
Stacking Interactions of Poly Para-Phenylene Vinylene Oligomers with Graphene and Single-Walled Carbon Nanotubes: A Molecular Dynamics Approach


 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Simulation Method
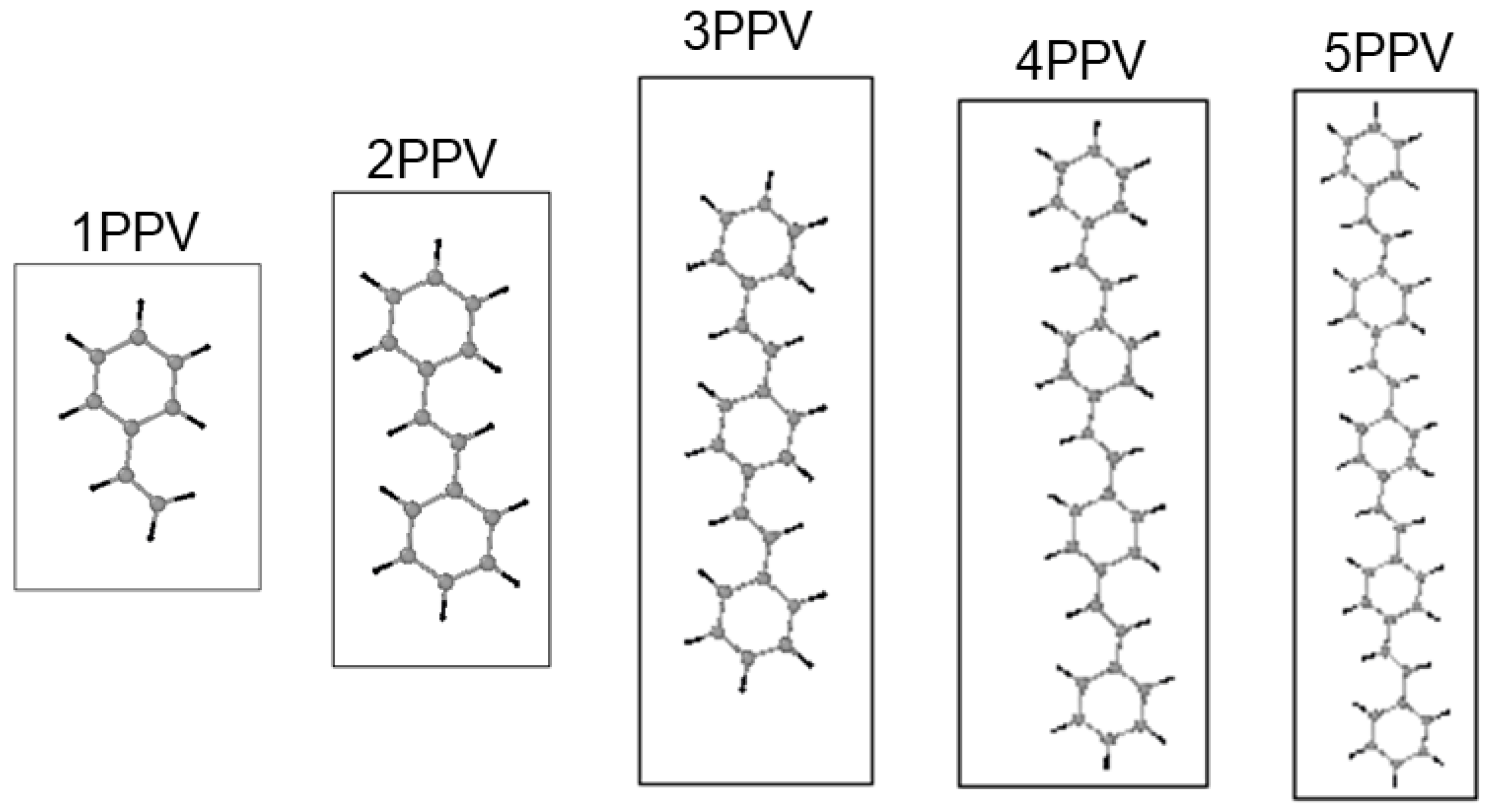
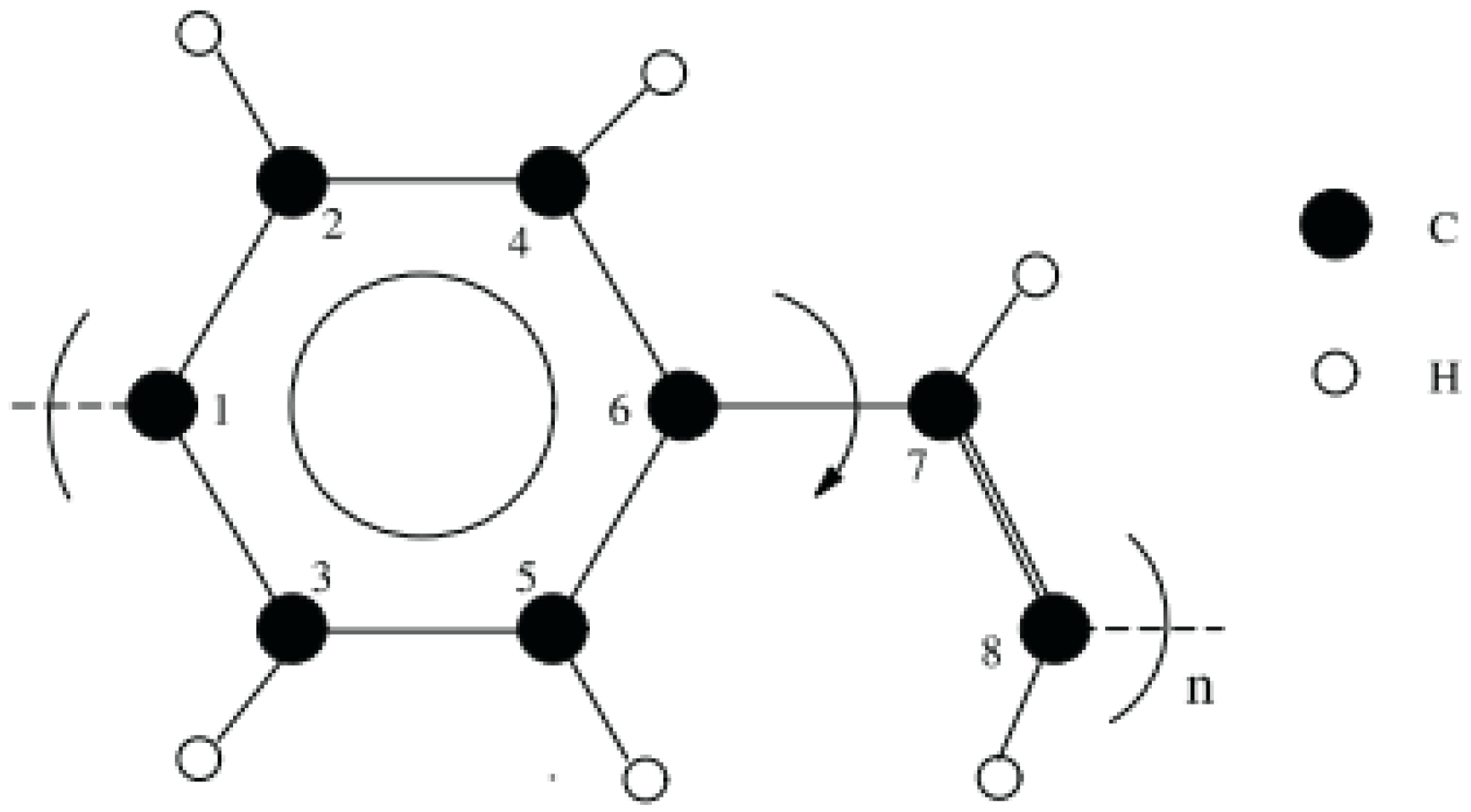
2.2. Simulation Models
3. Results
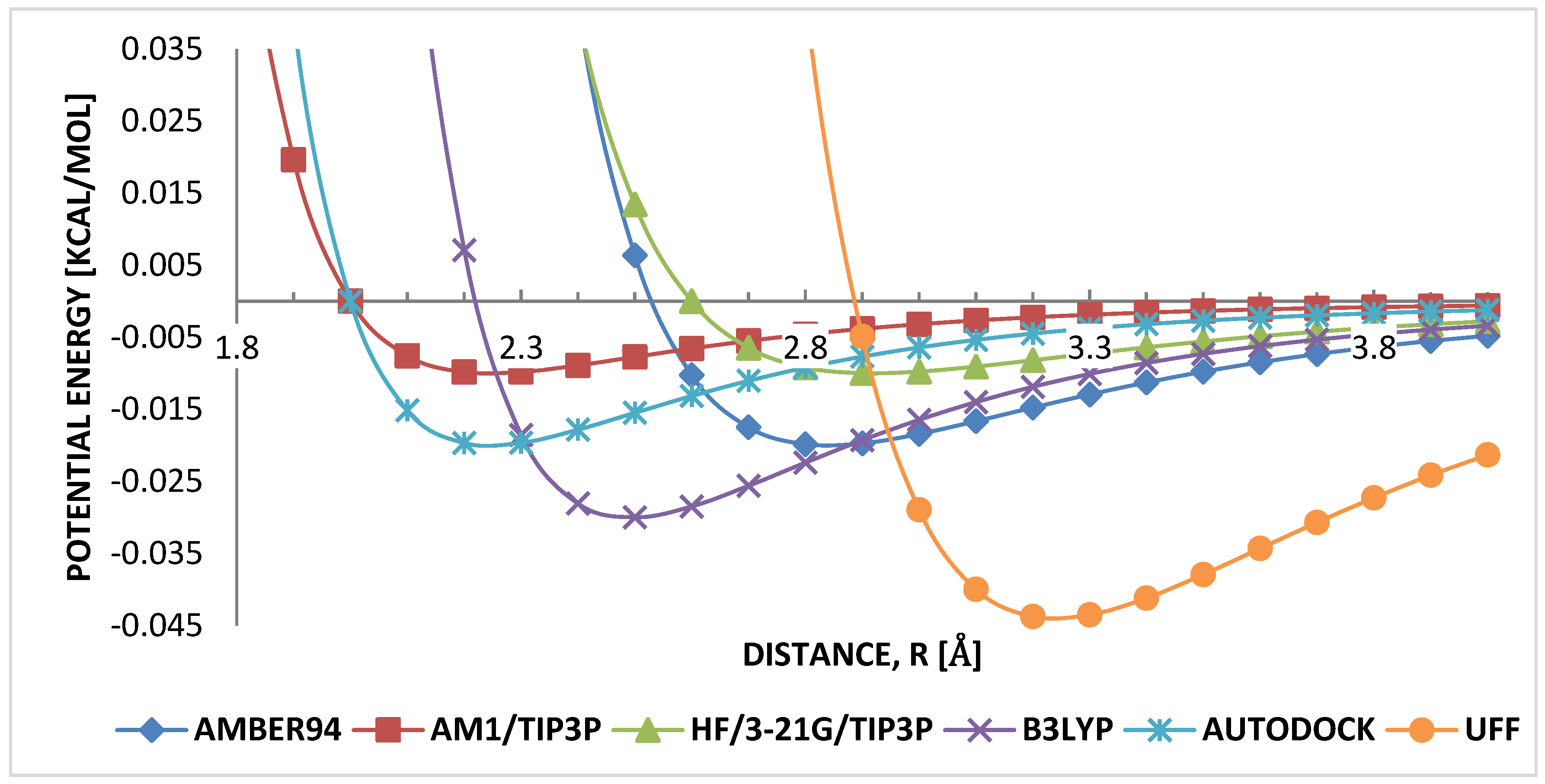
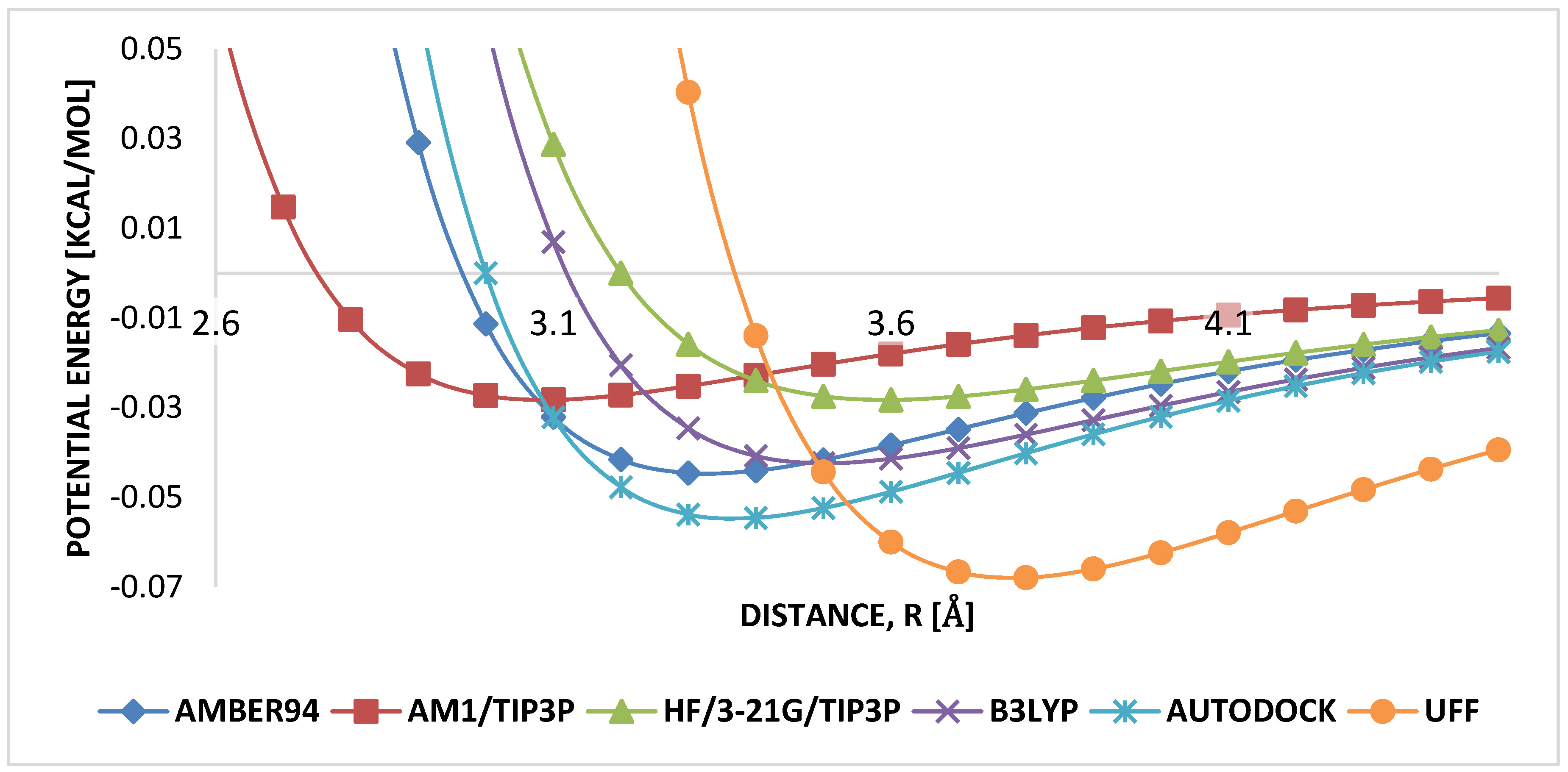
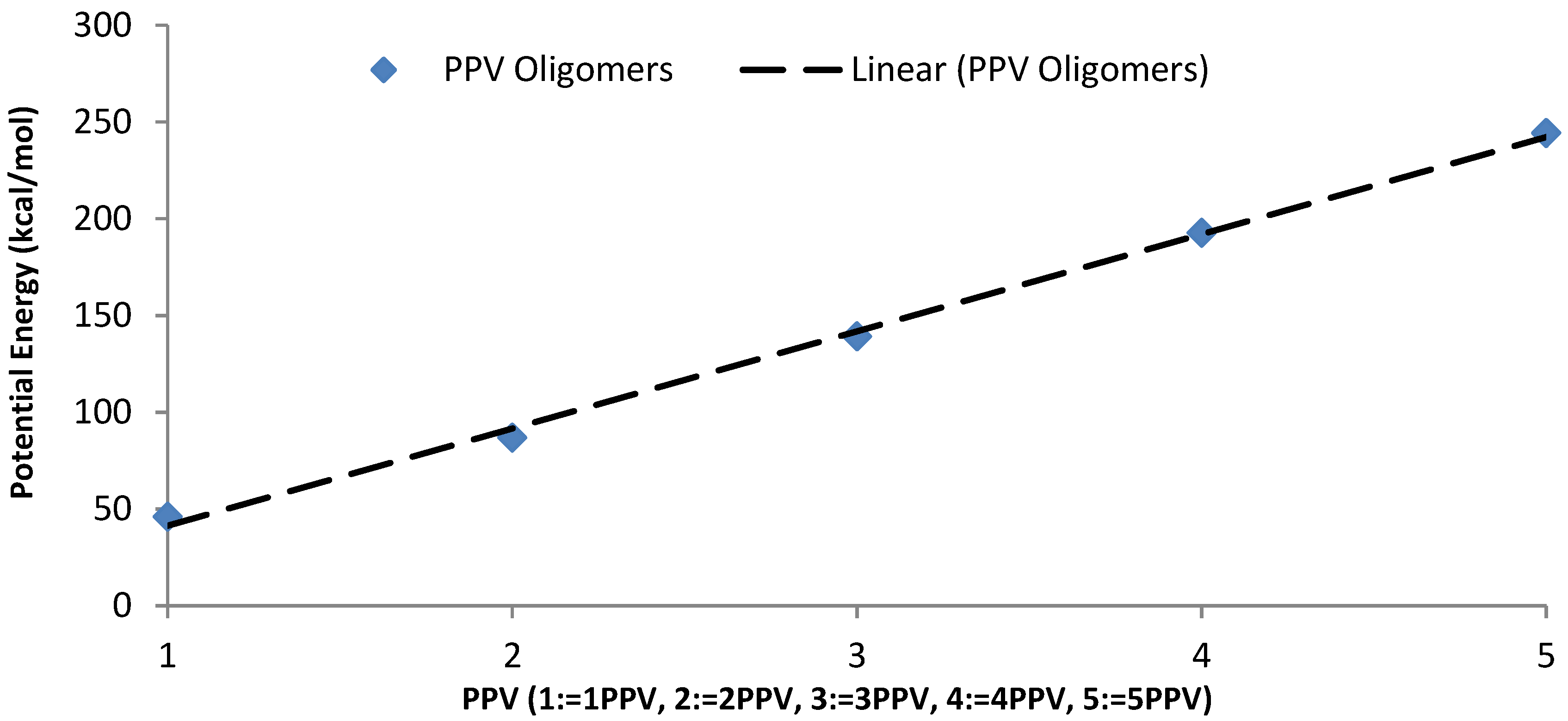
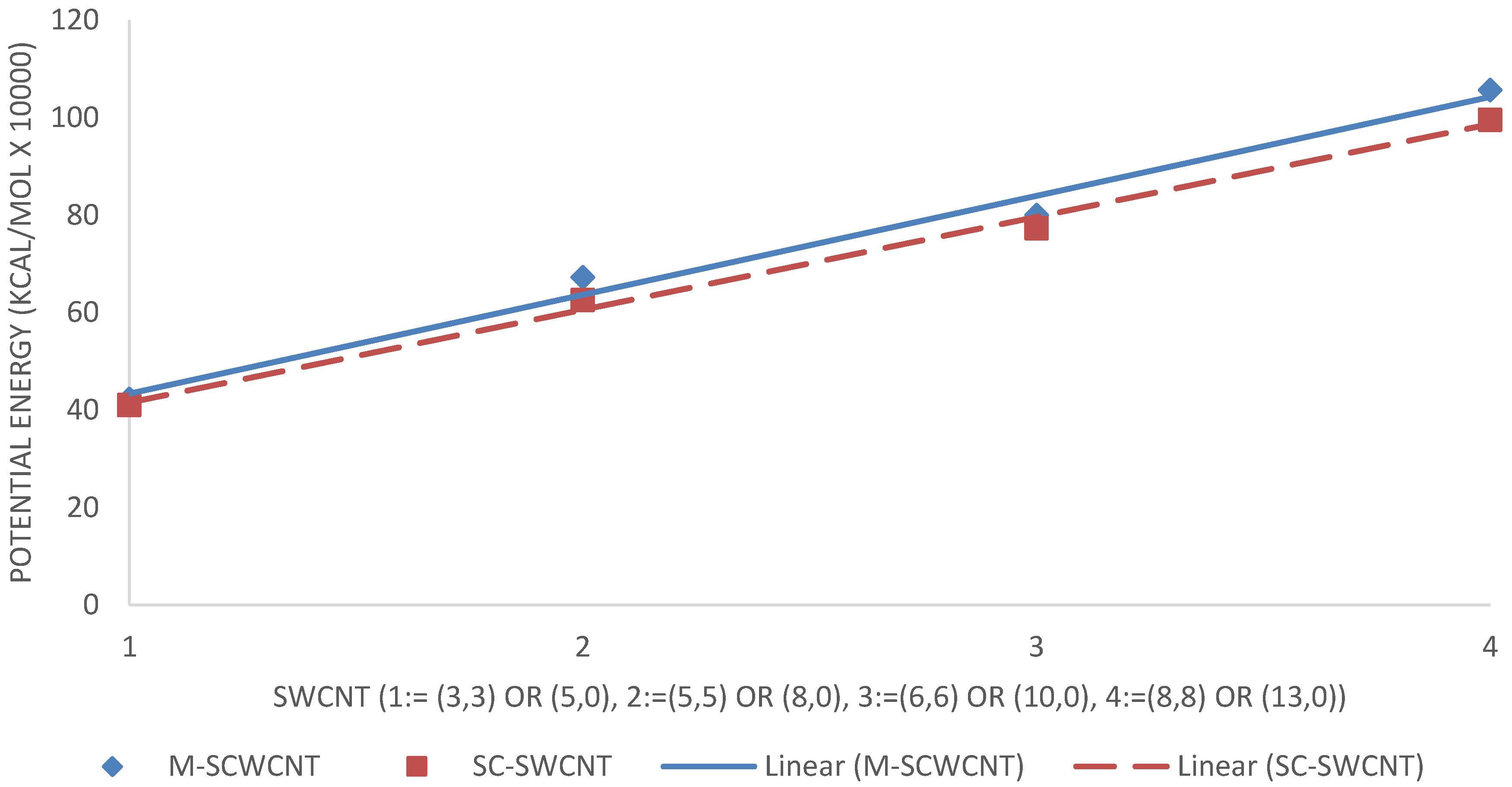
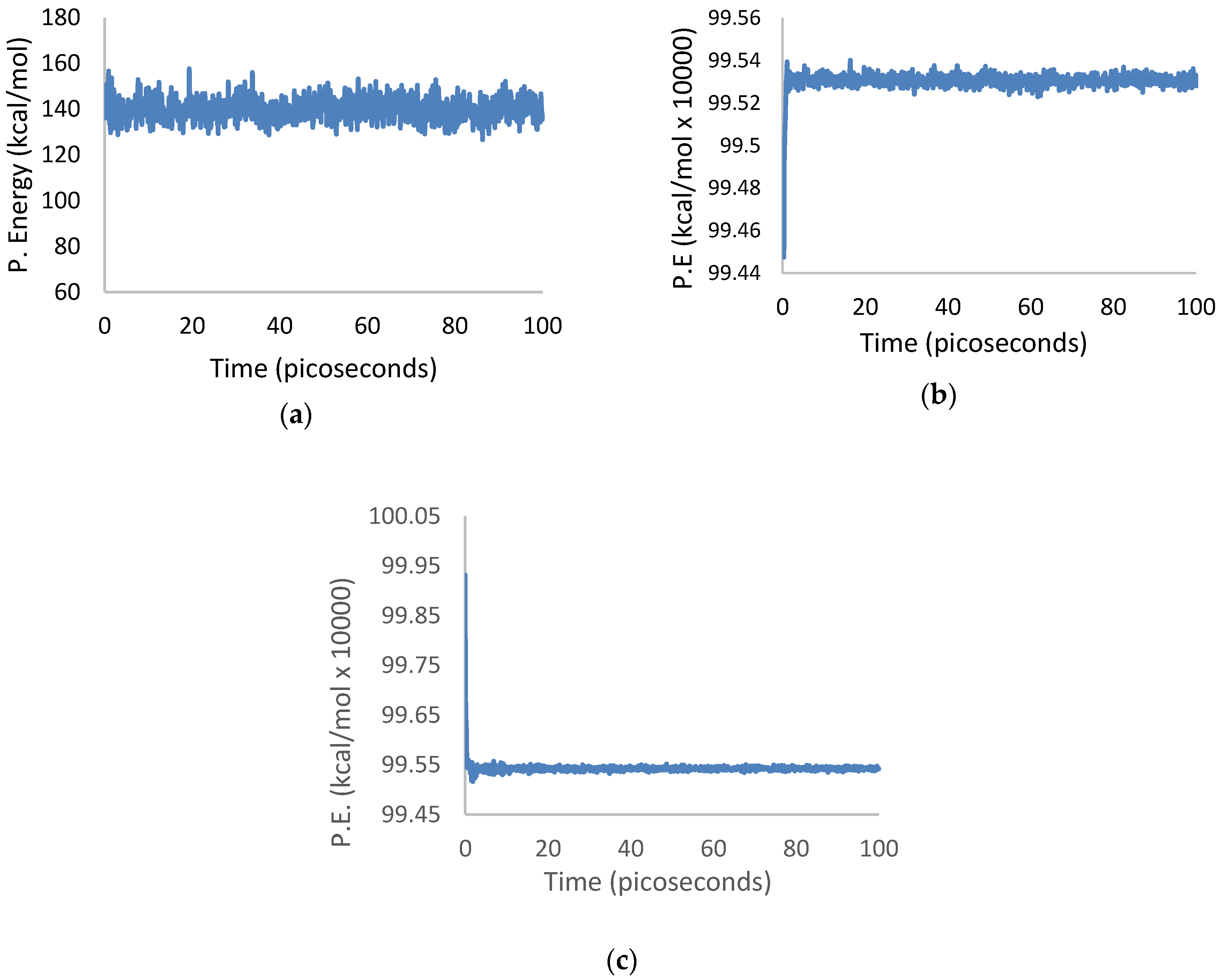
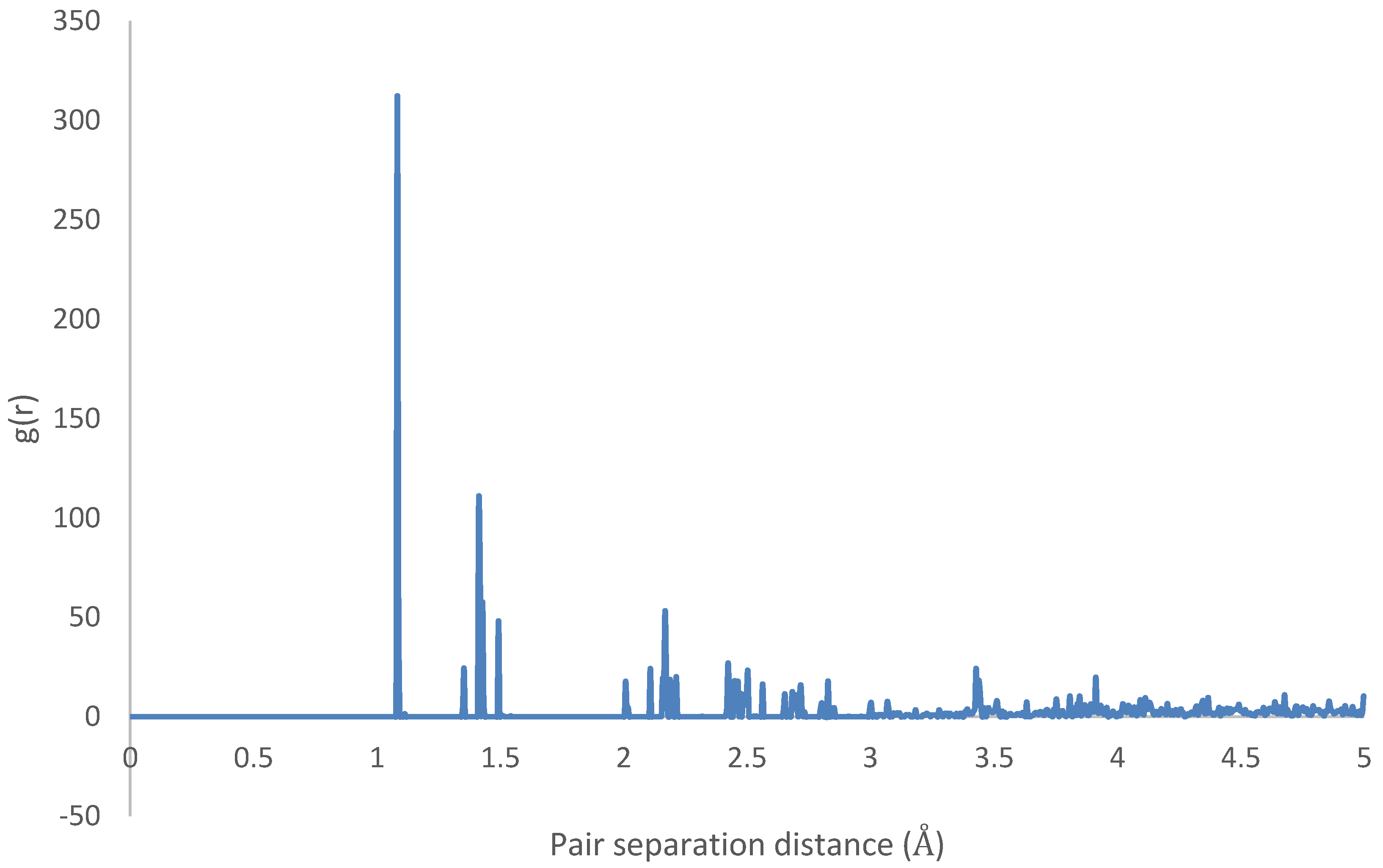
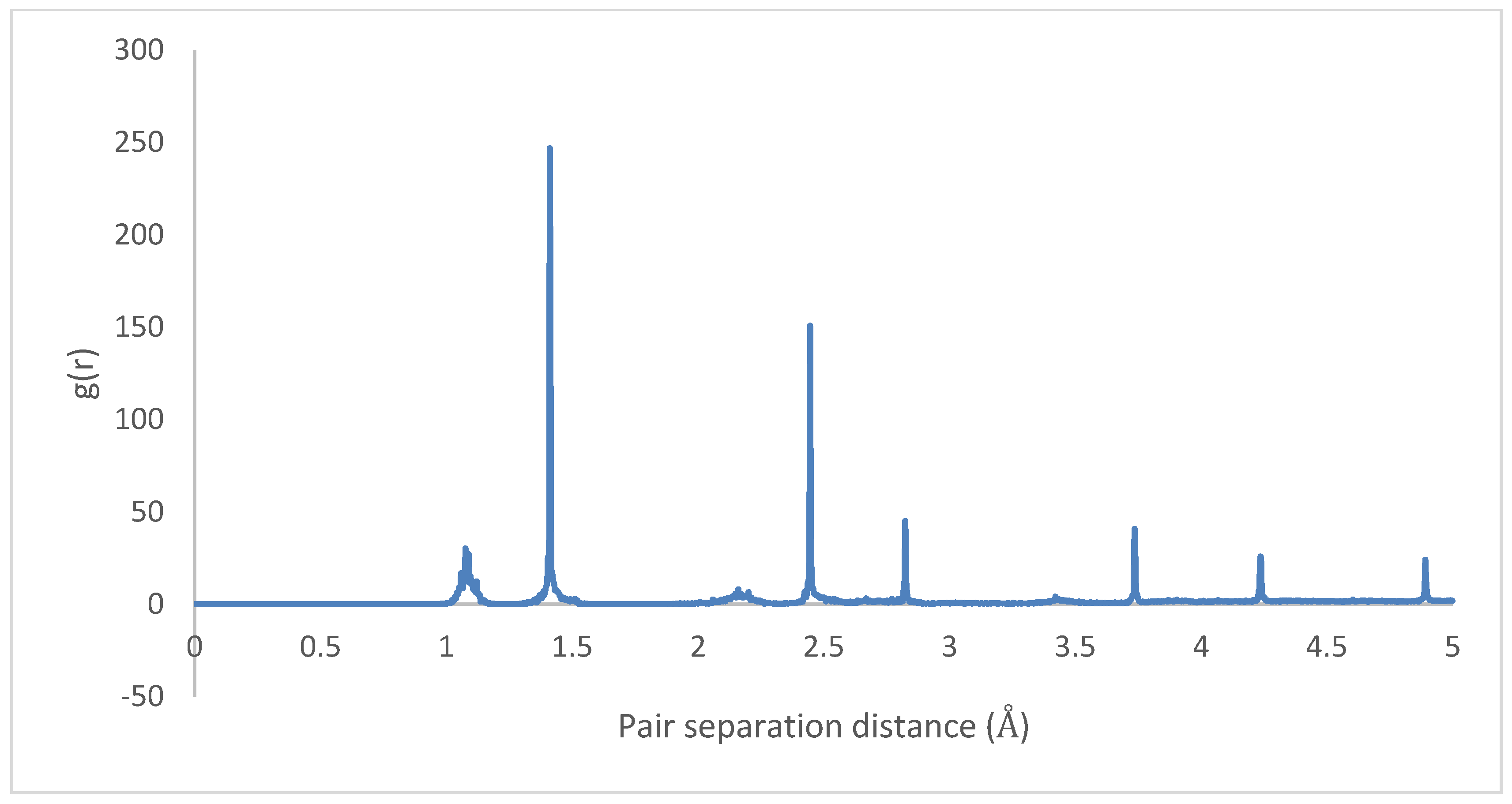
3.1. Geometry Optimization
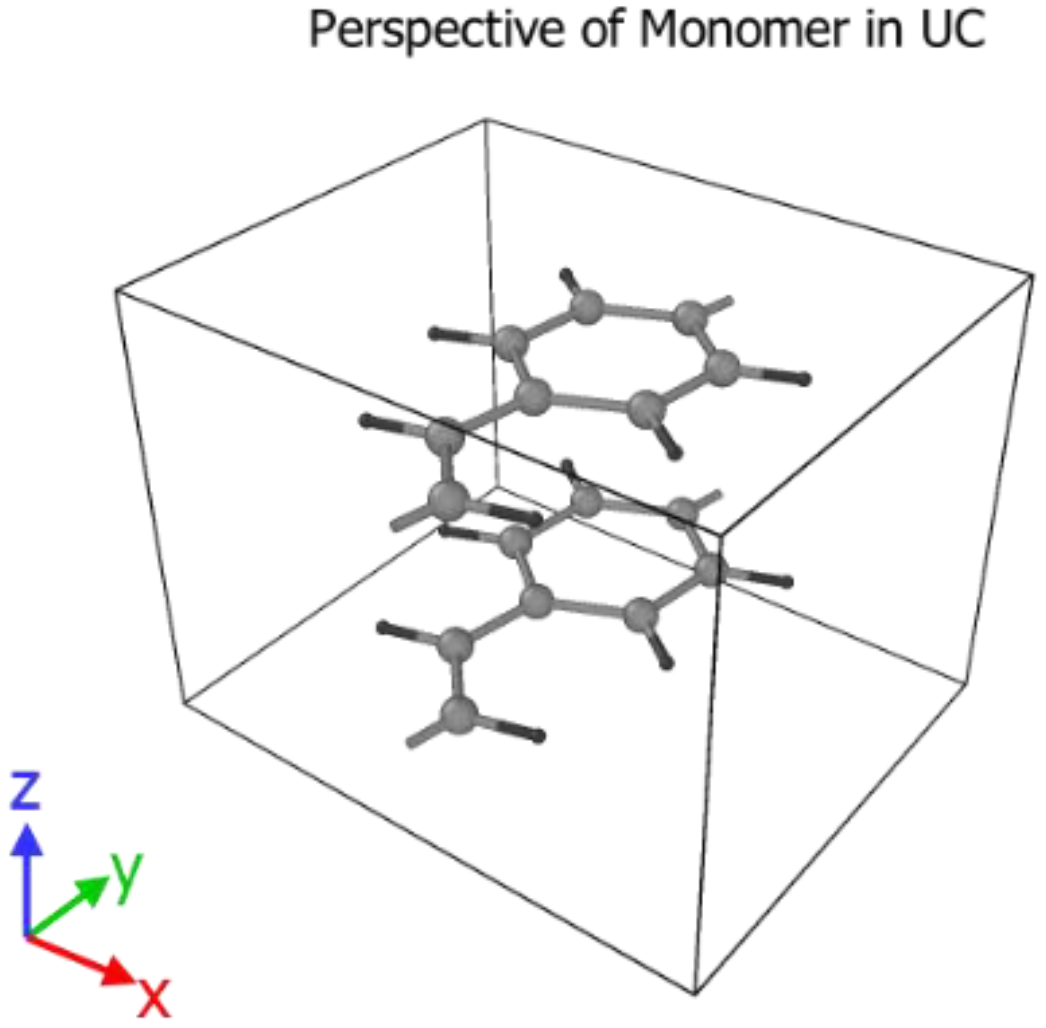
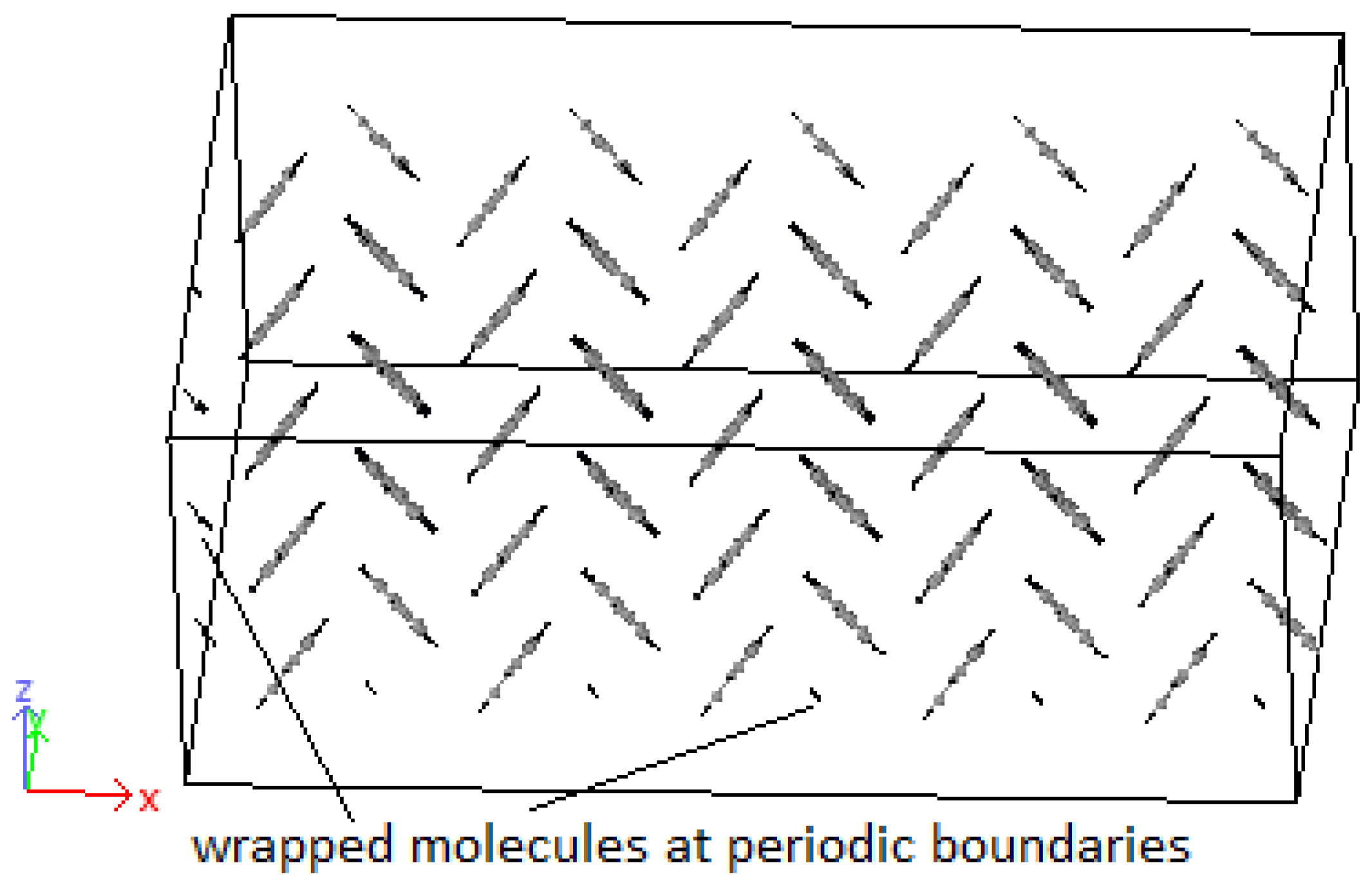
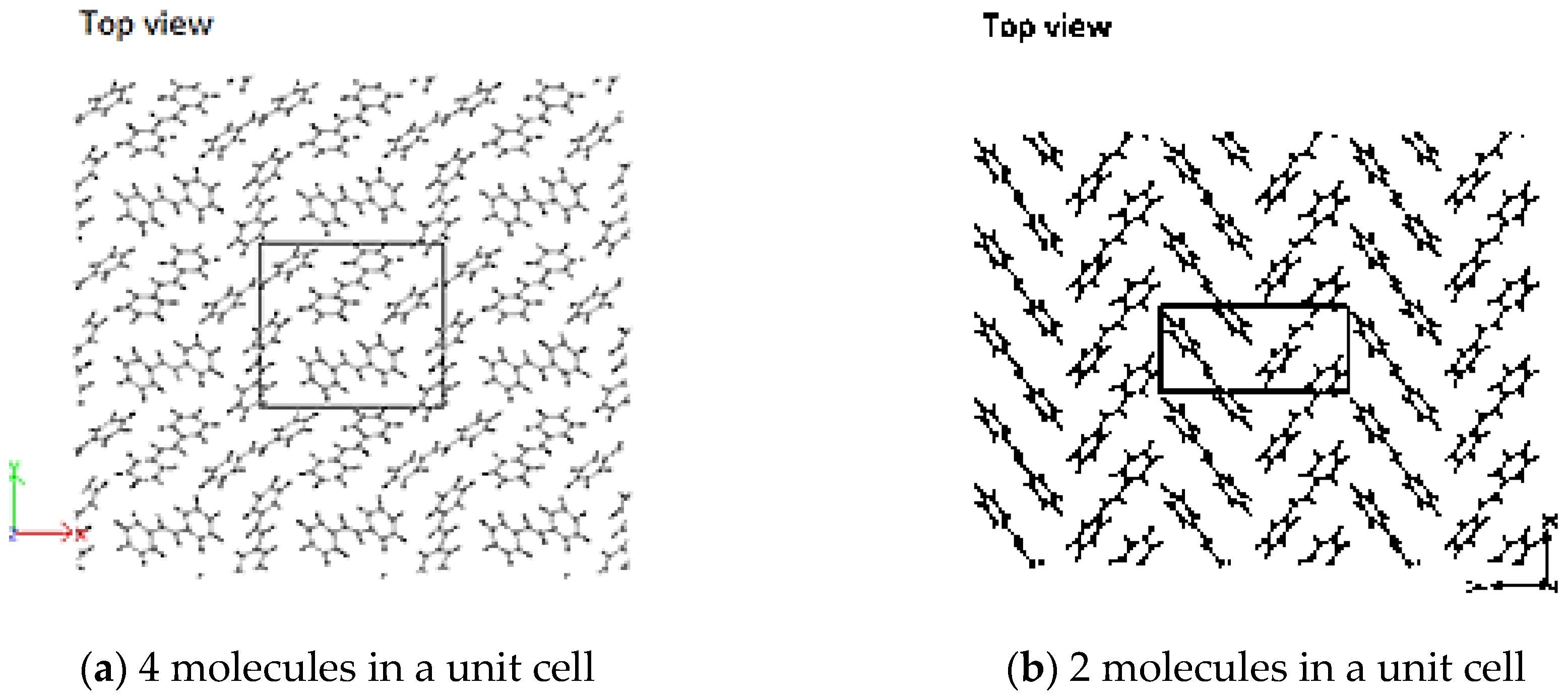
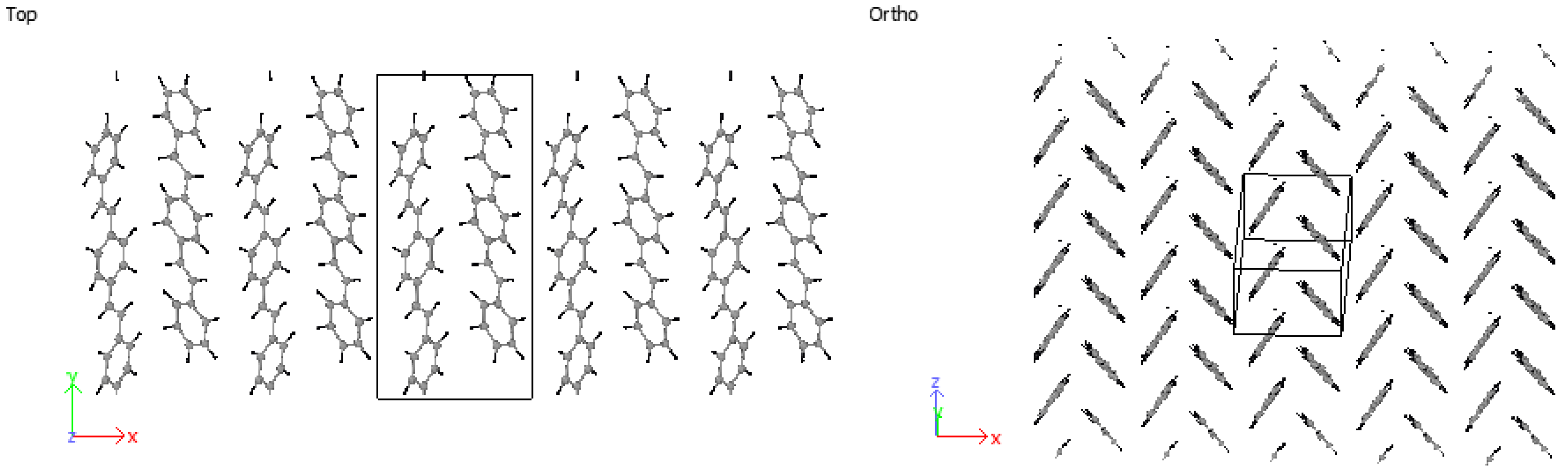
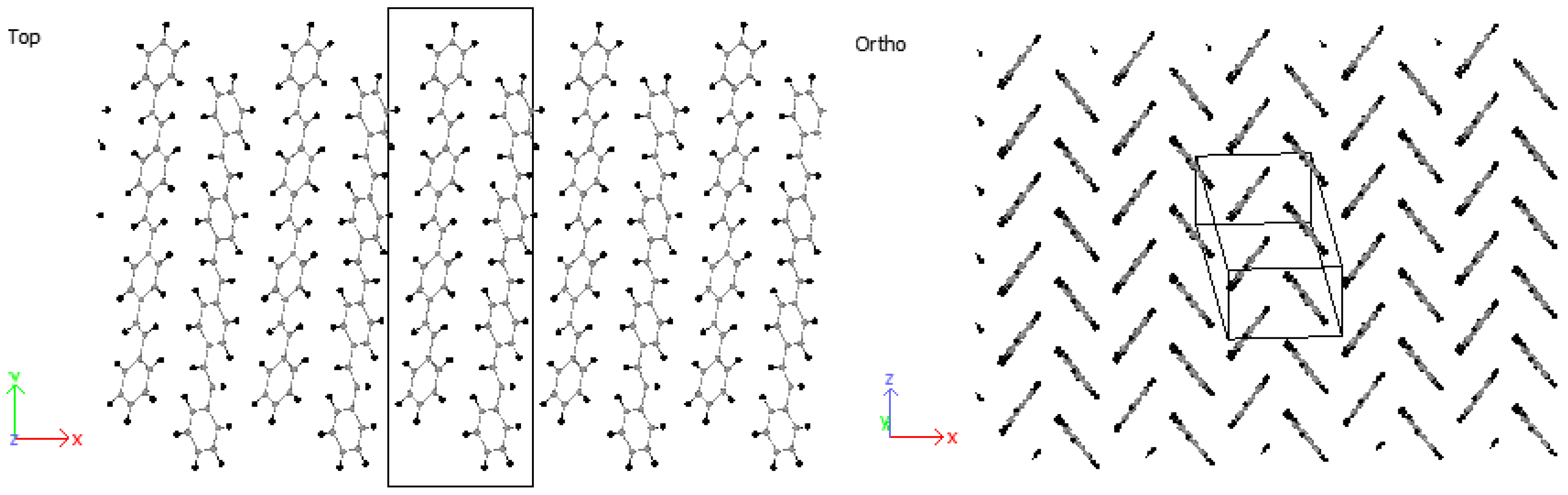
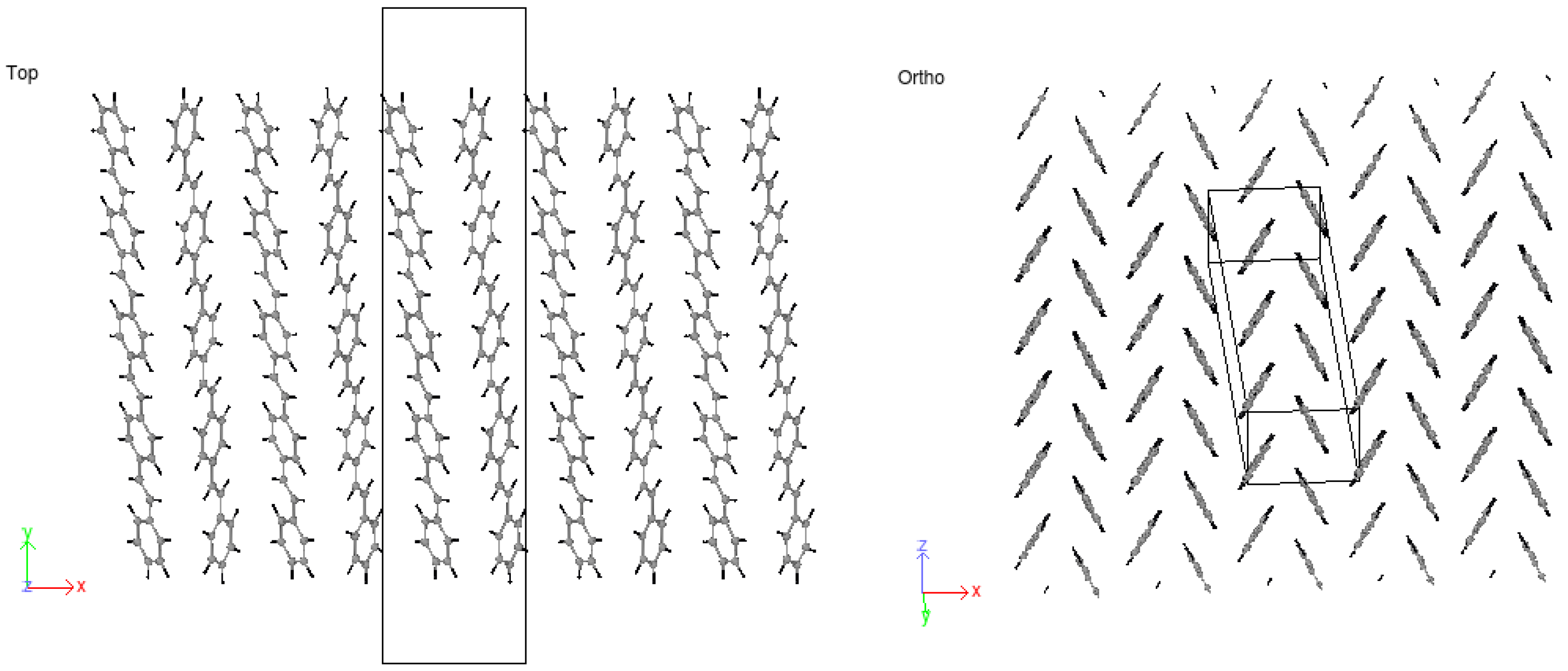
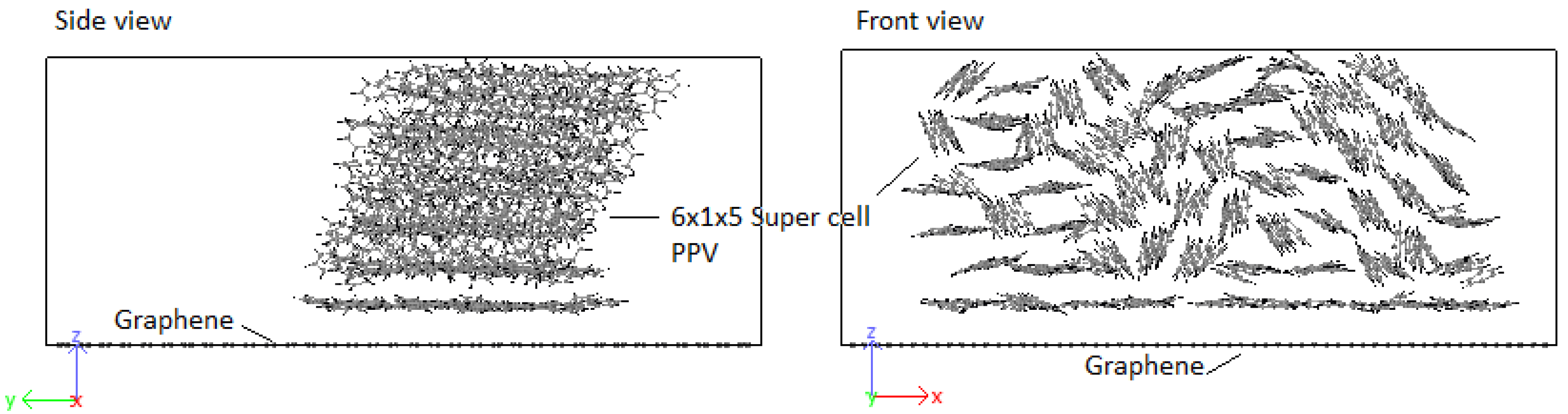
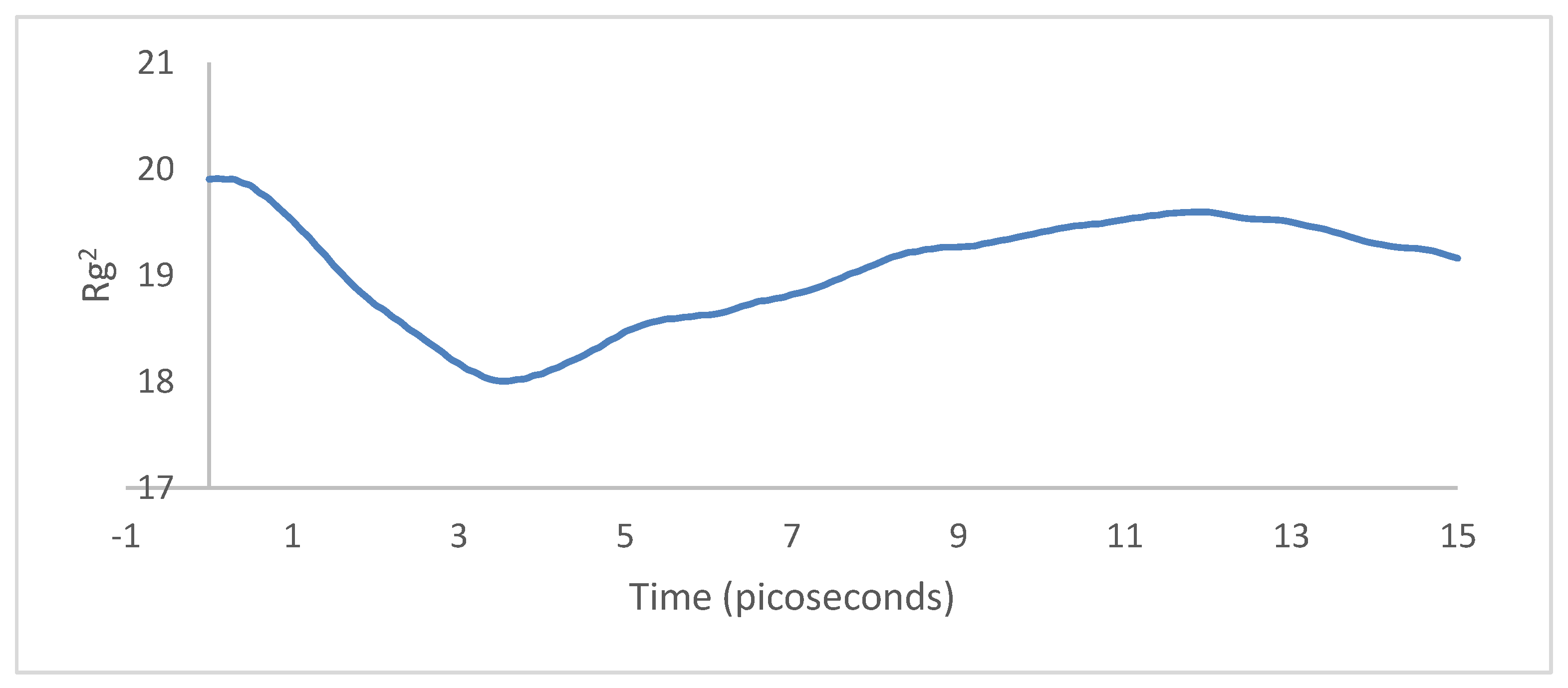
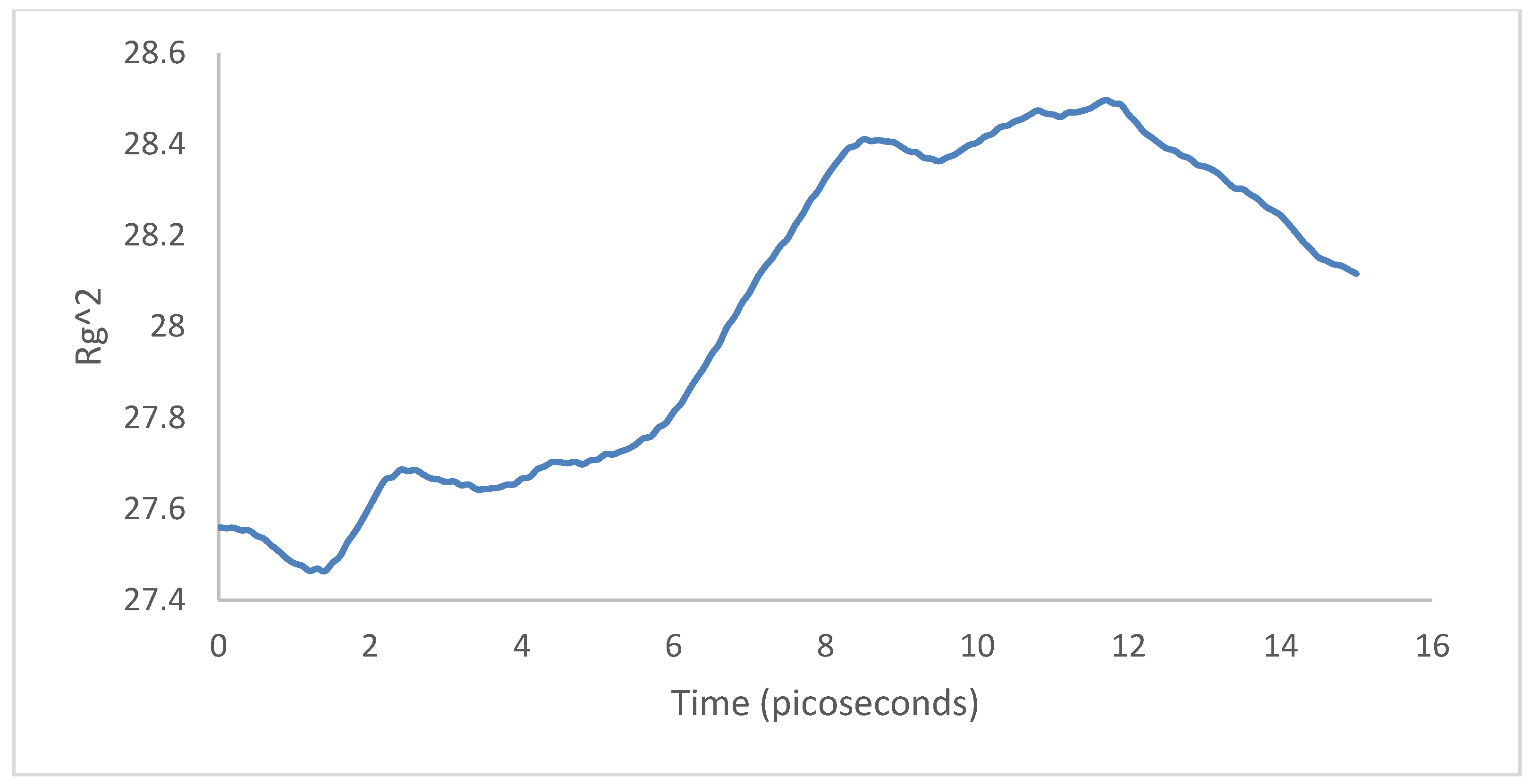
3.2. Crystallization
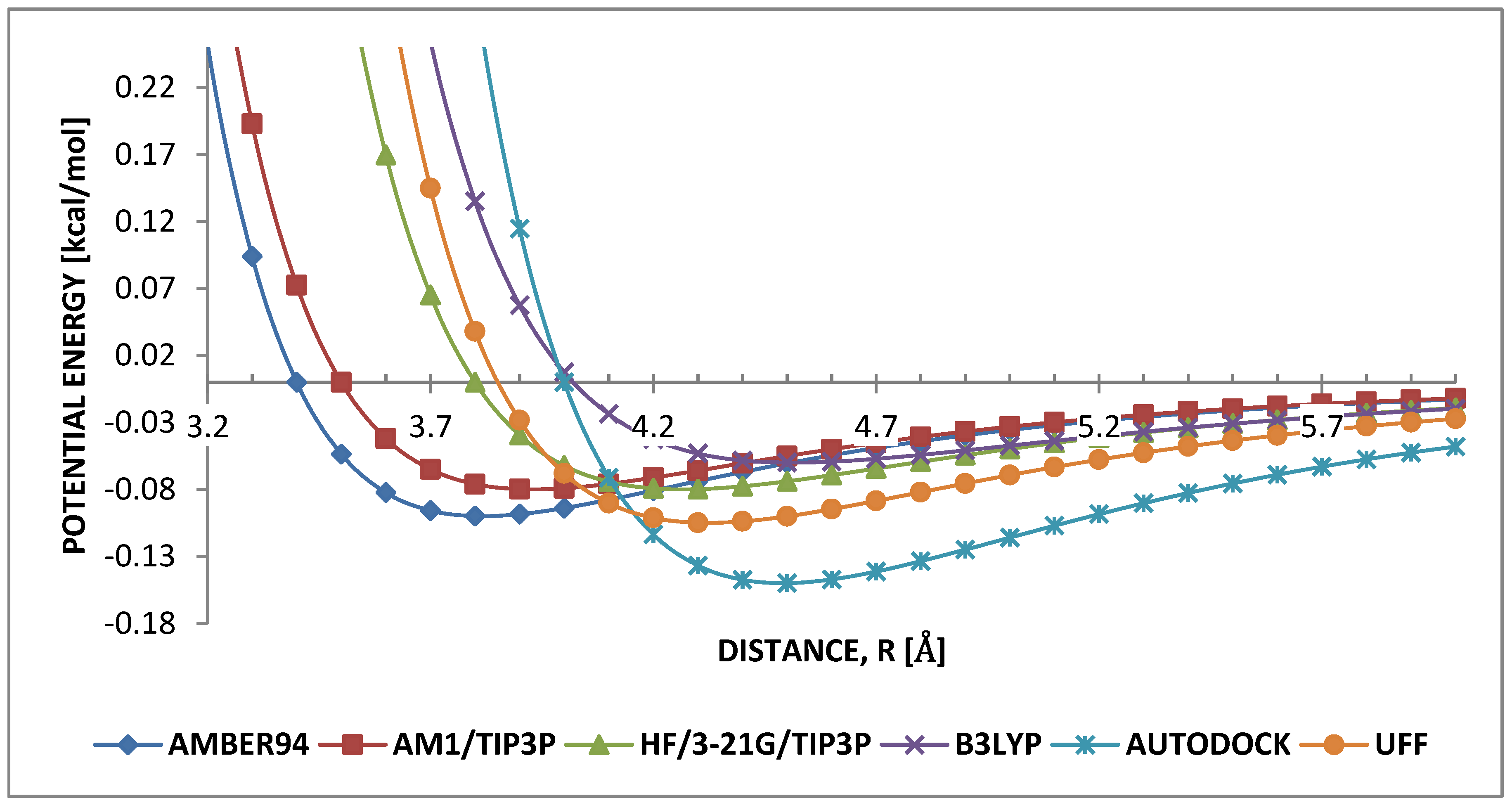
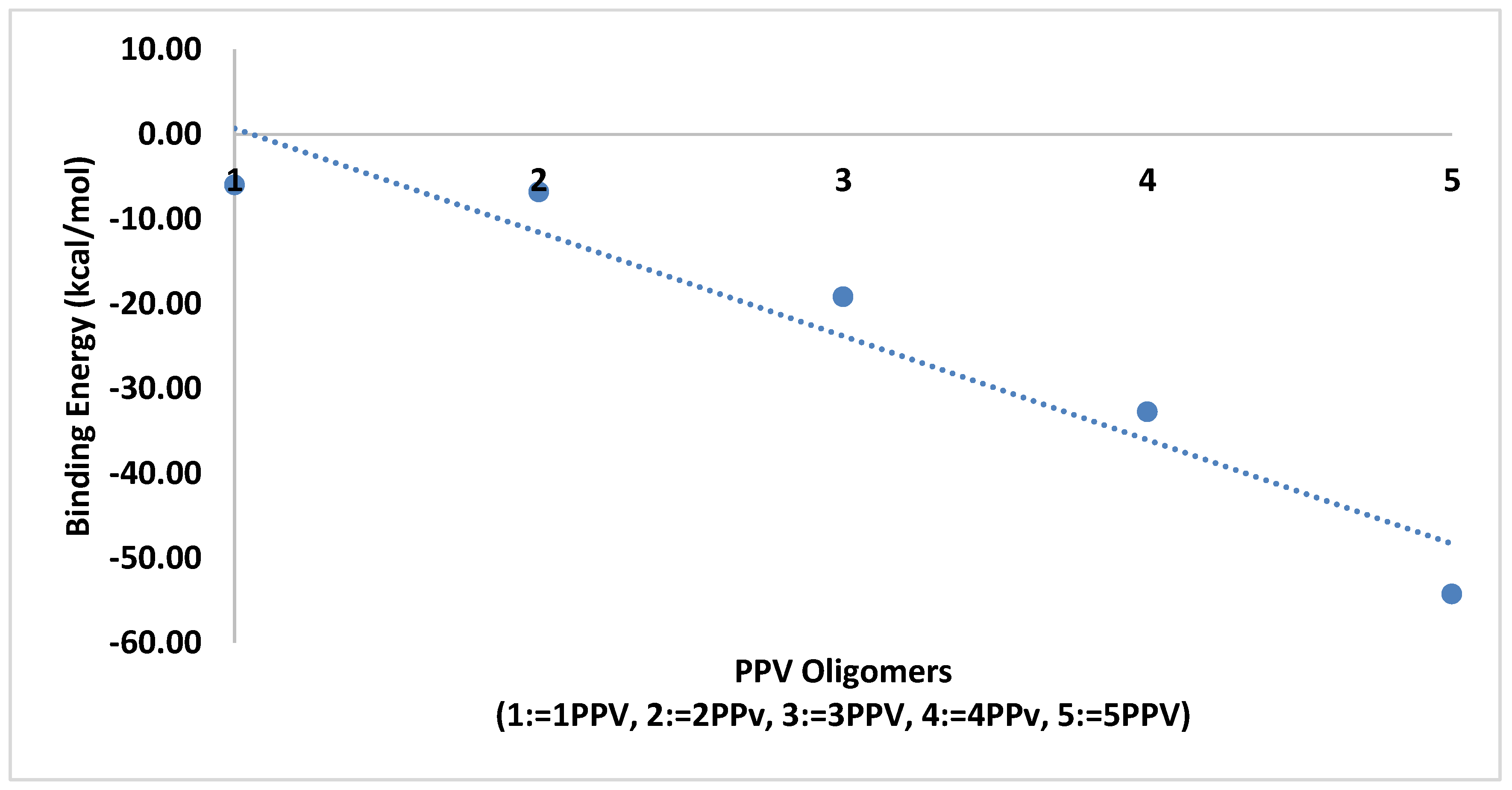
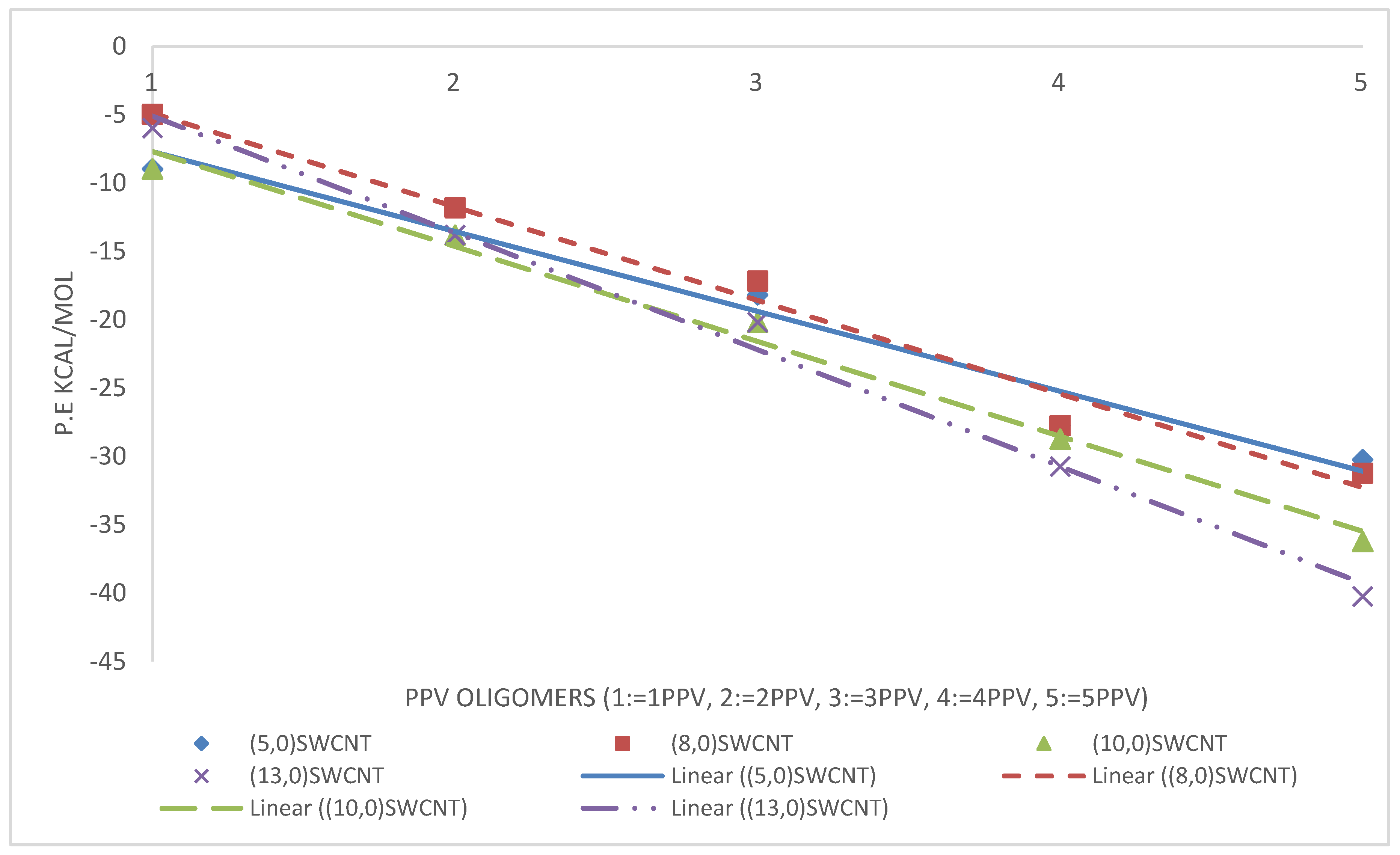
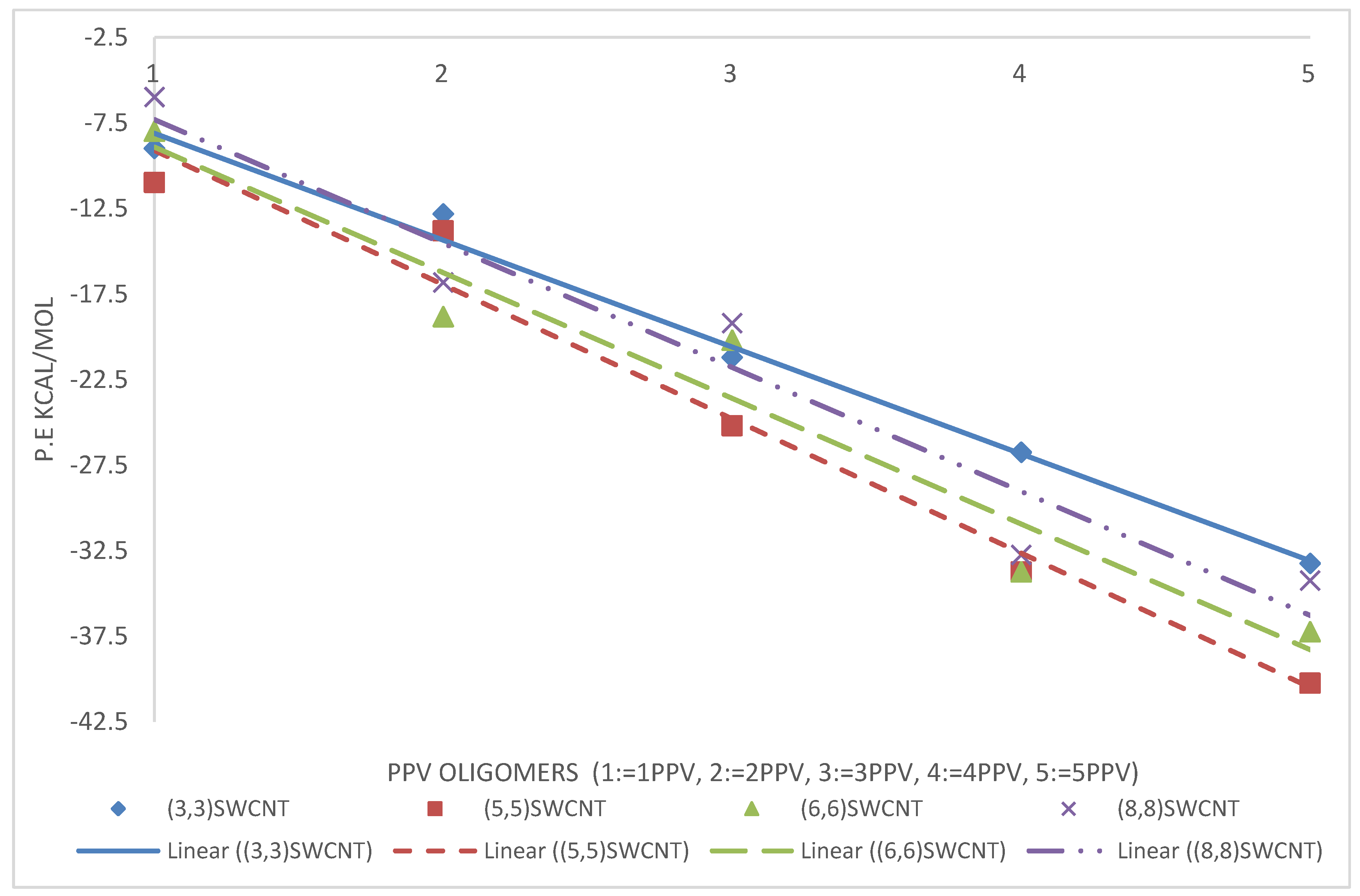
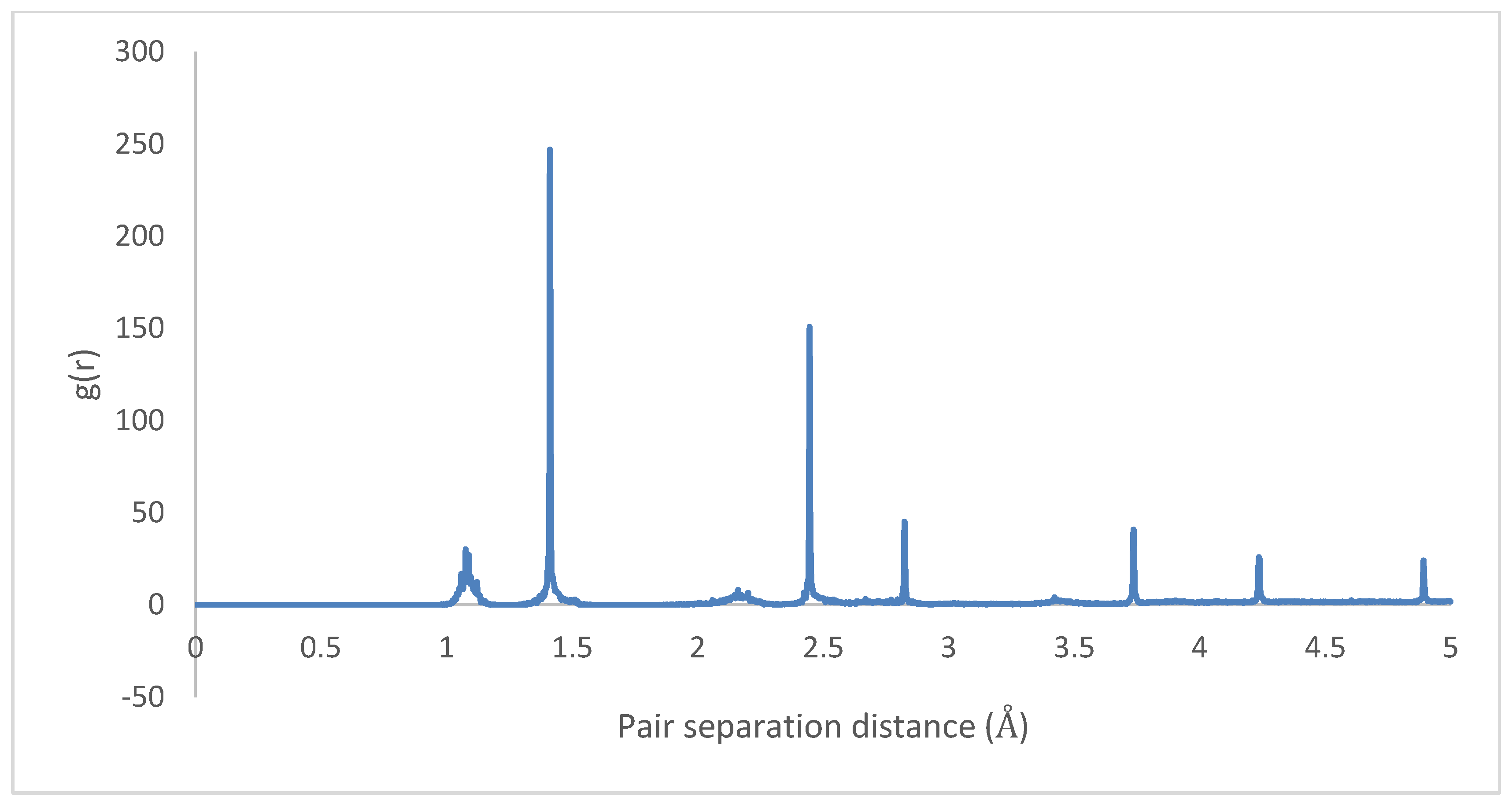
3.3. Intramolecular Interactions and Binding Energy
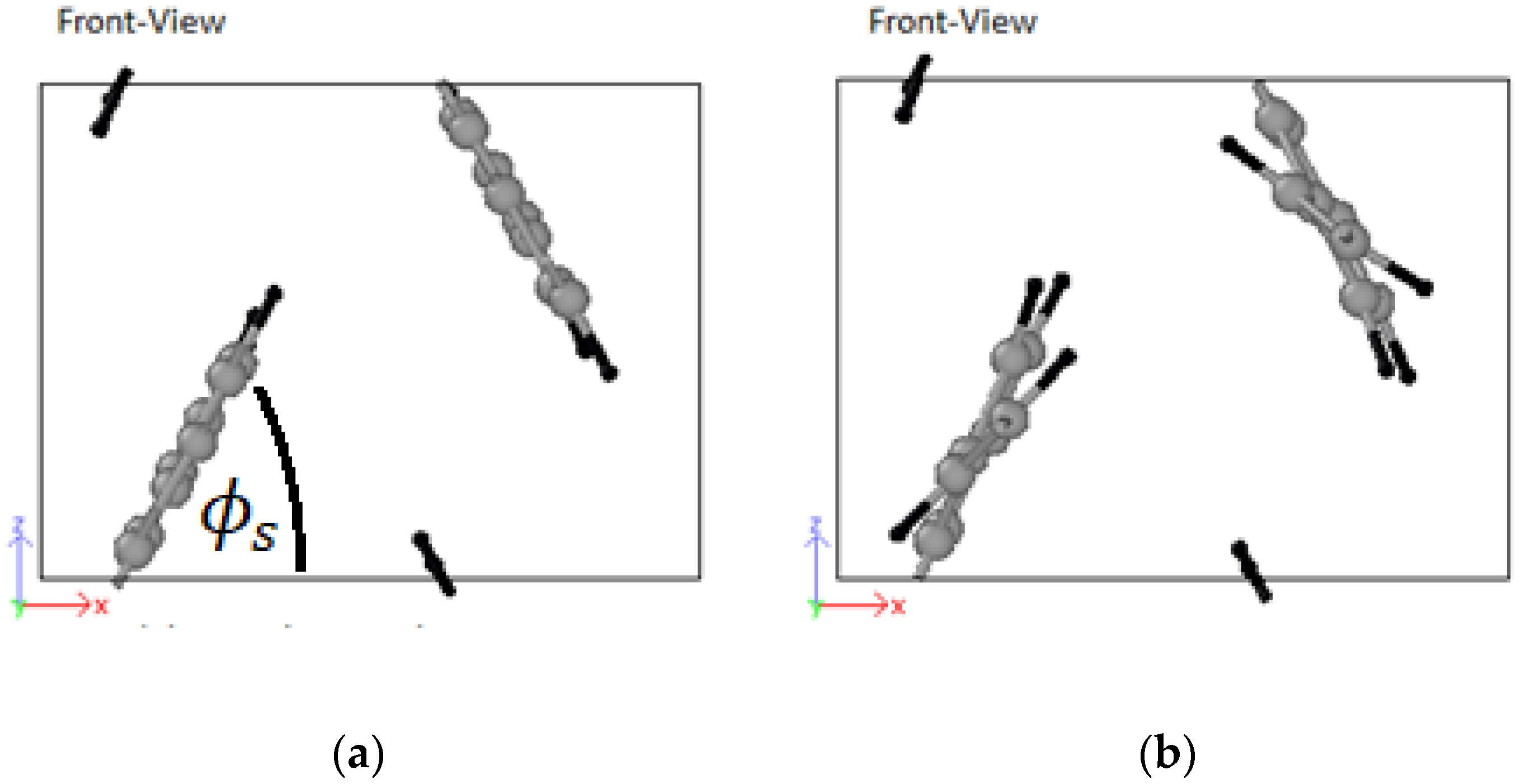
3.4. Structural Configurations
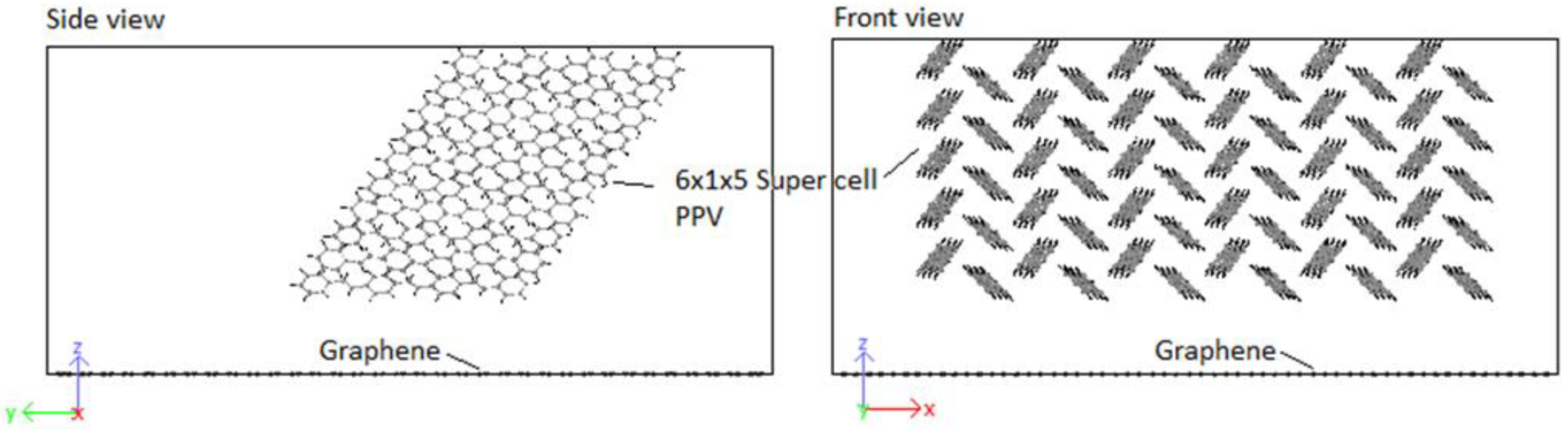
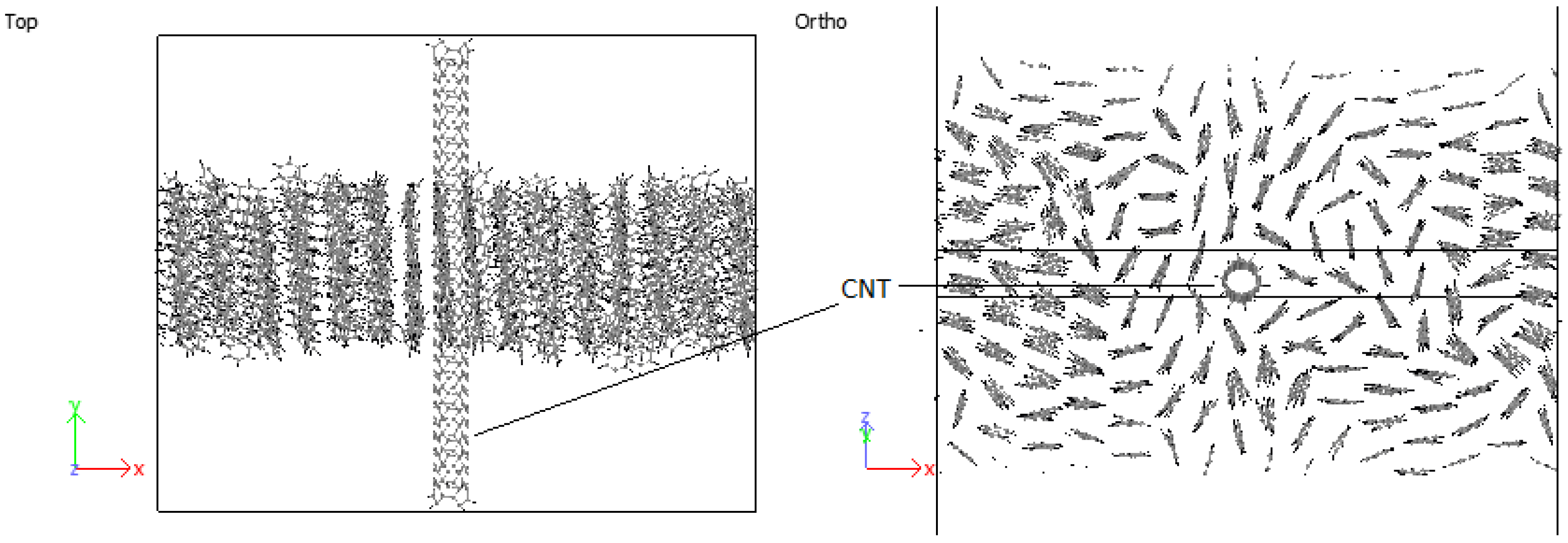
3.4.1. PPV/Graphene
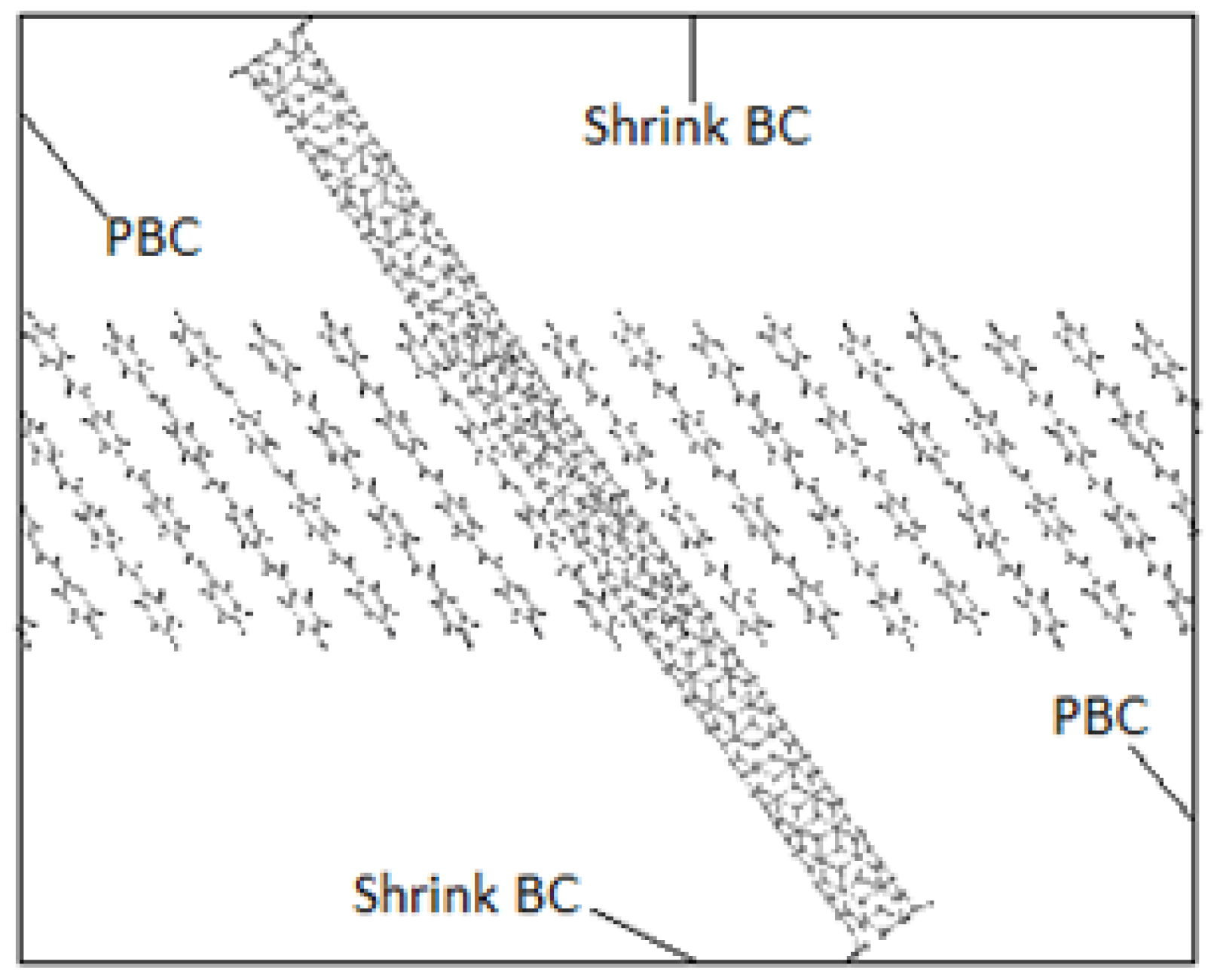
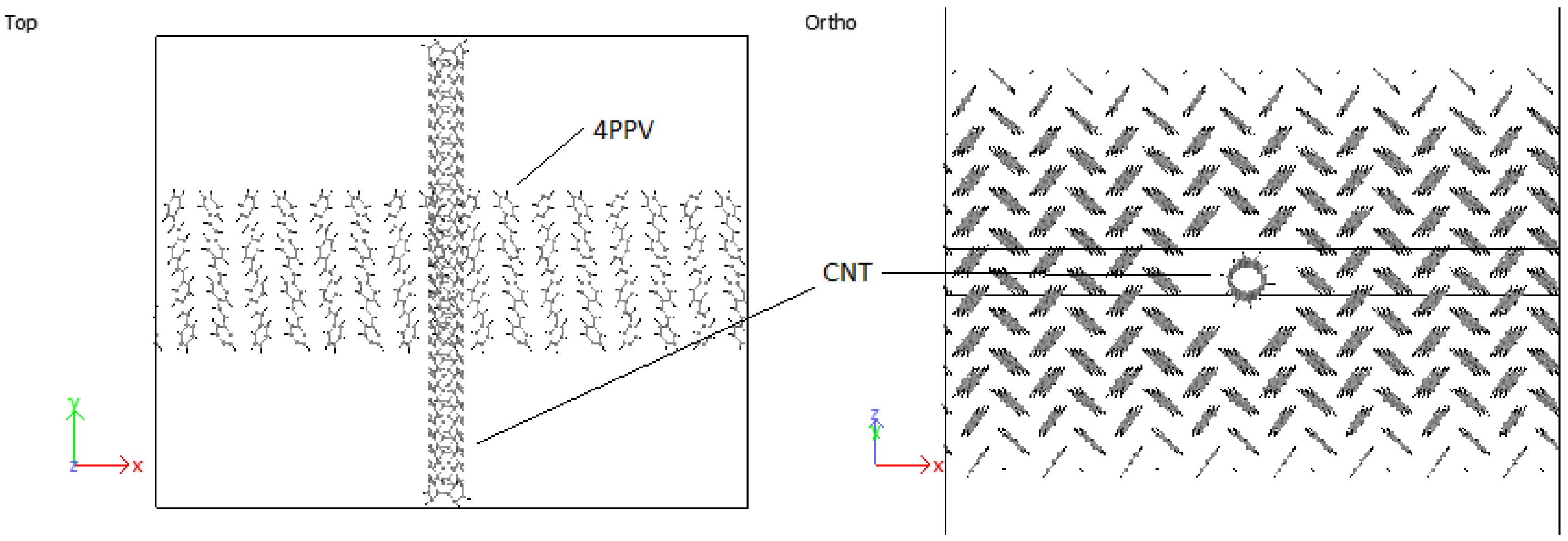
3.4.2. PPV/SWCNT
4. Discussion
4.1. Geometry Optimization
4.2. Crystallization
4.3. Intramolecular Interactions and Binding Energy
4.4. Structural Configurations
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pray, L. Organic Electronics for a Better Tomorrow: Innovation, Accessibility, Sustainability (A collaborative report from the Chemical Sciences and Society Summit). R. Soc. Chem. 2012, 9, 34. [Google Scholar]
- Ling, Y.; Kozakiewicz, P.; Blockhuys, F.; Biesemans, M.; Van Alsenoy, C.; Moons, H.; Goovaerts, E.; Willem, R.; Van Doorslaer, S. The solid-state organization of ‘self-doped’ PPV oligomers. Phys. Chem. Chem. Phys. 2011, 13, 18516–18522. [Google Scholar] [CrossRef]
- Segura, J.L.; Martín, N. Functionalized oligoarylenes as building blocks for new organic materials. J. Mater. Chem. 2000, 10, 2403–2435. [Google Scholar] [CrossRef]
