
Isoliquiritigenin Pretreatment Induces Endoplasmic Reticulum Stress-Mediated Hormesis and Attenuates Cisplatin-Induced Oxidative Stress and Damage in LLC-PK1 Cells



Abstract
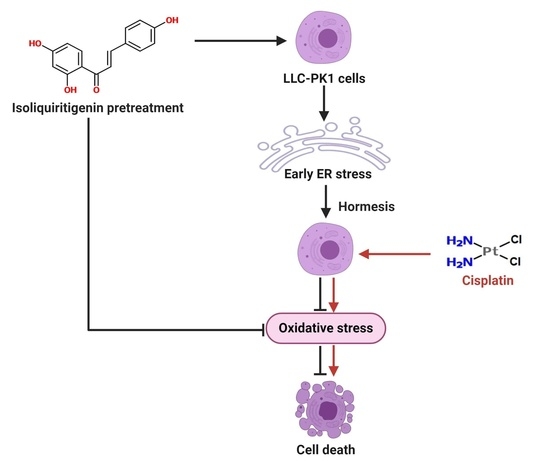
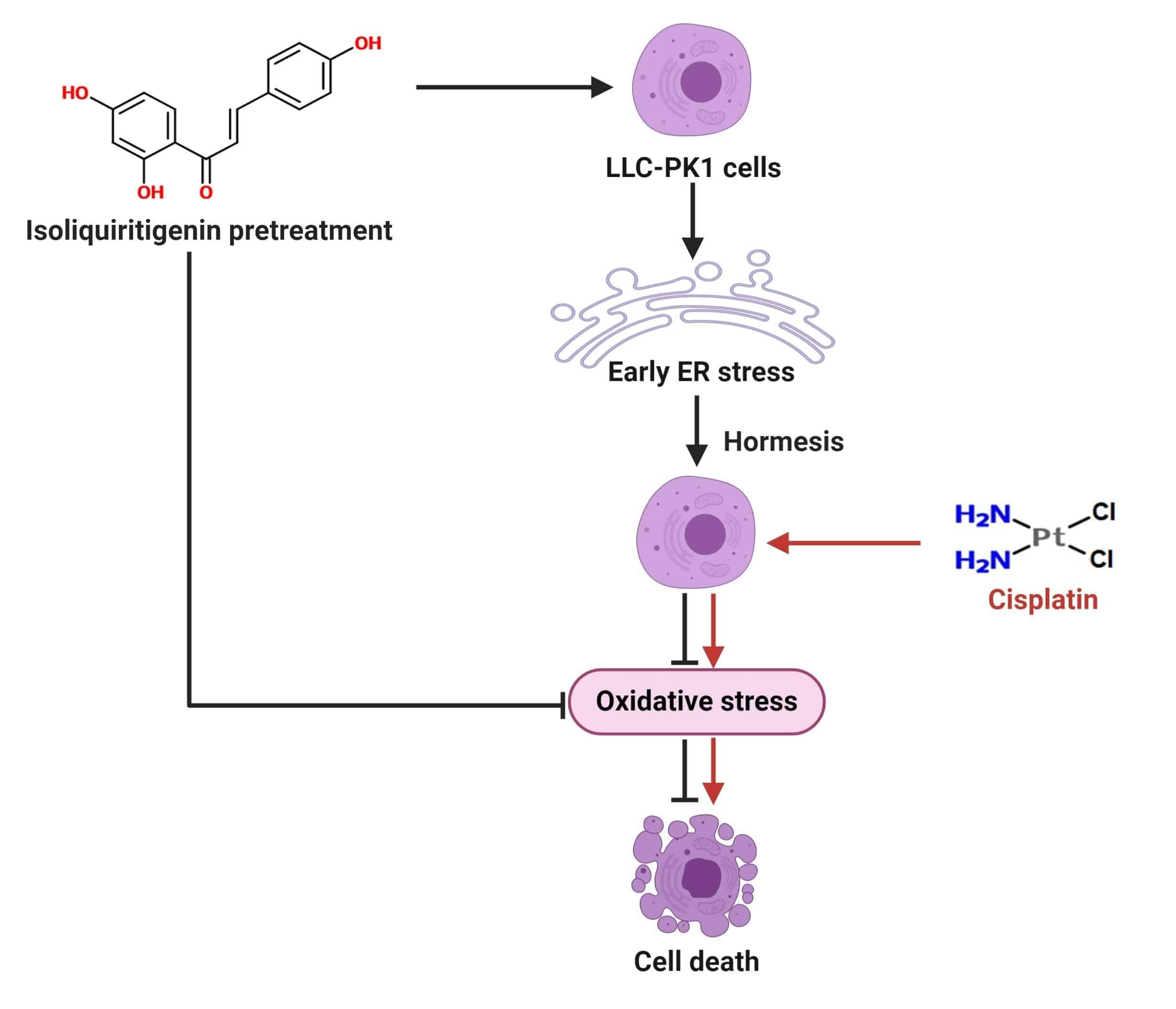
:
1. Introduction
2. Results
2.1. First Stage
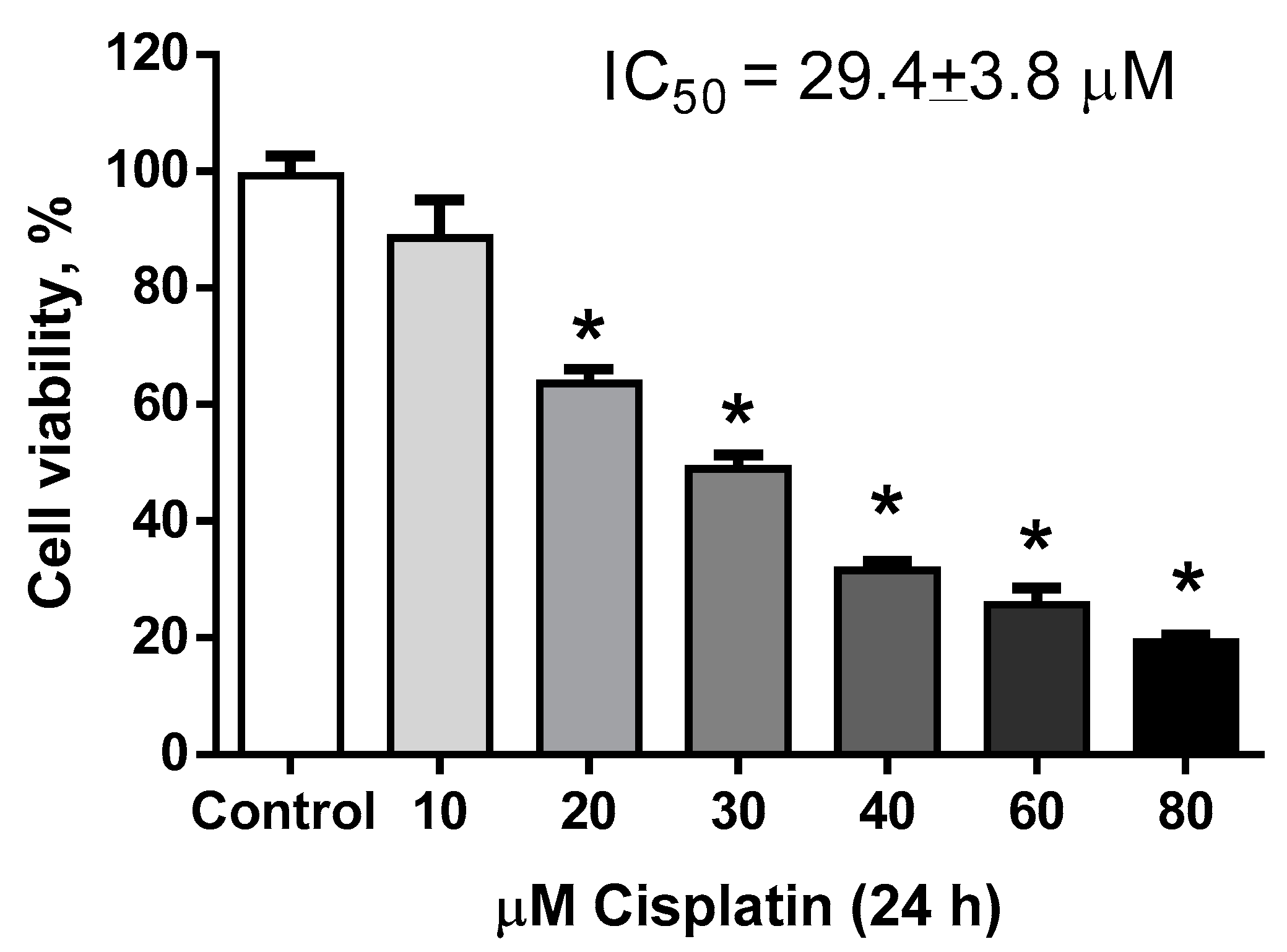
2.1.1. Estimation of Hormetic Zone of IsoLQ in CP-Induced Toxicity in LLC-PK1 Cells
2.1.2. Determination of IsoLQ Protective Concentration and Pretreatment Time against CP-Induced Toxicity in LLC-PK1 Cells
2.1.3. Pretreatment of IsoLQ Induces ER Stress
2.2. Second Stage
2.2.1. IsoLQ Pretreatment Enhanced ER Stress in CP-Induced Nephrotoxicity in LLC-PK1 Cells
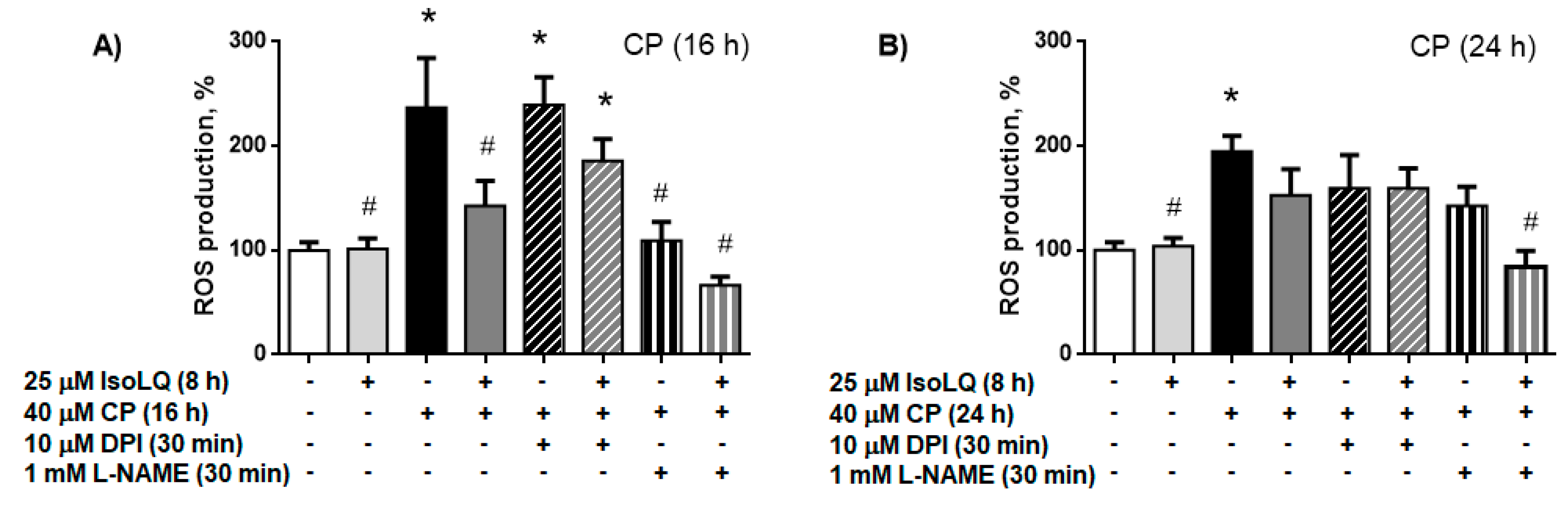
2.2.2. IsoLQ-Induced Hormesis Attenuates Oxidative Stress in CP-Treated LLC-PK1 Cells
Nitric Oxide Synthase (NOS) is the Source of ROS Production in LLC-PK1 Cells Pretreated with IsoLQ and Exposed to CP
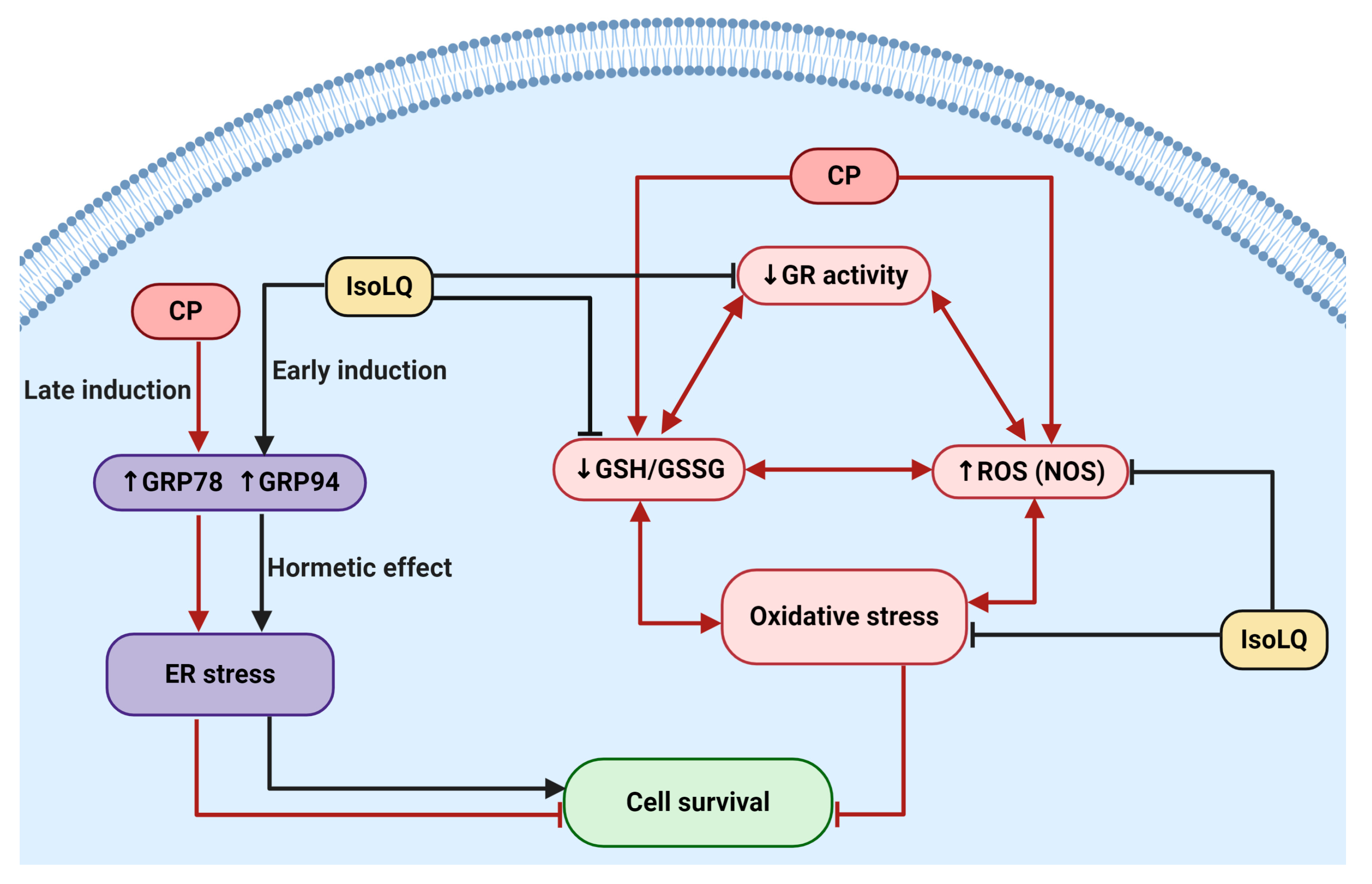
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
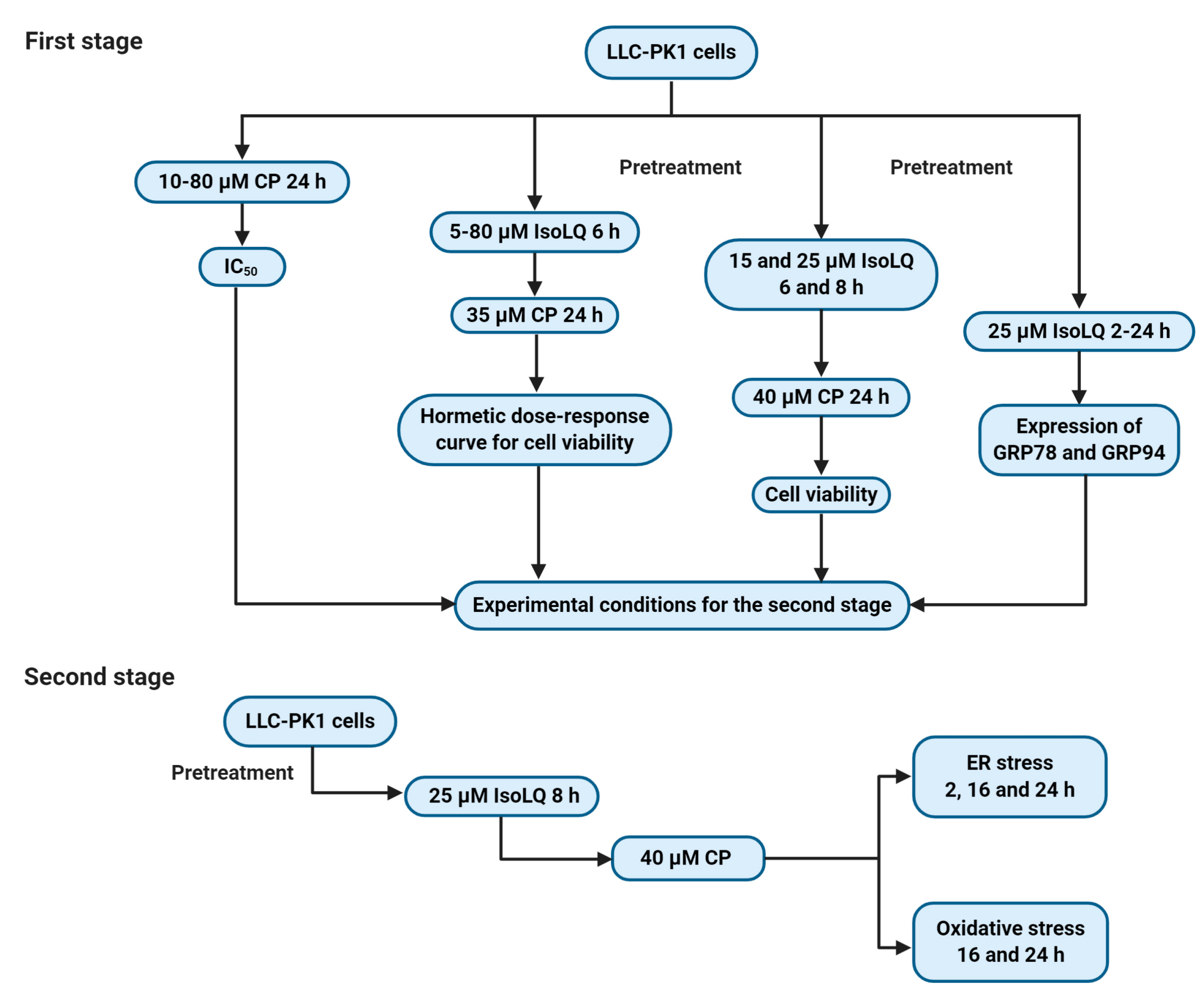
4.3. Experimental Design
4.3.1. First Stage
4.3.2. Second Stage
4.4. Cell Viability
4.5. Extraction of Total Fractions of Proteins for Western Blot
4.6. ER Stress Evaluation
Western Blot Analysis
4.7. Oxidative Stress Evaluation
4.7.1. Measurement of ROS Production
4.7.2. Measurement of GSH/GSSG Ratio
4.7.3. Free Thiols Levels
4.7.4. GR Activity
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, C.; Huang, S.; Chen, C.L.; Su, S.B.; Fang, D.D. Isoliquiritigenin Inhibits Ovarian Cancer Metastasis by Reversing Epithelial-to-Mesenchymal Transition. Molecules 2019, 24, 3725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Park, S.J.; Yun, K.J.; Cho, Y.W.; Park, H.J.; Lee, K.T. Isoliquiritigenin isolated from the roots of Glycyrrhiza uralensis inhibits LPS-induced iNOS and COX-2 expression via the attenuation of NF-κB in RAW 264.7 macrophages. Eur. J. Pharmacol. 2008, 584, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Chin, Y.W.; Bae, J.K.; Seo, J.; Choi, Y. Pharmacokinetics of Isoliquiritigenin and Its Metabolites in Rats: Low Bioavailability Is Primarily Due to the Hepatic and Intestinal Metabolism. Planta Med. 2013, 79, 1656–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalingam, M.; Kim, H.; Lee, Y.; Lee, Y.I. Phytochemical and Pharmacological Role of Liquiritigenin and Isoliquiritigenin From Radix Glycyrrhizae in Human Health and Disease Models. Front. Aging Neurosci. 2018, 10, 348. