
Small Molecules Targeting Biological Clock; A Novel Prospective for Anti-Cancer Drugs


Abstract
:1. Introduction
2. Interrelation of Circadian Clock Genes and Cell Cycle
3. Oncogenesis and Circadian Clock

{kind=link}
| Cancer Type | Deregulated Clock Genes | Results | References |
|---|---|---|---|
| Breast Cancer | PER1, PER2, PER3, CRY2 | Downregulation | [31,32,39] |
| CLOCK, TIM | Upregulation | ||
| Hepatocellular carcinoma (HCC) | PER2 | Downregulation | [33] |
| Chronic myeloid leukemia (CML) | PER2 | Downregulation | [34] |
| Pancreatic Cancer | PER2 | Downregulation | [36] |
| Colorectal cancer (CRC) | PER2, NPAS2 | Downregulation | [40] |
| Gastric Cancer | PER2, CRY1 | Upregulation | [37] |
| Head and squamous cell carcinoma (HNSCC) | PER1, PER2, PER3, BMAL1, CRY2 | Downregulation | [28] |
4. Chemoresistance and the Circadian Clock
5. Screening Method for Circadian Clock Modulators
6. Management of Clock-Related Diseases Using Small Molecules
7. Management of Cancers Using Small Molecules
8. Synthetic Anticancer Chronobiotics
8.1. REV-ERBs; RORs; CRY1/2
8.1.1. SR9009/SR9011
8.1.2. SR1078
8.1.3. ARN5817
8.1.4. MLN4924
8.1.5. KS15
8.2. Casein Kinase
8.2.1. Longdaysin
8.2.2. DK359
8.2.3. CX-4945
8.2.4. GO289
8.3. Cyclin-Dependant Kinase (CDK9)
LY2857785
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| LD | Light dark |
| SCN | Suprachiasmatic nucleus |
| HNSCC | Head and neck squamous cell carcinoma |
| CML | Chronic myeloid leukemia |
| CRC | Colorectal cancer |
| HCC | Hepatocellular carcinoma |
| GC | Gastric cancer |
References
- Hida, A.; Kitamura, S.; Mishima, K.T. Pathophysiology and pathogenesis of circadian rhythm sleep disorders. J. Physiol. Anthropol. 2012, 1, 7. [Google Scholar] [CrossRef] [Green Version]
- Young, M.W.; Kay, S.A. Time zones: Comparative genetics of circadian clocks. Nat. Rev. Genet. 2001, 2, 702–715. [Google Scholar] [CrossRef]
- Hatori, M.; Panda, S. The emerging roles of melanopsin in behavioral adaptation to light. Trends Mol. Med. 2010, 16, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Hastings, M.H.; Maywood, E.S.; Brancaccio, M. Generation of circadian rhythms in the suprachiasmatic nucleus. Nat. Rev. Neurosci. 2018, 8, 453–469. [Google Scholar] [CrossRef] [PubMed]
- Balsalobre, A. Clock genes in mammalian peripheral tissues. Cell Tissue Res. 2002, 309, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Reppert, S.M.; Weaver, D.R. Coordination of circadian timing in mammals. Nature 2002, 418, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Strayer, C.A.; Kay, S.A. The ins and outs of circadian regulated gene expression. Curr. Opin. Plant Biol. 1999, 2, 114–120. [Google Scholar] [CrossRef]
- Rahman, S.; Kraljević Pavelić, S.; Markova-Car, E. Circadian (De)regulation in Head and Neck Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 11, E2662. [Google Scholar] [CrossRef] [Green Version]
- Eide, E.J.; Kang, H.; Crapo, S.; Gallego, M.; Virshup, D.M. Casein kinase I in the mammalian circadian clock. Methods Enzymol. 2005, 393, 408–418. [Google Scholar]
- Markova-Car, E.P.; Jurišic, D.; Ilic, N.; Kraljevic Pavelic, S. Running for time: Circadian rhythms and melanoma. Tumour. Biol. 2014, 35, 8359–8368. [Google Scholar] [CrossRef]
- Takahashi, J.S.; Hong, H.K.; Ko, C.H.; McDearmon, E.L. The genetics of mammalian circadian order and disorder: Implications for physiology and disease. Nat. Rev. Genet. 2008, 9, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Bechtold, D.A.; Gibbs, J.E.; Loudon, A.S. Circadian dysfunction in disease. Trends Pharmacol. Sci. 2010, 31, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Greene, M.W. Circadian rhythms and tumor growth. Cancer Lett. 2012, 2, 115–123. [Google Scholar] [CrossRef]
- Gery, S.; Koeffler, H.P. Circadian rhythms and cancer. Cell Cycle. 2010, 6, 1097–1103. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.; Gery, S.; Dashti, A.; Yin, D.; Zhou, Y.; Gu, J.; Koeffler, H.P. A role for the clock gene per1 in prostate cancer. Cancer Res. 2009, 19, 7619–7625. [Google Scholar] [CrossRef] [Green Version]
- Lahti, T.A.; Partonen, T.; Kyyronen, P.; Kauppinen, T.; Pukkala, E. Night-time work predisposes to non-Hodgkin lymphoma. Int. J. Cancer 2008, 9, 2148–2151. [Google Scholar] [CrossRef]
