
Synthesis of Naphthoquinone Derivatives: Menaquinones, Lipoquinones and Other Vitamin K Derivatives



Abstract
:1. Introduction
1.1. Properties and Biological Function of Menaquinones
1.2. Synthetic Strategy for the Preparation of Menaquinones
2. Nucleophilic Ring Methods
2.1. Enolate Alkylation
2.2. Transmetalation
2.3. Friedel-Crafts Alkylation
2.4. Summary
3. Metal-Mediated and Radical Reactions
3.1. Cross-Coupling
3.2. Coordination Complex
3.3. Radical Reactions
3.3.1. Metal-Mediated Radical Reactions
3.3.2. Non-Metal-Mediated Radical Reactions
3.4. Summary
4. Electrophilic Ring Methods
4.1. 1,2-Addition versus 1,4-Addition
5. Pericyclic Reactions
5.1. Diels-Alder Reactions
5.2. Anionic Diels-Alder Reactions
5.3. [3,3] Sigmatropic Rearrangements-Cope
5.4. Summary
6. Homologation & Side Chain Extension Methods
6.1. Homologation
6.2. Side Chain Extensions
6.3. Summary
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kroppenstedt, R.M.; Mannheim, W. Lipoquinones in Members of the Family Pasteurellaceae. Int. J. Syst. Bacteriol. 1989, 39, 304–308. [Google Scholar] [CrossRef] [Green Version]
- Collins, M.D.; Jones, D. Distribution of Isoprenoid Quinone Structural Types in Bacteria and Their Taxonomic Implications. Microbiol. Rev. 1981, 45, 316–354. [Google Scholar] [CrossRef]
- Bergdoll, L.; Ten Brink, F.; Nitschke, W.; Picot, D.; Baymann, F. From low- to high-potential bioenergetic chains: Thermodynamic constraints of Q-cycle function. Biochim. Biophys. Acta 2016, 1857, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M.; Walker, C.; Epand, R.F.; Magarvey, N.A. Molecular mechanisms of membrane targeting antibiotics. Biochim. Biophys. Acta 2016, 1858, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Vilcheze, C.; Hartman, T.; Weinrick, B.; Jain, P.; Weisbrod, T.R.; Leung, L.W.; Freundlich, J.S.; Jacobs, W.R., Jr. Enhanced respiration prevents drug tolerance and drug resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2017, 114, 4495–4500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyay, A.; Fontes, F.L.; Gonzalez-Juarrero, M.; McNeil, M.R.; Crans, D.C.; Jackson, M.; Crick, D.C. Partial Saturation of Menaquinone in Mycobacterium tuberculosis: Function and Essentiality of a Novel Reductase, MenJ. ACS Cent. Sci. 2015, 1, 292–302. [Google Scholar] [CrossRef]
- Nakagawa, K.; Hirota, Y.; Sawada, N.; Yuge, N.; Watanabe, M.; Uchino, Y.; Okuda, N.; Shimomura, Y.; Suhara, Y.; Okano, T. Identification of UBIAD1 as a novel human menaquinone-4 biosynthetic enzyme. Nature 2010, 468, 117–121. [Google Scholar] [CrossRef]
- Mahdinia, E.; Demirci, A.; Berenjian, A. Production and application of menaquinone-7 (vitamin K2): A new perspective. World J. Microbiol. Biotechnol. 2017, 33, 1–7. [Google Scholar] [CrossRef]
- Tarento, T.D.C.; McClure, D.D.; Talbot, A.M.; Regtop, H.L.; Biffin, J.R.; Valtchev, P.; Dehghani, F.; Kavanagh, J.M. A potential biotechnological process for the sustainable production of vitamin K1. Crit. Rev. Biotechnol. 2019, 39, 1–19. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, D.; Ren, H. Economical production of vitamin K2 using crude glycerol from the by-product of biodiesel. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Dunphy, P.J.; Brodie, A.F. [233] The structure and function of quinones in respiratory metabolism. In Methods Enzymology; Elsevier: Amsterdam, The Netherlands, 1971; Volume 18, pp. 407–461. [Google Scholar]
- Koehn, J.T.; Magallanes, E.S.; Peters, B.J.; Beuning, C.N.; Haase, A.A.; Zhu, M.J.; Rithner, C.D.; Crick, D.C.; Crans, D.C. A Synthetic Isoprenoid Lipoquinone, Menaquinone-2, Adopts a Folded Conformation in Solution and at a Model Membrane Interface. J. Org. Chem. 2018, 83, 275–288. [Google Scholar] [CrossRef]
- Tetali, S.D. Terpenes and isoprenoids: A wealth of compounds for global use. Planta 2019, 249, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, V.F.; Ferreira, V.; Nicoletti, C.; Ferreira, P.; Futuro, D.; da Silva, F. Strategies for Increasing the Solubility and Bioavailability of Anticancer Compounds: β-Lapachone and Other Naphthoquinones. Curr. Pharm. Des. 2016, 22, 5899–5914. [Google Scholar] [CrossRef] [PubMed]
- Ingles, D.P.; Cruz Rodriguez, J.B.; Garcia, H. Supplemental Vitamins and Minerals for Cardiovascular Disease Prevention and Treatment. Curr. Cardiol. Rep. 2020, 22, 1–8. [Google Scholar] [CrossRef]
- Gengenbacher, M.; Kaufmann, S.H. Mycobacterium tuberculosis: Success through dormancy. FEMS Microbiol. Rev. 2012, 36, 514–532. [Google Scholar] [CrossRef] [Green Version]
- Bahuguna, A.; Rawat, D.S. An overview of new antitubercular drugs, drug candidates, and their targets. Med. Res. Rev. 2020, 40, 263–292. [Google Scholar] [CrossRef] [PubMed]
