
Naturally Occurring Calanolides: Occurrence, Biosynthesis, and Pharmacological Properties Including Therapeutic Potential


 ,
, 
Abstract
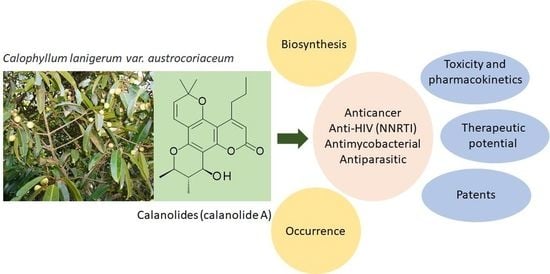
:
1. Introduction
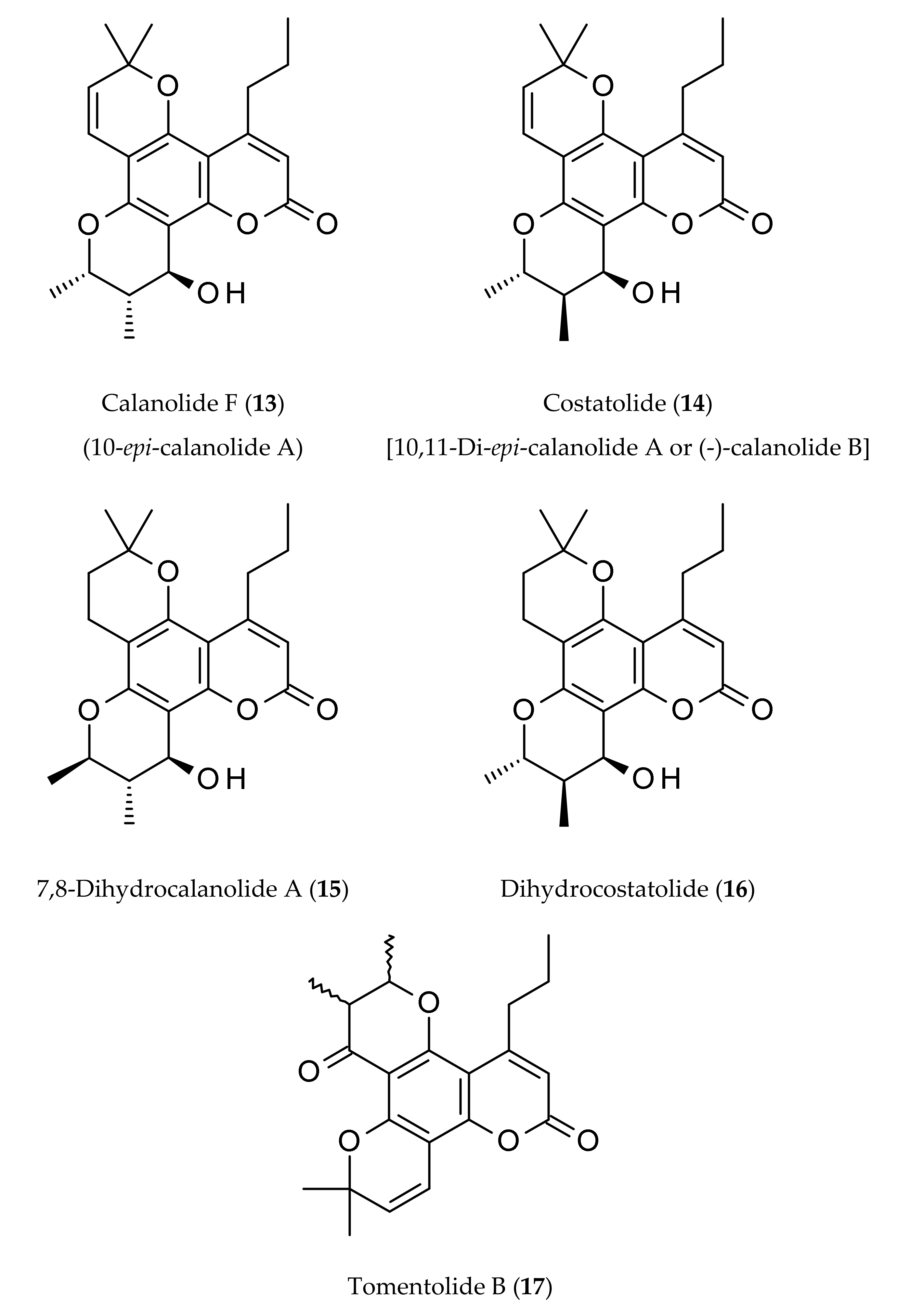
2. Occurrence
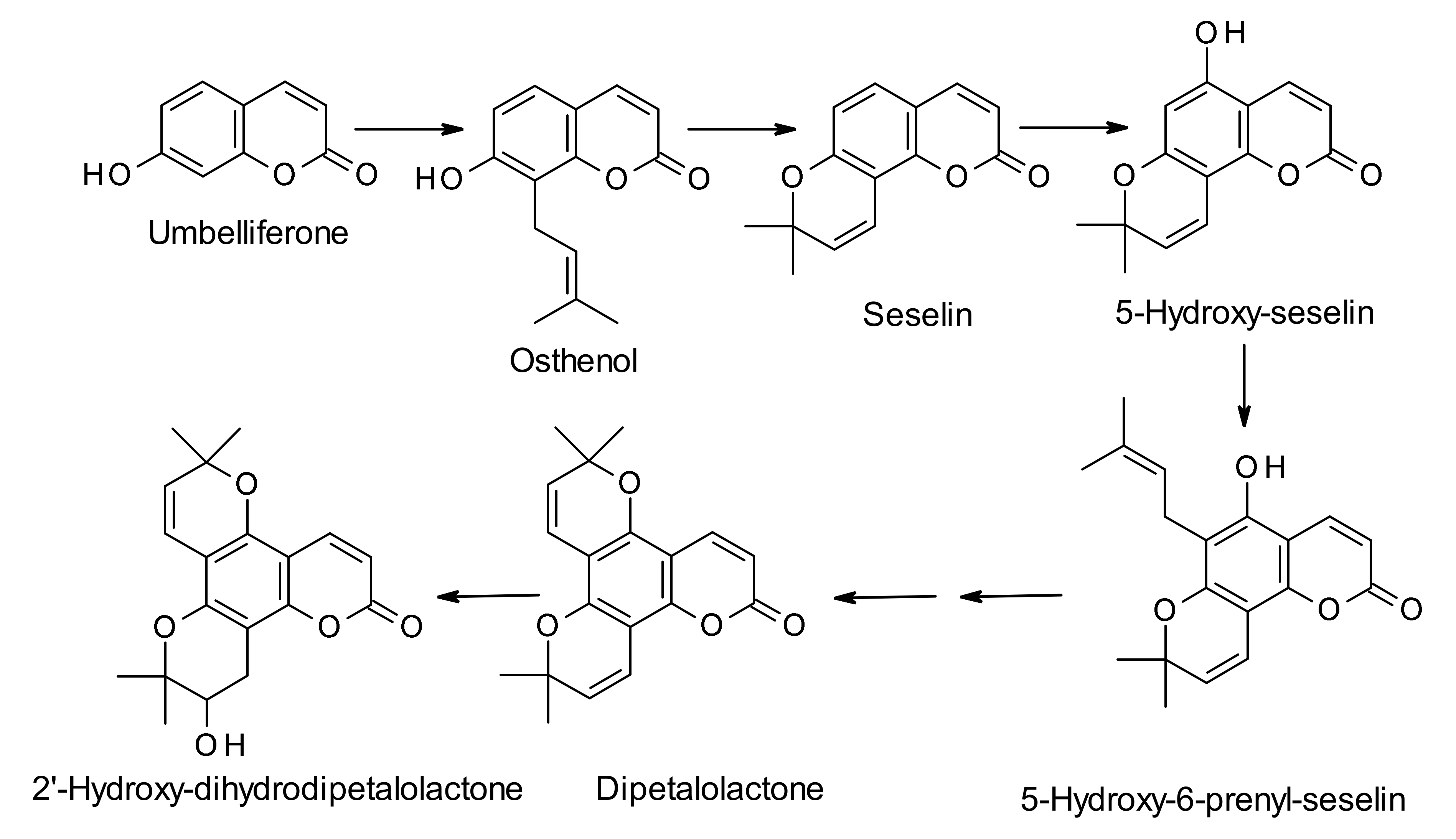
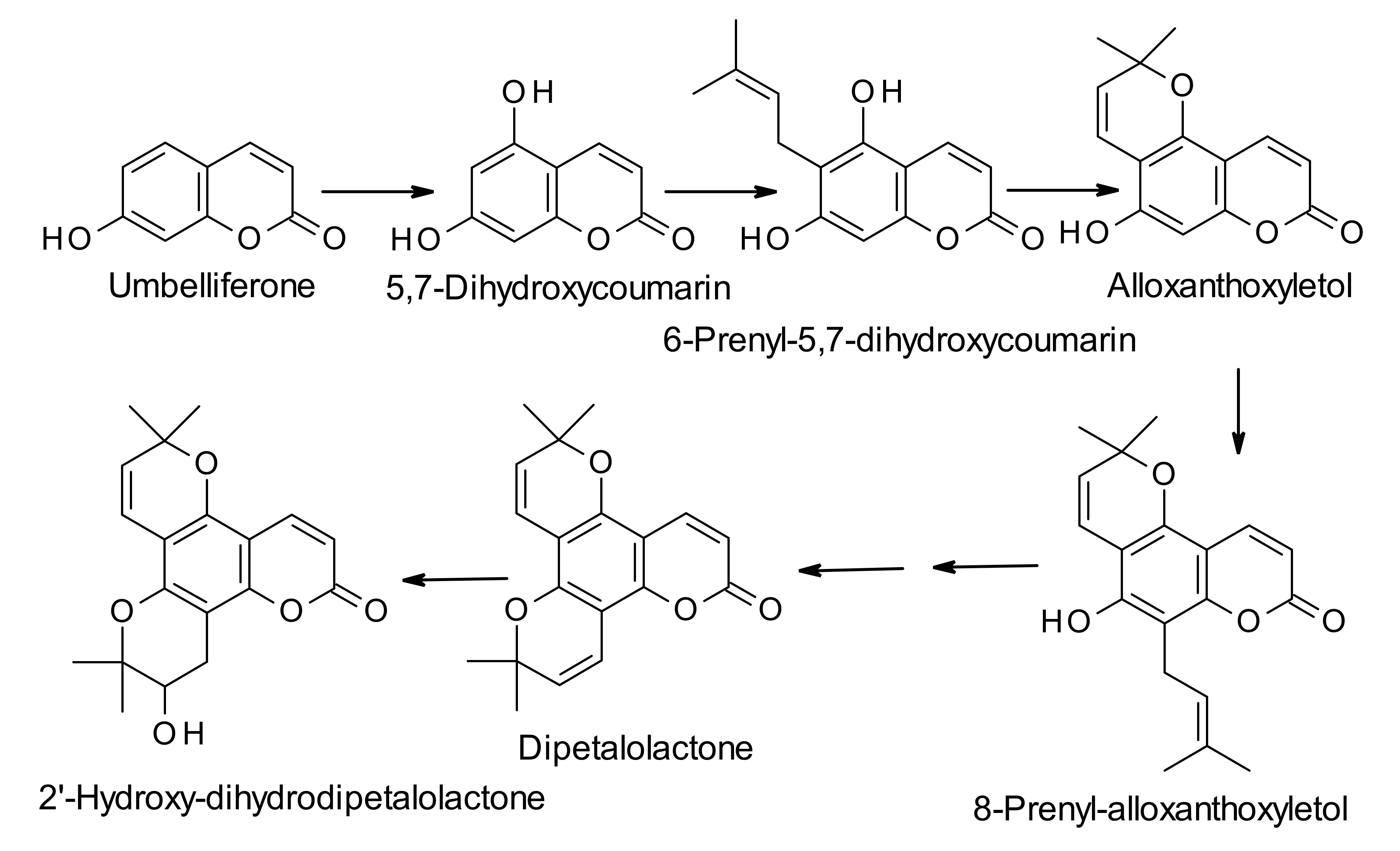
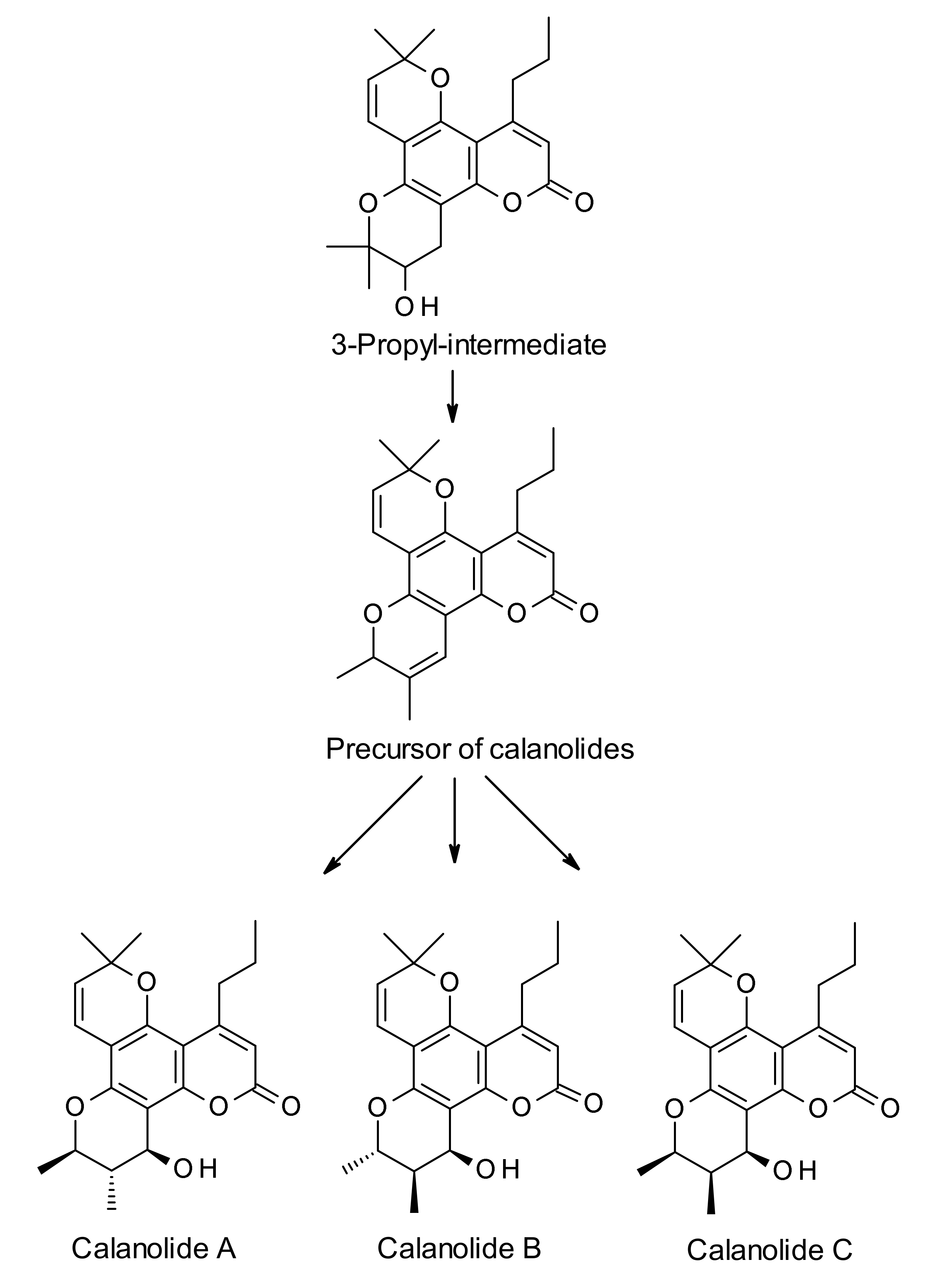
3. Biosynthesis
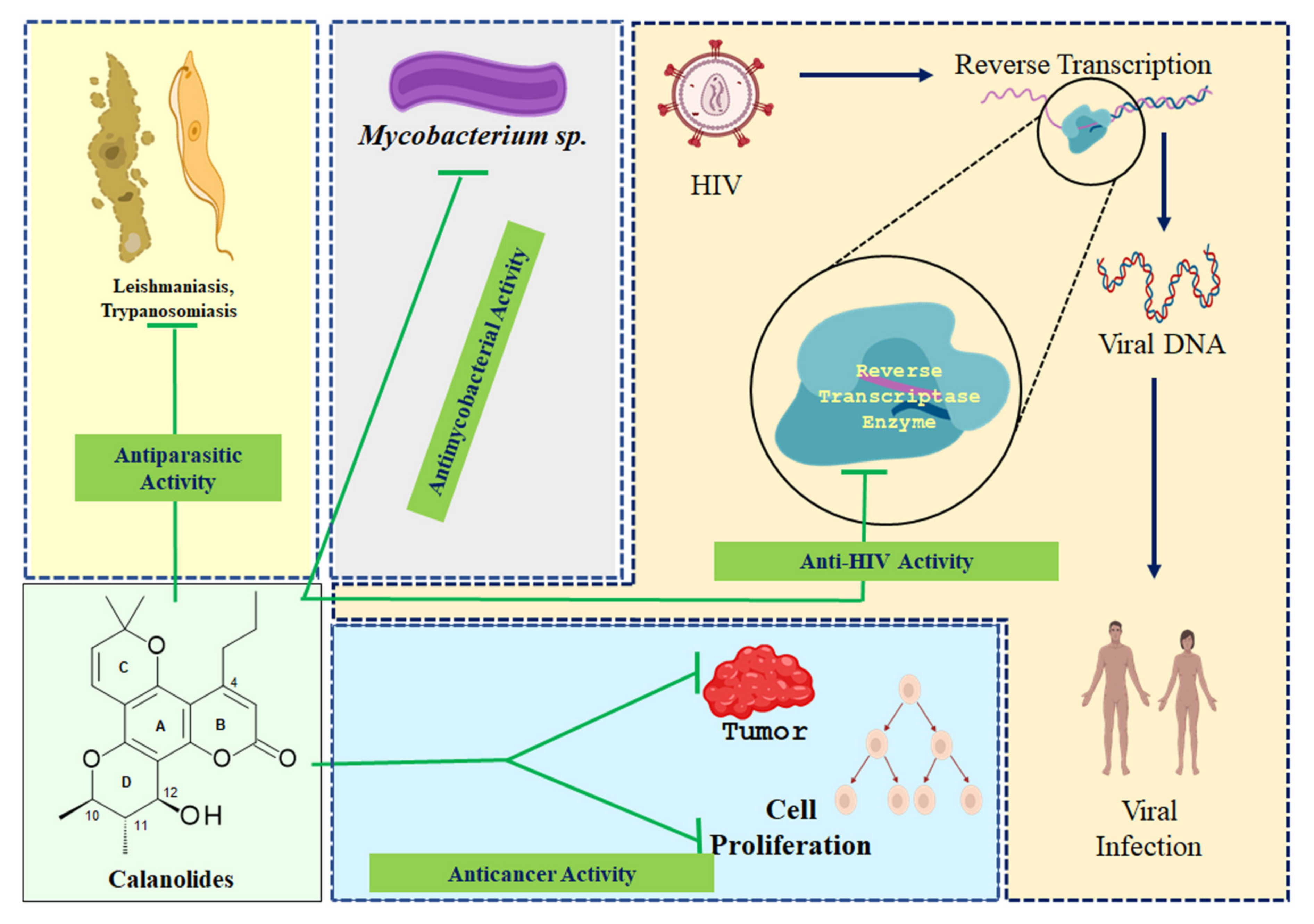
4. Pharmacological Properties
4.1. Anticancer Activity
4.2. Anti-HIV Activity
4.2.1. Activity Against Drug Resistant Strains of HIV-1
4.2.2. Calanolides in Anti-HIV-1 Combination Therapy
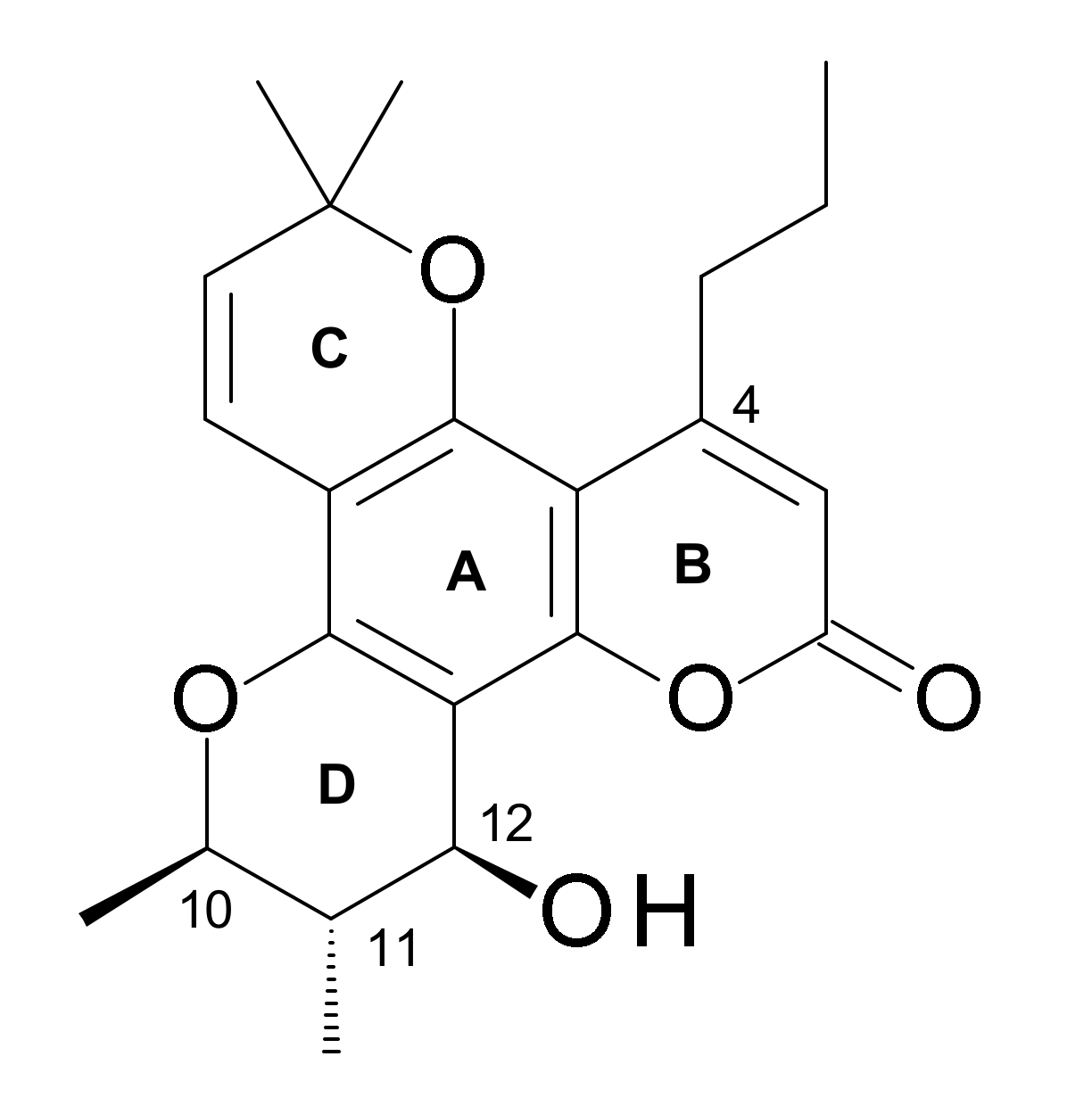
4.2.3. Structure-Activity-Relationships (SAR)
- (i)
- Δ11,12 Olefination diminishes activity.
- (ii)
- A C-12 hetero atom is essential for the activity.
- (iii)
- Relative potencies of C-12 ketone, thiol, azide, amine, and acetylated derivatives suggest stringent spatial and stereochemical requirements around C-12.
- (iv)
- The enantiomers of 12-oxocalanolide A, synthetic intermediates containing one fewer chiral center, still retain anti-HIV potency in the cytopathic assays.
- (v)
- The oxygen substituent can either be in the plane of the aromatic system or possess S configuration.
- (vi)
- Optical activity is important. For example, (+)-12-oxocalanolide A and (±)-12-oxocalanolide A have similar (but not same) anti-HIV activity, but (−)-12-oxocalanolide A is much less active.
- (vii)
- The racemic form, for example, (±)-12-oxocalanolide A, is more active than its pure (+)-enantiomer, (+)-12-oxocalanolide A, which suggests a possible synergistic effect in the combination of the two enantiomers.
- (viii)
- Hydrogenation at C-7 and C-8 of calanolides has little effect on the anti-HIV activity, e.g., the dihydro derivatives of calanolides A (1) and B (4) possess the same activity as the parent calanolides.
- (ix)
- Modifications at C-4 substituent can affect the anti-HIV activity of calanolides. For example, a methyl substituent at C-4 (as in cordatolides), instead of a propyl function as in calanolides reduces the anti-HIV potency.
- (x)
- Both the surface area of the substituted group attached on C-10, S-R3, and the distance between atoms O-13 and X-14 (O, N, S), L, of the calanolide analogues play important roles in determining the inhibitory activity of HIV-1 [56].
4.2.4. Mechanism of Action
4.3. Antimycobacterial Activity
4.4. Antiparasitic Activity
5. Toxicological Aspects Including Pharmacokinetics
6. Therapeutic Potential
7. Patents
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kashman, Y.; Gustafson, K.R.; Fuler, R.W.; Cardellina, J.H.; McMahon, J.B.; Currens, M.J.; Buckheit, R.W.; Hughes, S.H.; Cragg, G.M.; Boyd, M.R. HIV inhibitory natural products 7. The calanolides, a novel HIV-inhibitory class of coumarin derivatives from the tropical rain forest tree, Calophyllum lanigerum. J. Med. Chem. 1992, 35, 2735–2743. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. Plants as a source of anti-cancer and anti-HIV agents. Ann. Appl. Biol. 2003, 143, 127–133. [Google Scholar] [CrossRef]