- Nakanotani, H.; Saito, M.; Nakamura, H.; Adachi, C. Highly balanced ambipolar mobilities with intense electroluminescence in field-effect transistors based on organic single crystal oligo(p-phenylenevinylene) derivatives. Appl. Phys. Lett. 2009, 95, 33308. [Google Scholar] [CrossRef]
- Van Hutten, P.F.; Wildeman, J.; Meetsma, A.; Hadziioannou, G. Molecular Packing in Unsubstituted Semiconducting Phenylenevinylene Oligomer and Polymer. J. Am. Chem. Soc. 1999, 121, 5910–5918. [Google Scholar] [CrossRef]
- Group, F. Ultrafast Dynamics and Laser Action of Organic Semiconductors; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Jangid, N.K.; Chauhan, N.P.S.; Meghwal, K.; Punjabi, P.B. Conducting polymers and their applications. Res. J. Pharm. Biol. Chem. Sci. 2014, 5, 383–412. [Google Scholar]
- Cornil, J.; Beljonne, D.; Shuia, Z.; Hagler, T.W.; Campbell, I.; Bradley, D.D.C.; Müllen, K. Vibronic structure in the optical absorption spectra of phenylene vinylene oligomers: A joint experimental and theoretical study. Chem. Phys. Lett. 1995, 247, 425–432. [Google Scholar] [CrossRef]
- Zheng, G.; Clark, S.J.; Brand, S.; Abram, R.A. First-principles studies of the structural and electronic properties of poly-para-phenylene vinylene. J. Phys. Condens. Matter 2004, 16, 8609–8620. [Google Scholar] [CrossRef] [Green Version]
- Nelson, J. Polymer: Fullerene bulk heterojunction solar cells. Mater. Today 2011, 14, 462–470. [Google Scholar] [CrossRef]
- He, Y.; Cheng, N.; Xu, X.; Fu, J.; Wang, J. A high efficiency pure organic room temperature phosphorescence polymer PPV derivative for OLED. Org. Electron. 2019, 64, 247–251. [Google Scholar] [CrossRef]
- Varghese, S.; Park, S.K.; Casado, S.; Fischer, R.C.; Resel, R.; Milián-Medina, B.; Wannemacher, R.; Park, S.Y.; Gierschner, J. Stimulated Emission Properties of Sterically Modified Distyrylbenzene-Based H-Aggregate Single Crystals. J. Phys. Chem. Lett. 2013, 4, 1597–1602. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Turnbull, G.A.; Samuel, I.D.W. Organic Lasers: The Development of Luminescent Concentrators for Pumping Organic Semiconductor Lasers (Adv. Mater. 31/2009). Adv. Mater. 2009, 21. [Google Scholar] [CrossRef]
- Peters, M.; Seneca, S.; Hellings, N.; Junkers, T.; Ethirajan, A. Size-dependent properties of functional PPV-based conjugated polymer nanoparticles for bioimaging. Colloids Surf. B Biointerfaces 2018, 169, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Mullen, K.; Wegner, G. Electronic Materials: The Oligomer Approach; John Wiley & Sons: Hoboken, NJ, USA, 1998. [Google Scholar]
