How Do Aromatic Nitro Compounds React with Nucleophiles? Theoretical Description Using Aromaticity, Nucleophilicity and Electrophilicity Indices


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Aromaticity
2.2. Electrophilicity and Nucleophilicity
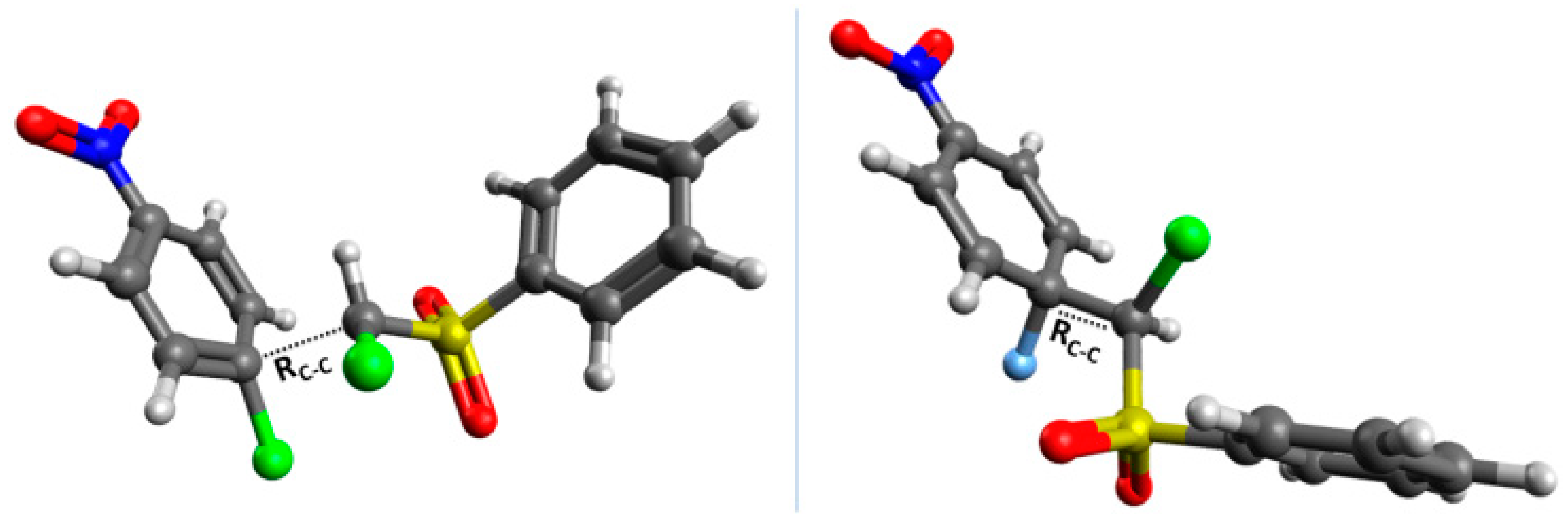
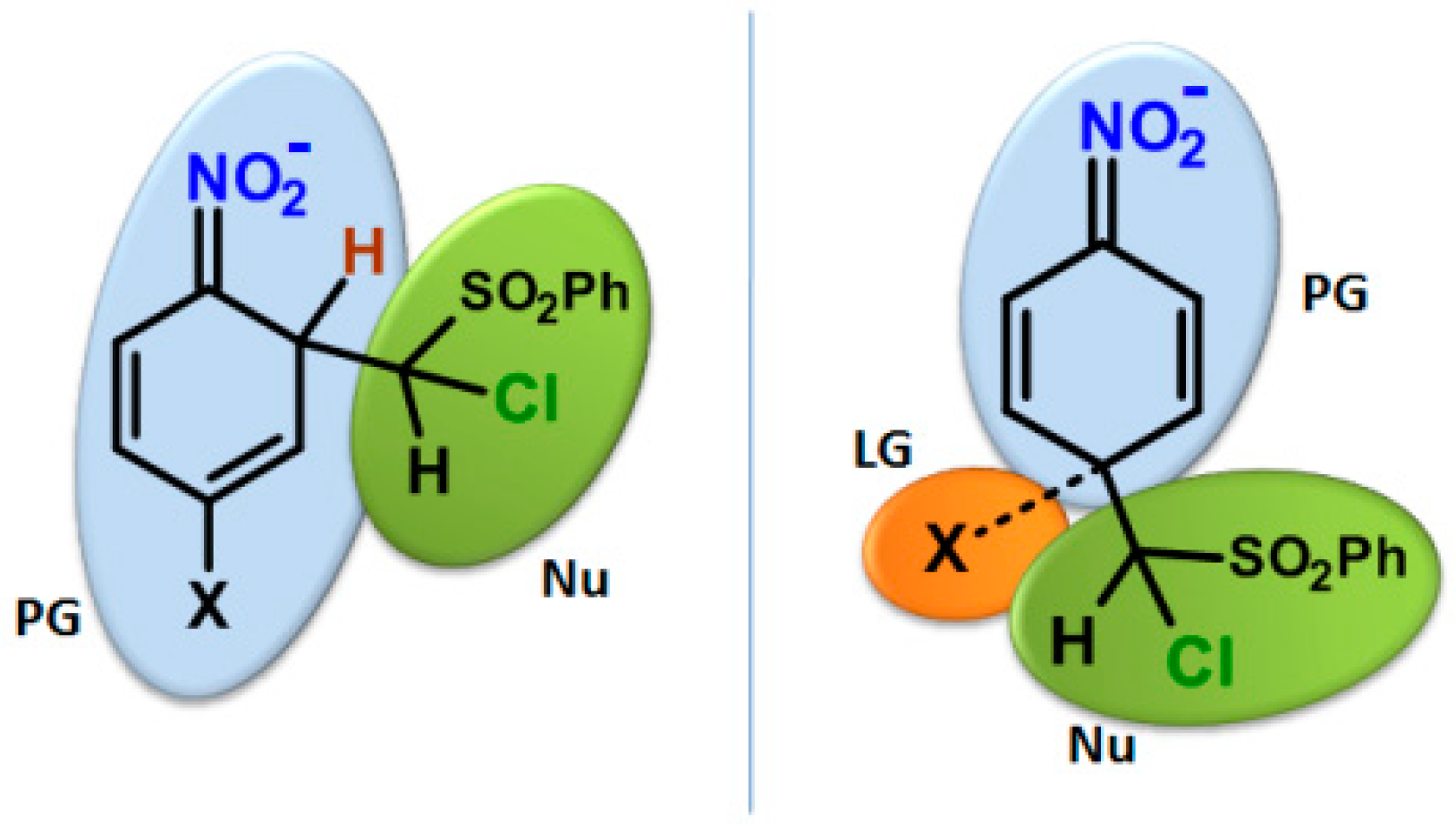
2.3. Kinetic Isotope Effect
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Miller, J. Aromatic Nucleophilic Substitution; Elsevier: New York, NY, USA, 1968. [Google Scholar]
- Caron, S.; Ghosh, A. Practical Synthetic Organic Chemistry; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2011; pp. 237–253. [Google Scholar]
- Crampton, M.R. Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Bunnett, J.F. Mechanism and Reactivity in Aromatic Nucleophilic Substitution Reactions. Quart. Rev. Chem. Soc. 1958, 12, 1–16. [Google Scholar] [CrossRef]
- Bunnett, J.F.; Zahler, R.E. Aromatic Nucleophilic Substitution Reactions. Chem. Rev. 1951, 49, 273–412. [Google Scholar] [CrossRef]
- Terrier, F. Modern Nucleophilic Aromatic Substitution; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Nudelman, N.S.; MacCormack, P. Theoretical Calculations of Chemical Interactions. Part 5. 1,1-Disubstituted 2,4,6-Trinitrocyclohexadienylide (’Meisenheimer’) Complexes. J. Chem. Soc. Perkin Trans. 2 1987, 227–229. [Google Scholar] [CrossRef]
- Dotterer, S.K.; Harris, R.L. Mndo Study of Nucleophilic Aromatic Substitution. J. Org. Chem. 1988, 53, 777–779. [Google Scholar] [CrossRef]
- Simkin, B.Y.; Gluz, E.B.; Glukhovtsev, M.N.; Minkin, V.I. Theoretical Study of Mechanisms of Aromatic Nucleophilic Substitution in the Gas Phase. J. Mol. Struct. Theochem. 1993, 284, 123–137. [Google Scholar] [CrossRef]
- Glukhovtsev, M.N.; Bach, R.D.; Laiter, S. Single-Step and Multistep Mechanisms of Aromatic Nucleophilic Substitution of Halobenzenes and Halonitrobenzenes with Halide Anions: Ab Initio Computational Study. J. Org. Chem. 1997, 62, 4036–4046. [Google Scholar] [CrossRef]
- Um, I.H.; Min, S.W.; Dust, J.M. Choice of Solvent (Mecn Vs H2o) Decides Rate-Limiting Step in Snar Aminolysis of 1-Fluoro-2,4-Dinitrobenzene with Secondary Amines: Importance of Brønsted-Type Analysis in Acetonitrile. J. Org. Chem. 2007, 72, 8797–8803. [Google Scholar] [CrossRef]