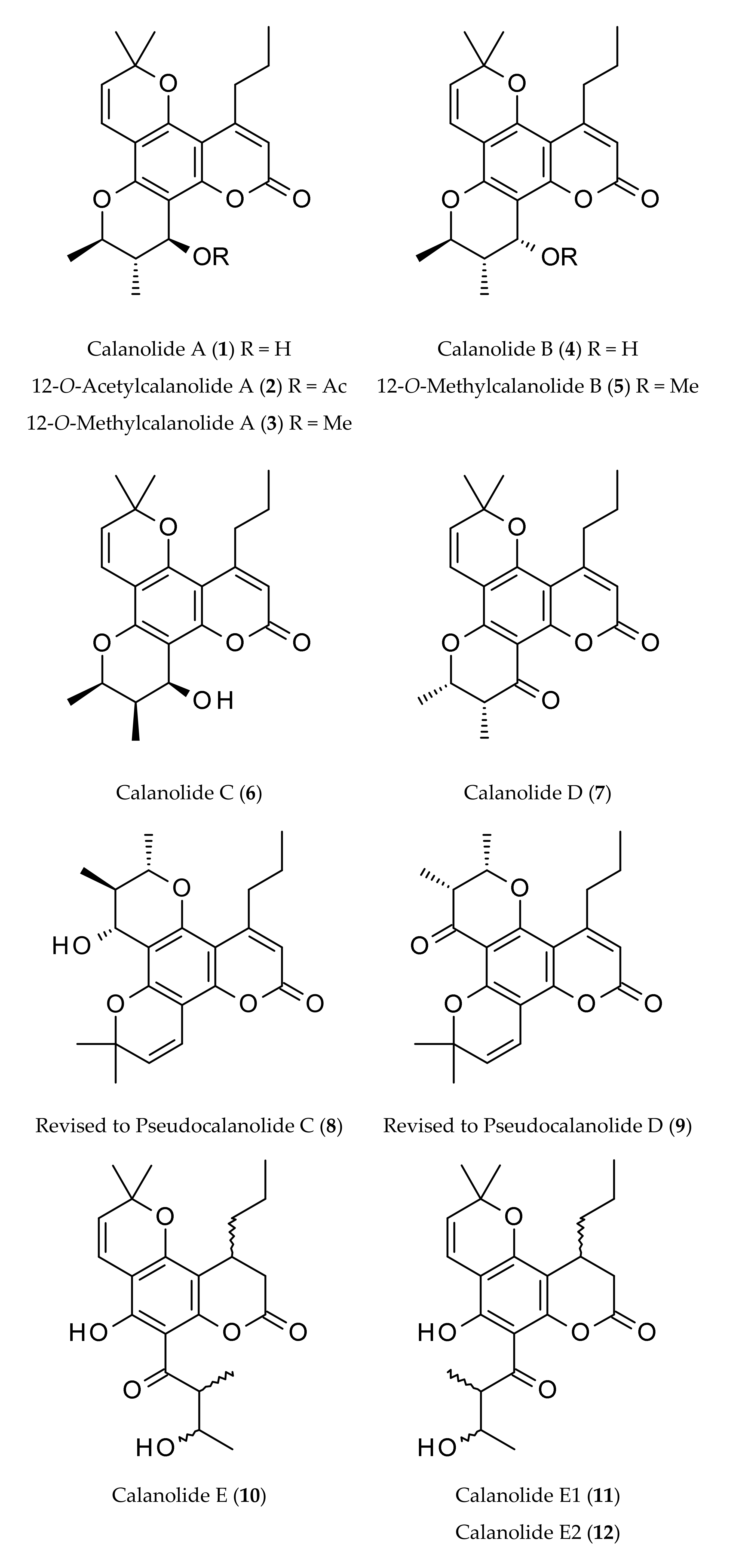
- McKee, T.C.; Cardellina, J.H.; Dreyer, G.B.; Boyd, M.R. The pseudocalanolides—Structure revision of Calanolide C. and Calanolide D. J. Nat. Prod. 1995, 58, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Reyes, M.; Basualdo, M.D.C.; Abe, F.; Jimenez-Estrada, M.; Soler, C.; Reyes-Chilpa, R. HIV-1 inhibitory compounds from Calophyllum brasiliense leaves. Biol. Pharm. Bull. 2004, 27, 1471–1475. [Google Scholar] [CrossRef] [Green Version]
- Bernabé-Antonio, A.; Estrada-Zúñiga, M.E.; Buendía-González, L.; Reyes-Chilpa, R.; Chávez-Ávila, V.M.; Cruz-Sosa, F. Production of anti-HIV-1 calanolides in a callus culture of Calophyllum brasiliense (Cambes). Plant Cell Tissue Organ Cult. 2010, 103, 33–40. [Google Scholar] [CrossRef]
- Gomez-Verjan, J.C.; Gonzalez-Sanchez, I.; Estrella-Parra, E.; Reyes-Chilpa, R. Trends in the chemical and pharmacological research on the tropical trees Calophyllum brasiliense and Calophyllum inophyllum, a global context. Scientometrics 2015, 105, 1019–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahmachari, G.; Jash, S.K. Naturally occurring calanolides: An update on their anti-HIV potential and total syntheses. Recent Patents Biotechnol. 2014, 8, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Brahmachari, G. Naturally occurring calanolides: Chemistry and biology. In Bioactive Natural Products: Chemistry and Biology; Brahmachari, G., Ed.; Wiley-VCH: Verlag, UK, 2014; pp. 349–374. [Google Scholar]
- McKee, T.C.; Covington, C.D.; Fuller, R.W.; Bokesch, H.R.; Young, S.; Cardellina, J.H.; Kadushin, M.R.; Soejarto, D.D.; Stevens, P.F.; Cragg, G.M.; et al. Pyranocoumarins from Tropical Species of the Genus Calophyllum: A Chemotaxonomic Study of Extracts in the National Cancer Institute Collection1. J. Nat. Prod. 1998, 61, 1252–1256. [Google Scholar] [CrossRef]
- Sunthitikawinsakul, A.; Kongkathip, N.; Kongkathip, B.; Phonnakhu, S.; Daly, J.W.; Spande, T.F.; Nimit, Y.; Napaswat, C.; Kasisit, J.; Yoosook, C. Anti-HIV limonoid: First isolation from Clausena excavate. Phytotherap. Res. 2003, 17, 1101–1103. [Google Scholar] [CrossRef]
- De Clercq, E. Current lead natural products for the chemotherapy of human immunodeficiency virus (HIV) infection. Med. Res. Rev. 2000, 20, 323–349. [Google Scholar] [CrossRef]
- Ishikawa, T. Anti-HIV-1 Active Calophyllum Coumarins: Distribution, Chemistry, and Activity. Heterocycles 2000, 53, 453. [Google Scholar] [CrossRef]