- Karamzad, N.; Maleki, V.; Carson-Chahhoud, K.; Azizi, S.; Sahebkar, A.; Gargari, B.P. A systematic review on the mechanisms of vitamin K effects on the complications of diabetes and pre-diabetes. Biofactors 2020, 46, 21–37. [Google Scholar] [CrossRef]
- Turner, M.E.; Adams, M.A.; Holden, R.M. The Vitamin K Metabolome in Chronic Kidney Disease. Nutrients 2018, 10, 1076. [Google Scholar] [CrossRef] [Green Version]
- Simes, D.C.; Viegas, C.S.B.; Araujo, N.; Marreiros, C. Vitamin K as a Diet Supplement with Impact in Human Health: Current Evidence in Age-Related Diseases. Nutrients 2020, 12, 138. [Google Scholar] [CrossRef] [Green Version]
- Schurgers, L.J.; Teunissen, K.J.; Hamulyak, K.; Knapen, M.H.; Vik, H.; Vermeer, C. Vitamin K-containing dietary supplements: Comparison of synthetic vitamin K1 and natto-derived menaquinone-7. Blood 2007, 109, 3279–3283. [Google Scholar] [CrossRef] [Green Version]
- Bentley, R.; Meganathan, R. Biosynthesis of Vitamin K (Menaquinone) in Bacteria. Microbiol. Rev. 1982, 46, 241–280. [Google Scholar] [CrossRef]
- Conly, J.; Stein, K. The production of menaquinones (vitamin K2) by intestinal bacteria and their role in maintaining coagulation homeostasis. Prog. Food Nutr. Sci. 1992, 16, 307–343. [Google Scholar] [PubMed]
- Kurosu, M.; Begari, E. Vitamin K2 in electron transport system: Are enzymes involved in vitamin K2 biosynthesis promising drug targets? Molecules 2010, 15, 1531–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cockayne, S.; Adamson, J.; Lanham-New, S.; Shearer, M.J.; Gilbody, S.; Torgerson, D.J. Vitamin K and the Prevention of Fractures. Arch. Intern. Med. 2006, 166, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Koehn, J.T.; Gonzalez-Juarrero, M.; Crans, D.C.; Crick, D.C. Mycobacterium tuberculosis survival in J774A.1 cells is dependent on MenJ moonlighting activity, not its enzymatic activity. ACS Infect. Dis. 2020. Accepted. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Siricilla, S.; Wan, B.; Crick, D.C.; Lenaerts, A.J.; Franzblau, S.G.; Kurosu, M. Discovery of selective menaquinone biosynthesis inhibitors against Mycobacterium tuberculosis. J. Med. Chem. 2012, 55, 3739–3755. [Google Scholar] [CrossRef]
- Abe, T.; Ozaki, S.; Ueda, D.; Sato, T. Insight into Isoprenoid Biosynthesis by Functional Analysis of Isoprenyl Diphosphate Synthases from Mycobacterium vanbaalenii and Mycobacterium tuberculosis. ChemBioChem 2020, 21, 1–9. [Google Scholar] [CrossRef]
- Prince, R.C.; Dutton, P.L.; Bruce, J.M. Menaquinones and plastoquinones in aprotic solvents. FEBS Lett. 1983, 160, 273–276. [Google Scholar] [CrossRef] [Green Version]
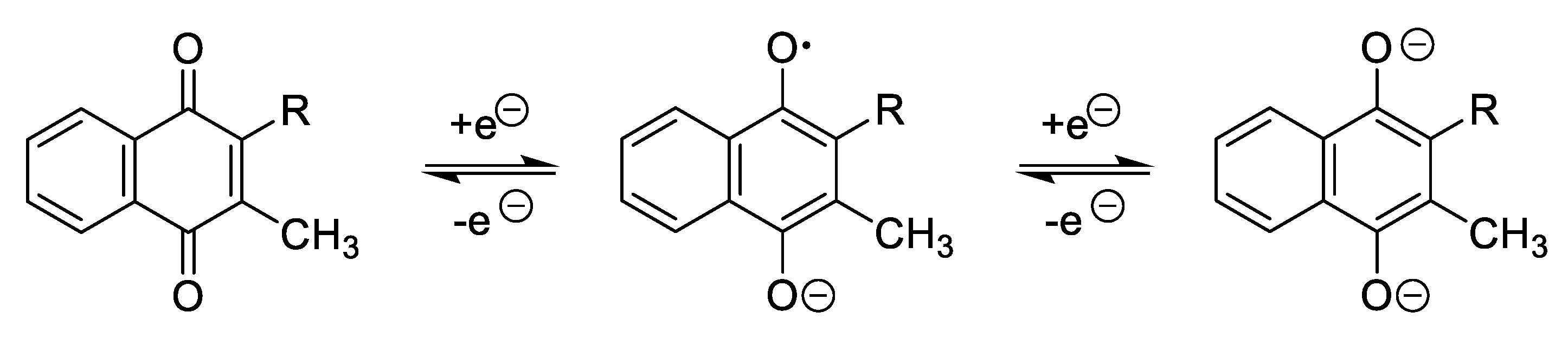
- Guin, P.S.; Das, S.; Mandal, P.C. Electrochemical Reduction of Quinones in Different Media: A Review. Int. J. Electrochem. 2011, 2011, 816202. [Google Scholar] [CrossRef] [Green Version]
- Dryhurst, G.; Kadish, K.M.; Scheller, F.; Renneberg, R. Biological Electrochemistry; Elsevier: Amsterdam, The Netherlands, 1982. [Google Scholar]
- Jaworski, J.S.; Leniewska, E.; Kalinowski, M.K. Solvent Effect on the Redox Potential of Quinone-Semiquinone Systems. J. Electroanal. Chem. Interfacial Electrochem 1979, 105, 329–334. [Google Scholar] [CrossRef]
- Gómez-Murcia, V.; Torrecillas, A.; de Godos, A.M.; Corbalán-García, S.; Gómez-Fernández, J.C. Both idebenone and idebenol are localized near the lipid–water interface of the membrane and increase its fluidity. Biochim. Biophys. Acta Biomembr. 2016, 1858, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Kaurola, P.; Sharma, V.; Vonk, A.; Vattulainen, I.; Rog, T. Distribution and dynamics of quinones in the lipid bilayer mimicking the inner membrane of mitochondria. Biochim. Biophys. Acta 2016, 1858, 2116–2122. [Google Scholar] [CrossRef] [PubMed]
- Quinn, P.J.; Esfahani, M.A. Ubiquinones have Surface-Active Properties Suited to Transport Electrons and Protons across Membranes. Biochem. J. 1980, 185, 715–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roche, Y.; Peretti, P.; Bernard, S. The Redox State Influences the Interactions of Ubiquinones with Phospholipid Bilayers: A DSC Study. J. Therm. Anal. Calorim. 2007, 89, 867–873. [Google Scholar] [CrossRef]
- Koehn, J.T.; Beuning, C.N.; Peters, B.J.; Dellinger, S.K.; Van Cleave, C.; Crick, D.C.; Crans, D.C. Investigating Substrate Analogues for Mycobacterial MenJ: Truncated and Partially Saturated Menaquinones. Biochemistry 2019, 58, 1596–1615. [Google Scholar] [CrossRef]
- Koehn, J.T.; Crick, D.C.; Crans, D.C. Synthesis and Characterization of Partially and Fully Saturated Menaquinone Derivatives. ACS Omega 2018, 3, 14889–14901. [Google Scholar] [CrossRef]