- Gierschner, J.; Lüer, L.; Milián-Medina, B.; Oelkrug, D.; Egelhaaf, H.-J. Highly Emissive H-Aggregates or Aggregation-Induced Emission Quenching? The Photophysics of All-Trans para-Distyrylbenzene. J. Phys. Chem. Lett. 2013, 4, 2686–2697. [Google Scholar] [CrossRef]
- Oelkrag, D.; Egelhaaf, H.-J.; Gierschner, J.; Tompert, A. Electronic deactivation in single chains, nano-aggregates and ultrathin films of conjugated oligomers. Synth. Met. 1996, 76, 249–253. [Google Scholar] [CrossRef]
- Oelkrug, D.; Gierschner, J.; Egelhaaf, H.J.; Lüer, L.; Tompert, A. Evolution of optical absorption from small oligomers to ideally conjugated PPV and MEH-PPV polymers. Synth. Met. 2001, 121, 1693–1694. [Google Scholar]
- Prasad, N.; Singh, I.; Kumari, A.; Madhwal, D.; Madan, S.; Dixit, S.K.; Bhatnagar, P.; Mathur, P. Improving the performance of MEH-PPV based light emitting diode by incorporation of graphene nanosheets. J. Lumin. 2015, 159, 166–170. [Google Scholar] [CrossRef]
- Liu, Z.; He, D.; Wang, Y.; Wu, H.; Wang, J. Effect of SPFGraphene dopant in MEH–PPV organic light-emitting devices. Synth. Met. 2010, 160, 1587–1589. [Google Scholar] [CrossRef]
- Kuo, J.-K.; Huang, P.-H.; Wu, W.-T.; Hsu, Y.-C. Molecular dynamics investigations on the interfacial energy and adhesive strength between C60-filled carbon nanotubes and metallic surface. Mater. Chem. Phys. 2014, 143, 873–880. [Google Scholar] [CrossRef]
- Sun, S.; Chen, S.; Weng, X.; Shan, F.; Hu, S. Effect of Carbon Nanotube Addition on the Interfacial Adhesion between Graphene and Epoxy: A Molecular Dynamics Simulation. Polymers 2019, 11, 121. [Google Scholar] [CrossRef] [Green Version]
- Stankovich, S.; Dikin, D.A.; Dommett, G.H.B.; Kohlhaas, K.M.; Zimney, E.J.; Stach, E.A.; Piner, R.D.; Nguyen, S.T.; Ruoff, R.S. Graphene-based composite materials. Nat. Cell Biol. 2006, 442, 282–286. [Google Scholar] [CrossRef]
- Guo, B.; Fang, L.; Zhang, B.; Gong, J.R. Graphene Doping: A Review. Insci. J. 2011, 1, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Yaya, A. Interactions Faibles Dans Les Nanosystemes Carbones. Ph.D. Thesis, Université de Nantes, Nantes, France, 2011. [Google Scholar]