- Ito, C.; Itoigawa, M.; Mishina, Y.; Filho, V.C.; Enjo, F.; Tokuda, H.; Nishino, H.; Furukawa, H. Chemical Constituents of Calophyllum brasiliense 2. Structure of Three New Coumarins and Cancer Chemopreventive Activity of 4-Substituted Coumarins. J. Nat. Prod. 2003, 66, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.G.; Gomes, K.S.; Costa-Silva, T.A.; Romanelli, M.M.; Tempone, A.G.; Sartorelli, P.; Lago, J.H.G. Calanolides E1 and E2, two related coumarins from Calophyllum brasiliense Cambess. (Clusiaceae), displayed in vitro activity against amastigote forms of Trypanosoma cruzi and Leishmania infantum. Nat. Prod. Res. 2020, 1–5. [Google Scholar] [CrossRef]
- Tee, K.H.; Ee, G.C.L.; Ismail, I.S.; Karunakaran, T.; Teh, S.S.; Jong, V.Y.M.; Nor, S.M.M. A new coumarin from stem bark of Calophyllum wallichianum. Nat. Prod. Res. 2018, 32, 2565–2570. [Google Scholar] [CrossRef]
- Singh, I.P.; Bharate, S.B.; Bhutani, K.K. Anti-HIV natural products. Curr. Sci. 2005, 89, 269–290. [Google Scholar]
- Gómez-Cansino, R.; Espitia-Pinzón, C.I.; Campos-Lara, M.G.; Guzmán-Gutiérrez, S.L.; Segura-Salinas, E.; Echeverría-Valencia, G.; Torras-Claveria, L.; Cuevas-Figueroa, X.M.; Reyes-Chilpa, R. Antimycobacterial and HIV-1 Reverse Transcriptase Activity of Julianaceae and Clusiaceae Plant Species from Mexico. Evidence-Based Complement. Altern. Med. 2015, 2015, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Zebadua, J.C.; Reyes-Chilpa, R.; Huerta-Reyes, M.; Castillo-Arellano, J.I.; Santillan-Hernandez, S.; Vazaquez-Astudillo, B.; Mendoza-Espinoza, J.A. The tropical tree Calophyllum brasiliense: A botanical, chemical and pharmacological review. Vita Rev. Facul. Quimica Farmaceut. 2014, 21, 126–145. [Google Scholar]
- Gustafson, K.R.; Bokesch, H.R.; Fuller, R.W.; Cardellina, J.H.; Kadushin, M.R.; Soejarto, D.D.; Boyd, M.R. Calanone, a novel coumarin from Calophyllum teysmannii. Tetrahedron Lett. 1994, 35, 5821–5824. [Google Scholar] [CrossRef]
- McKee, T.C.; Fuller, R.W.; Covington, C.D.; Cardellina, J.H.; Gulakowski, R.J.; Krepps, B.L.; McMahon, J.B.; Boyd, M.R. New Pyranocoumarins Isolated from Calophyllum lanigerum and Calophyllum teysmannii 1. J. Nat. Prod. 1996, 59, 754–758. [Google Scholar] [CrossRef]
- Zou, J.; Jin, D.; Chen, W.; Wang, J.; Liu, Q.; Zhu, X.; Zhao, W. Selective Cyclooxygenase-2 Inhibitors from Calophyllum membranaceum. J. Nat. Prod. 2005, 68, 1514–1518. [Google Scholar] [CrossRef]
- Ma, C.-H.; Chen, B.; Qi, H.-Y.; Li, B.-G.; Zhang, G.-L. Two Pyranocoumarins from the Seeds of Calophyllum polyanthum. J. Nat. Prod. 2004, 67, 1598–1600. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-J.; Xu, M.; Luo, S.-D.; Wang, H.-Y.; Xu, J.-C. Chemical constituents of Calophyllum polyanthum. Acta Botan. Yunnanica 2001, 23, 521–526. [Google Scholar]
- Spino, C.; Dodier, M.; Sotheeswaran, S. Anti-HIV coumarins from calophyllum seed oil. Bioorganic Med. Chem. Lett. 1998, 8, 3475–3478. [Google Scholar] [CrossRef]
