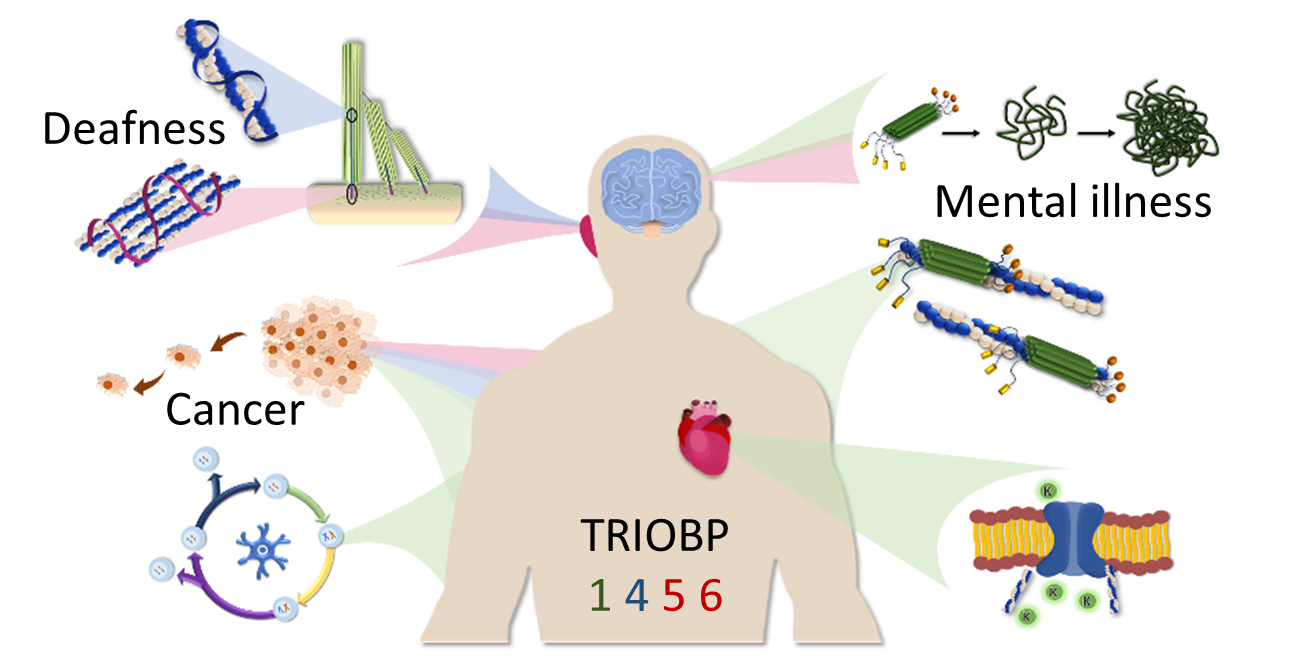
The TRIOBP Isoforms and Their Distinct Roles in Actin Stabilization, Deafness, Mental Illness, and Cancer


Abstract
:
1. Introduction
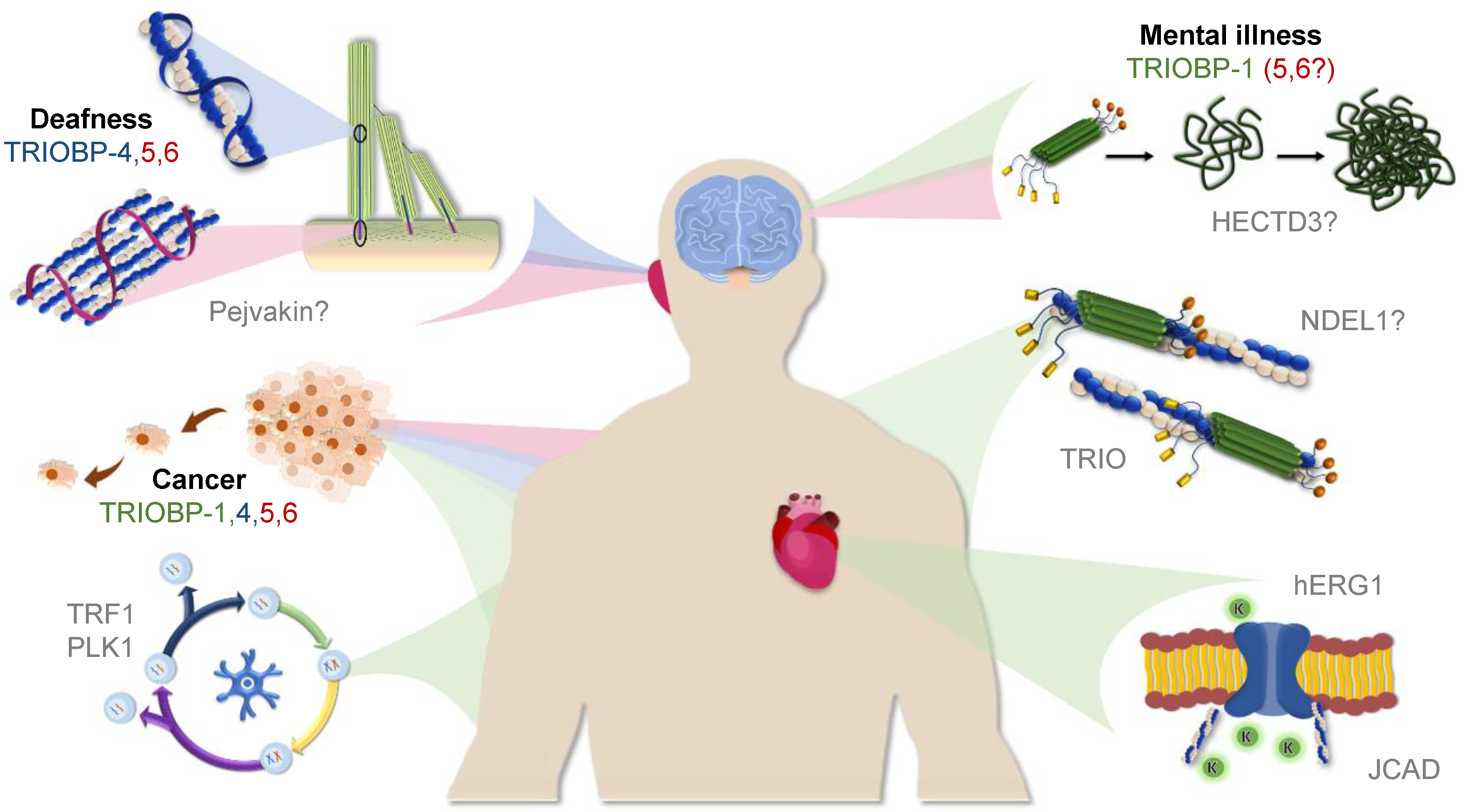
2. TRIOBP-1: A Structured Protein Implicated in Mental Illness and Cancer
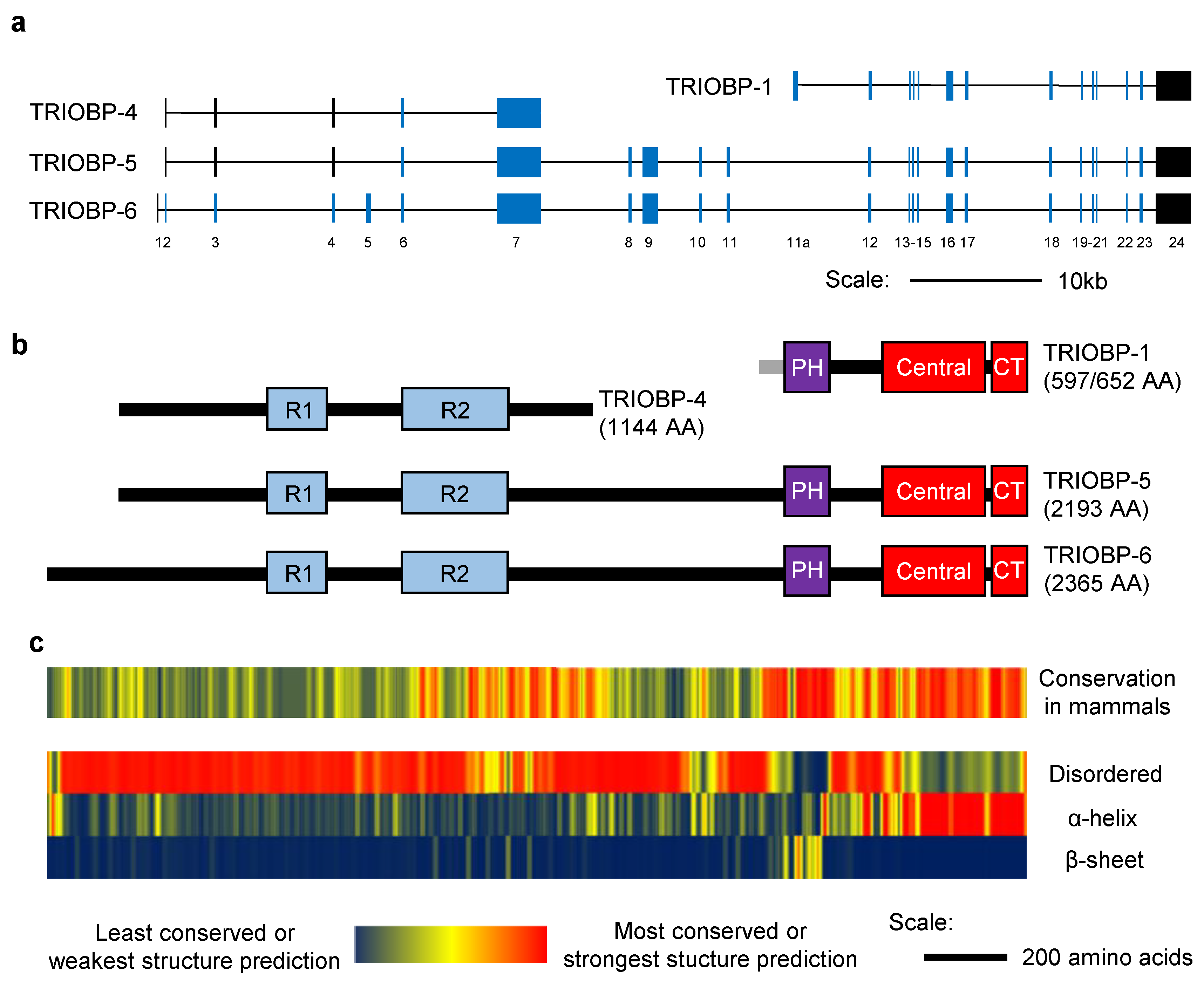
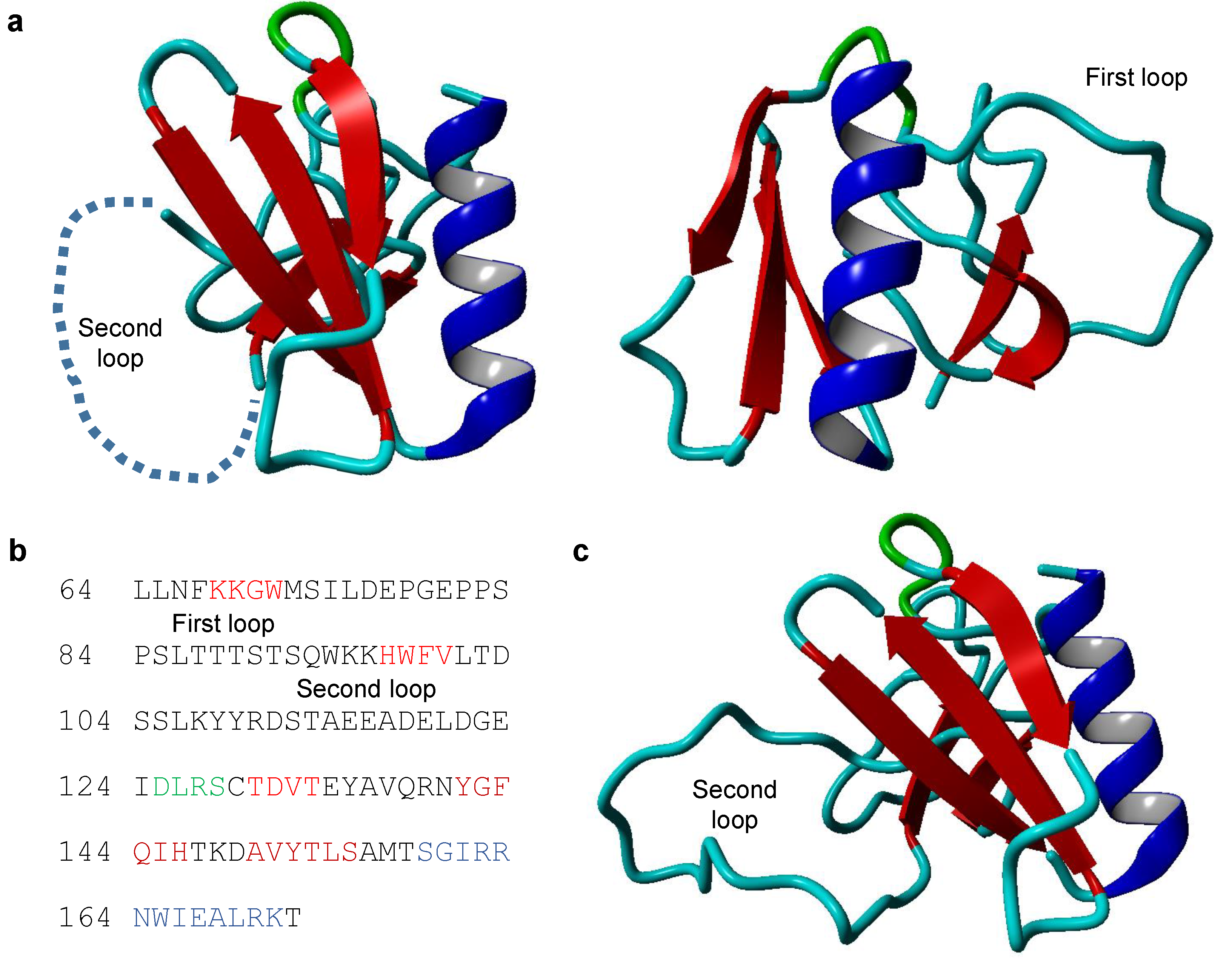
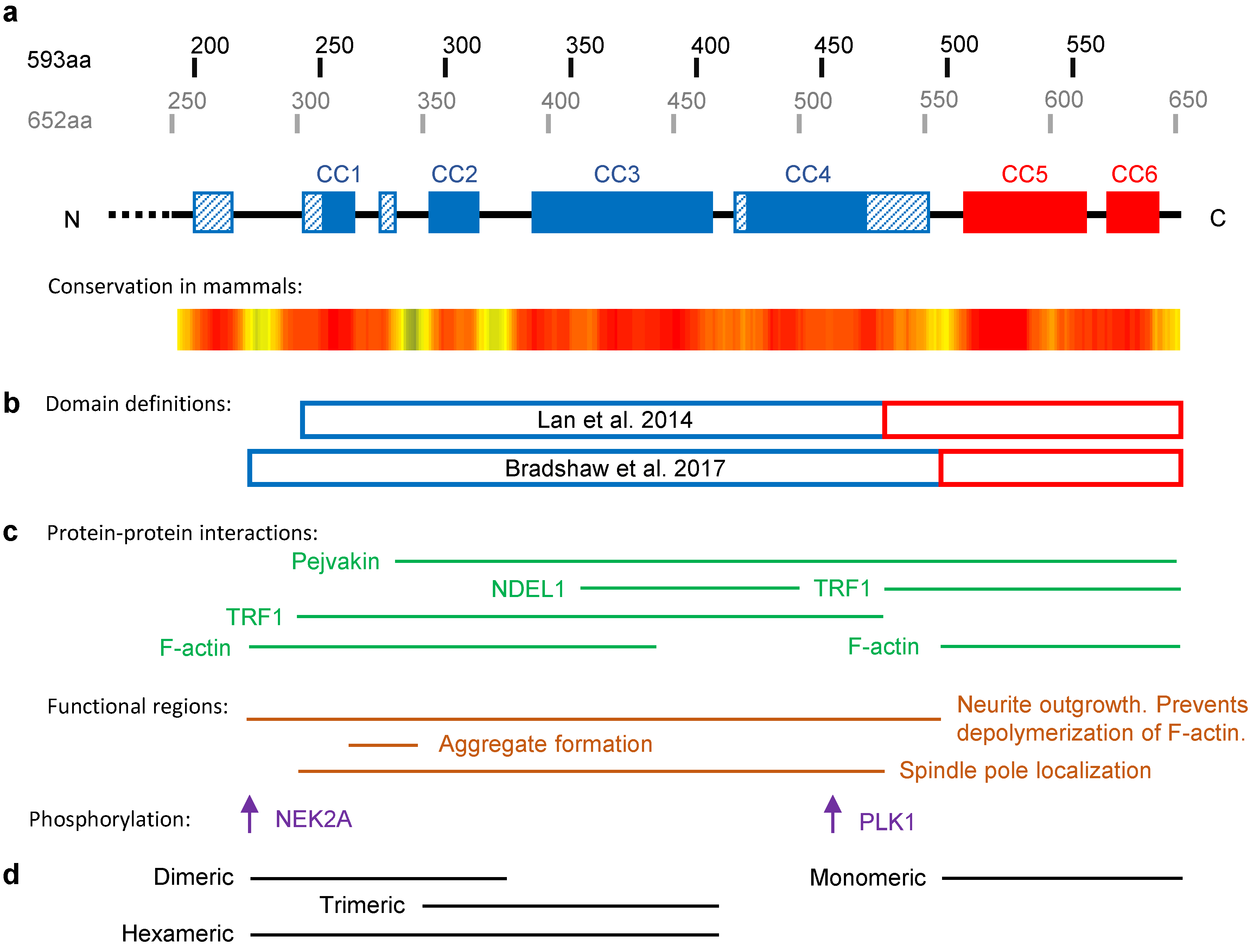
2.1. The Structure of TRIOBP-1
2.2. TRIOBP-1 as a Regulator of Actin Polymerization
2.3. TRIOBP-1 in the Cell Cycle
2.4. TRIOBP-1 in Mental Illness
2.5. TRIOBP-1 in Cancer
2.6. TRIOBP-1 in Other Diseases
3. TRIOBP-4: A Disordered Protein Implicated in Deafness
3.1. The Structure of TRIOBP-4
3.2. TRIOBP-4 as an Actin Bundling Protein in the Inner Ear
3.3. TRIOBP-4 in Hearing Loss
3.4. TRIOBP-4 in Cancer
3.5. TRIOBP-4 Mutations in Other Illnesses
4. Potential Significance of the Longer Splice Variants TRIOBP-5 and TRIOBP-6
4.1. The Structure of the Long Splice Variants
4.2. TRIOBP-5 in the Inner Ear and Deafness
4.3. Potential Significance for the Long Splice Variants in Other Processes and Diseases
5. Conclusions and Unanswered Questions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Straub, F.B. Actin II. In Muscular Contraction, Blood Coagulation; Szent-Györgyi, A., Ed.; S. Karger: Basel, Switzerland; New York, NY, USA, 1943; pp. 23–37. [Google Scholar]
- Dos Remedios, C.G.; Chhabra, D. Actin-Binding Proteins and Disease; Springer: New York, NY, USA, 2008. [Google Scholar]
- Seipel, K.; O’Brien, S.P.; Iannotti, E.; Medley, Q.G.; Streuli, M. Tara, a novel F-actin binding protein, associates with the Trio guanine nucleotide exchange factor and regulates actin cytoskeletal organization. J. Cell Sci. 2001, 114, 389–399. [Google Scholar]
- Riazuddin, S.; Khan, S.N.; Ahmed, Z.M.; Ghosh, M.; Caution, K.; Nazli, S.; Kabra, M.; Zafar, A.U.; Chen, K.; Naz, S.; et al. Mutations in TRIOBP, which encodes a putative cytoskeletal-organizing protein, are associated with nonsyndromic recessive deafness. Am. J. Hum. Genet. 2006, 78, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Shahin, H.; Walsh, T.; Sobe, T.; Abu Sa’ed, J.; Abu Rayan, A.; Lynch, E.D.; Lee, M.K.; Avraham, K.B.; King, M.-C.; Kanaan, M. Mutations in a novel isoform of TRIOBP that encodes a filamentous-actin binding protein are responsible for DFNB28 recessive nonsyndromic hearing loss. Am. J. Hum. Genet. 2006, 78, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Lee, H.; Kim, M.; Park, J.; Kim, S.-H.; Park, J. Emerging roles of TRIO and F-actin-binding protein in human diseases. Cell Commun. Signal. 2018, 16, 29. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.; Grishin, N.V. AL2CO: Calculation of positional conservation in a protein sequence alignment. Bioinformatics 2001, 17, 700–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradshaw, N.J.; Yerabham, A.S.K.; Marreiros, R.; Zhang, T.; Nagel-Steger, L.; Korth, C. An unpredicted aggregation-critical region of the actin-polymerizing protein TRIOBP-1/Tara, determined by elucidation of its domain structure. J. Biol. Chem. 2017, 292, 9583–9598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, T.; Yamazaki, Y.; Adachi, M.; Okawa, K.; Fort, P.; Uji, M.; Tsukita, S.; Tsukita, S. Tara up-regulates E-cadherin transcription by binding to the Trio RhoGEF and inhibiting Rac signaling. J. Cell Biol. 2011, 193, 319–332. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Zhu, Y.; Xu, L.; Yu, H.; Yu, J.; Liu, X.; Fu, C.; Wang, X.; Ke, Y.; Huang, H.; et al. The 68-kDa Telomeric Repeat binding Factor 1 (TRF1)-Associated Protein (TAP68) interacts with and recruits TRF1 to the spindle pole during mitosis. J. Biol. Chem. 2014, 289, 14145–14156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.K.; Johnson, A.C.; Roti Roti, E.C.; Liu, F.; Uelmen, R.; Ayers, R.A.; Baczko, I.; Tester, D.J.; Ackerman, M.J.; Trudeau, M.C.; et al. Localization and functional consequences of a direct interaction between TRIOBP-1 and hERG/KCNH2 proteins in the heart. J. Cell Sci. 2018, 131, jcs206730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Struct. Bioinform. 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [Green Version]
- Katsuno, T.; Belyantseva, I.A.; Cartagena-Rivera, A.X.; Ohta, K.; Crump, S.M.; Petralia, R.S.; Ono, K.; Tona, R.; Imtiaz, A.; Rehman, A.; et al. TRIOBP-5 sculpts stereocilia rootlets and stiffens supporting cells enabling hearing. JCI Insight 2019, 4, e128561. [Google Scholar] [CrossRef]
- Hong, J.-H.; Kwak, Y.; Woo, Y.; Park, C.; Lee, S.-A.; Lee, H.; Park, S.J.; Suh, Y.; Suh, B.K.; Goo, B.S.; et al. Regulation of the actin cytoskeleton by the Ndel1-Tara complex is critical for cell migration. Sci. Rep. 2016, 6, 31827. [Google Scholar] [CrossRef]
- Kazmierczak, M.; Kazmierczak, P.; Peng, A.W.; Harris, S.L.; Shah, P.; Puel, J.-L.; Lenoir, M.; Franco, S.J.; Schwander, M. Pejvakin, a candidate stereociliary rootlet protein, regulates hair cell function in a cell-autonomous manner. J. Neurosci. 2017, 37, 3447–3464. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Wang, C.; Lan, J.; Yu, J.; Jin, C.; Huang, H. Phosphorylation of Tara by Plk1 is essential for faithful chromosome segregation in mitosis. Exp. Cell Res. 2012, 318, 2344–2352. [Google Scholar] [CrossRef]
- Li, X.; Lan, J.; Zhu, Y.; Yu, J.; Dou, Z.; Huang, H. Expression, purification, and characterization of Tara, a novel telomere repeat-binding factor 1 (TRF1)-binding protein. Protein Expr. Purif. 2007, 55, 84–92. [Google Scholar] [CrossRef]