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Takasuka, N.; Iigo, M.; Baba, M.; Nishino, H.; Tsuda, H.; Okuyama, T. Isoliquiritigenin, a flavonoid from licorice, reduces prostaglandin E2 and nitric oxide, causes apoptosis, and suppresses aberrant crypt foci development. Cancer Sci. 2004, 95, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Tanemoto, R.; Okuyama, T.; Matsuo, H.; Okumura, T.; Ikeya, Y.; Nishizawa, M. The constituents of licorice (Glycyrrhiza uralensis) differentially suppress nitric oxide production in interleukin-1β-treated hepatocytes. Biochem. Biophys. Rep. 2015, 2, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Lu, Y.; Shi, P.; Zhang, H. Hydroxyl and hydroperoxyl radicals scavenging by isoliquiritigenin and liquiritigenin: A quantum chemical study. Struct. Chem. 2017, 28, 1181–1186. [Google Scholar] [CrossRef]
- Zhao, H.; Yuan, X.; Li, D.; Chen, H.; Jiang, J.; Wang, Z.; Sun, X.; Zheng, Q. Isoliquiritigen Enhances the Antitumour Activity and Decreases the Genotoxic Effect of Cyclophosphamide. Molecules 2013, 18, 8786–8798. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Londoño, A.P.; Bello-Alvarez, C.; Pedraza-Chaverri, J. Isoliquiritigenin pretreatment attenuates cisplatin induced proximal tubular cells (LLC-PK1) death and enhances the toxicity induced by this drug in bladder cancer T24 cell line. Food Chem. Toxicol. 2017, 109, 143–154. [Google Scholar] [CrossRef]
- Orlikova, B.; Tasdemir, D.; Golais, F.; Dicato, M.; Diederich, M. Dietary chalcones with chemopreventive and chemotherapeutic potential. Genes Nutr. 2011, 6, 125–147. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Huang, T.C.; Shieh, T.M.; Wu, C.H.; Lin, L.C.; Hsia, S.M. Isoliquiritigenin Induces Autophagy and Inhibits Ovarian Cancer Cell Growth. IJMS 2017, 18, 2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, F.W.; Yu, C.C.; Hsieh, P.L.; Liao, Y.W.; Lu, M.Y.; Chu, P.M. Targeting oral cancer stemness and chemoresistance by isoliquiritigenin-mediated GRP78 regulation. Oncotarget 2017, 8, 93912–93923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanazawa, M.; Satomi, Y.; Mizutani, Y.; Ukimura, O.; Kawauchi, A.; Sakai, T.; Baba, M.; Okuyama, T.; Nishino, H.; Miki, T. Isoliquiritigenin Inhibits the Growth of Prostate Cancer. Eur. Urol. 2003, 43, 580–586. [Google Scholar] [CrossRef]
- Si, L.; Yang, X.; Yan, X.; Wang, Y.; Zheng, Q. Isoliquiritigenin induces apoptosis of human bladder cancer T24 cells via a cyclin-dependent kinase-independent mechanism. Oncol. Lett. 2017, 14, 241–249. [Google Scholar] [CrossRef]
- Zhou, G.S.; Song, L.J.; Yang, B. Isoliquiritigenin inhibits proliferation and induces apoptosis of U87 human glioma cells in vitro. Mol. Med. Rep. 2013, 7, 531–536. [Google Scholar] [CrossRef]
- Lee, C.K.; Son, S.H.; Park, K.K.; Park, J.H.Y.; Lim, S.S.; Chung, W.Y. Isoliquiritigenin Inhibits Tumor Growth and Protects the Kidney and Liver Against Chemotherapy-Induced Toxicity in a Mouse Xenograft Model of Colon Carcinoma. J. Pharm. Sci 2008, 106, 444–451. [Google Scholar] [CrossRef] [Green Version]