- Yang, S.S.; Cragg, G.M.; Newman, A.D.J.; Bader, J.P. Natural Product-Based Anti-HIV Drug Discovery and Development Facilitated by the NCI Developmental Therapeutics Program. J. Nat. Prod. 2001, 64, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Buckheit, R.W.; White, E.L.; Fliakas-Boltz, V.; Russell, J.; Stup, T.L.; Kinjerski, T.L.; Osterling, M.C.; Weigand, A.; Bader, J.P. Unique Anti-Human Immunodeficiency Virus Activities of the Nonnucleoside Reverse Transcriptase Inhibitors Calanolide A, Costatolide, and Dihydrocostatolide. Antimicrob. Agents Chemother. 1999, 43, 1827–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahar, L.; Sarker, S.D. Chemistry for Pharmacy Students: General, Organic and Natural Product Chemistry, 2nd ed.; Wiley and Sons: Chichester, UK, 2019. [Google Scholar]
- Gómez-Robledo, H.-B.; Cruz-Sosa, F.; Bernabé-Antonio, A.; Guerrero-Analco, A.; Olivares, J.L.; Alonso-Sanchez, A.; Villafán, E.; Ibarra-Laclette, E. Identification of candidate genes related to calanolide biosynthesis by transcriptome sequencing of Calophyllum brasiliense (Calophyllaceae). BMC Plant Biol. 2016, 16, 177. [Google Scholar] [CrossRef] [Green Version]
- Castillo-Arellano, J.I.; Osuna-Fernández, H.R.; Mumbru-Massip, M.; Gómez-Cancino, R.; Reyes-Chilpa, R. The biosynthesis of pharmacologically active compounds in Calophyllum brasiliense seedlings is influenced by calcium and potassium under hydroponic conditions. Bot. Sci. 2019, 97, 89–99. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Crag, G.M.; Newman, D.J. Natural products drug discovery in the next millennium. Pharm. Biol. 2001, 39, 8–17. [Google Scholar]
- Wilson, E.O. What is nature worth? Wilson Quarter. 2002, 26, 36–37. [Google Scholar]
- Jaikumar, K.; Sheikh, N.M.M.; Anand, D.; Saravanan, P. Anticancer activity of Calophyllum inophyllum L. ethanolic leaf extract in MCF human breast cell lines. Int. J. Pharm. Sci. Res. 2016, 7, 3330–3335. [Google Scholar]
- Ito, C.; Murata, T.; Itoigawa, M.; Nakao, K.; Kaneda, N.; Furukawa, H. Apoptosis inducing activity of 4-substituted coumarins from Calophyllum brasiliense in human leukaemia HL-60 cells. J. Pharm. Pharmacol. 2006, 58, 975–980. [Google Scholar] [CrossRef]
- Omer, A.; Singh, P. An integrated approach of network-based systems biology, molecular docking, and molecular dynamics approach to unravel the role of existing antiviral molecules against AIDS-associated cancer. J. Biomol. Struct. Dyn. 2016, 35, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Hanna, L. Calanolide A: A natural non-nucleoside reverse transcriptase inhibitor. BETA Bull. Exp. Treat. AIDS Publ. San Francisco AIDS Found. 1999, 12, 8–9. [Google Scholar]
- Xu, Z.-Q.; Norris, K.J.; Weinberg, D.S.; Kardatzke, J.; Wertz, P.; Frank, P.; Flavin, M.T. Quantification of (+)-calanolide A, a novel and naturally occurring anti-HIV agent, by high-performance liquid chromatography in plasma from rat, dog and human. J. Chromatogr. B Biomed. Sci. Appl. 2000, 742, 267–275. [Google Scholar] [CrossRef]
- Boyer, P.L.; Currens, M.J.; McMahon, J.B.; Boyd, M.R.; Hughes, S.H. Analysis of nonnucleoside drug-resistant variants of human immunodeficiency virus type 1 reverse transcriptase. J. Virol. 1993, 67, 2412–2420. [Google Scholar] [CrossRef] [Green Version]