- Gauger, M.A.; Sancar, A. Cryptochrome, circadian cycle, cell cycle checkpoints, and cancer. Cancer Res. 2005, 65, 6828–6834. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Kettner, N.M. The circadian clock in cancer development and therapy. Prog. Mol. Biol. Transl. Sci. 2013, 119, 221–282. [Google Scholar]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Lee, C.C. The circadian clock: Pacemaker and tumour suppressor. Nat. Rev. Cancer 2003, 5, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Yamaguchi, S.; Mitsui, S.; Emi, A.; Shimoda, F.; Okamura, H. Control mechanism of the circadian clock for timing of cell division in vivo. Science 2003, 5643, 255–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Pelicano, H.; Liu, J.; Huang, P.; Lee, C. The circadian gene Period 2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 2002, 111, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Wood, P.A.; Ansell, C.M.; Quiton, D.F.; Oh, E.Y.; Du-Quiton, J.; Hrushesky, W.J. The circadian clock gene Per1 suppresses cancer cell proliferation and tumor growth at specific times of day. Chronobiol. Int. 2009, 7, 1323–1339. [Google Scholar] [CrossRef] [PubMed]
- Gery, S.; Komatsu, N.; Baldjyan, L.; Yu, A.; Koo, D.; Koeffler, H.P. The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Mol. Cell 2006, 3, 375–382. [Google Scholar] [CrossRef]
- Li, H.X. The role of circadian clock genes in tumors. Oncol. Targets Ther. 2019, 12, 3645–3660. [Google Scholar] [CrossRef] [Green Version]
- Cadenas, C.; van de Sandt, L.; Edlund, K.; Lohr, M.; Hellwig, B.; Marchan, R.; Schmidt, M.; Rahnenführer, J.; Oster, H.; Hengstler, J.G. Loss of circadian clock gene expression is associated with tumor progression in breast cancer. Cell Cycle 2014, 20, 3282–3291. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.M.; Lin, S.F.; Lu, C.T.; Lin, P.M.; Yang, M.Y. Altered expression of circadian clock genes in head and neck squamous cell carcinoma. Tumour. Biol. 2012, 33, 149–155. [Google Scholar] [CrossRef]
- Hansen, J. Increased breast cancer risk among women who work predominantly at night. Epidemiology 2001, 1, 74–77. [Google Scholar] [CrossRef]
- Schernhammer, E.S.; Laden, F.; Speizer, F.E.; Willett, W.C.; Hunter, D.J.; Kawachi, I.; Colditz, G.A. Rotating night shifts and risk of breast cancer in women participating in the nurses‘health study. J. Natl. Cancer Inst. 2001, 20, 1563–1568. [Google Scholar] [CrossRef] [Green Version]
- Davis, S.; Mirick, D.K.; Stevens, R.G. Night shift work, light at night, and risk of breast cancer. Natl. Cancer Inst. 2001, 20, 1557–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reszka, E.; Przybek, M. Circadian Genes in Breast Cancer. Adv. Clin. Chem. 2016, 75, 53–70. [Google Scholar] [PubMed]
- Lin, Y.M.; Chang, J.H.; Yeh, K.T.; Yang, M.Y.; Liu, T.C.; Lin, S.F.; Su, W.W.; Chang, J.G. Disturbance of circadian gene expression in hepatocellular carcinoma. Mol. Carcinog. 2008, 47, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.Y.; Yang, W.C.; Lin, P.M.; Hsu, J.F.; Hsiao, H.H.; Liu, Y.C.; Tsai, H.J.; Chang, C.S.; Lin, S.F. Altered expression of circadian clock genes in human chronic myeloid leukemia. J. Biol. Rhythms. 2011, 2, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.T.; Choo, K.B.; Hou, M.F.; Yeh, K.T.; Kuo, S.J.; Chang, J.G. Deregulated expression of the PER1, PER2 and PER3 genes in breast cancers. Carcinogenesis 2005, 7, 1241–1246. [Google Scholar] [CrossRef]
- Relles, D.; Sendecki, J.; Chipitsyna, G.; Hyslop, T.; Yeo, C.J.; Arafat, H.A. Circadian gene expression and clinicopathologic correlates in pancreatic cancer. J. Gastrointest. Surg. 2013, 3, 443–450. [Google Scholar] [CrossRef]
- Hu, M.; Yeh, K.; Lin, P.; Hsu, C.M.; Hsiao, H.H.; Liu, Y.C.; Lin, H.Y.; Lin, S.F.; Yang, M.Y. Deregulated expression of circadian clock genes in gastric cancer. BMC Gastroenterol. 2014, 14, 67. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Meng, X.; Wu, J.; Pan, C.; Ying, X.; Zhou, Y.; Liu, R.; Huang, W. Cryptochrome1 Overexpression correlates with tumor progression and poor prognosis in patients with colorectal cancer. PLoS ONE 2013, 4, e61679-10. [Google Scholar]