- Peng, F.; Du, Q.; Peng, C.; Wang, N.; Tang, H.; Xie, X.; Shen, J.; Chen, J. A Review: The Pharmacology of Isoliquiritigenin: A Review: The Pharmacology of ISL. Phytother. Res. 2015, 29, 969–977. [Google Scholar] [CrossRef]
- Wang, Z.F.; Liu, J.; Yang, Y.A.; Zhu, H.L. A Review: The Anti-inflammatory, Anticancer and Antibacterial Properties of Four Kinds of Licorice Flavonoids Isolated from Licorice. CMC 2020, 27, 1997–2011. [Google Scholar] [CrossRef]
- Chirino, Y.I.; Pedraza-Chaverri, J. Role of oxidative and nitrosative stress in cisplatin-induced nephrotoxicity. Exp. Toxicol. Pathol. 2009, 61, 223–242. [Google Scholar] [CrossRef]
- dos Santos, N.A.G.; Carvalho Rodrigues, M.A.; Martins, N.M.; dos Santos, A.C. Cisplatin-induced nephrotoxicity and targets of nephroprotection: An update. Arch. Toxicol. 2012, 86, 1233–1250. [Google Scholar] [CrossRef]
- Manohar, S.; Leung, N. Cisplatin nephrotoxicity: A review of the literature. J. Nephrol. 2018, 31, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin Nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancho-Martínez, S.M.; Prieto-García, L.; Prieto, M.; López-Novoa, J.M.; López-Hernández, F.J. Subcellular targets of cisplatin cytotoxicity: An integrated view. Pharmacol. Ther. 2012, 136, 35–55. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Sierra, T.; Eugenio-Pérez, D.; Sánchez-Chinchillas, A.; Pedraza-Chaverri, J. Role of food-derived antioxidants against cisplatin induced-nephrotoxicity. Food Chem. Toxicol. 2018, 120, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peres, L.A.B.; da Cunha Júnior, A.D. Acute nephrotoxicity of cisplatin: Molecular mechanisms. J. Bras. Nefrol. 2013, 35, 332–340. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signaling-from basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Braakman, I.; Hebert, D.N. Protein Folding in the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönthal, A.H. Endoplasmic Reticulum Stress: Its Role in Disease and Novel Prospects for Therapy. Scientifica 2012, 2012, 857516. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Groenendyk, J.; Michalak, M. Glycoprotein Quality Control and Endoplasmic Reticulum Stress. Molecules 2015, 20, 13689–13704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Cycle or a Double-Edged Sword? Antioxid. Redox Signal. 2007, 9, 2277–2294. [Google Scholar] [CrossRef] [Green Version]