- Hizi, A.; Tal, R.; Shaharabany, M.; Currens, M.J.; Boyd, M.R.; Hughes, S.H.; McMahon, J.B. Specific inhibition of the reverse transcriptase of human immunodeficiency virus type 1 and the chimeric enzymes of human immunodeficiency virus type 1 and type 2 by nonnucleoside inhibitors. Antimicrob. Agents Chemother. 1993, 37, 1037–1042. [Google Scholar] [CrossRef] [Green Version]
- Buckheit, R.W.; Fliakasboltz, V.; Yeagybargo, S.; Weislow, O.; Mayers, D.L.; Boyer, P.L.; Hughes, S.H.; Pan, B.C.; Chu, S.H.; Bader, J.P. Resistance to 1-[(2-hydroxyethoxy)methyl]-6-(phenylthiol)thymine derivatives is generated by mutations ad multiple sites in the HIV-1 reverse-transcriptase. Virology 1995, 210, 186–193. [Google Scholar] [CrossRef] [Green Version]
- Quan, Y.; Motakis, D.; Buckheit, R.; Xu, Z.-Q.; Flavin, M.T.; Parniak, M.A.; Wainberg, M.A. Sensitivity and resistance to (+)-calanolide A of wild-type and mutated forms of HIV-1 reverse transcriptase. Antivir. Ther. 1999, 4, 203–209. [Google Scholar]
- Xu, Z.-Q.; Hollingshead, M.G.; Borgel, S.; Elder, C.; Khilevich, A.; Flavin, M.T. In vivo anti-HIV activity of (+)-calanolide a in the hollow fiber mouse model. Bioorganic Med. Chem. Lett. 1999, 9, 133–138. [Google Scholar] [CrossRef]
- Xu, Z.-Q.; Flavin, M.T.; Jenta, T.R. Calanolides, the naturally occurring anti-HIV agents. Curr. Opin. Drug Discov. Dev. 2000, 3, 155–166. [Google Scholar]
- Buckheit, R.W.; Kinjerski, T.L.; Fliakasboltz, V.; Russell, J.D.; Stup, T.L.; Pallansch, L.A.; Brouwer, W.G.; Dao, D.C.; Harrison, W.A.; Schultz, R.J.; et al. Structure-activity and cross resistance evaluations of a series of human-deficiency-virus type-1 specific compounds related to oxanthin carboxanilide. Antimicrob. Agents Chemotherap. 1995, 39, 2718–2727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckheit, R.; Fliakas-Boltz, V.; Russell, J.; Snow, M.; Pallansch, L.; Yang, S.; Bader, J.; Khan, T.; Zanger, M. A Diarylsulphone Non-Nucleoside Reverse Transcriptase Inhibitor with a Unique Sensitivity Profile to Drug-Resistant Virus Isolates. Antivir. Chem. Chemother. 1996, 7, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Buckheit, J.R.W.; Russell, J.D.; Xu, Z.-Q.; Flavin, M. Anti-HIV-1 Activity of Calanolides Used in Combination with other Mechanistically Diverse Inhibitors of HIV-1 Replication. Antivir. Chem. Chemother. 2000, 11, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Budihas, S.R.; Gorshkova, I.; Gaidmakov, S.; Wamiru, A.; Bona, M.K.; Parniak, M.A.; Crouch, R.J.; McMahon, J.B.; Beutler, J.A.; le Grice, S.F.J.; et al. Selective inhibition of HIV-1 reverse transcriptase-associated ribonuclease H activity by hydroxylated tropolones. Nucleic Acid Res. 2005, 33, 1249–1256. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.Q.; Buckheit, R.W.; Stup, T.L.; Flavin, M.T.; Khilevich, A.; Rizzo, J.D.; Lin, L.; Zembower, D.E. In vitro anti-human deficiency virus (HIV) activity of the chromanone derivative, 12-oxocalanolide A, a novel NNRTI. Bioorg. Med. Chem. Lett. 1998, 8, 2179–2184. [Google Scholar] [PubMed]