- Mao, Y.; Fu, A.; Hoffman, A.E.; Jacobs, D.I.; Jin, M.; Chen, K.; Zhu, Y. The circadian gene CRY2 is associated with breast cancer aggressiveness possibly via epigenomic modifications. Tumour Biol. 2015, 5, 3533–3539. [Google Scholar] [CrossRef]
- Xue, X.; Liu, F.; Han, Y.; Li, P.; Yuan, B.; Wang, X.; Chen, Y.; Kuang, Y.; Zhi, Q.; Zhao, H. Silencing NPAS2 promotes cell growth and invasion in DLD-1 cells and correlated with poor prognosis of colorectal cancer. Biochem. Biophys. Res. Commun. 2014, 450, 1058–1062. [Google Scholar] [CrossRef]
- Yi, C.; Mu, L.; de la Longrais, I.A.; Sochirca, O.; Arisio, R.; Yu, H.; Hoffman, A.E.; Zhu, Y.; Katsaro, D. The circadian gene NPAS2 is a novel prognostic biomarker for breast cancer. Breast Cancer Res. Treat. 2010, 120, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.L.; Wu, M.W.; Sun, J.; Sun, Y.L.; Cai, Y.C.; Huang, Y.J.; Xian, L.J. Effects of the biological clock gene Bmal1 on tumour growth and anti-cancer drug activity. J. Biochem. 2010, 3, 319–326. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Z.; Mo, G.; Chen, H. Association between circadian gene CLOCK and cisplatin resistance in ovarian cancer cells: A preliminary study. Ondcol. Lett. 2018, 6, 8945–8950. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, T.; Izumi, H.; Uchiumi, T.; Nishio, K.; Arao, T.; Tanabe, M.; Uramoto, H.; Sugio, K.; Yasumoto, K.; Sasaguri, Y.; et al. Clock and ATF4 transcription system regulates drug resistance in human cancer cell lines. Oncogene 2007, 26, 4749–4760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Long, J.; Yu-xia, S.; Li-li, H.; Jia-hua, L. Circadian Gene CLOCK Affects Drug-Resistant Gene Expression and Cell Proliferation in Ovarian Cancer SKOV3/DDP Cell Lines Through Autophagy. Cancer Biother. Radiopharm. 2017, 32, 139–146. [Google Scholar]
- Fang, L.; Yang, Z.; Zhou, J.; Tung, J.Y.; Hsiao, C.D.; Wang, L.; Deng, Y.; Wang, P.; Wang., J.; Lee, M.H. Circadian Clock Gene CRY2 Degradation Is Involved in Chemoresistance of Colorectal Cancer. Mol. Cancer Ther. 2015, 6, 1476–1487. [Google Scholar] [CrossRef] [Green Version]
- Hong, Z.; Feng, Z.; Sai, Z.; Tao, S. PER3, a novel target of miR-103, plays a suppressive role in colorectal cancer in vitro. BMB Rep. 2014, 9, 500–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Yan, D.; Teng, M.; Fan, J.; Zhou, C.; Li, D.; Qiu, G.; Sun, X.; Li, T.; Xing, T.; et al. Reduced expression of PER3 is associated with incidence and development of colon cancer. Ann. Surg. Oncol. 2012, 9, 3081–3088. [Google Scholar] [CrossRef]
- Zhang, F.; Sun, H.; Zhang, S.; Yang, X.; Zhang, G.; Su, T. Overexpression of PER3 Inhibits Self-Renewal Capability and Chemoresistance of Colorectal Cancer Stem-Like Cells via Inhibition of Notch and β-Catenin Signaling. Oncol. Res. 2017, 5, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Tan, Y.; Liang, Y.; Li, Y.; Chen, L.; Wu, S.; Xu, W.; Wang, Y.; Zhao, W.; Wu, J. Per2 participates in AKT-mediated drug resistance in A549/DDP lung adenocarcinoma cells. Oncol. Lett. 2017, 13, 423–428. [Google Scholar] [CrossRef] [Green Version]
- Katamune, C.; Koyanagi, S.; Hashikawa, K.I.; Kusunose, N.; Akamine, T.; Matsunaga, N.; Ohdo, S. Mutation of the gene encoding the circadian clock component PERIOD2 in oncogenic cells confers chemoresistance by up-regulating the Aldh3a1 gene. J. Biol. Chem. 2019, 2, 547–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, A.; Katayose, Y.; Yabuuchi, S.; Yamamoto, K.; Mizuma, M.; Shirasou, S.; Onogawa, T.; Ohtsuka, H.; Yoshida, H.; Hayashi, H.; et al. Clock Gene Mouse Period2 Overexpression Inhibits Growth of Human Pancreatic Cancer Cells and Has Synergistic Effect with Cisplatin. Anticancer Res. 2009, 4, 1201–1209. [Google Scholar]
- Mitchell, M.I.; Engelbrecht, A.M. Circadian Rhythms and Breast Cancer: The Role of Per2 in Doxorubicin-Induced Cell Death. J. Toxicol. 2015, 2015, 392360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dakup, P.P.; Porter, K.I.; Little, A.A.; Gajula, R.P.; Zhang, H.; Skornyakov, E.; Kemp, M.G.; Van Dongen, H.P.A.; Gaddameedhi, S. The circadian clock regulates cisplatin-induced toxicity and tumor regression in melanoma mouse and human models. Oncotarget 2018, 9, 14524–14538. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Cheng, B.; Xie, M.; Chen, Y.; Zhao, J.; Zhou, X.; Chen, L. Circadian Clock Gene Bmal1 Inhibits Tumorigenesis and Increases Paclitaxel Sensitivity in Tongue Squamous Cell Carcinoma. Cancer Res. 2017, 2, 532–544. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Luo, H.; Yang, J.; Wu, W.; Chen, D.L.; Huang, P.; Xu, R.H. Overexpression of the Circadian Clock Gene Bmal1 Increases Sensitivity to Oxaliplatin in Colorectal Cancer. Clin. Cancer Res. 2014, 20, 1042–1052. [Google Scholar] [CrossRef] [Green Version]