- Bradshaw, N.J.; Bader, V.; Prikulis, I.; Lueking, A.; Müllner, S.; Korth, C. Aggregation of the protein TRIOBP-1 and its potential relevance to schizophrenia. PLoS ONE 2014, 9, e111196. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Lee, Y.J.; Park, S.W.; Kim, H.S.; Han, H.J. Caveolin-1 and Integrin β1 regulate embryonic stem cell proliferation via p38 MAPK and FAK in high glucose. J. Cell Physiol. 2011, 226, 1850–1859. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Ryu, J.M.; Jang, M.W.; Han, H.J. Interaction of profilin-1 and F-actin via a b-arrestin-1/JNK signaling pathway involved in prostaglandin E2-induced human mesenchymal stem cells migration and proliferation. J. Cell Physiol. 2011, 226, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Kim, M.O.; Ryu, J.M.; Han, H.J. Regulation of SGLT expression and localization through Epac/PKA-dependent caveolin-1 and F-actin activation in renal proximal tubule cells. Biochim. Biophys. Acta 2012, 1823, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, N.J.; Hayashi, M.A.F. NDE1 and NDEL1 from genes to (mal)functions: Parallel but distinct roles impacting on neurodevelopmental disorders and psychiatric illness. Cell Mol. Life Sci. 2017, 74, 1191–1210. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.; Kim, S.J.; Suh, B.K.; Kwak, Y.; Jung, H.-J.; Nhung, T.T.M.; Mun, D.J.; Hong, J.-H.; Noh, S.-J.; Kim, S.; et al. Sequential phosphorylation of NDEL1 by the DYRK2-GSK3b complex is critical for neuronal morphogenesis. eLife 2019, 8, e50850. [Google Scholar] [CrossRef] [PubMed]
- Mui, K.L.; Chen, C.S.; Assoian, R.K. The mechanical regulation of integrin–cadherin crosstalk organizes cells, signaling and forces. J. Cell Sci. 2016, 129, 1093–1100. [Google Scholar] [CrossRef] [Green Version]
- Kuo, J.-C.; Han, X.; Hsiao, C.-T.; Yates, J.R., III; Waterman, C.M. Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for β-Pix in negative regulation of focal adhesion maturation. Nat. Cell Biol. 2011, 13, 383–393. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Xu, Y.; Liu, P.; Zhang, S.; Liu, H.; Slavin, S.; Kumar, S.; Koroleva, M.; Luo, J.; Wu, X.; et al. The novel coronary artery disease risk gene JCAD/KIAA1462 promotes endothelial dysfunction and atherosclerosis. Eur. Heart J. 2019, 40, 2398–2408. [Google Scholar] [CrossRef]
- Yu, J.; Lan, J.; Zhu, Y.; Li, X.; Lai, X.; Xue, Y.; Jin, C.; Huang, H. The E3 ubiquitin ligase HECTD3 regulates ubiquitination and degradation of Tara. Biochem. Biophys. Res. Commun. 2008, 367, 805–812. [Google Scholar] [CrossRef]
- Lan, J.P.; Luo, Y.; Zhu, Y.Y.; Sun, J.; Lai, X.Y.; Li, J.Y.; Yu, J.; Shi, J.M.; Lin, M.F.; Huang, H. Isolation of Tara protein and its gene cloning. Zhejiang Da Xue Xue Bao Yi Xue Ban 2004, 33, 486–490. [Google Scholar]