- Sorbera, L.A.; Leeson, P.; Castaner, J. Calanolide A: Antiviral for AIDS, reverse transcriptase inhibitor. Drugs Future 1999, 24, 235–245. [Google Scholar] [CrossRef]
- Auwerx, J.; Rodríguez-Barrios, F.; Ceccherini-Silberstein, F.; San-Félix, A.; Velázquez, S.; de Clercq, E.; Camarasa, M.-J.; Perno, C.F.; Gago, F.; Balzarini, J. The Role of Thr139 in the Human Immunodeficiency Virus Type 1 Reverse Transcriptase Sensitivity to (+)-Calanolide A. Mol. Pharmacol. 2005, 68, 652–659. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Chilpa, R.; Huerta-Reyes, M. Natural compounds from plants of the Clausiaceae family: Type 1 human immunodeficiency virus inhibitors. Interciencia 2009, 34, 385–392. [Google Scholar]
- Cardellina, J.H.; Bokesch, H.R.; McKee, T.C.; Boyd, M.R. Resolution and comparative anti-HIV evaluation of the enantiomers of calanolides A and B. Bioorganic Med. Chem. Lett. 1995, 5, 1011–1014. [Google Scholar] [CrossRef]
- Galinis, D.L.; Fuller, R.W.; McKee, T.C.; Cardellina, J.H.; Gulakowski, R.J.; McMahon, J.B.; Boyd, M.R. Structure–Activity Modifications of the HIV-1 Inhibitors (+)-Calanolide A and (−)-Calanolide B1. J. Med. Chem. 1996, 39, 4507–4510. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T. Chemistry of Anti HIV-1 Active Calophyllum Coumarins. J. Synth. Org. Chem. Jpn. 1998, 56, 116–124. [Google Scholar] [CrossRef]
- Yu, D.; Suzuki, M.; Xie, L.; Morris-Natschke, S.L.; Lee, K.-H. Recent progress in the development of coumarin derivatives as potent anti-HIV agents. Med. Res. Rev. 2003, 23, 322–345. [Google Scholar] [CrossRef] [PubMed]
- Qiu, K.X.; Xie, H.D.; Guo, Y.P.; Huang, Y.; Liu, B.; Li, W. QSAR studies on the calanolide analogues as anti-HIV-1 agents. Chin. J. Struct. Chem. 2010, 29, 1477–1482. [Google Scholar]
- Peng, Z.-G.; Chen, H.-S.; Wang, L.; Liu, G. Anti-HIV activities of HIV-1 reverse transcriptase inhibitor racemic 11-demethyl-calanolide A. Acta Pharm. Sin. 2008, 43, 456–460. [Google Scholar]
- Sarker, S.D.; Nahar, L. An Introduction to Computational Phytochemistry. Comput. Phytochem. 2018, 1146, 1–41. [Google Scholar]
- Patel, R.D.; Kumar, S.P.; Patel, C.N.; Shankar, S.S.; Pandya, H.A.; Solanki, H. Parallel screening of drug-like natural compounds using Caco-2 cell permeability QSAR model with applicability domain, lipophilic ligand efficiency index and shape property: A case study of HIV-1 reverse transcriptase inhibitors. J. Mol. Struct. 2017, 1146, 80–95. [Google Scholar] [CrossRef]
- Currens, M.J.; Gulakowski, R.J.; Mariner, J.M.; Moran, R.A.; Buckheit, R.W.; Gustafson, K.R.; McMahon, J.B.; Boyd, M.R. Antiviral activity and mechanism of action of calanolide A against the human deficiency virus type-1. J. Pharmacol. Experim. Therapeut. 1996, 279, 645–651. [Google Scholar]