- Ozgur, R.; Uzilday, B.; Iwata, Y.; Koizumi, N.; Turkan, I. Interplay between the unfolded protein response and reactive oxygen species: A dynamic duo. J. Exp. Bot. 2018, 69, 3333–3345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Fermín, L.M.; Aparicio-Trejo, O.E.; Avila-Rojas, S.H.; Gómez-Sierra, T.; Martínez-Klimova, E.; Pedraza-Chaverri, J. Natural antioxidants’ effects on endoplasmic reticulum stress-related diseases. Food Chem. Toxicol. 2020, 138, 111229. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Yuan, J. Cellular response to endoplasmic reticulum stress: A matter of life or death. Cell Death Differ. 2006, 13, 363–373. [Google Scholar] [CrossRef]
- Inagi, R.; Kumagai, T.; Nishi, H.; Kawakami, T.; Miyata, T.; Fujita, T.; Nangaku, M. Preconditioning with Endoplasmic Reticulum Stress Ameliorates Mesangioproliferative Glomerulonephritis. JASN 2008, 19, 915–922. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, M. Endoplasmic reticulum stress and unfolded protein response in renal pathophysiology: Janus faces. Am. J. Physiol.-Ren. Physiol. 2008, 295, F323–F334. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Tan, P.; Yan, B.; Gao, R.; Zhao, J.; Wang, J.; Guo, J.; Li, N.; Ma, Z. ER stress and autophagy are involved in the apoptosis induced by cisplatin in human lung cancer cells. Oncol. Rep. 2016, 35, 2606–2614. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Tan, J.; Miao, Y.; Li, M.; Zhang, Q. Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress. J. Cell Physiol. 2017, 232, 2977–2984. [Google Scholar] [CrossRef]
- Mattson, M.P. Hormesis defined. Ageing Res. Rev. 2008, 7, 1–7. [Google Scholar] [CrossRef]
- Zimmermann, A.; Bauer, M.; Kroemer, G.; Madeo, F.; Carmona-Gutierrez, D. When less is more: Hormesis against stress and disease. MIC 2014, 1, 150–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollereau, B. Establishing Links between Endoplasmic Reticulum-Mediated Hormesis and Cancer. Mol. Cell. Biol. 2013, 33, 2372–2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollereau, B.; Manié, S.; Napoletano, F. Getting the better of ER stress. J. Cell Commun. Signal. 2014, 8, 311–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, F.Z.; Markus, M.A.; Morris, B.J. Hormesis as a pro-healthy aging intervention in human beings? Dose Response 2009, 8, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Breithaupt, H. Fierce creatures: Zoonoses, diseases that jump from animals to humans, are a growing health problem around the world. Understanding their causes and their effects on humans have therefore become an important topic for global public health. EMBO Rep. 2003, 4, 921–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, E.J. Hormesis: Why it is important to toxicology and toxicologists. Environ. Toxicol. Chem. 2008, 27, 1451. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Baldwin, L.A. The Dose Determines the Stimulation (and Poison): Development of a Chemical Hormesis Database. Int. J. Toxicol. 1997, 16, 545–559. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Baldwin, L.A. U-Shaped Dose-Responses in Biology, Toxicology, and Public Health. Annu. Rev. Public Health 2001, 22, 15–33. [Google Scholar] [CrossRef]