- Acevedo, O.; Jorgensen, V.L. Solvent Effects and Mechanism for a Nucleophilic Aromatic Substitution from Qm/Mm Simulations. Org. Lett. 2004, 6, 2881–2884. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.W.; Aptula, A.O.; Patlewicz, G.Y. Chemistry-Based Risk Assessment for Skin Sensitization: Quantitative Mechanistic Modeling for the Snar Domain. Chem. Res. Toxicol. 2011, 24, 1003–1011. [Google Scholar] [CrossRef]
- Imoto, M.; Matsui, Y.; Takeda, M.; Tamaki, A.; Taniguchi, H.; Mizuno, K.; Ikeda, H. A Probable Hydrogen-Bonded Meisenheimer Complex: An Unusually High Snar Reactivity of Nitroaniline Derivatives with Hydroxide Ion in Aqueous Media. J. Org. Chem. 2011, 76, 6356–6361. [Google Scholar] [CrossRef]
- Jose, K.B.; Cyriac, J.; Moolayil, J.T.; Sebastian, V.S.; George, M. The Mechanism of Aromatic Nucleophilic Substitution Reaction between Ethanolamine and Fluoro-Nitrobenzenes: An Investigation by Kinetic Measurements and Dft Calculations. J. Phys. Org. Chem. 2011, 24, 714–719. [Google Scholar] [CrossRef]
- Ormazábal-Toledo, R.; Contreras, R.; Campodónico, P.R. Reactivity Indices Profile: A Companion Tool of the Potential Energy Surface for the Analysis of Reaction Mechanisms. Nucleophilic Aromatic Substitution Reactions as Test Case. J. Org. Chem. 2013, 78, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Ormazabal-Toledo, R.; Contreras, R.; Tapia, R.A.; Campodonico, P.R. Specific Nucleophile-Electrophile Interactions in Nucleophilic Aromatic Substitutions. Org. Biomol. Chem. 2013, 11, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Contreras, R.; Andres, J.; Safont, V.S.; Campodonico, P.; Santos, J.G. A Theoretical Study on the Relationship between Nucleophilicity and Ionization Potentials in Solution Phase. J. Phys. Chem. A 2003, 107, 5588–5593. [Google Scholar] [CrossRef]
- Fernández, I.; Frenking, G.; Uggerud, E. Rate-Determining Factors in Nucleophilic Aromatic Substitution Reactions. J. Org. Chem. 2010, 75, 2971–2980. [Google Scholar] [CrossRef]
- Von Richter, V. Technical Translations. Chem. Ber. 1871, 4, 459. [Google Scholar]
- Davis, R.B.; Pizzini, L.C. Condensation of Aromatic Nitro Compounds with Acrylacetonitriles.1,2 Ii. Some P-Substituted Nitrobenzenes. J. Org. Chem. 1960, 25, 1884–1888. [Google Scholar] [CrossRef]
- Mąkosza, M.; Staliński, K. Oxidative Nucleophilic Substitution of Hydrogen in Nitroarenes. Chem. Eur. J. 1997, 3, 2025–2031. [Google Scholar] [CrossRef]