- Veenstra, S.; Van Hutten, P.; Post, A.; Wang, Y.; Hadziioannou, G.; Jonkman, H. Structural studies on thin films of an unsubstituted oligo(para-phenylenevinylene). Thin Solid Films 2002, 422, 104–111. [Google Scholar] [CrossRef] [Green Version]
- Ishii, B.H.; Sugiyama, K.; Ito, E.; Seki, K. Energy Level Alignment and Interfacial.pdf. Adv. Mater. 1999, 11, 605–625. [Google Scholar] [CrossRef]
- Hill, I.G.; Mäkinen, A.J.; Kafafi, Z.H. Initial stages of metal/organic semiconductor interface formation. J. Appl. Phys. 2000, 88, 889–895. [Google Scholar] [CrossRef] [Green Version]
- Viau, L.; Maury, O.; Le Bozec, H. New 4,4′-oligophenylenevinylene functionalized-[2,2′]-bipyridyl chromophores: Synthesis, optical and thermal properties. Tetrahedron Lett. 2004, 45, 125–128. [Google Scholar] [CrossRef]
- Zheng, G.; Clark, S.J.; Tulip, P.R.; Brand, S.; Abram, R.A. Ab initio dynamics study of poly-para-phenylene vinylene. J. Chem. Phys. 2005, 123, 24904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardozo, T.M.; Aquino, A.J.A.; Barbatti, M.; Borges, I.; Lischka, H. Absorption and Fluorescence Spectra of Poly(p-phenylenevinylene) (PPV) Oligomers: An ab Initio Simulation. J. Phys. Chem. A 2014, 119, 1787–1795. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef] [Green Version]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Gime, X.; Bofill, J.M.; Gonza, J. Algorithm to Evaluate Rate Constants for Polyatomic Chemical Reactions. II. Applications. J. Comput. Chem. 2007, 28, 2111–2121. [Google Scholar]
- Steinbach, P.J.; Brooks, B.R. New spherical-cutoff methods for long-range forces in macromolecular simulation. J. Comput. Chem. 1994, 15, 667–683. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- E Tuckerman, M.; Alejandre, J.; López-Rendón, R.; Jochim, A.L.; Martyna, G.J. A Liouville-operator derived measure-preserving integrator for molecular dynamics simulations in the isothermal–isobaric ensemble. J. Phys. A Math. Gen. 2006, 39, 5629–5651. [Google Scholar] [CrossRef]
- Shinoda, W.; Shiga, M.; Mikami, M. Rapid estimation of elastic constants by molecular dynamics simulation under constant stress. Phys. Rev. B 2004, 69, 16–18. [Google Scholar] [CrossRef]
- Dullweber, A.; Leimkuhler, B.; McLachlan, R. Symplectic splitting methods for rigid body molecular dynamics. J. Chem. Phys. 1997, 107, 5840–5851. [Google Scholar] [CrossRef] [Green Version]