- Lushchak, V.I. Dissection of the Hormetic Curve: Analysis of Components and Mechanisms. Dose-Response 2014, 12. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Mattson, M.P. How does hormesis impact biology, toxicology, and medicine? NPJ Aging Mech. Dis. 2017, 3, 13. [Google Scholar] [CrossRef] [Green Version]
- Foufelle, F.; Fromenty, B. Role of endoplasmic reticulum stress in drug-induced toxicity. Pharm. Res. Perspect 2016, 4, e00211. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R. Endoplasmic reticulum stress as a progression factor for kidney injury. Curr. Opin. Pharmacol. 2010, 10, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Peyrou, M.; Cribb, A.E. Effect of endoplasmic reticulum stress preconditioning on cytotoxicity of clinically relevant nephrotoxins in renal cell lines. Toxicol. In Vitro 2007, 21, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Liu, R.; Qu, Q. Role of endoplasmic reticulum stress on cisplatin resistance in ovarian carcinoma. Oncol. Lett. 2017, 13, 1437–1443. [Google Scholar] [CrossRef] [Green Version]
- Mohan, I.K.; Khan, M.; Shobha, J.C.; Naidu, M.U.R.; Prayag, A.; Kuppusamy, P.; Kutala, V.K. Protection against cisplatin-induced nephrotoxicity by Spirulina in rats. Cancer Chemother. Pharm. 2006, 58, 802–808. [Google Scholar] [CrossRef]
- Zou, P.; Song, J.; Jiang, B.; Pei, F.; Chen, B.; Yang, X.; Liu, G.; Hu, Z. Epigallocatechin-3-gallate protects against cisplatin nephrotoxicity by inhibiting the apoptosis in mouse. Int. J. Clin. Exp. Pathol. 2014, 7, 4607–4616. [Google Scholar]
- Khan, S.A.; Priyamvada, S.; Khan, W.; Khan, S.; Farooq, N.; Yusufi, A.N.K. Studies on the protective effect of green tea against cisplatin induced nephrotoxicity. Pharmacol. Res. 2009, 60, 382–391. [Google Scholar] [CrossRef]
- Sun, C.; Zhang, H.; Ma, X.; Zhou, X.; Gan, L.; Liu, Y.; Wang, Z. Isoliquiritigenin Enhances Radiosensitivity of HepG2 Cells via Disturbance of Redox Status. Cell Biochem. Biophys. 2013, 65, 433–444. [Google Scholar] [CrossRef]
- Bhakta-Guha, D.; Efferth, T. Hormesis: Decoding Two Sides of the Same Coin. Pharmaceuticals 2015, 8, 865–883. [Google Scholar] [CrossRef]
- Cox, L.A. (Tony) Hormesis for Fine Particulate Matter (PM 2.5). Dose-Response 2012, 10. [Google Scholar] [CrossRef]
- Cybulsky, A.V. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat. Rev. Nephrol. 2017, 13, 681–696. [Google Scholar] [CrossRef]
