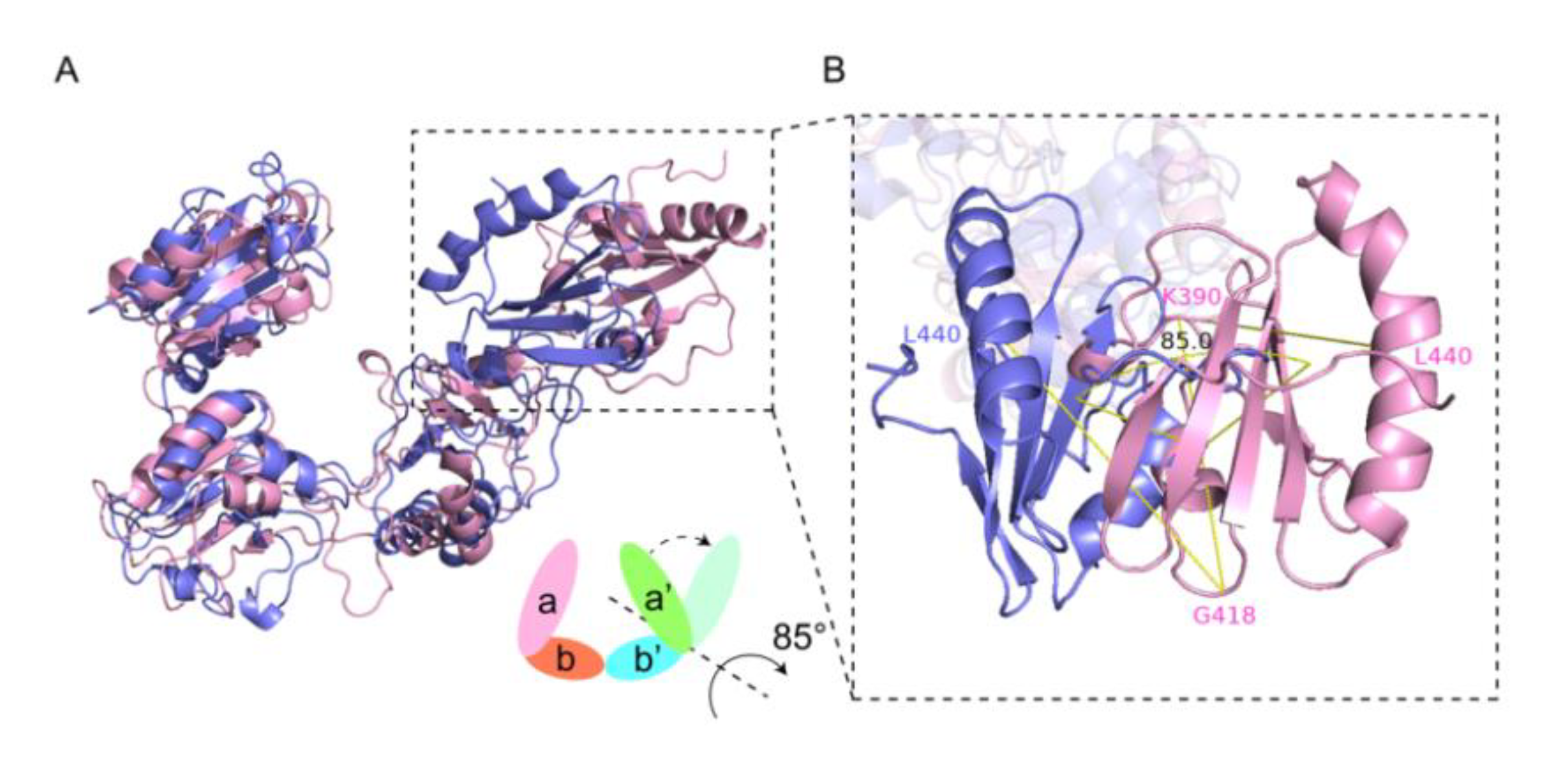
2.1. Homology Comparison, Structure Simulation and Redox Conformations of wPDI
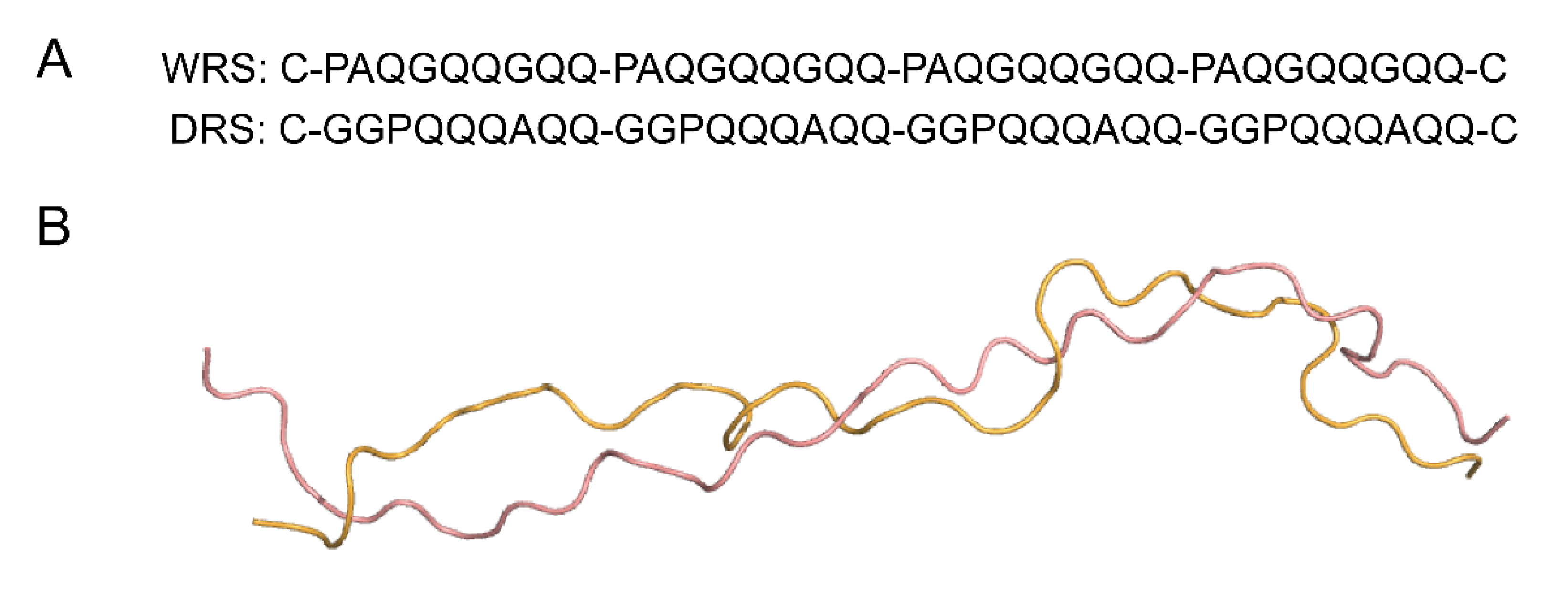
2.2. Structure Simulation and Comparison between Various Repetitive Sequences
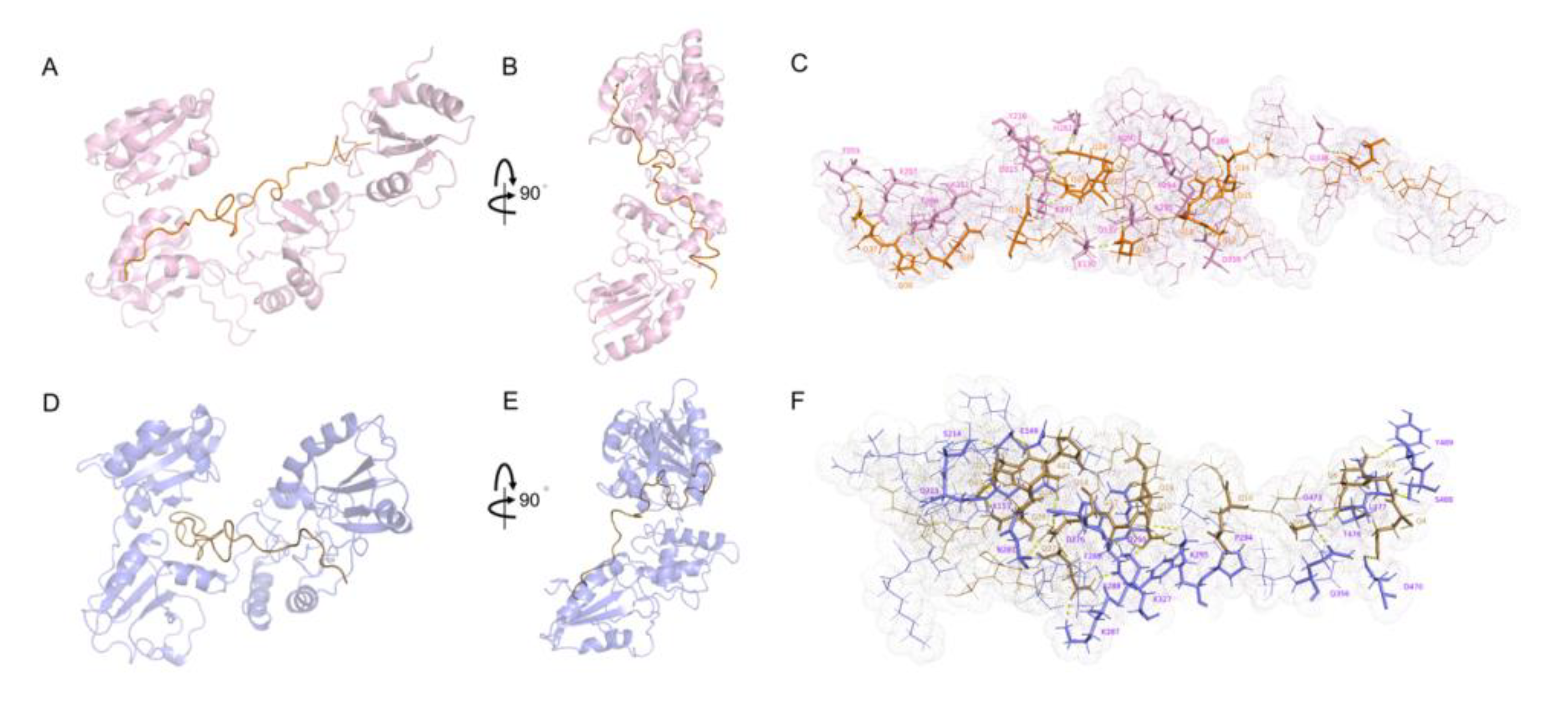
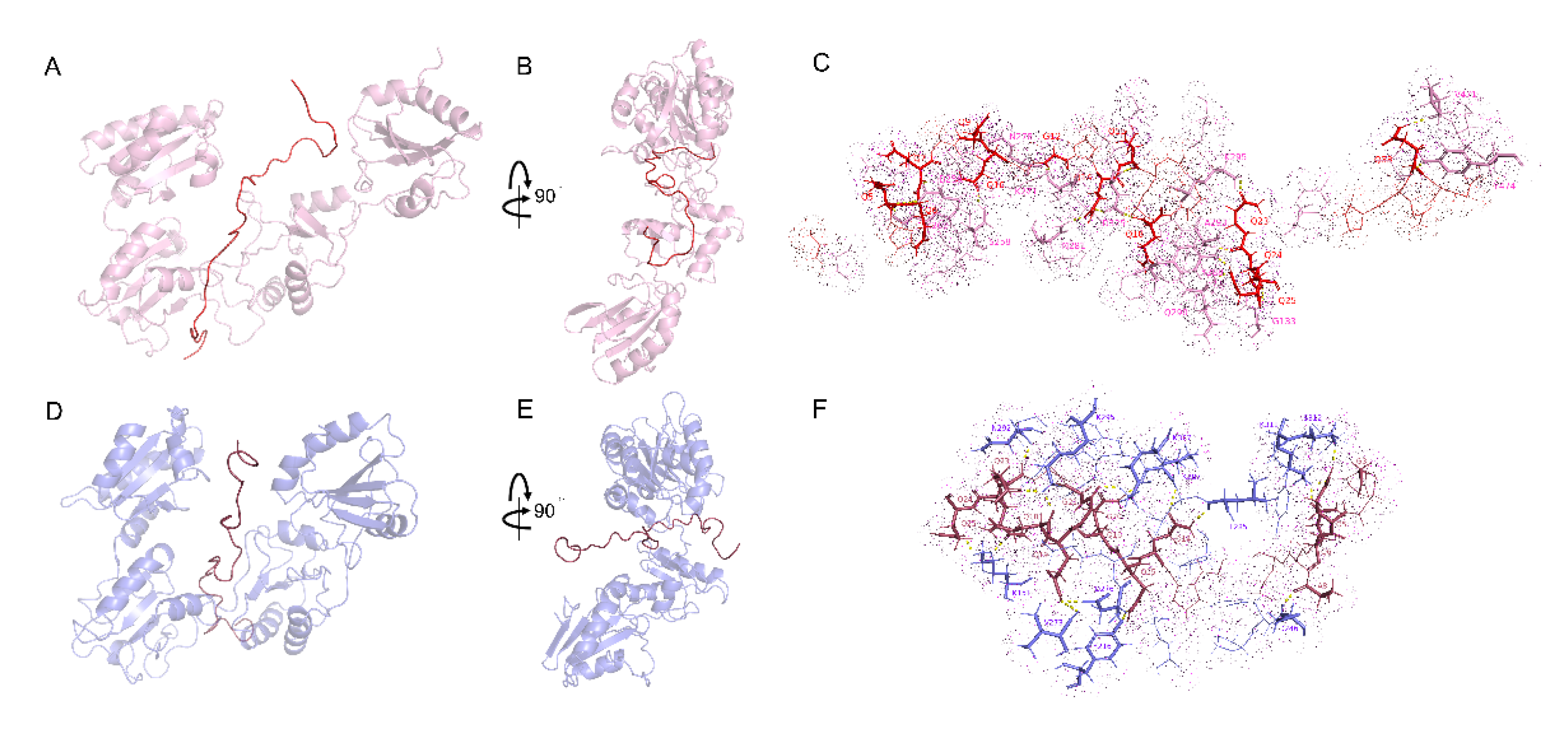
2.3. Interactions between WRS and wPDIO/R
2.4. Interactions between DRS and wPDIO/R
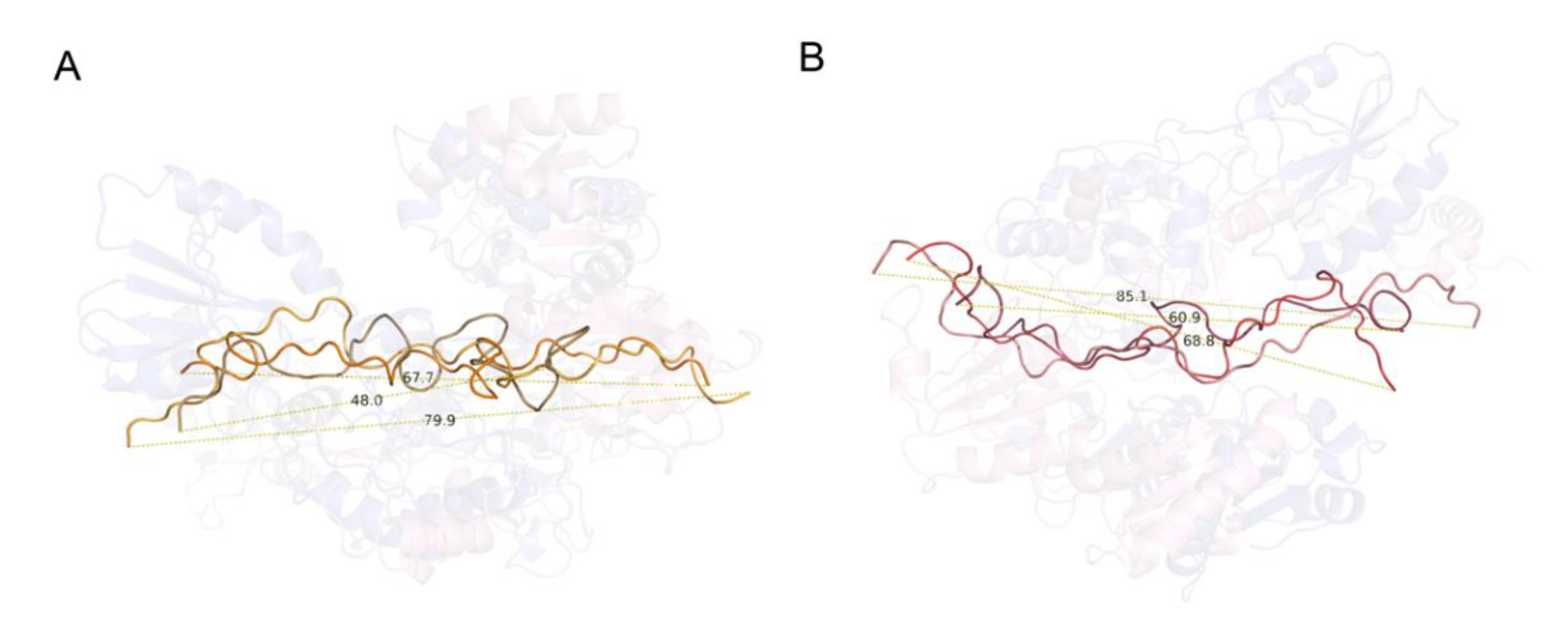
2.5. wPDI-Mediated Folding of the Glutenin Repetitive Sequence
3. Discussion
4. Materials and Methods
4.1. Homology Comparison
4.2. Structural Simulation of wPDI and Glutenin Repetitive Sequence
4.3. MD Simulation of the Solution Structures and Interaction between wPDIO/R and the Repetitive Sequences
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Payne, P.I.; Nightingale, M.A.; Krattiger, A.F.; Holt, L.M. The relationship between HMW glutenin subunit composition and the bread-making quality of British-grown wheat varieties. J. Sci. Food Agric. 1987, 40, 51–65. [Google Scholar] [CrossRef]
- Don, C.; Lichtendonk, W.J.; Plijter, J.J.; Hamer, R.J. Understanding the link between GMP and dough: From glutenin particles in flour towards developed dough. J. Cereal Sci. 2003, 38, 157–165. [Google Scholar] [CrossRef]
- Popineau, Y.; Cornec, M.; Lefebvre, J.; Marchylo, B. Influence of high Mr glutenin subunits on glutenin polymers and rheological properties of glutens and gluten subfractions of near-isogenic lines of wheat sicco. J. Cereal Sci. 1994, 19, 231–241. [Google Scholar] [CrossRef]