- Gorbacheva, V.Y.; Kondratov, R.V.; Zhang, R.; Cherukuri, S.; Godkov, A.V.; Takahashi, J.S.; Antoch, M.P. Circadian sensitivity to the chemotherapeutic agent cyclophosphamide depends on the functional status of the CLOCK/BMAL1 transactivation complex. Proc. Natl. Acad. Sci. USA 2005, 9, 3407–3412. [Google Scholar] [CrossRef] [Green Version]
- Korkmaz, T.; Aygenli, F.; Emisoglu, H.; Ozcelik, G.; Canturk, A.; Yilmaz, S.; Ozturk, N. Opposite Carcinogenic Effects of Circadian Clock Gene BMAL1. Sci. Rep. 2018, 8, 16023. [Google Scholar] [CrossRef] [Green Version]
- Burgermeister, E.; Battaglin, F.; Eladly, F.; Wu, W.; Herweck, F.; Schulte, N.; Betge, J.; Härtel, N.; Kather, J.N.; Weis, C.A.; et al. Aryl hydrocarbon receptor nuclear translocator-like (ARNTL/BMAL1) is associated with bevacizumab resistance in colorectal cancer via regulation of vascular endothelial growth factor A. EBioMedicine 2019, 45, 139–154. [Google Scholar] [CrossRef] [Green Version]
- Wallach, T.; Kramer, A. Chemical chronobiology: Toward drugs manipulating time. FEBS Lett. 2015, 589, 1530–1538. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Yoo, S.H.J.; Takahashi, J.S. Development and therapeutic potential of small-molecule modulators of circadian systems. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 231–252. [Google Scholar] [CrossRef] [PubMed]
- Antoch, M.P.; Kondratov, R.V. Pharmacological Modulators of the Circadian Clock as Potential Therapeutic Drugs: Focus on Genotoxic/Anticancer Therapy. Handb. Exp. Pharmacol. 2013, 217, 289–309. [Google Scholar]
- Chen, Z.; Yoo, S.H.; Takahashi, J.S. Small molecule modifiers of circadian clocks. Cell Mol. Life Sci. 2013, 16, 2985–2998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.; Hirota, T. Pharmacological Interventions to Circadian Clocks and Their Molecular Bases. J. Mol. Biol. 2020, 432, 3498–3514. [Google Scholar] [CrossRef]
- Son, G.H.; Chung, S.; Ramirez, V.D. Pharmacological Modulators of Molecular Clock and their Therapeutic Potentials in Circadian Rhythm-Related Diseases. Med. Chem. 2016, 6, 12. [Google Scholar] [CrossRef]
- Chang, M.R.; He, Y.; Khan, T.M.; Kuruvilla, D.S.; Garcia-Ordonez, R.; Corzo, C.A.; Unger, T.J.; White, D.W.; Khan, S.; Lin, L.; et al. Antiobesity Effect of a Small Molecule Repressor of RORγ. Mol. Pharmacol. 2015, 1, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Hirota, T.; Lee, J.W.; St John, P.C.; Sawa, M.; Iwaisako, K.; Noguchi, T.; Pongsawakul, P.Y.; Sonntag, T.; Welsh, D.K.; Brenner, D.A.; et al. Identification of small molecule activators of cryptochrome. Science 2012, 6098, 1094–1097. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Chen, Z. Molecular Targets for Small-Molecule Modulators of Circadian Clocks. Curr. Drug Metab. 2016, 5, 503–512. [Google Scholar] [CrossRef] [Green Version]
- Yu, E.A.; Weaver, D.R. Disrupting the circadian clock: Gene-specific effects on aging, cancer, and other phenotypes. Aging 2011, 3, 479–493. [Google Scholar] [CrossRef] [Green Version]
- Sulli, G.; Manoogian, E.N.C.; Taub, P.R.; Panda, S. Training the Circadian Clock, Clocking the Drugs, and Drugging the Clock to Prevent, Manage, and Treat Chronic Diseases. Trends Pharmacol. Sci. 2018, 9, 812. [Google Scholar] [CrossRef]