- Dufey, E.; Sepúlveda, D.; Rojas-Rivera, D.; Hetz, C. Cellular Mechanisms of Endoplasmic Reticulum Stress Signaling in Health and Disease. 1. An overview. Am. J. Physiol. Cell Physiol. 2014, 307, C582–C594. [Google Scholar] [CrossRef]
- Hiramatsu, N.; Chiang, W.-C.; Kurt, T.D.; Sigurdson, C.J.; Lin, J.H. Multiple Mechanisms of Unfolded Protein Response–Induced Cell Death. Am. J. Pathol. 2015, 185, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Urra, H.; Dufey, E.; Lisbona, F.; Rojas-Rivera, D.; Hetz, C. When ER stress reaches a dead end. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2013, 1833, 3507–3517. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; MacDonald, N.; Collins, J.; Cribb, A. Cytoprotection Following Endoplasmic Reticulum Stress Protein Induction in Continuous Cell Lines. Basic Clin. Pharmacol. Toxicol. 2004, 94, 124–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prachasilchai, W.; Sonoda, H.; Yokota-Ikeda, N.; Oshikawa, S.; Aikawa, C.; Uchida, K.; Ito, K.; Kudo, T.; Imaizumi, K.; Ikeda, M. A protective role of unfolded protein response in mouse ischemic acute kidney injury. Eur. J. Pharmacol. 2008, 592, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Guo, M.; Jiang, W.; Dong, H.; Han, Y.; An, X.F.; Zhang, J. Endoplasmic reticulum stress and its effects on renal tubular cells apoptosis in ischemic acute kidney injury. Ren. Fail. 2016, 38, 831–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.; Shu, S.; Guo, C.; Tang, C.; Dong, Z. Endoplasmic reticulum stress in ischemic and nephrotoxic acute kidney injury. Ann. Med. 2018, 50, 381–390. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Targeting endoplasmic reticulum stress in metabolic disease. Expert Opin. Ther. Targets 2013, 17, 437–448. [Google Scholar] [CrossRef]
- Halliwell, B. Biochemistry of oxidative stress. Biochem. Soc. Trans. 2007, 35, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- Small, D.M.; Coombes, J.S.; Bennett, N.; Johnson, D.W.; Gobe, G.C. Oxidative stress, anti-oxidant therapies and chronic kidney disease: Oxidative stress and CKD. Nephrology 2012, 17, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Bhandary, B.; Marahatta, A.; Kim, H.-R.; Chae, H.-J. An Involvement of Oxidative Stress in Endoplasmic Reticulum Stress and Its Associated Diseases. IJMS 2012, 14, 434–456. [Google Scholar] [CrossRef] [PubMed]
- Goligorsky, M.S. Oxidative Stress and the Kidney: Riding on the Curve of Hormesis. Antioxid. Redox Signal. 2016, 25, 117–118. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Jin, X.; Willmore, W.G. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2014, 2, 123–139. [Google Scholar] [CrossRef] [Green Version]