- Gilbert, S.M.; Wellner, N.; Belton, P.; Greenfield, J.A.; Siligardi, G.; Shewry, P.R.; Tatham, A.S. Expression and characterisation of a highly repetitive peptide derived from a wheat seed storage protein. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. Enzym. 2000, 1479, 135–146. [Google Scholar] [CrossRef]
- Shewry, P.; Tatham, A. Disulphide bonds in wheat gluten proteins. J. Cereal Sci. 1997, 25, 207–227. [Google Scholar] [CrossRef]
- Belitz, H.-D.; Wieser, H.; Köhler, P. Disulfidbindungen im Weizenkleber: Weitere Cystinpeptide aus HMW- und LMW-Untereinheiten von Glutenin und aus γ-Gliadinen. Z. Für Lebensm. Unters. Forsch. 1993, 196, 239–247. [Google Scholar] [CrossRef]
- Anjum, F.M.; Khan, M.R.; Din, A.; Saeed, M.; Pasha, I.; Arshad, M.U. Wheat gluten: High molecular weight glutenin subunits—Structure, genetics, and relation to dough elasticity. J. Food Sci. 2007, 72, R56–R63. [Google Scholar] [CrossRef]
- Sugiyama, T.; Rafalski, A.; Peterson, D.; Söll, D. A wheat HMW glutenin subunit gene reveals a highly repeated structure. Nucl. Acids Res. 1985, 13, 8729–8737. [Google Scholar] [CrossRef] [Green Version]
- Anderson, O.D.; Greene, F.C. The characterization and comparative analysis of high-molecular-weight glutenin genes from genomes A and B of a hexaploid bread wheat. Theor. Appl. Genet. 1989, 77, 689–700. [Google Scholar] [CrossRef]