- Malykhin, E.V.; Kolesnichenko, G.A.; Shteingarts, V.D. Reaction of aromatic-compounds with nucleophilic-reagents in the liquid ammonium medium. 5. mechanism and hydroxylation orientation of para-substituted nitrobenzene with potassium hydroxide. Zh. Org. Khim. 1985, 21, 1150–1159. [Google Scholar]
- Goliński, J.; Makosza, M. “Vicarious” Nucleophilic Substitution of Hydrogen in Aromatic Nitro Compounds. Tetrahedron Lett. 1978, 19, 3495–3498. [Google Scholar] [CrossRef]
- Mąkosza, M.; Winiarski, J. Vicarious Nucleophilic Substitution of Hydrogen. Acc. Chem. Res. 1987, 20, 282–289. [Google Scholar] [CrossRef]
- Mąkosza, M. Nucleophilic Substitution of Hydrogen in Nitroarenes: A New Chapter of Aromatic Chemistry. Synthesis 2011, 2011, 2341–2356. [Google Scholar] [CrossRef]
- Mąkosza, M. Nucleophilic Substitution of Hydrogen in Electron-Deficient Arenes, a General Process of Great Practical Value. Chem. Soc. Rev. 2010, 39, 2855–2868. [Google Scholar] [CrossRef] [PubMed]
- Chupakhin, O.N.; Charushin, V.N.; Van der Plas, H.C. Nucleophilic Aromatic Substitution of Hydrogen; Academic Press: New York, NY, USA, 1994. [Google Scholar]
- Mąkosza, M.; Wojciechowski, K. Nucleophilic Substitution of Hydrogen in Heterocyclic Chemistry. Chem. Rev. 2004, 104, 2631–2666. [Google Scholar] [CrossRef]
- Antoniak, D.; Barbasiewicz, M. Corey–Chaykovsky Cyclopropanation of Nitronaphthalenes: Access to Benzonorcaradienes and Related Systems. Org. Lett. 2019, 21, 9320–9325. [Google Scholar] [CrossRef]
- Khutorianskyi, V.V.; Klepetářová, B.; Beier, P. Vicarious Nucleophilic Chloromethylation of Nitroaromatics. Org. Lett. 2019, 21, 5443–5446. [Google Scholar] [CrossRef]
- Brześkiewicz, J.; Loska, R.; Mąkosza, M. A-Chlorobenzylation of Nitroarenes Via Vicarious Nucleophilic Substitution with Benzylidene Dichloride: Umpolung of the Friedel–Crafts Reaction. J. Org. Chem. 2018, 83, 8499–8508. [Google Scholar] [CrossRef]
- Mąkosza, M. Reactions of Nucleophiles with Nitroarenes: Multifacial and Versatile Electrophiles. Chem. Eur. J. 2014, 20, 5536–5545. [Google Scholar] [CrossRef]
- Ormazábal-Toledo, R.; Richter, S.; Robles-Navarro, A.; Maulén, B.; Matute, R.A.; Gallardo-Fuentes, S. Meisenheimer Complexes as Hidden Intermediates in the Aza-Snar Mechanism. Org. Biomol. Chem. 2020, 18, 4238–4247. [Google Scholar] [CrossRef]