- Upadhyay, A.; Kumar, S.; Rooker, S.A.; Koehn, J.T.; Crans, D.C.; McNeil, M.R.; Lott, J.S.; Crick, D.C. Mycobacterial MenJ: An Oxidoreductase Involved in Menaquinone Biosynthesis. ACS Chem. Biol. 2018, 13, 2498–2507. [Google Scholar] [CrossRef]
- Ishihara, M.; Sakagami, H. QSAR of Molecular Structure and Cytotoxic Activity of Vitamin K2 Derivatives with Concept of Absolute Hardness. Anticancer Res. 2007, 27, 4059–4064. [Google Scholar]
- Siricilla, S.; Mitachi, K.; Skorupinska-Tudek, K.; Swiezewska, E.; Kurosu, M. Biosynthesis of a water-soluble lipid I analogue and a convenient assay for translocase I. Anal. Biochem. 2014, 461, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Daines, A.M.; Payne, R.J.; Humphries, M.E.; Abell, A.D. The Synthesis of Naturally Occurring Vitamin K and Vitamin K Analogues. Curr. Org. Chem. 2003, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Rüttimann, A. Recent Advances in the Synthesis of K-Vitamins. Chimia 1986, 40, 290–306. [Google Scholar]
- Snyder, C.D.; Rapoport, H. Synthesis of menaquinones. J. Am. Chem. Soc. 1974, 96, 8046–8054. [Google Scholar] [CrossRef] [PubMed]
- Tabushi, I.; Fujita, K.; Kawakubo, H. One-step preparation of vitamin K1 or K2 analogs by cyclodextrin inclusion catalysis. J. Am. Chem. Soc. 1977, 99, 6456–6457. [Google Scholar] [CrossRef] [PubMed]
- Tabushi, I.; Yamamura, K.; Fujita, K.; Kawakubo, H. Specific inclusion catalysis by beta.-cyclodextrin in the one-step preparation of vitamin K1 or K2 analogs. J. Am. Chem. Soc. 1979, 101, 1019–1026. [Google Scholar] [CrossRef]
- Garcias, X.; Ballester, P.; Capo, M.; Saá, J.M. 2.DELTA.-Stereocontrolled Entry to (E)- or (Z)-Prenyl Aromatics and Quinones. Synthesis of Menaquinone-4. J. Org. Chem. 1994, 59, 5093–5096. [Google Scholar] [CrossRef]
- Ballester, P.; Capo, M.; Garcias, X.; Saá, J.M. Synthesis of prenylated quinones by the oxidative degradation approach. Birch vs vinylogous Birch hydrogenolysis (BIHY vs VIBIHY) in controlling 2.DELTA. stereochemistry of the prenyl chain. J. Org. Chem. 1993, 58, 328–334. [Google Scholar] [CrossRef]
- Ballester, P.; Saá, J.; Capo, M. Prenylation of Aromatics. Lithium-Ammonia Reduction of 0-Silylated Tertiary Cinnamyl Alcohols. Tetrehedron Lett. 1990, 31, 1339–1342. [Google Scholar] [CrossRef]
- Raynolds, P.W.; Manning, M.J.; Swenton, J.S. Utilization of quinone bisacetal organocopper compounds in the synthesis of isoprenoid systems. ChemComm 1977, 14, 499–500. [Google Scholar] [CrossRef]
- Chenard, B.L.; Manning, M.J.; Raynolds, P.W.; Swenton, J.S. Organocopper chemistry of quinone bisketals. Application to the synthesis of isoprenoid quinone systems. J. Org. Chem. 1980, 45, 378–384. [Google Scholar] [CrossRef]
- Lindlar, H. Verfahren zur Herstellung von Kondensationsprodukten. U.S. Patent 320,582, 12 August 1953. [Google Scholar]
- Isler, O.; Doebel, K. Synthesen in der Vitamin-K-Reihe I. Über totalsynthetisches Vitamin K 1. Helv. Chim. Acta 1954, 37, 225–233. [Google Scholar] [CrossRef]
- Hirschmann, R.; Miller, R.; Wendler, N.L. The Synthesis of Vitamin K1. J. Am. Chem. Soc. 1954, 76, 4592–4594. [Google Scholar] [CrossRef]
- Fieser, L.F. Synthesis of Vitamin K 1. J. Am. Chem. Soc. 1939, 61, 3467–3475. [Google Scholar] [CrossRef]
- Fieser, L.F.; Campbell, W.P.; Fry, E.M. Synthesis of Quinones Related to Vitamins K 1 and K 2. J. Am. Chem. Soc. 1939, 61, 2206–2218. [Google Scholar] [CrossRef]
- Fieser, L.F.; Campbell, W.P.; Fry, E.M.; Gates, M.D. Synthetic Approach to Vitamin K1. J. Am. Chem. Soc. 1939, 61, 2559. [Google Scholar] [CrossRef]
- Almquist, H.J.; Klose, A.A. Synthetic and Natural Antihemorrhagic Compounds. J. Am. Chem. Soc. 1939, 61, 2557–2558. [Google Scholar] [CrossRef]
- Binkley, S.B.; Cheney, L.C.; Holcomb, W.F.; McKee, R.W.; Thayer, S.A.; MacCorquodale, D.W.; Doisy, E.A. The Constitution and Synthesis of Vitamin K1. J. Am. Chem. Soc. 1939, 61, 2558–2559. [Google Scholar] [CrossRef]
- Klose, A.A.; Almquist, H.J. Synthesis of Vitamink K1. J. Biol. Chem. 1940, 132, 469–471. [Google Scholar]
- Isler, O. Process for Producing Condensation Products of 1,4-Naphthohydroquinones. U.S. Patent 2325681, 3 August 1943. [Google Scholar]
- Tishler, M.; Fieser, L.F.; Wendler, N.L. Nature of the By-product in the Synthesis of Vitamin K 1. J. Am. Chem. Soc. 1940, 62, 1982–1991. [Google Scholar] [CrossRef]
- Schmid, R.; Antoulas, S.; Rüttimann, A.; Schmid, M.; Vecchi, M.; Weiserb, H. Synthesis of All Four Stereoisomers of (E)-Vitamin KT (Phylloquinone), Analysis of Their Diastereoisomeric and Enantiomeric Purities and Determination of Their Biopotencies. Helv. Chim. Acta 1990, 73, 1276–1299. [Google Scholar] [CrossRef]