- Miles, M.J.; Carr, H.J.; McMaster, T.C.; I’Anson, K.J.; Belton, P.S.; Morris, V.J.; Field, J.M.; Shewry, P.R.; Tatham, A.S. Scanning tunneling microscopy of a wheat seed storage protein reveals details of an unusual supersecondary structure. Proc. Natl. Acad. Sci. USA 1991, 88, 68–71. [Google Scholar] [CrossRef] [Green Version]
- Tatham, A.S.; Drake, A.F.; Shewry, P.R. Conformational studies of synthetic peptides corresponding to the repetitive regions of the high molecular weight (HMW) glutenin subunits of wheat. J. Cereal Sci. 1990, 11, 189–200. [Google Scholar] [CrossRef]
- Kasarda, D.D.; King, G.; Kumosinski, T.F. Comparison of spiral structures in wheat high molecular weight glutenin subunits and elastin by molecular modeling. In Molecular Modeling; American Chemical Society: Washington, DC, USA, 1994; Volume 576, pp. 209–220. [Google Scholar]
- Zhang, J.R.; Liu, Y.; Lin, G.; He, G.Y. Relationship between the sequence and structure of wheat high molecular weight glutenin subunits and processing quality. China Biotechnol. 2006, 26, 103–110. [Google Scholar]
- Wellner, N.; Marsh, J.T.; Savage, A.W.J.; Halford, N.G.; Shewry, P.R.; Mills, E.N.C.; Belton, P.S. Comparison of repetitive sequences derived from high molecular weight subunits of wheat glutenin, an elastomeric plant protein. Biomacromolecules 2006, 7, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Belton, P. Mini Review: On the elasticity of wheat gluten. J. Cereal Sci. 1999, 29, 103–107. [Google Scholar] [CrossRef]