- Leliveld, S.R.; Bader, V.; Hendriks, P.; Prikulis, I.; Sajnani, G.; Requena, J.R.; Korth, C. Insolubility of Disrupted-in-Schizophrenia 1 disrupts oligomer-dependent interactions with Nuclear Distribution Element 1 and is associated with sporadic mental disease. J. Neurosci. 2008, 28, 3839–3845. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, N.J.; Korth, C. Protein misassembly and aggregation as potential convergence points for non-genetic causes of chronic mental illness. Mol. Psychiatry 2019, 24, 936–951. [Google Scholar] [CrossRef]
- Bader, V.; Tomppo, L.; Trossbach, S.V.; Bradshaw, N.J.; Prikulis, I.; Leliveld, S.R.; Lin, C.-Y.; Ishizuka, K.; Sawa, A.; Ramos, A.; et al. Proteomic, genomic and translational approaches identify CRMP1 for a role in schizophrenia and its underlying traits. Hum. Mol. Genet. 2012, 21, 4406–4418. [Google Scholar] [CrossRef]
- Maycox, P.R.; Kelly, F.; Taylor, A.; Bates, S.; Reid, J.; Logendra, R.; Barnes, M.R.; Larminie, C.; Jones, N.; Lennon, M.; et al. Analysis of gene expression in two large schizophrenia cohorts identifies multiple changes associated with nerve terminal function. Mol. Psychiatry 2009, 14, 1083–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennah, W.; Tomppo, L.; Hiekkalinna, T.; Palo, O.M.; Kilpinen, H.; Ekelund, J.; Tuulio-Henriksson, A.; Silander, K.; Partonen, T.; Paunio, T.; et al. Families with the risk allele of DISC1 reveal a link between schizophrenia and another component of the same molecular pathway, NDE1. Hum. Mol. Genet. 2007, 6, 453–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennah, W.; Porteous, D. The DISC1 pathway modulates expression of neurodevelopmental, synaptogenic and sensory perception genes. PLoS ONE 2009, 4, e4906. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, N.J.; Ukkola-Vuoti, L.; Pankakoski, M.; Zheutlin, A.B.; Ortega-Alonso, A.; Torniainen-Holm, M.; Sinha, V.; Therman, S.; Paunio, T.; Suvisaari, J.; et al. The NDE1 genomic locus affects treatment of psychiatric illness through gene expression changes related to MicroRNA-484. Open Biol. 2017, 7, 170153. [Google Scholar] [CrossRef]
- Knight, H.M.; Maclean, A.; Irfan, M.; Naeem, F.; Cass, S.; Pickard, B.S.; Muir, W.J.; Blackwood, D.H.R.; Ayub, M. Homozygosity mapping in a family presenting with schizophrenia, epilepsy and hearing impairment. Eur. J. Hum. Genet. 2008, 16, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Sugaya, M.; Takenoyama, M.; Shigematsu, Y.; Baba, T.; Fukuyama, T.; Nagata, Y.; Mizukami, M.; So, T.; Ichiki, Y.; Yasuda, M.; et al. Identification of HLA-A24 restricted shared antigen recognized by autologous cytotoxic T lymphocytes from a patient with large cell carcinoma of the lung. Int. J. Cancer 2007, 120, 1055–1062. [Google Scholar] [CrossRef]
- Lee, H.; Kim, M.; Park, J.; Tran, Q.; Hong, Y.; Cho, H.; Park, S.; Hong, S.; Brazil, D.P.; Kim, S.-H.; et al. The roles of TRIO and F-actin-binding protein in glioblastoma cells. Mol. Med. Rep. 2018, 17, 4540–4546. [Google Scholar] [CrossRef] [PubMed]