- Scheiermann, C.; Kunisaki, Y.; Frenette, P.S. Circadian control of the immune system. Nat. Rev. Immunol. 2013, 13, 190–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delezie, J.; Dumont, S.; Dardente, H.; Oudart, H.; Gr-echez-Cassiau, A.; Klosen, P.; Teboul, M.; Delaunay, F.; Pévet, P.; Challet, E. The nuclear receptor REV-ERBα is required for the daily balance of carbohydrate and lipid metabolism. FASEB J. 2012, 8, 3321–3335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solt, L.A.; Wang, Y.; Banerjee, S.; Hughes, T.; Kojetin, D.J.; Lundasen, T.; Shin, Y.; Liu, J.; Cameron, M.D.; Noel, R.; et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature 2012, 7396, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Woldt, E.; Sebti, Y.; Solt, L.A.; Duhem, C.; Lancel, S.; Eeckhoute, J.; Hesselink, M.K.; Paquet, C.; Delhaye, S.; Shin, Y.; et al. Rev-erb-a modulates skeletal muscle oxidative capacity by regulating mitochondrial biogenesis and autophagy. Nat. Med. 2013, 8, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Grant, D.; Yin, L.; Collins, J.L.; Parks, D.J.; Orband-Miller, L.A.; Wisely, G.B.; Joshi, S.; Lazar, M.A.; Willson, T.M.; Zuercher, W.J. GSK4112, a small molecule chemical probe for the cell biology of the nuclear heme receptor Rev-erbα. ACS Chem. Biol. 2010, 5, 925–932. [Google Scholar] [CrossRef]
- Hong, C.; Tontonoz, P. Liver X receptors in lipid metabolism: Opportunities for drug discovery. Nat. rev. Drug Discov. 2014, 13, 433–444. [Google Scholar] [CrossRef]
- Steffensen, K.R.; Jakobsson, T.; Gustafsson, J.A. Targeting liver X receptors in inflammation. Expert Opin. Ther. Targets 2013, 17, 977–990. [Google Scholar] [CrossRef]
- Trump, R.P.; Bresciani, S.; Cooper, A.W.; Tellam, J.P.; Wojno, J.; Blaikley, J.; Orband-Miller, L.A.; Kashatus, J.A.; Boudjelal, M.; Dawson, H.C.; et al. Optimized chemical probes for REV-ERBα. J. Med. Chem. 2013, 56, 4729–4737. [Google Scholar] [CrossRef]
- Sulli, G.; Rommel, A.; Wang, X.; Kolar, M.J.; Puca, F.; Saghatelian, A.; Plikus, M.V.; Verma, I.M.; Panda, S. Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature 2018, 7688, 351–355. [Google Scholar] [CrossRef]
- Wagner, P.M.; Monjes, N.M.; Guido, M.E. Chemotherapeutic Effect of SR9009, a REV-ERB Agonist, on the Human Glioblastoma T98G Cells. ASN Neuro 2019, 11, 1759091419892713. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Zhang, W.; Ye, W.; Wang, H.; Zhang, Q.; Shen, J.; Hong, Q.; Li, X.; Wen, G.; Wei, T.; et al. SR9009 induces a REV-ERB dependent anti-small-cell lung cancer effect through inhibition of autophagy. Theranostics 2020, 10, 4466–4480. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Solt, L.A.; Conkright, J.J.; Wang, Y.; Istrate, M.A.; Busby, S.A.; Garcia-Ordonez, R.D.; Burris, T.P.; Griffin, P.R. The benzenesulfonamide T0901317 is a novel ROR alpha/gamma Inverse Agonist. Mol. Pharmacol. 2010, 77, 228–236. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Kumar, N.; Nuhant, P.; Cameron, M.D.; Istrate, M.A.; Roush, W.R.; Griffin, P.R.; Burris, T.P. Identification of SR1078, a synthetic agonist for the orphan nuclear receptors RORα and RORγ. ACS Chem. Biol. 2010, 11, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; McAvoy, S.; Kuhn, R.; Smith, D.I. RORA, a large common fragile site gene, is involved in cellular stress response. Oncogene 2006, 25, 2901–2908. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Solt, L.A.; Kojetin, D.J.; Burris, T.P. Regulation of p53 stability and apoptosis by a ROR agonist. PLoS ONE 2012, 4, e34921. [Google Scholar] [CrossRef] [Green Version]
- De Mei, C.; Ercolani, L.; Parodi, C.; Veronesi, M.; Lo Vecchio, C.; Bottegoni, G.; Torrente, E.; Scarpelli, R.; Marotta, R.; Ruffili, R.; et al. Dual inhibition of REV-ERBβ and autophagy as a novel pharmacological approach to induce cytotoxicity in cancer cells. Oncogene 2015, 34, 2597–2608. [Google Scholar] [CrossRef] [PubMed]
- Checinska, A.; Soengas, M.S. The gluttonous side of malignant melanoma: Basic and clinical implications of macroautophagy. Pigment Cell Melanoma Res. 2011, 24, 1116–1132. [Google Scholar] [CrossRef]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell. Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Cheong, H.; Lu, C.; Lindsten, T.; Thompson, C.B. Therapeutic targets in cancer cell metabolism and autophagy. Nat. Biotechnol. 2012, 30, 671–678. [Google Scholar] [CrossRef]
- Green, D.R.; Levine, B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014, 157, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- Carew, J.S.; Kelly, K.R.; Nawrocki, S.T. Autophagy as a target for cancer therapy: New developments. Cancer Manag. Res. 2012, 4, 357–365. [Google Scholar]