- Ooms, N.; Delcour, J.A. How to impact gluten protein network formation during wheat flour dough making. Curr. Opin. Food Sci. 2019, 25, 88–97. [Google Scholar] [CrossRef]
- Joye, I.J.; Lagrain, B.; Delcour, J.A. Use of chemical redox agents and exogenous enzymes to modify the protein network during breadmaking—A review. J. Cereal Sci. 2009, 50, 11–21. [Google Scholar] [CrossRef]
- Shanmugavel, V.; Santhi, K.K.; Kurup, A.H.; Kalakandan, S.K.; Anandharaj, A.; Rawson, A. Potassium bromate: Effects on bread components, health, environment and method of analysis: A review. Food Chem. 2020, 311, 125964. [Google Scholar] [CrossRef]
- Olofinnade, A.T.; Adeyeba, A.; Onaolapo, A.Y.; Onaolapo, O.J. An assessment of the effects of azodicarbonamide-containing diet on neurobehaviour, brain antioxidant status and membrane lipid peroxidation status in rats. Central Nerv. Syst. Agents Med. Chem. 2020, 20, 49–57. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.; Wang, L. Protein disulfide–isomerase, a folding catalyst and a redox-regulated chaperone. Free. Radic. Biol. Med. 2015, 83, 305–313. [Google Scholar] [CrossRef]
- Wilkinson, B.; Gilbert, H.F. Protein disulfide isomerase. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2004, 1699, 35–44. [Google Scholar] [CrossRef]