- Thompson, M.A.; Zerner, M.C.; Fajer, J. A theoretical examination of the electronic structure and excited states of the bacteriochlorophyll b dimer from Rhodopseudomonas viridis. J. Phys. Chem. 1991, 95, 5693–5700. [Google Scholar] [CrossRef]
- Glendening, E.D.; Feller, D.; Thompson, M.A. An Ab Initio Investigation of the Structure and Alkali Metal Cation Selectivity of 18-Crown-6. J. Am. Chem. Soc. 1994, 116, 10657–10669. [Google Scholar] [CrossRef]
- Thompson, M.A.; Schenter, G.K. Excited States of the Bacteriochlorophyll b Dimer of Rhodopseudomonas viridis: A QM/MM Study of the Photosynthetic Reaction Center That Includes MM Polarization. J. Phys. Chem. 1995, 99, 6374–6386. [Google Scholar] [CrossRef]
- Thompson, M.A. QM/MMpol: A Consistent Model for Solute/Solvent Polarization. Application to the Aqueous Solvation and Spectroscopy of Formaldehyde, Acetaldehyde, and Acetone. J. Phys. Chem. 1996, 100, 14492–14507. [Google Scholar] [CrossRef]
- Rappe, A.K.; Colwell, K.S.; Casewit, C.J. Application of a universal force field to metal complexes. Inorg. Chem. 1993, 32, 3438–3450. [Google Scholar] [CrossRef]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Rappe, A.K.; Goddard, W.A. Charge equilibration for molecular dynamics simulations. J. Phys. Chem. 1991, 95, 3358–3363. [Google Scholar] [CrossRef]
- Casewit, C.J.; Colwell, K.S.; Rappe, A.K. Application of a universal force field to organic molecules. J. Am. Chem. Soc. 1992, 114, 10035–10046. [Google Scholar] [CrossRef]
- Casewit, C.J.; Colwell, K.S.; Rappe, A.K. Application of a universal force field to main group compounds. J. Am. Chem. Soc. 1992, 114, 10046–10053. [Google Scholar] [CrossRef]
- Ajori, S.; Ansari, R.; Haghighi, S. A molecular dynamics study on the buckling behavior of cross-linked functionalized carbon nanotubes under physical adsorption of polymer chains. Appl. Surf. Sci. 2018, 427, 704–714. [Google Scholar] [CrossRef]
- Ansari, R.; Rouhi, S.; Ajori, S. On the Interfacial Properties of Polymers/Functionalized Single-Walled Carbon Nanotubes. Braz. J. Phys. 2016, 46, 361–369. [Google Scholar] [CrossRef]
- Zaminpayma, E.; Mirabbaszadeh, K. Interaction between single-walled carbon nanotubes and polymers: A molecular dynamics simulation study with reactive force field. Comput. Mater. Sci. 2012, 58, 7–11. [Google Scholar] [CrossRef]
- Capaz, R.B.; Caldas, M.J. Ab initio calculations of structural and dynamical properties of poly(p-phenylene) and poly(p-phenylene vinylene). Phys. Rev. B 2003, 67, 205205. [Google Scholar] [CrossRef]