- Yoshizawa, T.; Toyofuku, H.; Tachibana, K.; Kuroda, T. Regioselective Polyprenyl Rearrangement of Polyprenyl 2,3,4,5-Tetrasubstituted Phenyl Ethers Promoted by Boron Trifluoride. Chem. Lett. 1982, 11, 1131–1134. [Google Scholar] [CrossRef]
- Min, J.-H.; Lee, J.-S.; Yang, J.-D.; Koo, S. The Friedel−Crafts Allylation of a Prenyl Group Stabilized by a Sulfone Moiety: Expeditious Syntheses of Ubiquinones and Menaquinones. J. Org. Chem. 2003, 68, 7925–7927. [Google Scholar] [CrossRef] [PubMed]
- Coman, S.M.; Parvulescu, V.I.; Wuttke, S.; Kemnitz, E. Synthesis of Vitamin K 1 and K 1 -Chromanol by Friedel–Crafts Alkylation in Heterogeneous Catalysis. ChemCatChem 2010, 2, 92–97. [Google Scholar] [CrossRef]
- Wuttke, S.; Coman, S.M.; Scholz, G.; Kirmse, H.; Vimont, A.; Daturi, M.; Schroeder, S.L.M.; Kemnitz, E. Novel Sol-Gel Synthesis of Acidic MgF2−x(OH)x Materials. Chem. Eur. J. 2008, 14, 11488–11499. [Google Scholar] [CrossRef] [PubMed]
- Scholz, G.; Stosiek, C.; Feist, M.; Kemnitz, E. Magnesium Hydroxide Fluorides-New Materials with Adjustable Composition and Properties. Eur. J. Inorg. Chem. 2012, 2012, 2337–2340. [Google Scholar] [CrossRef]
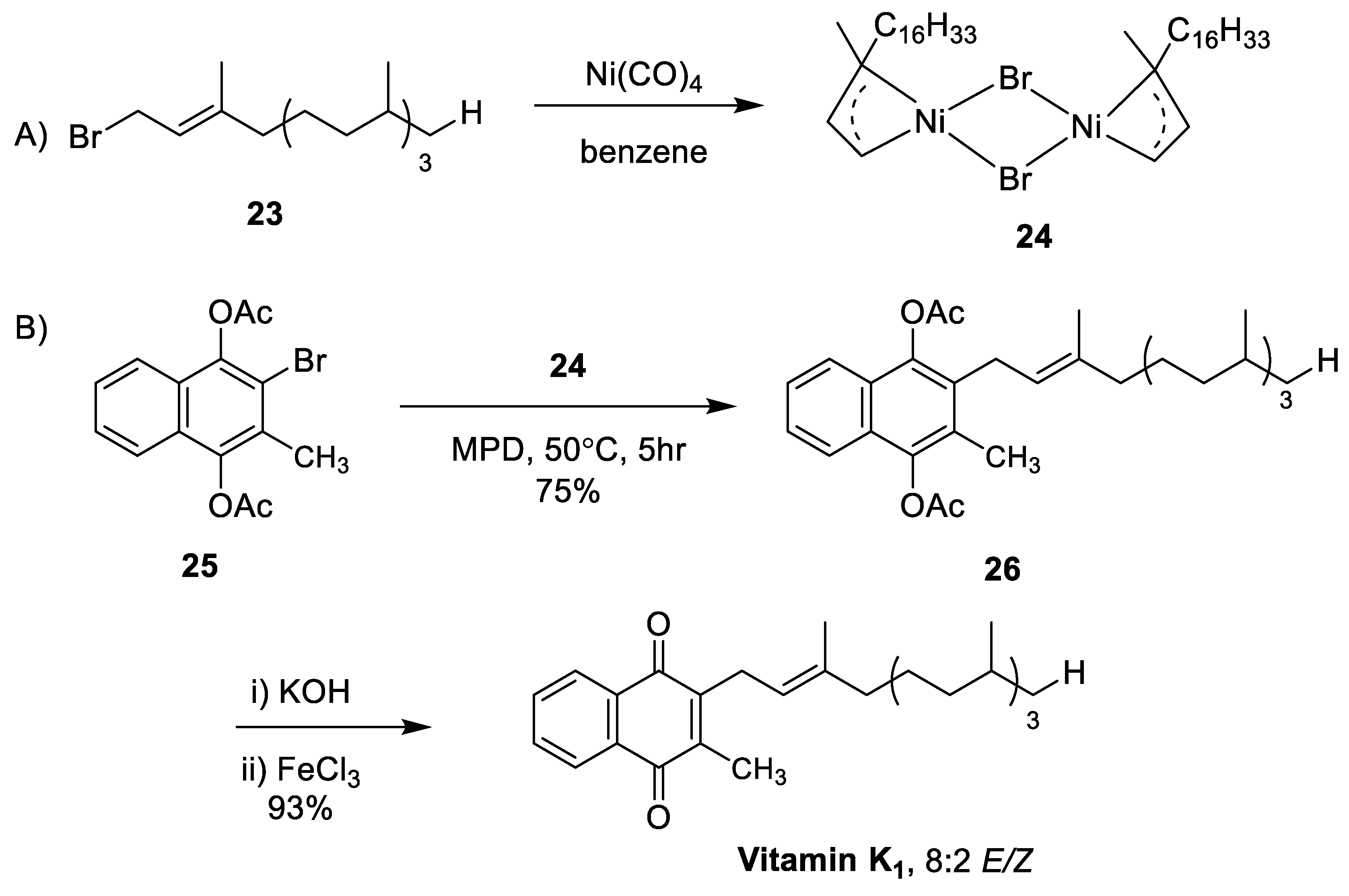
- Sato, K.; Inoue, S.; Saito, K. A New Synthesis of Vitamin K via π-Allylnickel Intermediates. J. Chem. Soc. Perkin Trans. I 1973, 2289–2293. [Google Scholar] [CrossRef]
- Godschalx, J.P.; Stille, J.K. Cross-coupling reaction of allyl bromides with organotin reagents catalyzed by zinc chloride. Tetrahedron Lett. 1983, 24, 1905–1908. [Google Scholar] [CrossRef]
- Liebeskind, L.S.; Foster, B.S. Stannylquinones. Synthesis and utilization as quinone carbanion synthetic equivalents. J. Am. Chem. Soc. 1990, 112, 8612–8613. [Google Scholar] [CrossRef]
- Dötz, K.H. Carbenkomplexe in der Organischen Synthese. Angew. Chem. 1984, 96, 573–594. [Google Scholar] [CrossRef]
- Dötz, K.H.; Pruskil, I.; Muhlemeier, I. Vitamin-Synthesen mit Carben-Komplexen, II. Carbonyl(carben)-Komplex-induzierte Synthese von Vitaminen der K1- und K2-Reihe. Chem. Ber. 1982, 115, 1278. [Google Scholar]
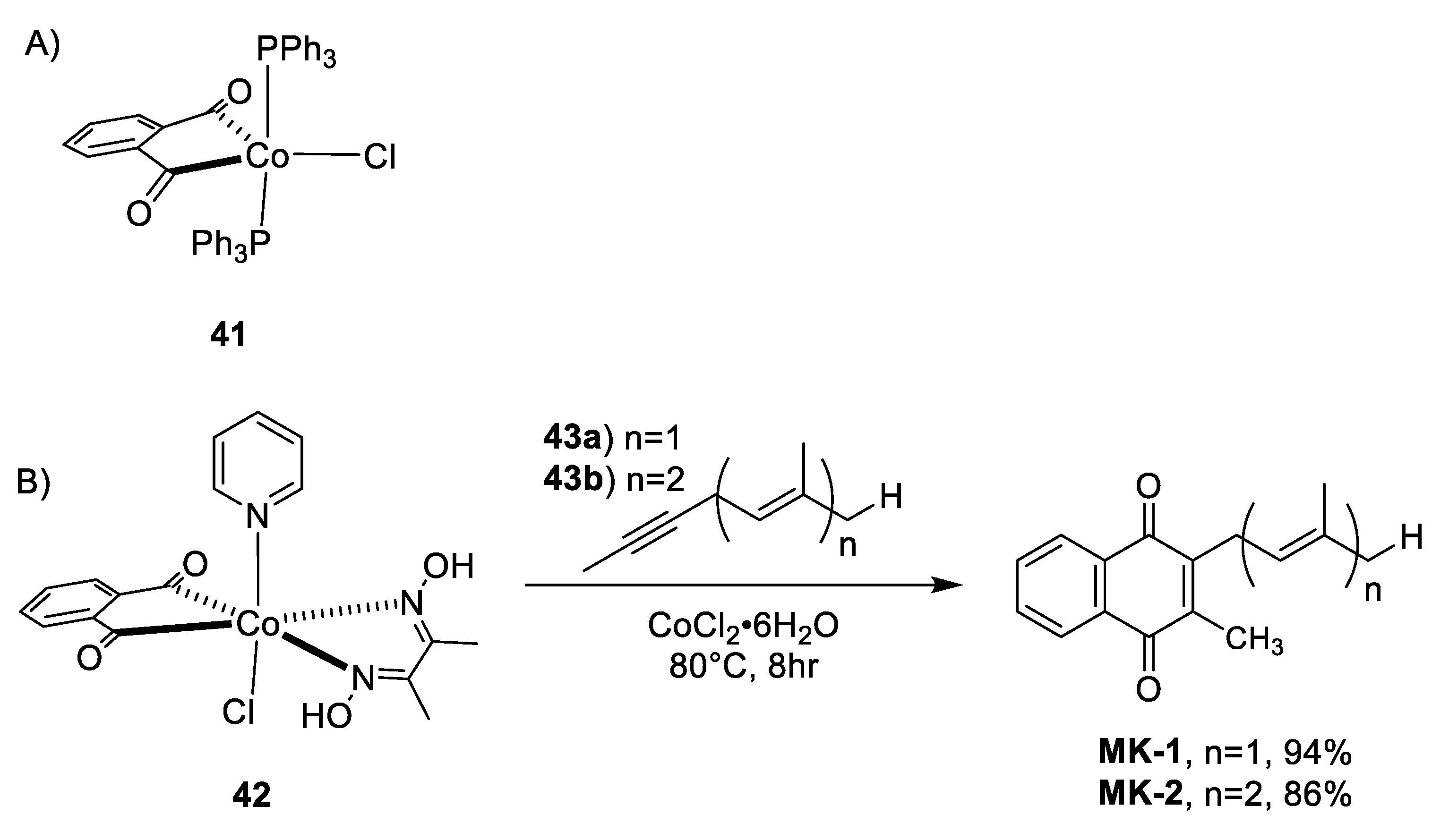
- Liebeskind, L.S.; Baysdon, S.L.; South, M.S. An organotransition-metal synthesis of naphthoquinones. J. Am. Chem. Soc. 1980, 102, 7397–7398. [Google Scholar] [CrossRef]
- Liebeskind, L.S.; Baysdon, S.L.; Goedken, V.; Chidambaram, R. Phthaloylcobalt complexes in synthesis. Ligand modifications leading to a practical naphthoquinone synthesis. Organometallics 1986, 5, 1086–1092. [Google Scholar] [CrossRef]
- Jacobsen, N.; Torssell, K. Radikalische Alkylierung von Chinonen: Erzeugung von Radikalen in Redoxreaktionen. Liebigs Ann. 1972, 763, 135–147. [Google Scholar] [CrossRef]
- Jacobsen, N.; Torssell, K. Synthesis of Naturally Occuring Quinones: Alkylation with the Silver Ion-Peroxydisulphite-Carboxylic Acid System. Acta Chem. Scand. 1973, 27, 3211–3216. [Google Scholar] [CrossRef] [Green Version]
- Yamago, S.; Hashidume, M.; Yoshida, J.-I. Radical-Mediated Synthesis of Substituted Quinones with Organotellurium Compounds. Chem. Lett. 2000, 29, 1234–1235. [Google Scholar] [CrossRef]
- Yamago, S.; Hashidume, M.; Yoshida, J.-I. A new synthetic route to substituted quinones by radical-mediated coupling of organotellurium compounds with quinones. Tetrahedron 2002, 58, 6805–6813. [Google Scholar] [CrossRef]
- Coppa, F.; Fontana, F.; Minisci, F.; Nogueira, M.C.; Vismara, E. Homolytic Alkylation of Naphthoquinone and Methyl-Naphthoquinone. Enthalpic, Steric, and Polar Effects. Tetrahedron 1991, 47, 7347–7352. [Google Scholar] [CrossRef]
- Naruta, Y. Regio- and stereoselective synthesis of coenzymes Qn (n = 2–10), vitamin K, and related polyprenylquinones. J. Org. Chem. 1980, 45, 4097–4104. [Google Scholar] [CrossRef]
- Naruta, Y. Allylation of quinones with allyltin reagents. J. Am. Chem. Soc. 1980, 102, 3774–3783. [Google Scholar] [CrossRef]
- Maruyama, K.; Naruta, Y. Synthesis of naturally occurring quinones. Part 3. Allylation of quinones with allyltin reagents. New synthesis of coenzyme Q1 and plastoquinone-1. J. Org. Chem. 1978, 43, 3796–3798. [Google Scholar] [CrossRef]
- Araki, S.; Katsumura, N.; Butsugan, Y. Allylation of quinones by allylic indium reagents. J. Organomet. Chem. 1991, 415, 7–24. [Google Scholar] [CrossRef]
- Evans, D.A.; Hoffman, J.M. Regiospecific quinone isoprenylation. Examples of remarkably facile [3,3] sigmatropic processes. J. Am. Chem. Soc. 1976, 98, 1983–1984. [Google Scholar] [CrossRef]
- Troll, T.; Schmid, K. Darstellung und reaktionen von 1,3-bis-(trimethylsiloxy)-isobenzofuranen. Tetrahedron Lett. 1984, 25, 2981–2984. [Google Scholar] [CrossRef]