- Pirneskoski, A.; Klappa, P.; Williamson, R.A.; Alanen, H.I.; Salo, K.E.H.; Kivirikko, K.I.; Freedman, R.B.; Ruddock, L.; Lobell, M.; Byrne, L. Molecular characterization of the principal substrate binding site of the ubiquitous folding catalyst protein disulfide isomerase. J. Biol. Chem. 2003, 279, 10374–10381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klappa, P.; Ruddock, L.W.; Darby, N.J.; Freedman, R.B. The b’ domain provides the principal peptide-binding site of protein disulfide isomerase but all domains contribute to binding of misfolded proteins. EMBO J. 1998, 17, 927–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darby, N.J.; Creighton, T.E. Functional properties of the individual thioredoxin-like domains of protein disulfide isomerase. Biochemistry 1995, 34, 11725–11735. [Google Scholar] [CrossRef]
- Song, J.-L.; Wang, C.-C. Chaperone-like activity of protein disulfide-isomerase in the refolding of rhodanese. JBIC J. Biol. Inorg. Chem. 2008, 231, 312–316. [Google Scholar] [CrossRef]
- Liu, G.; Wang, J.; Hou, Y.; Huang, Y.; Li, C.-Z.; Li, L.; Hu, S.-Q. Improvements of modified wheat protein disulfide isomerases with chaperone activity only on the processing quality of flour. Food Bioprocess Technol. 2016, 10, 568–581. [Google Scholar] [CrossRef]