- Ha, M.H.; Nguyen, V.T.; Nguyen, K.Q.C.; Cheah, E.L.C.; Heng, P.W.S. Antimicrobial activity of Calophyllum inophyllum crude extracts obtained by pressurised liquid extraction. Asian J. Trad. Med. 2009, 4, 141–146. [Google Scholar]
- Alkhamaiseh, S.I.; Taher, M.; Ahmad, F. The Phytochemical Contents and Antimicrobial Activities of Malaysian Calophyllum rubiginosum. Am. J. Appl. Sci. 2011, 8, 201–205. [Google Scholar] [CrossRef]
- Saravanan, R.; Dhachinamoorthi, D.; Senthikumar, K.; Thamizhvanan, K. Antimicrobial activity of various extracts from various parts of Calophyllum inophyllum L. J. Appl. Pharm. Sci. 2011, 1, 102–106. [Google Scholar]
- Adewuyi, A.; Fasusi, O.H.; Oderinde, R.A. Antibacterial activities of acetonides prepared from the seed oils of Calophyllum inophyllum and Pterocarpus osun. J. Acute Med. 2014, 4, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Léguillier, T.; Lecsö-Bornet, M.; Lemus, C.; Rousseau-Ralliard, D.; Lebouvier, N.; Hnawia, E.; Nour, M.; Aalbersberg, W.; Ghazi, K.; Raharivelomanana, P.; et al. The Wound Healing and Antibacterial Activity of Five Ethnomedical Calophyllum inophyllum Oils: An Alternative Therapeutic Strategy to Treat Infected Wounds. PLoS ONE 2015, 10, e0138602. [Google Scholar] [CrossRef]
- Kudera, T.; Rondevaldova, J.; Kant, R.; Umar, M.; Skřivanová, E.; Kokoska, L. In vitro growth-inhibitory activity of Calophyllum inophyllum ethanol leaf extract against diarrhoea-causing bacteria. Trop. J. Pharm. Res. 2017, 16, 2207. [Google Scholar] [CrossRef] [Green Version]
- Oo, W.M. Pharmacological properties of Calophyllum inophyllum—Updated review. Int. J. Photochem. Photobiol. 2018, 2, 28–32. [Google Scholar]
- Xu, Z.Q.; Barrow, W.W.; Suling, W.J.; Westbrook, L.; Barrow, E.; Lin, Y.M.; Flavin, M.T. Anti-HIV natural product (+)-calanolide A is active against both drug-susceptible and drug-resistant strains of Mycobacterium tuberculosis. Bioorg. Med. Chem. 2004, 12, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Bueno, J.; Coy, E.D.; Stashenko, E. Antimycobacterial natural products—An opportunity for the Colombian biodiversity. Rev. Espanola Quimiot. 2011, 24, 175–183. [Google Scholar]
- Xu, Z.-Q.; Lin, Y.M.; Flavin, M.T. Method for Treating and Preventing Mycobacterium infections. U.S. Patent 6,268,393, 31 July 2001. [Google Scholar]
- Souto, E.B.; Dias-Ferreira, J.; Craveiro, S.A.; Severino, P.; Sanchez-Lopez, E.; Garcia, M.L.; Silva, A.M.; Souto, S.B.; Mahant, S. Therapeutic Interventions for Countering Leishmaniasis and Chagas’s Disease: From Traditional Sources to Nanotechnological Systems. Pathogens 2019, 8, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismail, F.M.D.; Nahar, L.; Zhang, K.; Sarker, S.D. Antimalarial and antiparasitic natural products. In Medicinal Natural Products—A Disease-Focused Approach; Sarker, S.D., Nahar, L., Eds.; Elsevier: London, UK, 2020; pp. 115–151. [Google Scholar]