- Błaziak, K.; Danikiewicz, W.; Mąkosza, M. How Does Nucleophilic Aromatic Substitution Really Proceed in Nitroarenes? Computational Prediction and Experimental Verification. J. Am. Chem. Soc. 2016, 138, 7276–7281. [Google Scholar] [CrossRef]
- Mąkosza, M. How Does Nucleophilic Aromatic Substitution in Nitroarenes Really Proceed: General Mechanism. Synthesis 2017, 49, 3247–3254. [Google Scholar] [CrossRef]
- Makosza, M. Nucleophilic substitution in nitroarenes: A general corrected mechanism. ChemTexts 2019, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Lemek, T.; Mąkosza, M.; Stephenson, D.S.; Mayr, H. Direct Observation of the Intermediate in Vicarious Nucleophilic Substitutions of Hydrogen. Angew. Chem. Int. Ed. 2003, 42, 2793–2795. [Google Scholar] [CrossRef] [PubMed]
- Kruszewski, J.; Krygowski, T.M. Definition of Aromaticity Basing on the Harmonic Oscillator Model. Tetrahedron Lett. 1972, 13, 3839–3842. [Google Scholar] [CrossRef]
- Gwaltney, S.R.; Rosokha, S.V.; Head-Gordon, M.; Kochi, J.K. Charge-Transfer Mechanism for Electrophilic Aromatic Nitration and Nitrosation Via the Convergence of (Ab Initio) Molecular-Orbital and Marcus−Hush Theories with Experiments. J. Am. Chem. Soc. 2003, 125, 3273–3283. [Google Scholar] [CrossRef]
- Mąkosza, M.; Mortier, J. The Discovery and Development of Metal-Free Arylation Reactions with Unsymmetrical Diaryliodonium Salts; Mortier, J., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016; Chapter 11; pp. 269–298. [Google Scholar]
- Makosza, M.; Stalinski, K.; Klepka, C. Oxidative Nucleophilic Substitution of Hydrogen in Nitrobenzene with 2-Phenylpropionitrile Carbanion and Potassium Permanganate Oxidant. Chem. Commun. 1996, 837–838. [Google Scholar] [CrossRef]
- Wróbel, Z.; Mąkosza, M. New Simple Synthesis of N-Hydroxy 2-Vinylindoles. Synlett 1993, 1993, 597–598. [Google Scholar] [CrossRef]
- Mąkosza, M. Current Trends in Organic Synthesis, Proceedings of the Fourth International Conference on Organic Synthesis, Tokyo, Japan, 22–27 August 1982; Nozaki, H., Ed.; Pergamon: Oxford, UK, 1983; pp. 401–412. [Google Scholar] [CrossRef]
- Wróbel, Z.; Kwast, A. Simple Synthesis of N-Aryl-2-Nitrosoanilines in the Reaction of Nitroarenes with Aniline Anion Derivatives. Synthesis 2010, 2010, 3865–3872. [Google Scholar] [CrossRef]