- Zhao, C.; Luo, Z.; Li, M.; Gao, J.; Liang, Z.; Sun, S.; Wang, X.; Yang, D. Wheat protein disulfide isomerase improves bread properties via different mechanisms. Food Chem. 2020, 315, 126242. [Google Scholar] [CrossRef]
- Watanabe, E.; Bell, A.; Brockway, B. The effect of protein disulphide isomerase on dough rheology assessed by fundamental and empirical testing. Food Chem. 1998, 61, 481–486. [Google Scholar] [CrossRef]
- Anderson, O.D.; Bekes, F.; D’Ovidio, R. Effects of specific domains of high-molecular-weight glutenin subunits on dough properties by an in vitro assay. J. Cereal Sci. 2011, 54, 280–287. [Google Scholar] [CrossRef]
- Wang, C.; Li, W.; Ren, J.; Fang, J.; Ke, H.; Gong, W.; Feng, W.; Wang, C.-C. Structural insights into the redox-regulated dynamic conformations of human protein disulfide isomerase. Antioxid. Redox Signal. 2013, 19, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Karim, H.A.A.; Tayapiwatana, C.; Nimmanpipug, P.; Zain, S.M.; Rahman, N.A.; Lee, V.S. Peptide docking of HIV-1 p24 with single chain fragment variable (scFv) by CDOCKER algorithm. In Proceedings of the 3rd International Conference on Fundamental and Applied Sciences (ICFAS 2014): Innovative Research in Applied Sciences for a Sustainable Future, Kuala Lumpur, Malaysia, 3–5 June 2014; pp. 98–102. [Google Scholar] [CrossRef]
- Denisov, A.Y.; Määttänen, P.; Dabrowski, C.; Kozlov, G.; Thomas, D.Y.; Gehring, K. Solution structure of the bb′ domains of human protein disulfide isomerase. FEBS J. 2009, 276, 1440–1449. [Google Scholar] [CrossRef]