- Ichiki, Y.; Hanagiri, T.; Takenoyama, M.; Baba, T.; Nagata, Y.; Mizukami, M.; So, T.; Sugaya, M.; Yasuda, M.; Uramoro, H.; et al. Differences in sensitivity to tumor-specific CTLs between primary and metastatic esophageal cancer cell lines derived from the same patient. Surg. Today 2012, 42, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Wang, S.; Gunther, L.K.; Kitajiri, S.-I.; Li, C.; Sakamoto, T. The actin-bundling protein TRIOBP-4 and -5 promotes the motility of pancreatic cancer cells. Cancer Lett. 2015, 356, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, K.; Mei, Y.; Huang, X.; Li, Z.; Yang, Q.; Yang, H. Sp1 suppresses miR-3178 to promote the metastasis invasion cascade via upregulation of TRIOBP. Mol. Ther. Nucleic Acids 2018, 12, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fardaei, M.; Sarrafzadeh, S.; Ghafouri-Fard, S.; Miryounesi, M. Autosomal Recessive Nonsyndromic Hearing Loss: A Case Report with a Mutation in TRIOBP Gene. Int. J. Mol. Cell Med. 2015, 4, 245–247. [Google Scholar]
- Zou, S.; Mei, X.; Yang, W.; Zhu, R.; Yang, T.; Hu, H. Whole-exome sequencing identifies rare pathogenic and candidate variants in sporadic Chinese Han deaf patients. Clin. Genet. 2020, 97, 352–356. [Google Scholar] [CrossRef]
- Bao, J.; Bielski, E.; Bachhawat, A.; Taha, D.; Gunther, L.K.; Thirumurugan, K.; Kitajiri, S.-I.; Sakamoto, T. R1 motif is the major actin-binding domain of TRIOBP-4. Biochemistry 2013, 52, 5256–5264. [Google Scholar] [CrossRef]
- Kitajiri, S.-i.; Sakamoto, T.; Belyantseva, I.A.; Goodyear, R.J.; Stepanyan, R.; Fujiwara, I.; Bird, J.E.; Riazuddin, S.; Riazuddin, S.; Ahmed, Z.M.; et al. Actin-bundling protein TRIOBP forms resilient rootlets of hair cell stereocilia essential for hearing. Cell 2010, 141, 786–798. [Google Scholar] [CrossRef] [Green Version]
- Tekin, A.M.; de Ceulaer, G.; Govaerts, P.; Bayazit, Y.; Wuyts, W.; Van de Heyning, P.; Topsakal, V. A New Pathogenic Variant in the TRIOBP Associated with Profound Deafness Is Remediable with Cochlear Implantation. Audiol. Neurotol. 2020. [Google Scholar] [CrossRef]
- Hoffmann, T.J.; Keats, B.J.; Yoshikawa, N.; Schaefer, C.; Risch, N.; Lustig, L.R. A Large Genome-Wide Association Study of Age-Related Hearing Impairment Using Electronic Health Records. PLoS Genet. 2016, 12, e1006371. [Google Scholar] [CrossRef] [Green Version]
- Wells, H.R.R.; Freidin, M.B.; Abidin, F.N.Z.; Payton, A.; Dawes, P.; Munro, K.J.; Morton, C.C.; Moore, D.R.; Dawson, S.J.; Williams, F.M.K. GWAS Identifies 44 Independent Associated Genomic Loci for Self-Reported Adult Hearing Difficulty in UK Biobank. Am. J. Hum. Genet. 2019, 105, 788–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, D.; Tekin, D.; Bademci, G.; Foster, J.; Cengiz, F.B.; Kannan-Sundhari, A.; Guo, S.; Mittal, R.; Zou, B.; Grati, M.; et al. Spectrum of DNA variants for non-syndromic deafness in a large cohort from multiple continents. Hum. Genet. 2016, 35, 953–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Yu, L.; Wang, Y.; Shang, W.; Xie, Y.; Wang, X.; Han, F. A novel mutation in TRIOBP gene leading to congenital deafness in a Chinese family. BMC Med. Genet. 2020, 21, 121. [Google Scholar] [CrossRef]