- Büchi, G.H.; Rüttimann, A. Process for the Manufacture of Quinone Derivatives. U.S. Patent 4,603,223, 29 July 1986. [Google Scholar]
- Tso, H.-H.; Chen, Y.-J. A convenient one-flask synthesis of vitamin K. J. Chem. Res. Synop. 1995, 3, 104–105. [Google Scholar]
- Mal, D.; Ghosh, K.; Jana, S. Synthesis of Vitamin K and Related Naphthoquinones via Demethoxycarbonylative Annulations and a Retro-Wittig Rearrangement. Org. Lett. 2015, 17, 5800–5803. [Google Scholar] [CrossRef] [PubMed]
- Lipshutz, B.H.; Kim, S.-K.; Mollard, P. An Expeditious Route to COQn, Vitamins K1 and K2, and Related Allylated para-Quinones Utilizing Ni(0) Catalysis. Tetrahedron 1998, 54, 1241–1253. [Google Scholar] [CrossRef]
- Van Horn, D.E.; Negishi, E. Selective carbon-carbon bond formation via transition metal catalysts. 8. Controlled carbometalation. Reaction of acetylenes with organoalane-zirconocene dichloride complexes as a route to stereo- and regio-defined trisubstituted olefins. J. Am. Chem. Soc. 1978, 100, 2252–2254. [Google Scholar] [CrossRef]
- Negishi, E.; Van Horn, D.E.; Yoshida, T. Controlled carbometalation. 20. Carbometalation reaction of alkynes with organoalene-zirconocene derivatives as a route to stereo-and regiodefined trisubstituted alkenes. J. Am. Chem. Soc. 1985, 107, 6639–6647. [Google Scholar] [CrossRef]
- Matsushita, H.; Negishi, E. Palladium-catalyzed Synthesis of 1,4-Diene by Allylation of Alkenylalanes: α-Farnesene. Org. Synth. 1984, 62, 31–37. [Google Scholar]
- Negishi, E. Bimetallic catalytic systems containing Ti, Zr, Ni, and Pd. Their applications to selective organic syntheses. Pure Appl. Chem. 1981, 53, 2333–2356. [Google Scholar] [CrossRef]
- Mehta, D.S.; Mohan, P.; Shastri, M.; Reid, T. Method of Preparation of Stereospecific Quinone Derivatives. U.S. Patent 9,464,021, 11 October 2016. [Google Scholar]
- Masaki, Y.; Hasimoto, K.; Sakuma, K.; Kaji, K. Synthetic Studies on Isoprenoidquinones. I. A Facile Regio- and Stereocontrolled Synthesis. Chem. Pharm. Bull. 1984, 32, 3952–3958. [Google Scholar] [CrossRef] [Green Version]
- Masaki, Y.; Hasimoto, K.; Kaji, K. Synthetic Studies of Isoprenoidquinones II. Syntheses of Ubiquinone-10, Phylloquinone, and Menaquinone-4 by a Chain-Extending Method Utilizing Terminally Functionalized Isoprenoidhydroquinones. Chem. Pharm. Bull. 1984, 32, 3959–3967. [Google Scholar] [CrossRef] [Green Version]
- Chadar, D.; Camilles, M.; Patil, R.; Khan, A.; Weyhermüller, T.; Salunke-Gawali, S. Synthesis and characterization of n-alkylamino derivatives of vitamin K3: Molecular structure of 2-propylamino-3-methyl-1,4-naphthoquinone and antibacterial activities. J. Mol. Struct. 2015, 1086, 179–189. [Google Scholar] [CrossRef]
- Kathawate, L.; Joshi, P.V.; Dash, T.K.; Pal, S.; Nikalje, M.; Weyhermüller, T.; Puranik, V.G.; Konkimalla, V.B.; Salunke-Gawali, S. Reaction between lawsone and aminophenol derivatives: Synthesis, characterization, molecular structures and antiproliferative activity. J. Mol. Struct. 2014, 1075, 397–405. [Google Scholar] [CrossRef]
- Zacconi, F.C.; Nuñez, O.N.; Cabrera, A.L.; Valenzuela, L.M.; del Valle, J.M.; de la Fuente, J.C. Synthesis and solubility measureme11nt in supercritical carbon dioxide of two solid derivatives of 2-methylnaphthalene-1,4-dione (menadione): 2-(Benzylamino)-3-methylnaphthalene-1,4-dione and 3-(phenethylamino)-2-methylnaphthalene-1,4-dione. J. Chem. Thermodyn. 2016, 103, 325–332. [Google Scholar] [CrossRef]






































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acid Catalyst | % Yield 1 |
|---|---|
| KHSO4 | 55% |
| Oxalic Acid | N/A 2 |
| Duolite C-60 | 8% |
| BF3∙OEt2 | 66.5% |
| Lewis Acid 1 | % of 20 (E/Z) |
|---|---|
| BF3∙OEt2 | 0 (-) |
| MgBr2 | 0 (-) |
| TiCl4 | - 2(-) |
| FeCl3 | 55 (4:1) |
| Et2AlCl | 56 (7:1) |
| SnCl4 | 56 (E) |
| ZnBr2 | 60 (7:1) |
| ZnCl2 | 67 (7:1) |
| AlCl3 | 72 (E) |
| Catalyst | % of Vitamin K1 | % of K1 Chromanol | % of C2 Product |
|---|---|---|---|
| MgF2-40 | 21.2 | 5.9 | 58.8 |
| MgF2-57 | 26.5 | 21.0 | 43.7 |
| MgF2-71 | 15.6 | 20.8 | 52.9 |
| MgF2-87 1 | 0 | 0 | 0 |
| AlF2-50 | 7.6 | 42 | 41.2 |
| Methods | Advantages | Disadvantages |
|---|---|---|
| Section 2.1. Enolate Alkylations | ||
| Snyder and Rapoport Enolate Alkylation [44] | -Stereoretention of α-isoprene double bond (97% E-alkene) -3 step synthesis (not including starting material) | -Low yields (20–45%) -C2 alkylation competition via Friedel-Crafts alkylation -Unviable synthesis of starting material |