- Koivunen, P.; Salo, K.E.H.; Myllyharju, J.; Ruddock, L.W.; Collins, R.E.; Tachibana, M.; Tamaru, H.; Smith, K.M.; Jia, D.; Zhang, X.; et al. Three binding sites in protein-disulfide isomerase cooperate in collagen prolyl 4-hydroxylase tetramer assembly. J. Biol. Chem. 2004, 280, 5227–5235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, L.J.; Sidhu, A.; Wallis, A.K.; Ruddock, L.; Freedman, R.B.; Howard, M.J.; Williamson, R.A. Mapping of the ligand-binding site on the b′ domain of human PDI: Interaction with peptide ligands and the x-linker region. Biochem. J. 2009, 423, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serve, O.; Kamiya, Y.; Maeno, A.; Nakano, M.; Murakami, C.; Sasakawa, H.; Yamaguchi, Y.; Harada, T.; Kurimoto, E.; Yagi-Utsumi, M.; et al. Redox-dependent domain rearrangement of protein disulfide isomerase coupled with exposure of its substrate-binding hydrophobic surface. J. Mol. Biol. 2010, 396, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Xue, Y.; Ma, Y. Enhancing the thermostability of α-glucosidase from Thermoanaerobacter tengcongensis MB4 by single proline substitution. J. Biosci. Bioeng. 2010, 110, 12–17. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; Beer, T.A.P.D.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The PyMOL Molecular Graphics System; Version 1.8; Schrodinger, LLC: New York, NY, USA, 2015.