- Diaz-Horta, O.; Duman, D.; Foster, J.; Sırmacı, A.; Gonzalez, M.; Mahdieh, N.; Fotouhi, N.; Bonyadi, M.; Cengiz, F.B.; Menendez, I.; et al. Whole-exome sequencing efficiently detects rare mutations in autosomal recessive nonsyndromic hearing loss. PLoS ONE 2012, 7, e50628. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Guo, L.; Ji, H.; Sun, S.; Chai, R.; Wang, L.; Li, H. Genetic testing for sporadic hearing loss using targeted massively parallel sequencing identifies 10 novel mutations. Clin. Genet. 2015, 87, 588–593. [Google Scholar] [CrossRef]
- Bitarafan, F.; Seyedena, S.Y.; Mahmoudi, M.; Garshasbi, M. Identification of novel variants in Iranian consanguineous pedigrees with nonsyndromic hearing loss by next-generation sequencing. J. Clin. Lab. Anal. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pollak, A.; Lechowicz, U.; Pieńkowski, V.A.M.; Stawiński, P.; Kosińska, J.; Skarżyński, H.; Ołdak, M.; Płoski, R. Whole exome sequencing identifies TRIOBP pathogenic variants as a cause of postlingual bilateral moderate-to-severe sensorineural hearing loss. BMC Med. Genet. 2017, 18, 142. [Google Scholar] [CrossRef] [PubMed]
- Shang, H.; Yan, D.; Tayebi, N.; Saeidi, K.; Sahebalzamani, A.; Feng, Y.; Blanton, S.; Liu, X. Targeted next-generation sequencing of a deafness gene panel (MiamiOtoGenes) analysis in families unsuitable for linkage analysis. BioMed Res. Int. 2018, 2018, 3103986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesdorp, M.; van de Kamp, J.M.; Hensen, E.F.; Schraders, M.; Oostrik, J.; Yntema, H.G.; Feenstra, I.; Admiraal, R.J.C.; Kunst, H.P.M.; Tekin, M.; et al. Broadening the phenotype of DFNB28: Mutations in TRIOBP are associated with moderate, stable hereditary hearing impairment. Hear. Res. 2017, 347, 56–62. [Google Scholar] [CrossRef]
- Thutkawkorapin, J.; Picelli, S.; Kontham, V.; Liu, T.; Nilsson, D.; Lindblom, A. Exome sequencing in one family with gastric- and rectal cancer. BMC Genet. 2016, 17, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Pardeshi, L.A.; Rong, X.; Li, E.; Wong, K.H.; Peng, Y.; Xu, R.-H. Novel variants identified in multiple sclerosis patients from southern China. Front. Neurol. 2018, 9, 582. [Google Scholar] [CrossRef] [PubMed]