- Torrente, E.; Parodi, C.; Ercolani, L.; De Mei, C.; Ferrari, A.; Scarpelli, R.; Grimaldi, B. Synthesis and in Vitro Anticancer Activity of the First Class of Dual Inhibitors of REV-ERBβ and Autophagy. J. Med. Chem. 2015, 15, 5900–5915. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, J.; Deng, Z.; Liu, H.; Mao, W.; Jiang, F.; Xia, Z.; Li, J.D. Circadian clock components RORα and Bmal1 mediate the anti-proliferative effect of MLN4924 in osteosarcoma cells. Oncotarget 2016, 40, 66087–66099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visconti, R.; Della Monica, R.; Palazzo, L.; D’Alessio, F.; Raia, M.; Improta, S.; Villa, M.R.; Del Vecchio, L.; Grieco, D. The Fcp1-Wee1-Cdk1 axis affects spindle assembly checkpoint robustness and sensitivity to antimicrotubule cancer drugs. Cell Death Differ. 2015, 22, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Hogenesch, J.B.; Gu, Y.Z.; Jain, S.; Bradfield, C.A. The basic-helix-loop-helix-PAS orphan MOP3 forms transcriptionally active complexes with circadian and hypoxia factors. Proc. Natl. Acad. Sci. USA 1998, 95, 5474–5479. [Google Scholar] [CrossRef] [Green Version]
- Takahata, S.; Sogawa, K.; Kobayashi, A.; Ema, M.; Mimura, J.; Ozaki, N.; Fujii-Kuriyama, Y. Transcriptionally active heterodimer formation of an Arnt-like PAS protein, Arnt3, with HIF-1a, HLF, and Clock. Biochem. Biophys. Res. Commun. 1998, 248, 789–794. [Google Scholar] [CrossRef]
- Gekakis, N.; Staknis, D.; Nguyen, H.B.; Davis, F.C.; Wilsbacher, L.D.; King, D.P.; Takahashi, J.S.; Weitz, C.J. Role of the CLOCK protein in the mammalian circadian mechanism. Science 1998, 280, 1564–1569. [Google Scholar] [CrossRef]
- Chun, S.K.; Jang, J.; Chung, S.; Yun, H.; Kim, N.J.; Jung, J.W.; Son, G.H.; Suh, Y.G.; Kim, K. Identification and validation of cryptochrome inhibitors that modulate the molecular circadian clock. ACS Chem. Biol. 2014, 3, 703–710. [Google Scholar] [CrossRef]
- Nangle, S.N.; Rosensweig, C.; Koike, N.; Tei, H.; Takahashi, J.S.; Green, C.B.; Zheng, N. Molecular assembly of the period-cryptochrome circadian transcriptional repressor complex. elife 2014, 3, e03674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, S.K.; Chung, S.; Kim, H.D.; Lee, J.H.; Jang, J.; Kim, J.; Kim, D.; Son, G.H.; Oh, Y.J.; Suh, Y.G.; et al. A synthetic cryptochrome inhibitor induces anti-proliferative effects and increases chemoensitivity in human breast cancer cells. Biochem. Biophys. Res. Commun. 2015, 2, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Chung, S.; Choi, Y.; Lim, H.Y.; Son, Y.; Chun, S.K.; Son, G.H.; Kim, K.; Suh, Y.G.; Jung, J.W. The cryptochrome inhibitor KS15 enhances E-box-mediated transcrition by disrupting the feedback action of a circadian transcription-repressor complex. Life Sci. 2018, 200, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Eide, E.J.; Woolf, M.F.; Kang, H.; Woolf, P.; Hurst, W.; Camacho, F.; Vielhaber, E.L.; Giovanni, A.; Virshup, D.M. Control of mammalian circadian rhythm by CKIε-regulated proteasome mediated PER2 degradation. Mol. Cell. Biol. 2005, 7, 2795. [Google Scholar] [CrossRef] [Green Version]
- Etchegaray, J.P.; Machida, K.K.; Noton, E.; Constance, C.M.; Dallmann, R.; Di Napoli, M.N.; DeBruyne, J.P.; Lambert, C.M.; Yu, E.A.; Reppert, S.M.; et al. Casein kinase 1δ regulates the pace of the mammalian circadian clock. Mol. Cell. Biol. 2009, 14, 3853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isojima, Y.; Nakajima, M.; Ukai, H.; Fujishima, H.; Yamada, R.G.; Masumoto, K.H.; Kiuchi, R.; Ishida, M.; Ukai-Tadenuma, M.; Minami, Y.; et al. CKIe/d-dependent phosphorylation is a temperature-insensitive, period-determiningprocess in the mammalian circadian clock. Proc. Natl. Acad. Sci. USA 2009, 106, 15744–15749. [Google Scholar] [CrossRef] [Green Version]
- Hirota, T.; Lewis, W.G.; Liu, A.C.; Lee, J.W.; Schultz, P.G.; Kay, S.A. A chemical biology approach reveals period shortening of the mammalian circadian clock by specific inhibition of GSK-3beta. Proc. Natl. Acad. Sci. USA 2008, 52, 20746–20751. [Google Scholar] [CrossRef] [Green Version]
- Hirota, T.; Lee, J.W.; Lewis, W.G.; Zhang, E.E.; Breton, G.; Liu, X.; Garcia, M.; Peters, E.C.; Etchegaray, J.P.; Traver, D.; et al. High-throughput chemical screen identifies a novel potent modulator of cellular circadian rhythms and reveals CKIα as a clock regulatory kinase. PLoS Biol. 2010, 12, e1000559. [Google Scholar] [CrossRef] [Green Version]