- Ursini, F.; Maiorino, M.; Forman, H.J. Redox homeostasis: The Golden Mean of healthy living. Redox Biol. 2016, 8, 205–215. [Google Scholar] [CrossRef]
- Xiong, Y.; Uys, J.D.; Tew, K.D.; Townsend, D.M. S-Glutathionylation: From Molecular Mechanisms to Health Outcomes. Antioxid. Redox Signal. 2011, 15, 233–270. [Google Scholar] [CrossRef] [Green Version]
- Haynes, C.M.; Titus, E.A.; Cooper, A.A. Degradation of Misfolded Proteins Prevents ER-Derived Oxidative Stress and Cell Death. Mol. Cell 2004, 15, 767–776. [Google Scholar] [CrossRef]
- Townsend, D.M. Metabolism of Cisplatin to a Nephrotoxin in Proximal Tubule Cells. J. Am. Soc. Nephrol. 2003, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Birk, J.; Meyer, M.; Aller, I.; Hansen, H.G.; Odermatt, A.; Dick, T.P.; Meyer, A.J.; Appenzeller-Herzog, C. Endoplasmic reticulum: Reduced and oxidized glutathione revisited. J. Cell Sci. 2013, 126, 1604–1617. [Google Scholar] [CrossRef] [Green Version]
- Ognjanović, B.I.; Djordjević, N.Z.; Matić, M.M.; Obradović, J.M.; Mladenović, J.M.; Štajn, A.Š.; Saičić, Z.S. Lipid Peroxidative Damage on Cisplatin Exposure and Alterations in Antioxidant Defense System in Rat Kidneys: A Possible Protective Effect of Selenium. IJMS 2012, 13, 1790–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uzilday, B.; Ozgur, R.; Sekmen, A.H.; Turkan, I. Endoplasmic reticulum stress regulates glutathione metabolism and activities of glutathione related enzymes in Arabidopsis. Funct. Plant. Biol. 2018, 45, 284. [Google Scholar] [CrossRef] [PubMed]
- Zaazaa, A.M.; Motelp, B.A.A.E.; Aniss, N.N.D. Potential Protective Role of Rutin and Alpha-lipoic Acid Against Cisplatin-induced Nephrotoxicity in Rats. Pak. J. Biol. Sci. 2019, 22, 361–371. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 Is a Direct PERK Substrate and Effector of PERK-Dependent Cell Survival. MCB 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Li, W.; Su, Z.; Kong, A.N.T. The complexity of the Nrf2 pathway: Beyond the antioxidant response. J. Nutr. Biochem. 2015, 26, 1401–1413. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zheng, Z.; Hu, S.; Ru, X.; Fan, Z.; Qiu, L.; Zhang, Y. Unification of Opposites between Two Antioxidant Transcription Factors Nrf1 and Nrf2 in Mediating Distinct Cellular Responses to the Endoplasmic Reticulum Stressor Tunicamycin. Antioxidants 2019, 9, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thongnuanjan, P.; Soodvilai, S.; Chatsudthipong, V.; Soodvilai, S. Fenofibrate reduces cisplatin-induced apoptosis of renal proximal tubular cells via inhibition of JNK and p38 pathways. J. Toxicol. Sci. 2016, 41, 339–349. [Google Scholar] [CrossRef] [Green Version]