- Schoonen, M.; Smuts, I.; Louw, R.; Elson, J.L.; van Dyk, E.; Jonck, L.-M.; Rodenburg, R.J.T.; van der Westhuizen, F.H. Panel-based nuclear and mitochondrial next-generation sequencing outcomes of an ethnically diverse pediatric patient cohort with mitochondrial disease. J. Mol. Diagn. 2019, 21, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Pacentine, I.; Chatterjee, P.; Barr-Gillespie, P.G. Stereocilia Rootlets: Actin-Based Structures That Are Essential for Structural Stability of the Hair Bundle. Int. J. Mol. Sci. 2020, 21, 324. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
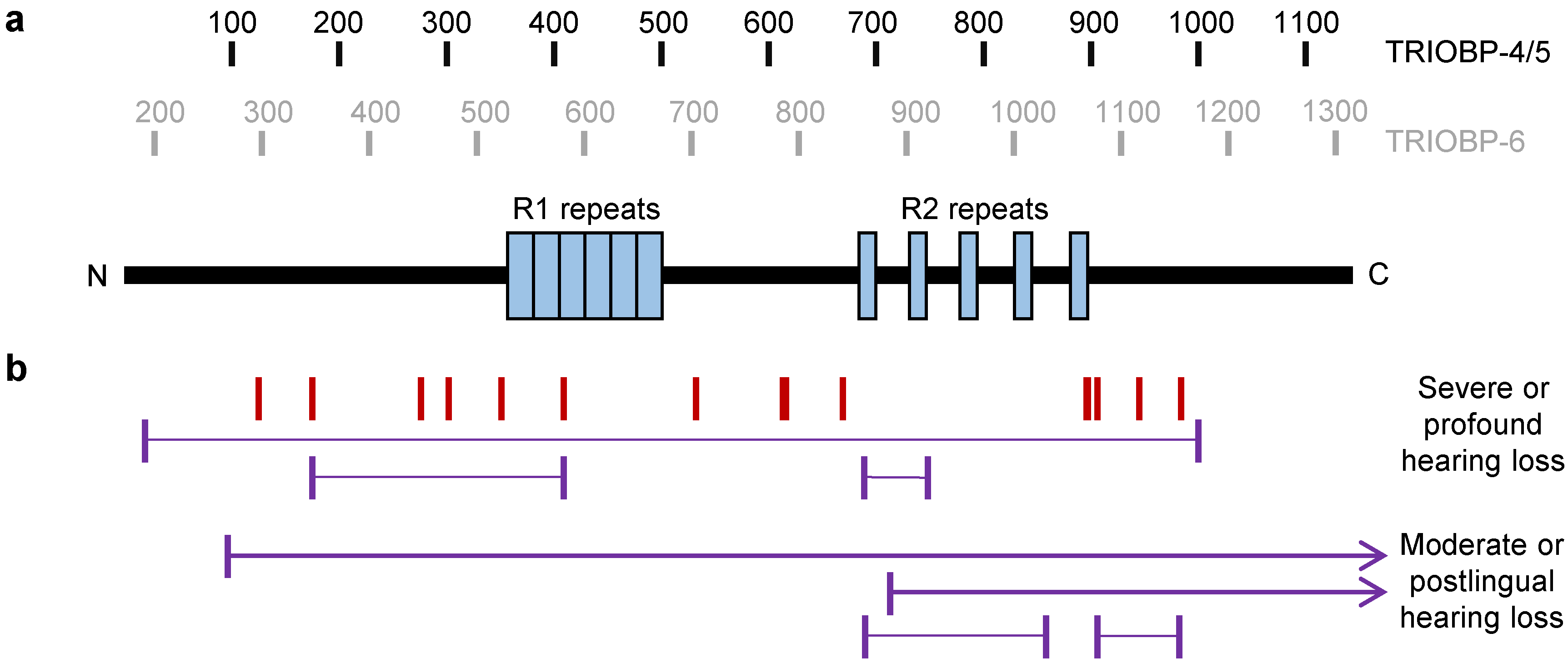
| Mutation 1 | Type 2 | Zygosity 3 | Isoforms (Location) 4 | Origin 5 of Proband(s) | Ref (s) |
|---|---|---|---|---|---|
| Severe to Profound Hearing Loss | |||||
| p.P191Rfs*50 | FS | CHT (p.P1172Cfs*13) | 4, 5, 6 | South Africa | [54] |
| p.Q297* | NON | HM | 4, 5, 6 | India | [4] |
| p.R347* | NON | HM CHT (p.Q581*) | 4, 5, 6 | Palestinian Palestinian | [5] [5] |
| p.R448* | NON | HM | 4, 5, 6 | China, Afghan | [51,55] |
| p.R474* | NON | HM 7 | 4, 5, 6 | Pakistan 7 | [50] |
| p.R523* | NON | HM 7 | 4, 5, 6 | Pakistan 7 | [50] |
| p.Q581* | NON | HM CHT (p.R347*) CHT (p.G1019R) | 4, 5, 6 (R1) | Palestinian Palestinian Palestinian | [5] [5] [5] |
| p.Q740* | NON | HM 7 | 4, 5, 6 | Pakistan 7 | [50] |
| p.R785Sfs*50 | FS | HM | 4, 5, 6 | Turkey | [56] |
| p.R788* | NON | HM | 4, 5, 6 | Pakistan | [4] |
| p.R841* | NON | HM | 4, 5, 6 | Turkey | [54] |
| p.R861* | NON | CHT (p.R920*) | 4, 5, 6 (R2) | China | [57] |
| p.R920* | NON | CHT (p.R861*) | 4, 5, 6 (R2) | China | [57] |
| p.G1019R | MIS | CHT (p.Q581*) | 4, 5, 6 (R2) | Palestinian | [5] |
| p.I1065V | MIS | CHT (p.R1982H) | 4, 5, 6 (R2) | China | [48] |
| p.R1068* | NON | HM | 4, 5, 6 (R2) | Pakistan, Iran | [4,58] |
| p.D1069fs*12 | FS | HM | 4, 5, 6 (R2) | India | [4] |
| p.R1078Pfs*6 | FS | HM | 4, 5, 6 (R2) | India | [4] |
| p.R1117* | NON | HM | 4, 5, 6 | India | [4] |
| p.E1156* | NON | HM7 | 4, 5, 6 | Pakistan 7 | [50] |
| p.P1172Cfs*13 | FS | CHT (p.R191Rfs*50) | 4, 5, 6 | South Africa | [54] |
| p.R1982H | MIS | CHT (p.I1065V) | 1, 5, 6 | China | [48] |
| p.S2121L | MIS | HM | 1, 5, 6 (Centr.) | Iran | [47] |
| Moderate or Postlingual Hearing Loss 6 | |||||
| p.Q268Lfs*432 | FS | CHT (p.G1672*) | 4, 5, 6 | Poland | [59] |
| p.R861* | NON | CHT(p.P1030Lfs*183) | 4, 5, 6 (R2) | USA, Iran | [54,60] |
| pR885Afs*120 | FS | CHT (p.G1672*) | 4, 5, 6 | Netherlands | [61] |
| p.P1030Lfs*183 | FS | CHT (p.R861*) | 4, 5, 6 (R2) | USA, Iran | [54,60] |
| p.R1078Pfs*6 | FS | CHT (p.L1154Afs*29) | 4, 5, 6 (R2) | Netherlands | [61] |
| p.M1151V | MIS | CHT (p.P1396R) | 4, 5, 6 | China | [57] |
| p.L1154Afs*29 | FS | CHT (R1078Pfs*6) | 4, 5, 6 | Netherlands | [61] |
| p.P1396R | MIS | CHT (p.M1151V) | 5, 6 | China | [57] |
| p.G1672* | NON | CHT (p.Q268Lfs*432) CHT (pR885Afs*120) | 5, 6 | Poland Netherlands | [59] [61] |
| Function or Phenotype | TRIOBP-1 | TRIOBP-4 | TRIOBP-5/6 1 | Ref. |