- Fabian, M.A.; Biggs, W.H., 3rd; Treiber, D.K.; Atteridge, C.E.; Azimioara, M.D.; Benedetti, M.G.; Carter, T.A.; Ciceri, P.; Edeen, P.T.; Floyd, M.; et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [Google Scholar] [CrossRef]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef]
- Takahashi, J.S.; Zatz, M. Regulation of circadian rhythmicity. Science 1982, 217, 1104–1111. [Google Scholar] [CrossRef]
- Vitaterna, M.H.; Ko, C.H.; Chang, A.M.; Buhr, E.D.; Fruechte, E.M.; Schook, A.; Antoch, M.P.; Turek, F.W.; Takahashi, J.S. The mouse Clock mutation reduces circadian pacemaker amplitude and enhances efficacy of resetting stimuli and phase-response curve amplitude. Proc. Natl. Acad. Sci. USA 2006, 103, 9327–9332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Yoo, S.H.; Park, Y.S.; Kim, K.H.; Wei, S.; Buhr, E.; Ye, Z.Y.; Pan, H.L.; Takahashi, J.S. Identification of diverse modulators of central and peripheral circadian clocks by high-throughput chemical screening. Proc. Natl. Acad. Sci. USA 2012, 1, 101–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolarski, D.; Sugiyama, A.; Breton, G.; Rakers, C.; Ono, D.; Schulte, A.; Tama, F.; Itami, K.; Szymanski, W.; Hirota, T.; et al. Controlling the Circadian Clock with Hugh Temporal Resolution through Photodosing. J. Am. Chem. Soc. 2019, 40, 15784–15791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, J.S.; Litchfield, D.W. Too much of a good thing: The role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochem. Biophys. Acta 2008, 1784, 33–47. [Google Scholar] [CrossRef]
- Ruzzene, M.; Pinna, L.A. Addiction to protein kinase CK2: A common denominator of diverse cancer cells? Biochim. Biophys. Acta 2010, 1804, 499–504. [Google Scholar] [CrossRef]
- Guerra, B.; Issinger, O.G. Protein kinase CK2 in human diseases. Curr. Med. Chem. 2008, 15, 1870–1886. [Google Scholar] [CrossRef]
- Solimini, N.L.; Luo, J.; Elledge, S.J. Non-oncogene addiction and the stress phenotype of cancer cells. Cell 2007, 130, 986–988. [Google Scholar] [CrossRef] [Green Version]
- Kramerov, A.A.; Saghizadeh, M.; Caballero, S.; Shaw, L.C.; Calzi, S.L.; Bretner, M.; Montenarh, M.; Pinna, L.A.; Grant, M.B.; Ljubimov, A.V. Inhibition of protein kinase CK2 suppresses angiogenesis and hematopoietic stem cell recruitment to retinal neovascularization sites. Mol. Cell Biochem. 2008, 316, 177–186. [Google Scholar] [CrossRef]
- Mottet, D.; Ruys, S.P.; Demazy, C.; Raes, M.; Michiels, C. Role for casein kinase 2 in the regulation ofHIF-1 activity. Int. J. Cancer 2005, 117, 764–774. [Google Scholar] [CrossRef]
- Maier, B.; Wendt, S.; Vanselow, J.T.; Wallach, T.; Reischl, S.; Oehmke, S.; Schlosser, A.; Kramer, A. A large-scale functional RNAi screen reveals a role for CK2 in the mammalian circadian clock. Genes Dev. 2009, 23, 708–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, Y.; Akashi, M.; Matsuda, M.; Goto, K.; Miyata, Y.; Node, K.; Nishida, E. Involvement of the protein kinase CK2 in the regulation of mammalian circadian rhythms. Sci. Signal. 2009, 2, ra26. [Google Scholar] [CrossRef]
- Tamaru, T.; Hirayama, J.; Isojima, Y.; Nagai, K.; Norioka, S.; Takamatsu, K.; Sassone-Corsi, P. CK2 phosphorylates BMAL1 to regulate the mammalian clock. Nat. Struct. Mol. Biol. 2009, 16, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945 an orally bioavailable selective inhibitor of proteine kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 24, 10288–10298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshima, T.; Niwa, Y.; Kuwata, K.; Srivastava, A.; Hyoda, T.; Tsuchiya, Y.; Kumagai, M.; Tsuyuguchi, M.; Tamaru, T.; Sugiyama, A.; et al. Cell-based screen identifies a new potent and highly selective CK2 inhibitor for modulation of circadian rhythms and cancer cell growth. Sci. Adv. 2019, 1, eaau9060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Zhao, X.; Hirota, T.; Han, X.; Cho, H.; Chong, L.W.; Lamia, K.; Liu, S.; Atkins, A.R.; Banayo, E.; Liddle, C.; et al. Circadian amplitude regulation via FBXW7-targeted REV-ERBalpha degradation. Cell 2016, 165, 1644–1657. [Google Scholar] [CrossRef] [Green Version]