| Tabushi et al. β-cyclodextrin inclusion catalyst [45,46] | -Regiocontrol via sterically hindered nature of β-cyclodextrin -Menadione is the only byproduct -1 step synthesis | -Low yields (40% with inclusion catalyst) -Competition between C3 alkylation and C3 protonation -Only synthesized MK-1 |
| Section 2.2. Transmetalations | ||
| Snyder and Rapoport Grignard reaction [44] | -Regiocontrol through lithium-bromide exchange -Stereoretention of the α-isoprene double bond (≥95%) -Alkylation step is high yielding (>95%) -3 step synthesis (not including starting material) | -Need to prepare starting material 6 |
| Saá and coworkers BIHY Reduction [47,48,49] | -Stereoretention of the α-isoprene double bond during BIHY reduction | -Moderate yields for nucleophilic addition (58–65%) and BIHY reduction (53–70%) -5 step synthesis (not including starting material) |
| Swenton and coworkers Electrolysis & Lithium Organocuprate [50,51] | -Unique use of electrolysis as a protection method -High yields for all reported steps (≥85%) -Regiocontrol through lithium bromide exchange -Stereoretention of α-isoprene double bond (<5% Z-alkene estimated) -Deprotection of bisketals to menaquinone ring structure via hydrolysis, no oxidation required | -Lithium organocuprate nucleophile only used one of two bisketal rings—poor atom economy -Difficult purification because of unreacted starting materials -5 step synthesis (not including starting material) |
| Section 2.3. Friedel-Crafts Alkylation | ||
| Hirschmann et al. Friedel-Crafts Alkylation Lewis Acid Analysis [54] | -Favors C3 alkylation over C2 due to monoacetate 14 -Avoided formation of undesired byproducts (phytadiene and chromanol) -Monoacetate 14 was the only recoverable byproduct -2 step synthesis (not including starting material) | -Low to moderate yields (8–66.5%) depending on acid catalyst used (Table 1) -Stereoretention of the α-isoprene double bond was not discussed |
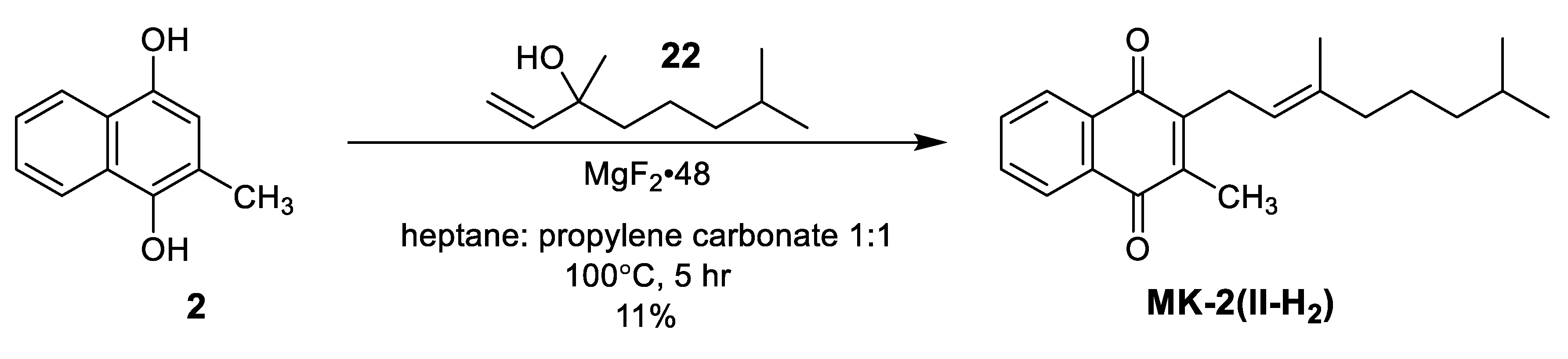
| Schmid et al. Intramolecular Friedel-Crafts [63] | -Features unique intramolecular Friedel-Crafts alkylation at C3 position -High yields (76–96.5%) throughout all steps -Stereoretention of α-isoprene double bond -3 step synthesis (not including starting material) | -Need to prepare starting material 22 |
| Min et al. Friedel-Crafts Alkylation Lewis Acid Analysis [65] | -Stereoretention of α-isoprene double bond with AlCl3 -Produced a functional handle for chain extension methods -1 step synthesis (not including starting material) | -Low to moderate yields (0–72%) depending on Lewis acid used |
| Coman et al. [66] and Koehn et al. [38] Heterogenous Lewis Acid Catalysts | -Predicted industrial benefit to replace BF3∙OEt2 -Performed without protecting groups, but could benefit from them -1 step synthesis (not including starting material) | -Universally low yields (0–26.5%) -Poor regiocontrol to prevent C2 alkylation -Difficult purification -Synthesis of partly hydroxylated metal fluorides requires the use of dangerous aqueous HF [67,68] |

| Additive (1 Equiv) | GC Yield % of 44 | ||
|---|---|---|---|
| 2 h | 5 h | 18 h | |
| None | 25 | 52 | 77 |
| AgBF4 | 80 | 82 | - |
| BF3∙OEt2 | 74 | 79 | 82 |
| SnCl2 | 41 | 39 | 70 |
| CoCl2∙6H2O | 59 | 83 | 91 |
| CoCl2(anhyd) | 61 | 86 | 86 |
| p-CH3PhSO3H | 14 | 31 | 76 |
| CH3CO2H | 23 | 47 | 74 |
| Methods | Advantages | Disadvantages |
|---|---|---|
| Section 3.1. Cross-Coupling | ||
| Sato et al. π-Allylnickel Cross Coupling [69] | -No coordination complex synthesis required-π-allyl complex is formed in situ -Moderate to high yields (52–93%) across the syntheses -E/Z ratio of the α-isoprene double bond (7:3 E/Z for MK-9) -3 step synthesis (not including starting material) | -The yields drop at the cross-coupling, especially for the much longer prenyl side chains, MK-9 (52%) -Authors note E/Z ratio is tunable depending on the solvent, but the yields drop as a result |
| Stille et al. Aryl Stannane Cross Coupling [70] | -High yields (77%) for the formation of the arylstannane -The regiochemistry of the system is controlled by transmetalation at C3 position -Allylic transposition was not observed in analogous syntheses of myrcene [70] | -Low yield for cross-coupling (40% over two steps) -Requires the use of t-BuLi -5 step synthesis (not including starting material) |
| Section 3.2. Coordination Complex | ||
| Liebeskind and Foster Ring Expansion to Stille Coupling [71] | -Stille coupling achieved high yields (90%) -3 step synthesis (not including starting material) | Low yield for key Liebeskind-Moore rearrangement (49% over two steps) -Only synthesized MK-1 |