- Clarke, J.M.; Gillings, M.R.; Altavilla, N.; Beattie, A.J. Potential problems with fluorescein diacetate assays of cell viability when testing natural products for antimicrobial activity. J. Microbiol. Methods 2001, 46, 261–267. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Rahman, I.; Kode, A.; Biswas, S.K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 2006, 1, 3159–3165. [Google Scholar] [CrossRef]
- Ellman, G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef]
- Carlberg, I.; Mannervik, B. Purification and characterization of the flavoenzyme glutathione reductase from rat liver. J. Biol. Chem. 1975, 250, 5475–5480. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Control | 25 μM IsoLQ (8 h) | 40 μM CP | 25 μM IsoLQ (8 h) + 40 μΜ CP | ||
|---|---|---|---|---|---|---|
| Time CP Treatment | 0 h | 0 h | 16 h | 24 h | 16 h | 24 h |
| ROS production, % | 100 ± 4.5 | 100.1 ± 6.7 **,# | 236.5 ± 36.2 * | 206.8 ± 24.5 * | 147.9 ± 18.2 *,** | 152.3 ± 24.5 * |
| GSH/GSSG | 5.92 ± 0.35 | ND | 1.67 ± 0.19 * | 1.49 ± 0.16 * | 2.14 ± 0.13 * | 3.22 ± 0.44 *,# |
| Free thiols (nmol/mg protein) | 143.9 ± 2.9 | 138.1 ± 13.6 # | 115.1 ± 3.6 | 85.1 ± 4.6 * | 148.9 ± 27.1 | 204.9 ± 28.7 # |
| GR activity (U/mg protein) | 0.015 ± 0.001 | 0.014 ± 0.001 # | 0.010 ± 0.001 * | 0.006 ± 0.001 * | 0.011 ± 0.001 * | 0.014 ± 0.001 # |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Sierra, T.; Medina-Campos, O.N.; Solano, J.D.; Ibarra-Rubio, M.E.; Pedraza-Chaverri, J. Isoliquiritigenin Pretreatment Induces Endoplasmic Reticulum Stress-Mediated Hormesis and Attenuates Cisplatin-Induced Oxidative Stress and Damage in LLC-PK1 Cells. Molecules 2020, 25, 4442. https://doi.org/10.3390/molecules25194442
Gómez-Sierra T, Medina-Campos ON, Solano JD, Ibarra-Rubio ME, Pedraza-Chaverri J. Isoliquiritigenin Pretreatment Induces Endoplasmic Reticulum Stress-Mediated Hormesis and Attenuates Cisplatin-Induced Oxidative Stress and Damage in LLC-PK1 Cells. Molecules. 2020; 25(19):4442. https://doi.org/10.3390/molecules25194442
Chicago/Turabian StyleGómez-Sierra, Tania, Omar Noel Medina-Campos, José D. Solano, María Elena Ibarra-Rubio, and José Pedraza-Chaverri. 2020. "Isoliquiritigenin Pretreatment Induces Endoplasmic Reticulum Stress-Mediated Hormesis and Attenuates Cisplatin-Induced Oxidative Stress and Damage in LLC-PK1 Cells" Molecules 25, no. 19: 4442. https://doi.org/10.3390/molecules25194442
APA StyleGómez-Sierra, T., Medina-Campos, O. N., Solano, J. D., Ibarra-Rubio, M. E., & Pedraza-Chaverri, J. (2020). Isoliquiritigenin Pretreatment Induces Endoplasmic Reticulum Stress-Mediated Hormesis and Attenuates Cisplatin-Induced Oxidative Stress and Damage in LLC-PK1 Cells. Molecules, 25(19), 4442. https://doi.org/10.3390/molecules25194442