- Swain, C.G.; Scott, C.B. Quantitative Correlation of Relative Rates. Comparison of Hydroxide Ion with Other Nucleophilic Reagents toward Alkyl Halides, Esters, Epoxides and Acyl Halides1. J. Am. Chem. Soc. 1953, 75, 141–147. [Google Scholar] [CrossRef]
- Edwards, J.O. Correlation of Relative Rates and Equilibria with a Double Basicity Scale. J. Am. Chem. Soc. 1954, 76, 1540–1547. [Google Scholar] [CrossRef]
- Ritchie, C.D. Nucleophilic Reactivities toward Cations. Acc. Chem. Res. 1972, 5, 348–354. [Google Scholar] [CrossRef]
- Ritchie, C.D. Cation–Anion Combination Reactions. 26. A Review. Can. J. Chem. 1986, 64, 2239–2250. [Google Scholar] [CrossRef]
- Lucius, R.; Mayr, H. Constant Selectivity Relationships of Addition Reactions of Carbanions. Angew. Chem. Int. Ed. 2000, 39, 1995–1997. [Google Scholar] [CrossRef]
- Mayr, H. Cc Bond Formation by Addition of Carbenium Ions to Alkenes: Kinetics and Mechanism. Angew. Chem. Int. Ed. Engl. 1990, 29, 1371–1384. [Google Scholar] [CrossRef]
- Mayr, H.; Bug, T.; Gotta, M.F.; Hering, N.; Irrgang, B.; Janker, B.; Kempf, B.; Loos, R.; Ofial, A.R.; Remennikov, G.; et al. Reference Scales for the Characterization of Cationic Electrophiles and Neutral Nucleophiles. J. Am. Chem. Soc. 2001, 123, 9500–9512. [Google Scholar] [CrossRef]
- Mayr, H.; Müller, K.H.; Rau, D. Comparison of the Electrophilicities of Cationic Metal Π Complexes and of Ordinary Carbenium Ions. Angew. Chem. Int. Ed. Engl. 1993, 32, 1630–1632. [Google Scholar] [CrossRef]
- Mayr, H.; Ofial, A.R. Electrophilicities of Iminium Ions. Tetrahedron Lett. 1997, 38, 3503–3506. [Google Scholar] [CrossRef]
- Mayr, H.; Ofial, A.R.; Sauer, J.; Schmied, B. [2++4] Cycloadditions of Iminium Ions − Concerted or Stepwise Mechanism of Aza Diels−Alder Reactions? Eur. J. Org. Chem. 2000, 2000, 2013–2020. [Google Scholar] [CrossRef]
- Roth, M.; Mayr, H. The Coexistence of the Reactivity–Selectivity Principle and of Linear Free Energy Relationships: A Diffusion Clock for Determining Carbocation Reactivities. Angew. Chem. Int. Ed. Engl. 1995, 34, 2250–2252. [Google Scholar] [CrossRef]
- Roy, R.K.; Krishnamurti, S.; Geerlings, P.; Pal, S. Local Softness and Hardness Based Reactivity Descriptors for Predicting Intra- and Intermolecular Reactivity Sequences: Carbonyl Compounds. J. Phys. Chem. A 1998, 102, 3746–3755. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Maiti, B. Reactivity Dynamics in Atom−Field Interactions: A Quantum Fluid Density Functional Study. J. Phys. Chem. A 2001, 105, 169–183. [Google Scholar] [CrossRef]