|---|---|---|---|---|
| Protein Structure | ||||
| Principle secondary structure | Helical | Disordered | Disordered | [3,49] |
| Contains… | ||||
| …repeat domains R1 and R1 | No | Yes | Yes | [49] |
| …PH domain | Yes | No | Yes | [3,12] |
| …coiled-coil domain | Yes | No | Yes | [12,19] |
| General function | ||||
| Interacts with F-actin | Yes | Yes | Yes | [3,18,50] |
| Prevents actin depolymerization | Yes | No (?) | ? | [12] |
| Actin bundling activity | No (?) | Yes | Yes (?) | [18,50] |
| Affects the actin cytoskeleton | Yes | Yes (?) | Yes | [3,45] |
| Roles in adhesion receptors | Yes | ? | ? | [13,30] |
| Implicated in cellular migration | Yes | Yes | Yes | [19,45] |
| Role in cell cycle progression | Yes | ? | ? | [21] |
| The brain and mental illness | ||||
| Expressed in the brain | Yes | No | Yes | [4,5] |
| Involved in neurite outgrowth | Yes | No | ? | [28] |
| Insoluble (aggregating) in brains of schizophrenia patients | Yes | No (?) | ? | [23] |
| Can aggregate in neurons | Yes | No | Yes | [23] |
| Inner ear and deafness | ||||
| Expressed in inner ear | Yes | Yes | Yes | [4,5] |
| Expressed in stereocilia | Yes | Yes 2 | Yes 2 | [18,20,50] |
| Required in stereocilia for | ||||
| rootlet formation | No | Yes | No | [50] |
| Initial bundling of actin | No | Yes | No | [50] |
| Sculpting and maintenance | No | No | Yes | [18] |
| Mouse knockout causes deafness? | (Knockout is lethal) | Yes 3 (profound) | Yes (progressive) | [18,50] |
| Mutations in human hearing loss | No (?) | Yes 3 | Yes | Table 1 |
| Cancer | ||||
| Upregulated in cancer cells? | Many | Specific | Specific | [43,45] |
| Potential role in metastasis? | Yes | Yes | Yes | [45,46] |
| Role in the heart | ||||
| Expressed in the heart | Yes | No | No | [4,5] |
| Function with hERG | Yes | No | No | [15] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaharija, B.; Samardžija, B.; Bradshaw, N.J. The TRIOBP Isoforms and Their Distinct Roles in Actin Stabilization, Deafness, Mental Illness, and Cancer. Molecules 2020, 25, 4967. https://doi.org/10.3390/molecules25214967
Zaharija B, Samardžija B, Bradshaw NJ. The TRIOBP Isoforms and Their Distinct Roles in Actin Stabilization, Deafness, Mental Illness, and Cancer. Molecules. 2020; 25(21):4967. https://doi.org/10.3390/molecules25214967
Chicago/Turabian StyleZaharija, Beti, Bobana Samardžija, and Nicholas J. Bradshaw. 2020. "The TRIOBP Isoforms and Their Distinct Roles in Actin Stabilization, Deafness, Mental Illness, and Cancer" Molecules 25, no. 21: 4967. https://doi.org/10.3390/molecules25214967
APA StyleZaharija, B., Samardžija, B., & Bradshaw, N. J. (2020). The TRIOBP Isoforms and Their Distinct Roles in Actin Stabilization, Deafness, Mental Illness, and Cancer. Molecules, 25(21), 4967. https://doi.org/10.3390/molecules25214967