- Finder, C.J.; Newton, M.G.; Allinger, N.L. An improved structure of trans-stilbene. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1974, 30, 411–415. [Google Scholar] [CrossRef]
- Weiner, P.K.; Kollman, P.A. AMBER: Assisted model building with energy refinement. A general program for modeling molecules and their interactions. J. Comput. Chem. 1981, 2, 287–303. [Google Scholar] [CrossRef]
- Gao, J. In Modeling the Hydrogen Bond. Am. Chem. Soc. 1994. [Google Scholar] [CrossRef] [Green Version]
- Gao, J. Parameters for a Combined Ab Initio Quantum Mechanical and Molecular Basis Set. J. Comput. Chem. 1996, 17, 386–395. [Google Scholar]
- Freindorf, M.; Shao, Y.; Furlani, T.R.; Kong, J. Lennard-Jones parameters for the combined QM/MM method using the B3LYP/6-31+G*/AMBER potential. J. Comput. Chem. 2005, 26, 1270–1278. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Huey, R.; Hart, W.E.; Halliday, S.; Belew, R.; Olson, A.J. User’s Guide Autodock 3.0.5. Methodol. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spano, F.C. The fundamental photophysics of conjugated oligomer herringbone aggregates. J. Chem. Phys. 2003, 118, 981–994. [Google Scholar] [CrossRef]
- Yang, M.; Koutsos, V.; Zaiser, M. Interactions between Polymers and Carbon Nanotubes: A Molecular Dynamics Study. J. Phys. Chem. B 2005, 109, 10009–10014. [Google Scholar] [CrossRef]
- Yaya, A.; Impellizzeri, A.; Massuyeau, F.; Duvail, J.; Briddon, P.; Ewels, C. Mapping the stacking interaction of triphenyl vinylene oligomers with graphene and carbon nanotubes. Carbon 2019, 141, 274–282. [Google Scholar] [CrossRef]
- Martens, J.; Halliday, D.; Marseglia, E.; Bradley, D.D.C.; Friend, R.; Burn, P.L.; Holmes, A. Structural order in poly(p-phenylene vinylene). Synth. Met. 1993, 55, 434–439. [Google Scholar] [CrossRef]
- Moon, Y.B.; Rughooputh, S.D.D.V.; Heeger, A.J.; Patil, A.O.; Wudl, F. X-ray scattering study of the coversion of poly(p-phenylene vinylene) precursor to the conjugated polymer. Synth. Met. 1989, 29, E79–E84. [Google Scholar] [CrossRef]
- Chen, D.; Winokur, M.; Masse, M.; E Karasz, F. A structural study of poly(p-phenylene vinylene). Polymer 1992, 33, 3116–3122. [Google Scholar] [CrossRef]
- Gagnon, D.R. Chemical, structural and electrical characterization of poly(p-phenylene vinylene). Ph.D. Thesis, University of Massachusetts, Amherst, MA, USA, 1986. [Google Scholar]