| Dötz et al. Chromium Complex Carbonylation [72,73] | -No coordination complex synthesis required -E/Z ratio of the α-isoprene double bonds was retained throughout the synthesis -The regiochemistry of the system is controlled by the alkynes 37a and 37b -Cr(CO)6 is recyclable | -Known adverse health effects related to hexavalent chromium -No yields reported in Rüttimann’s 1986 review [43] -5 step synthesis (not including starting material) |
| Liebeskind et al. Cobalt Complex Cycloaddition [75] | -High yields (>86%) -Simple coordination complex synthesis required using commercially available materials -The regiochemistry of the system is controlled by the alkynes 43a and 43b -1 step synthesis (not including catalyst) | -The authors did not address α-isoprene double bonds isomerization |
| Section 3.3. Radical Reactions | ||
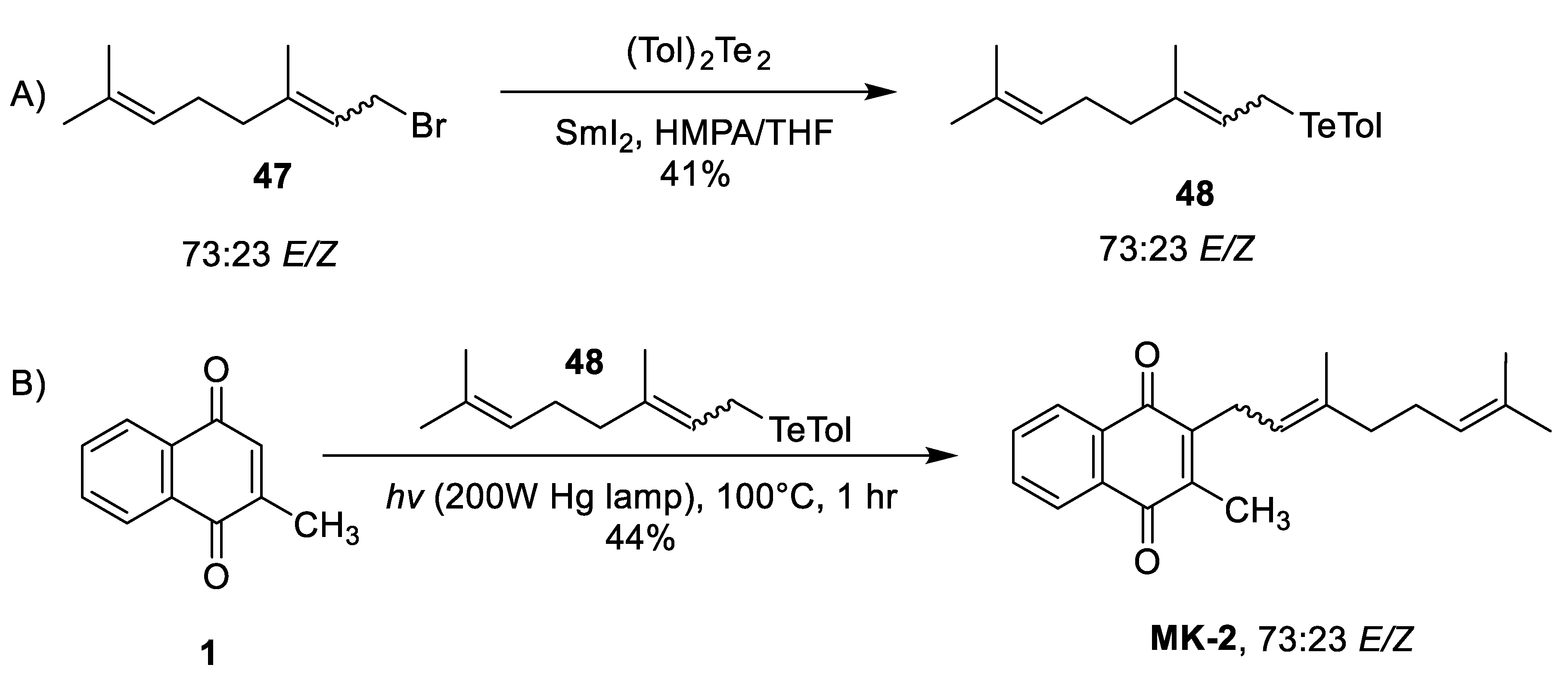
| Jacobsen & Torssell Radical Decarboxylation [77] | -Moderate yields (70%) -Regiocontrolled through aryl hydrogen abstraction -Selective for γ,γ-alkene product of MK-1 -1 step synthesis | -Only synthesized MK-1 |
| Yamago et al. Radical Organotelluride [78,79] | -Regiocontrolled through aryl hydrogen abstraction -Stereoretention of the α-isoprene double bond across all steps -2 step synthesis | -Low yields for both formation of tolyltelluride and radical coupling (~40%) -Known adverse health effects related to working with tellurium and tellurium compounds |
| Coppa et al. [80] & Koehn et al. [38] Benzoyl Peroxide Initiated Radical Alkylation | -Moderate to high yields of straight chain alkyl iodides (68–93%) [80] -1 step synthesis (not including starting material) | -Koehn et al. reported very low yields (17%) for this transformation with a branched alkane -Substantial α-isoprene double bond isomerism -Competing reactions interfere with C3-alkylated product (C3-C3, and C2 alkylation) |
| Methods | Advantages | Disadvantages |
|---|---|---|
| Section 4.1. 1,2-Addition vs. 1,4-Addition | ||
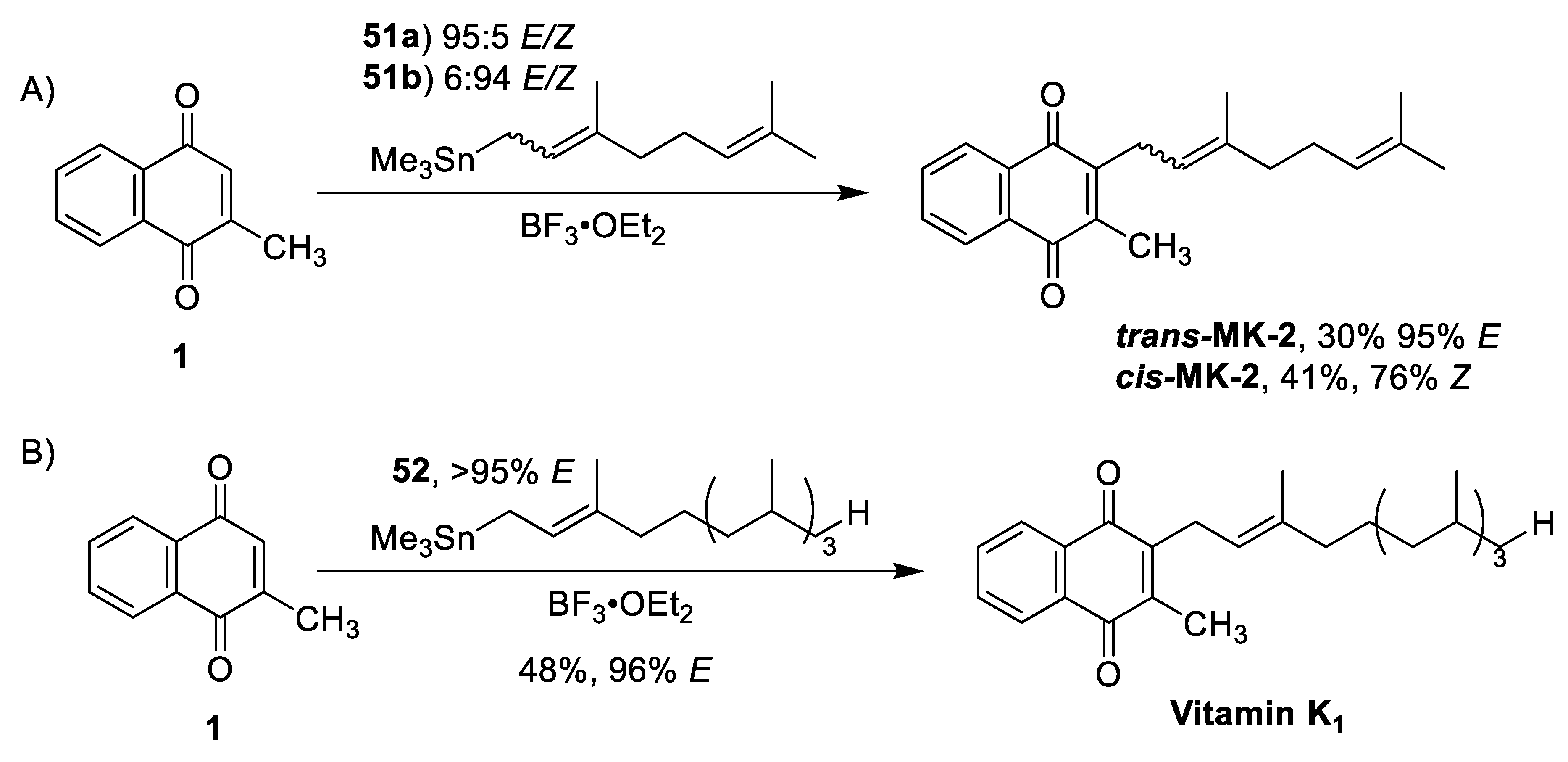
| Naruta and Maruyuma. Organostannane Michael Addition [81,82,83] | -Stereoretention of the α-isoprene double bond -1 step synthesis (not including starting materials) | -Low yields for both formations (30–48%) -Prominent competition between C2 and C3 alkylation |
| Methods | Advantages | Disadvantages |
|---|---|---|
| Section 5.1. Diels-Alder | ||
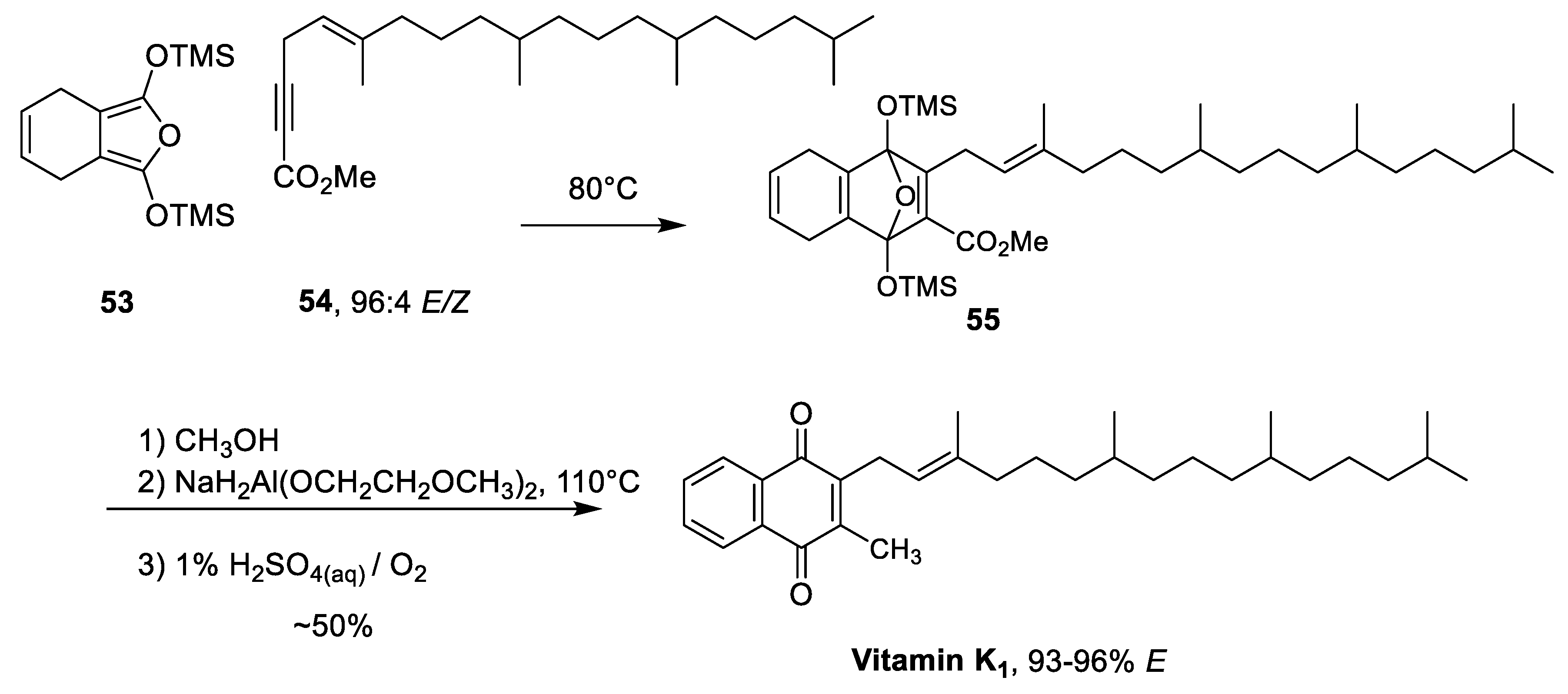
| Rüttimann et al. Diels-Alder Reaction inspired by Troll & Schmid [43] | -High regiocontrol through the symmetry of dihydroisobenzofurane diene -Stereoretention of α-isoprene double bond (≥ 93%) | -Overall low yields (~50% over four steps) -Synthesis of starting materials -4 step synthesis (not including starting material) |
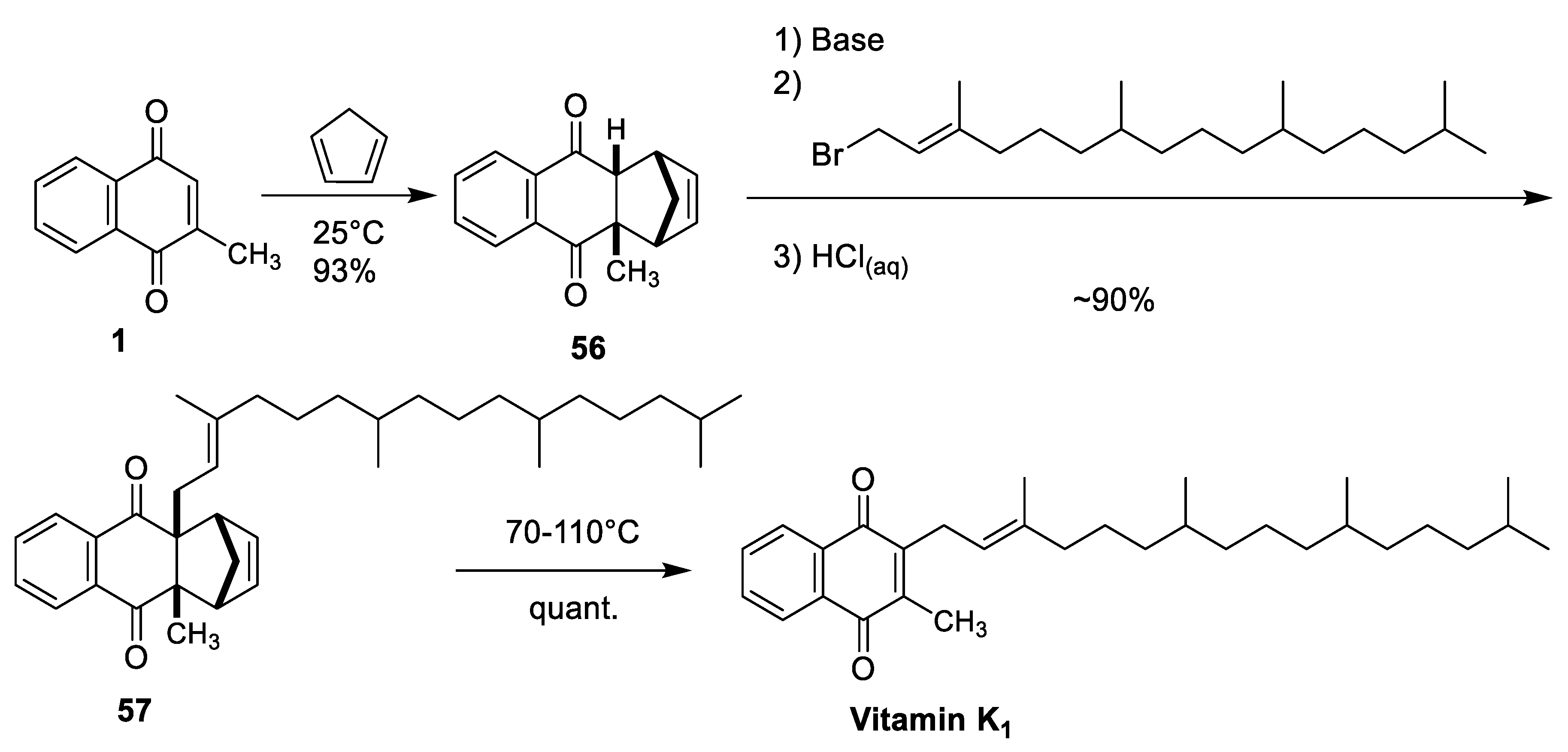
| Rüttimann et al. Auxiliary-Directed Diels-Alder [43] | -Uses commercially available starting materials (menadione and cyclopentadiene) -High regiocontrol through adduct 66 -Stereoretention of α-isoprene double bond -Cyclopentadiene can be recycled -High yields throughout the synthesis (≥90%) | -Slight competition between C-alkylation and O-alkylation -5 step synthesis |
| Section 5.2. Anionic Diels-Alder | ||
| Tso and Chen Anionic Diels-Alder [88] | -One-pot synthesis -High regiocontrol through asymmetry of dienophile -Stereoretention of α-isoprene double bond across all steps (>98%) -3 step synthesis (not including starting material) | -Moderate yields (60–64%) -Requires the synthesis of starting materials |
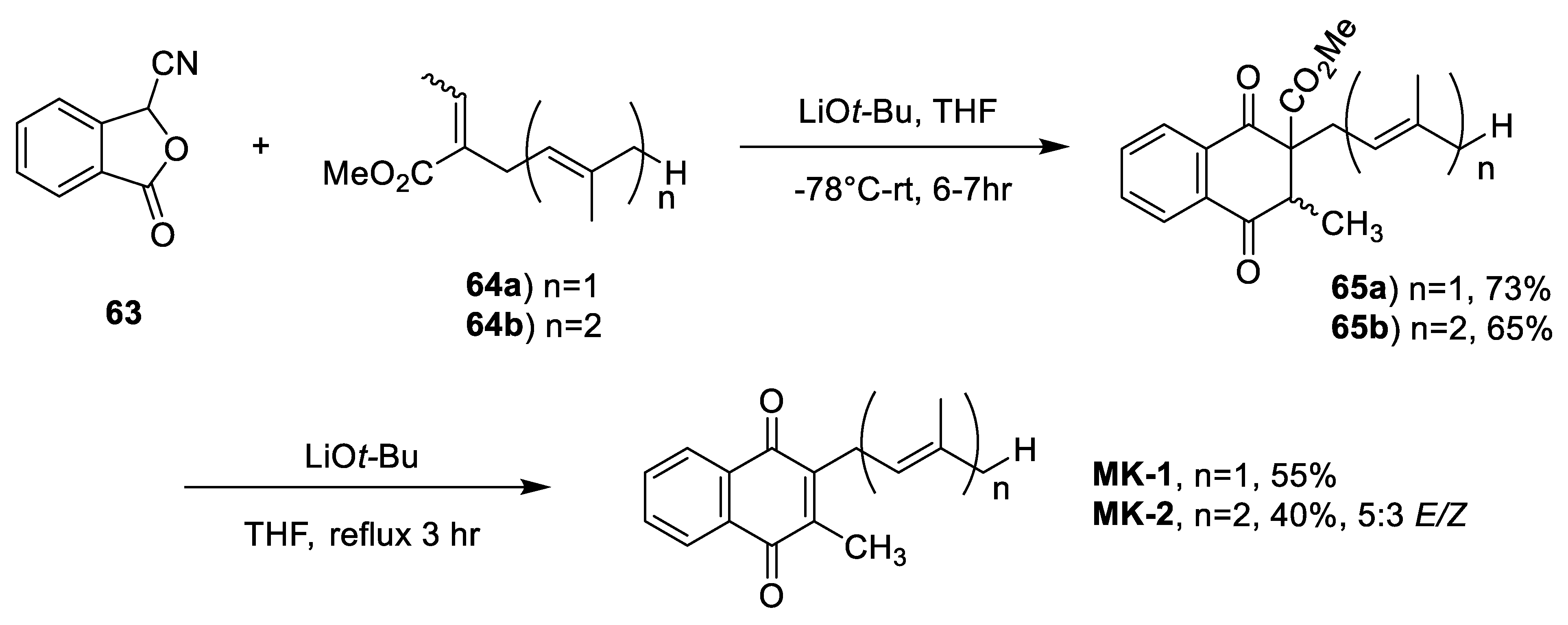
| Mal et al. Anionic Diels-Alder with Improved Atom Economy [89] | -Improved atom economy -High regiocontrol through asymmetry of dienophile -2 step synthesis (not including starting material) | -Low to moderate yields (40–73%) -5:3 E/Z ratio -Unclear if it is due to stereochemistry of starting material or caused by the reaction |
| Section 5.3. [3,3] Sigmatropic Rearrangements- Cope | ||
| Evans and Hoffmann Grignard-Promoted Cope Rearrangement [85] | -Regiocontrol achieved through protected naphthoquinone -Cope rearrangement to achieve C3 alkylation -Moderate yields (71% over two steps) -2 step synthesis (not including starting material) | -No consideration of the isomerization of the isoprene double bond |
| Araki et al. Organoindium-Promoted Cope Rearrangement [84] | -Regiocontrol achieved through less hindered 1,2-addition of organoindium reagent -No protecting groups required -Cope rearrangement to achieve C3 alkylation -Moderate yields (67% over two steps) -2 step synthesis (not including starting material) | No stereoretention observed in Cope rearrangement |
| Methods | Advantages | Disadvantages |
|---|---|---|
| Section 6.1. Homologation | ||