- Gaucher, J.; Montellier, E.; Sassone-Corsi, P. Molecular cogs: Interplay between circadian clock and cell cycle. Trends Cell Biol. 2018, 28, 368–379. [Google Scholar] [CrossRef]
- Ou, J.; Li, H.; Qiu, P.; Li, Q.; Chang, H.C.; Tang, Y.C. CDK9 modulates circadian clock by attenuating REV-ERBα activity. Biochem. Biophys. Res. Commun. 2019, 4, 967–973. [Google Scholar] [CrossRef]
- Gressel, S.; Schwalb, B.; Decker, T.M.; Qin, W.; Leonhardt, H.; Eick, D.; Cramer, P. CDK9-dependent RNA polymerase II pausing controls transcription initiation. elife 2017, 6, e29736. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Lach, R.; Heesom, K.J.; Valekunja, U.K.; Encheva, V.; Snijders, A.P.; Reddy, A.B. Phenotypic proteomic profiling identifies a landscape of targets for circadian clock-modulating compounds. Life Sci. Alliance 2019, 6, e201900603. [Google Scholar] [CrossRef] [PubMed] [Green Version]

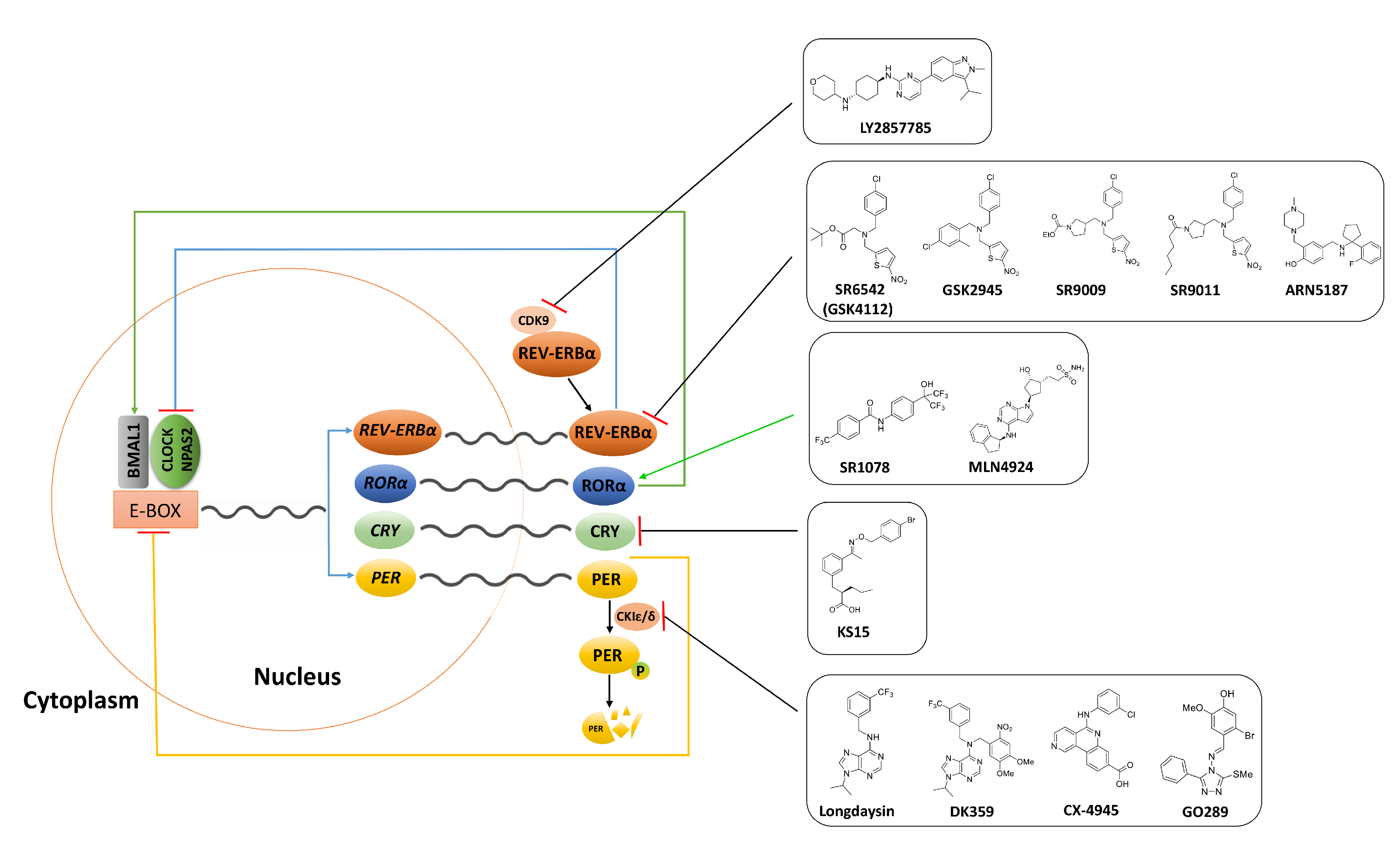
| Synthetic Anticancer Chronobiotics | |||||
|---|---|---|---|---|---|
| (A) REV-ERBs; RORs; CRY1/2 | |||||
| Compound | Chemical Structure | Function | Mechanism | System | Reference |
| SR6542 (GSK4112) |  | REV-ERB agonist | Inhibition of Bmal1 expression | HepG2 liver cell line | [75] |
| GSK2945 |  | REV-ERB agonist | 1000-fold selectivity for REV-ERB versus LXRα | HepG2 liver cell line | [78] |
| SR9009 |  | REV-ERB agonist | Circadian clock repressor Cytotoxic properties to a range of cancers e.g., brain, leukemia, breast, colon; in vivo inhibition of in vivo glioblastoma growth | Human gliobalstoma T98G cells; HepG2 cells | [73,79,80,81] |
| SR9011 |  | REV-ERB agonist | |||
| SR1078 |  | ROR agonist | Activation of target gene expression, such as G6Pase and fibroblast growth factor 21; inhibition of HepG2 and Hep3B hepatoma cell growth both in vitro and in vivo | HepG2 cells and mouse liver | [83] |
| ARN5187 |  | REV-ERBα/β agonist REV-ERBα IC50: 790 nM REV-ERBβ IC50: 560 nM; Inhibitor of autophagy | Apoptotic induction/anticancer activity; blocking later stages of autophagy | Breast cancer BT-474 cells | [86] |
| MLN4924 |  | RORα stabilization | Transactivation of Bmal1; induction of G2/M cell cycle arrest and apoptosis | U2OS osteosarcoma cells | [95] |
| KS15 |  | CRY inhibitor | Activation and enhancement of CLOCK-BMAL1-evoked E-box mediated transcription | MCF-7 breast cancer cell line | [100,102,103] |
| (B) Casein Kinase | |||||
| Longdaysin |  | CKIα/δ inhibitor IC50: CKIα 5.6 μM IC50: CKIδ 8.8 μM | The period-lengthening effect | Cells, tissues, and zebrafish in vivo | [108] |
| DK359 (caged longdaysin) |  | Inactive against CKIα/δ | Temporal control of the clock period | Cells, tissues, and zebrafish in vivo | [114] |
| CX-4945 (silmitasertib) |  | CK2 inhibitor IC50: CK2 1 nM | Broad antiproliferative activity; supression of angiogenesis; decreased phosphorylation of Akt substrate p21 | Panel of cancer cell lines; human umbilical vein endothelial cell (HUVEC); murine xenograft models | [124] |
| GO289 |  | CK2 inhibitor IC50: 7 nM | Inhibition of multiple phosphorylation sites on clock proteins | Human U2OS osteosarcoma cells | [125] |
| (C) Cyclin Dependent Kinase | |||||
| LY2857785 |  | CDK9 inhibitor IC50: 11 nM | Enhancement of REV-ERBα expression; liberation of REV-ERBα from CDK9; tuning RORE-mediated transcriptional activity | mPer2Luc mouse embryonic fibroblast (MEF) cell line | [130] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, S.; Wittine, K.; Sedić, M.; Markova-Car, E.P. Small Molecules Targeting Biological Clock; A Novel Prospective for Anti-Cancer Drugs. Molecules 2020, 25, 4937. https://doi.org/10.3390/molecules25214937
Rahman S, Wittine K, Sedić M, Markova-Car EP. Small Molecules Targeting Biological Clock; A Novel Prospective for Anti-Cancer Drugs. Molecules. 2020; 25(21):4937. https://doi.org/10.3390/molecules25214937
Chicago/Turabian StyleRahman, Sadia, Karlo Wittine, Mirela Sedić, and Elitza P. Markova-Car. 2020. "Small Molecules Targeting Biological Clock; A Novel Prospective for Anti-Cancer Drugs" Molecules 25, no. 21: 4937. https://doi.org/10.3390/molecules25214937
APA StyleRahman, S., Wittine, K., Sedić, M., & Markova-Car, E. P. (2020). Small Molecules Targeting Biological Clock; A Novel Prospective for Anti-Cancer Drugs. Molecules, 25(21), 4937. https://doi.org/10.3390/molecules25214937