- Granier, T.; Thomas, E.L.; Gagnon, D.R.; Karasz, F.E.; Lenz, R.W. Structure investigation of poly(p-phenylene vinylene). J. Polym. Sci. Part B Polym. Phys. 1986, 24, 2793–2804. [Google Scholar] [CrossRef]
































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Approximate Diameter () | Metallic SWCNTs | Semiconductor SWCNTs | ||||||
|---|---|---|---|---|---|---|---|---|
| n | m | Actual Diameter () | Length () | n | m | Actual Diameter () | Length () | |
| 4 | 3 | 3 | 4.01 | 5 | 0 | 3.98 | ||
| 6 | 5 | 5 | 6.75 | 8 | 0 | 6.27 | ||
| 8 | 6 | 6 | 8.14 | 10 | 0 | 7.83 | ||
| 10 | 8 | 8 | 10.85 | 13 | 0 | 10.11 | ||
| Parameters | UFF Stilbene (This Work) | UFF 3PPV (This Work) | Isolated Chain [9] | Isolated Chain [54] | Experiment [55] | Styrene [9] |
|---|---|---|---|---|---|---|
| Bond Lengths (Angstroms) | ||||||
| C2–C4 | 1.398 | 1.398 | 1.375 | 1.383 | 1.387 | 1.381 |
| C1–C2 | 1.398 | 1.398 | 1.401 | 1.411 | 1.397 | 1.384 |
| C1–C3 | 1.398 | 1.398 | 1.403 | 1.414 | 1.394 | 1.395 |
| C6–C7 | 1.464 | 1.467 | 1.451 | 1.443 | 1.469 | 1.449 |
| C7–C8 | 1.323 | 1.323 | 1.349 | 1.361 | 1.318 | 1.329 |
| C2–H | 1.085 | 1.085 | 1.096 | 1.103 | 0.93 | 1.095 |
| C3–H | 1.085 | 1.085 | 1.096 | 1.104 | 1.02 | 1.096 |
| C7–H | 1.085 | 1.085 | 1.101 | 1.108 | 1.00 | 1.098 |
| Bond Angles (Degrees) | ||||||
| C2–C1–C3 | 120.0 | 120.0 | 116.7 | 117.3 | 117.8 | 119.6 |
| C1–C2–C4 | 120.0 | 120.0 | 120.9 | 121.1 | 121.4 | 119.7 |
| C1–C3–C5 | 120.0 | 120.0 | 122.4 | 121.6 | 120.6 | 120.7 |
| C6–C7–C8 | 119.7 | 119.6 | 127 | 126.3 | 126.7 | 128 |
| C1–C2–H | 119.9 | 119.9 | 119.9 | 119.4 | 117.2 | 120.2 |
| C1–C3–H | 119.9 | 119.9 | 117.9 | 118.7 | 119 | 119.6 |
| C6–C7–H | 120.8 | 120.8 | 114.6 | 115 | 116.1 | 113.5 |
| (a) THEORETICAL | ||||||||
|---|---|---|---|---|---|---|---|---|
| B3LYP/6-31+G*/AMBER [59] | AUTODOCK [60] | HF/3-21G/TIP3P [58] | AM1/TIP3P [57] | |||||
| Sigma | Epsilon | Sigma | Epsilon | Sigma | Epsilon | Sigma | Epsilon | |
| C…C | 4.02 | 0.06 | 4.00 | 0.150 | 3.80 | 0.08 | 3.50 | 0.08 |
| H…H | 2.22 | 0.03 | 2.00 | 0.020 | 2.60 | 0.01 | 2.00 | 0.01 |
| C…H | 3.12 | 0.04243 | 3 | 0.05477 | 3.2 | 0.02828 | 2.75 | 0.02828 |
| (b) EXPERIMENTAL | ||||||||
| AMBER-94 [56] | UFF [47] | |||||||
| Sigma | Epsilon | Sigma | Epsilon | |||||
| C…C | 3.40 | 0.10 | 3.851 | 0.105 | ||||
| H…H | 2.53 | 0.02 | 2.886 | 0.044 | ||||