| Lipshutz et al. Homologation to Negishi Cross-Coupling [90] | -High yields throughout the synthesis (87–93%) -Method is applicable to a wide scope of benzo- and naphthoquinones -Stereochemistry of the α-isoprene double bond is defined by the configuration of the organoalane -Regiocontrolled by the installation of the chloromethyl group at the C3 position -No extraneous coordination complex synthesis required -3 step synthesis (including starting material) | -Requires the use of hydrogen chloride gas |
| Mehta et al. Stereoselective Alkene Syntheses [95] | -High yields throughout the synthesis for all reported steps (80–95%) -Strict use of stereoselective alkene syntheses -Methodology is applicable to full side chain extensions and smaller segments | -Requires the use of protecting groups and oxidation manipulations -11 step synthesis (not including starting material) |
| Section 6.2. Side Chain Extension | ||
| Masaki et al. Tosylate Substitution [96,97] | -Moderate to high yields throughout the synthesis (68–90%) Stereoretention of the α-isoprene double bond with minor isomerization (5–7%) Methodology is applicable to full side chain extensions and smaller segments | -4 step synthesis (not including starting material) |
| Schmid et al. Organocuprate Substitution [63] | -Achieved regio- and stereocontrol using isoprene oxide in a 1,4-addition -Stereoretention of the α-isoprene double bond (97:3) -Iterative methodology | -Low to moderate yields (51–79%) for alkylation step -4 step synthesis (not including starting material) |
| Strategy- | Advantages | Disadvantages |
|---|---|---|
| Nucleophilic Ring2.2. Transmetalation Swenton and coworkers Electrolysis and Lithium Organocuprate [50,51] | -Unique use of electrolysis as a protection method -High yields for all reported steps (≥85%) -Regiocontrol through lithium bromide exchange -Stereoretention of α-isoprene double bond (<5% Z alkene estimated) -Deprotection of bisketals to menaquinone ring structure via hydrolysis, no oxidation required | -Lithium organocuprate nucleophile only used one of two bisketal rings—poor atom economy -Difficult purification because of unreacted starting materials -5 step synthesis (not including starting material) |
| Metal-Mediated 3.2. Coordination Complex Liebeskind et al. Cobalt Complex Cycloaddition [75] | -High yields (>86%) -Simple coordination complex synthesis required using commercially available materials -The regiochemistry of the system is controlled by the alkynes 43a and 43b -1 step synthesis (not including catalyst) | -The authors did not address α-isoprene double bonds isomerization |
| Electrophilic Ring 4.1. 1,2- vs. 1,4-Addition Naruta and Maruyuma Organostannane Michael Addition [81,83] | -Stereoretention of the α-isoprene double bond | -Low yields for both formations (30–48%) -Prominent competition between C2 and C3 alkylation |
| Pericyclic 5.1. Diels-Adler Rüttimann et al. Auxiliary-Directed Diels-Alder [43] | -Uses commercially available starting materials (menadione and cyclopentadiene) -High regiocontrol through adduct 66 -Stereoretention of α-isoprene double bond -Cyclopentadiene can be recycled -High yields throughout the synthesis (≥90%) | -Slight competition between C-alkylation and O-alkylation |
| Homologation & Side Chain Extensions 6.1. Homologation Lipshutz et al. Homologation to Negishi Cross-Coupling [90] | -High yields throughout the synthesis (87–93%) -Method is applicable to a wide scope of benzo- and naphthoquinones -Stereochemistry of the α-isoprene double bond is defined by the configuration of the organoalane -Regiocontrolled by the installation of the chloromethyl group at the C3 position -No extraneous coordination complex synthesis required | -Requires the use of hydrogen chloride gas |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braasch-Turi, M.; Crans, D.C. Synthesis of Naphthoquinone Derivatives: Menaquinones, Lipoquinones and Other Vitamin K Derivatives. Molecules 2020, 25, 4477. https://doi.org/10.3390/molecules25194477
Braasch-Turi M, Crans DC. Synthesis of Naphthoquinone Derivatives: Menaquinones, Lipoquinones and Other Vitamin K Derivatives. Molecules. 2020; 25(19):4477. https://doi.org/10.3390/molecules25194477
Chicago/Turabian StyleBraasch-Turi, Margaret, and Debbie C. Crans. 2020. "Synthesis of Naphthoquinone Derivatives: Menaquinones, Lipoquinones and Other Vitamin K Derivatives" Molecules 25, no. 19: 4477. https://doi.org/10.3390/molecules25194477
APA StyleBraasch-Turi, M., & Crans, D. C. (2020). Synthesis of Naphthoquinone Derivatives: Menaquinones, Lipoquinones and Other Vitamin K Derivatives. Molecules, 25(19), 4477. https://doi.org/10.3390/molecules25194477