- Ormazabal-Toledo, R.; Contreras, R. Philicity and Fugality Scales for Organic Reactions. Adv. Chem. 2014, 2014, 13. [Google Scholar] [CrossRef]
- Błażej, S.; Mąkosza, M. Substituent Effects on the Electrophilic Activity of Nitroarenes in Reactions with Carbanions. Chem. Eur. J. 2008, 14, 11113–11122. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Persson, J.; Matsson, O. Use of Fluorine Kinetic Isotope Effects in the Study of Steric Effects in Nucleophilic Aromatic Substitution Reactions. J. Org. Chem. 1998, 63, 9348–9350. [Google Scholar] [CrossRef]
- Matsson, O.; Persson, J.; Axelsson, B.S.; Laangstroem, B. Fluorine Kinetic Isotope Effects. J. Am. Chem. Soc. 1993, 115, 5288–5289. [Google Scholar] [CrossRef]
- Mąkosza, M.; Lemek, T.; Kwast, A.; Terrier, F. Elucidation of the Vicarious Nucleophilic Substitution of Hydrogen Mechanism Via Studies of Competition between Substitution of Hydrogen, Deuterium, and Fluorine. J. Org. Chem. 2002, 67, 394–400. [Google Scholar] [CrossRef]
- Makosza, M.; Staliski, K. Oxidative Nucleophilic Substitution of Hydrogen with 2-Phenylpropanenitrile Carbanion in Heterocyclic Nitroarenes. Synthesis 1998, 11, 1631–1634. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Błaziak, K.; Danikiewicz, W.; Mąkosza, M. How Do Aromatic Nitro Compounds React with Nucleophiles? Theoretical Description Using Aromaticity, Nucleophilicity and Electrophilicity Indices. Molecules 2020, 25, 4819. https://doi.org/10.3390/molecules25204819
Błaziak K, Danikiewicz W, Mąkosza M. How Do Aromatic Nitro Compounds React with Nucleophiles? Theoretical Description Using Aromaticity, Nucleophilicity and Electrophilicity Indices. Molecules. 2020; 25(20):4819. https://doi.org/10.3390/molecules25204819
Chicago/Turabian StyleBłaziak, Kacper, Witold Danikiewicz, and Mieczysław Mąkosza. 2020. "How Do Aromatic Nitro Compounds React with Nucleophiles? Theoretical Description Using Aromaticity, Nucleophilicity and Electrophilicity Indices" Molecules 25, no. 20: 4819. https://doi.org/10.3390/molecules25204819
APA StyleBłaziak, K., Danikiewicz, W., & Mąkosza, M. (2020). How Do Aromatic Nitro Compounds React with Nucleophiles? Theoretical Description Using Aromaticity, Nucleophilicity and Electrophilicity Indices. Molecules, 25(20), 4819. https://doi.org/10.3390/molecules25204819