| C…H | 2.965 | 0.04472 | 3.3685 | 0.06797 | ||||
| PPV | |||
|---|---|---|---|
| Monomer | |||
| 2PPV [55] | |||
| 3PPV [61] | |||
| 4PPV | |||
| 5PPV [26] |
| This Work | This Work | Yaya et al. [63] |
|---|---|---|
| 3PPV-(13,0) | ~−22.5 kcal/mol | −22.3687 kcal/mol (−0.97 eV) |
| 3PPV-(11,0) | ---- | −21.6769 kcal/mol (−0.94 eV) |
| 3PPV-(10,0) | ~−22 kcal/mol | ---- |
| 3PPV-(9,0) | ---- | −21.4463 kcal/mol (−0.93 eV) |
| 3PPV-(8,0) | ~−18 kcal/mol | ---- |
| 3PPV-(7,0) | ---- | −20.0627 kcal/mol (−0.87 eV) |
| 3PPV-(5,0) | ~−19 kcal/mol | ---- |
| This Work | This Work | Yaya et al. [63] |
|---|---|---|
| 3PPV-(10,10) | ---- | −23.2911 kcal/mol (−1.01 eV) |
| 3PPV-(8,8) | ~−22 kcal/mol | −22.3687 kcal/mol (−0.97 eV) |
| 3PPV-(6,6) | ~−23 kcal/mol | −21.4463 kcal/mol (−0.93 eV) |
| 3PPV-(5,5) | ~−25 kcal/mol | ---- |
| 3PPV-(4,4) | ---- | −20.5239 kcal/mol (−0.89 eV) |
| 3PPV-(3,3) | ~−21 kcal/mol | ---- |
Sample Availability: Data on PPV, SWCNTs and Graphene used for the computational calculations are available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dagadu, N.A.; Ajori, S.; Bensah, Y.D.; Kan-Dapaah, K.; Armah, S.K.; Onwona-Agyeman, B.; Yaya, A. Stacking Interactions of Poly Para-Phenylene Vinylene Oligomers with Graphene and Single-Walled Carbon Nanotubes: A Molecular Dynamics Approach. Molecules 2020, 25, 4812. https://doi.org/10.3390/molecules25204812
Dagadu NA, Ajori S, Bensah YD, Kan-Dapaah K, Armah SK, Onwona-Agyeman B, Yaya A. Stacking Interactions of Poly Para-Phenylene Vinylene Oligomers with Graphene and Single-Walled Carbon Nanotubes: A Molecular Dynamics Approach. Molecules. 2020; 25(20):4812. https://doi.org/10.3390/molecules25204812
Chicago/Turabian StyleDagadu, Nii Amah, Shahram Ajori, Yaw Delali Bensah, Kwabena Kan-Dapaah, Stephen Kofi Armah, Boateng Onwona-Agyeman, and Abu Yaya. 2020. "Stacking Interactions of Poly Para-Phenylene Vinylene Oligomers with Graphene and Single-Walled Carbon Nanotubes: A Molecular Dynamics Approach" Molecules 25, no. 20: 4812. https://doi.org/10.3390/molecules25204812
APA StyleDagadu, N. A., Ajori, S., Bensah, Y. D., Kan-Dapaah, K., Armah, S. K., Onwona-Agyeman, B., & Yaya, A. (2020). Stacking Interactions of Poly Para-Phenylene Vinylene Oligomers with Graphene and Single-Walled Carbon Nanotubes: A Molecular Dynamics Approach. Molecules, 25(20), 4812. https://doi.org/10.3390/molecules25204812