- Sarkar, T.K.; Yang, X. Application of the conjugate gradient and steepest descent for computing the eigenvalues of an operator. Signal Process. 1989, 17, 31–38. [Google Scholar] [CrossRef]
- Yu, P.; Zhou, F.; Yang, D. Curdlan conformation change during its hydrolysis by multi-domain β-1,3-glucanases. Food Chem. 2019, 287, 20–27. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, J.; Yu, P.; Liang, H.; Fu, J.; Luo, Z.; Yang, D. The wPDI Redox Cycle Coupled Conformational Change of the Repetitive Domain of the HMW-GS 1Dx5—A Computational Study. Molecules 2020, 25, 4393. https://doi.org/10.3390/molecules25194393
Gao J, Yu P, Liang H, Fu J, Luo Z, Yang D. The wPDI Redox Cycle Coupled Conformational Change of the Repetitive Domain of the HMW-GS 1Dx5—A Computational Study. Molecules. 2020; 25(19):4393. https://doi.org/10.3390/molecules25194393
Chicago/Turabian StyleGao, Jihui, Peixuan Yu, Hongrui Liang, Jiahui Fu, Ziyue Luo, and Dong Yang. 2020. "The wPDI Redox Cycle Coupled Conformational Change of the Repetitive Domain of the HMW-GS 1Dx5—A Computational Study" Molecules 25, no. 19: 4393. https://doi.org/10.3390/molecules25194393
APA StyleGao, J., Yu, P., Liang, H., Fu, J., Luo, Z., & Yang, D. (2020). The wPDI Redox Cycle Coupled Conformational Change of the Repetitive Domain of the HMW-GS 1Dx5—A Computational Study. Molecules, 25(19), 4393. https://doi.org/10.3390/molecules25194393