- Creagh, T.; Ruckle, J.L.; Tolbert, D.T.; Giltner, J.; Eiznhamer, D.A.; Dutta, B.; Flavin, M.T.; Xu, Z.-Q. Safety and Pharmacokinetics of Single Doses of (+)-Calanolide A, a Novel, Naturally Occurring Nonnucleoside Reverse Transcriptase Inhibitor, in Healthy, Human Immunodeficiency Virus-Negative Human Subjects. Antimicrob. Agents Chemother. 2001, 45, 1379–1386. [Google Scholar] [CrossRef] [Green Version]
- César, G.-Z.J.; Gil-Alfonso, M.-G.; Marius, M.-M.; Elizabeth, E.-M.; Ángel, C.-B.M.; Maira, H.-R.; Guadalupe, C.-L.M.; Manuel, J.-E.; Ricardo, R.-C. Inhibition of HIV-1 reverse transcriptase, toxicological and chemical profile of Calophyllum brasiliense extracts from Chiapas, Mexico. Fitoterapia 2011, 82, 1027–1034. [Google Scholar] [CrossRef]
- Eiznhamer, D.A.; Creagh, T.; Ruckle, J.L.; Tolbert, D.T.; Giltner, J.; Dutta, B.; Flavin, M.T.; Jenta, T.; Xu, Z.-Q. Safety and Pharmacokinetic Profile of Multiple Escalating Doses of (+)-Calanolide A, a Naturally Occurring Nonnucleoside Reverse Transcriptase Inhibitor, in Healthy HIV-Negative Volunteers. HIV Clin. Trials 2002, 3, 435–450. [Google Scholar] [CrossRef]
- Newman, R.A.; Chen, W.; Madden, T.L. Pharmaceutical Properties of Related Calanolide Compounds with Activity against Human Immunodeficiency Virus. J. Pharm. Sci. 1998, 87, 1077–1080. [Google Scholar] [CrossRef] [PubMed]
- Usach, I.; Melis, V.; Peris, J.E. Non-nucleoside reverse transcriptase inhibitors: A review on pharmacokinetics, pharmacodynamics, safety and tolerability. J. Int. AIDS Soc. 2013, 16, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.P.; Bodiwala, H.S. Recent advances in anti-HIV natural products. Nat. Prod. Rep. 2010, 27, 1781–1800. [Google Scholar] [CrossRef]
- Buckheit, R.W. Non-nucleoside reverse transcriptase inhibitors: Perspectives on novel therapeutic compounds and strategies for the treatment of HIV infection. Expert Opin. Investig. Drugs 2001, 10, 1423–1442. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. The Safety and Effectiveness of (+)-Calanolide A in HIV-Infected Patients Who Have Never Taken Anti-HIV Drugs. NIH US National Library of Medicine. 2005. Available online: https://clinicaltrials.gov/ct2/show/NCT00005120 (accessed on 21 August 2020).
- Zhang, L.L.; Xue, H.; Li, L.; Lu, X.F.; Chen, Z.W.; Liu, G. HPLC enantioseparation, absolute configuration determination and anti-HIV activity of (+/-)-F19 enantiomers. Yaoxue Xuebao 2015, 50, 733–737. [Google Scholar]
- Wu, X.; Zhang, Q.; Guo, J.; Jia, Y.; Zhang, Z.; Zhao, M.; Yang, Y.; Wang, B.; Hu, J.; Sheng, L.; et al. Metabolism of F18, a Derivative of Calanolide A, in Human Liver Microsomes and Cytosol. Front. Pharmacol. 2017, 8, 479. [Google Scholar] [CrossRef] [Green Version]
- Xue, H.; Lu, X.; Zheng, P.; Liu, L.; Han, C.; Hu, J.; Liu, Z.; Ma, T.; Li, Y.; Wang, L.; et al. Highly Suppressing Wild-Type HIV-1 and Y181C Mutant HIV-1 Strains by 10-Chloromethyl-11-demethyl-12-oxo-calanolide A with Druggable Profile. J. Med. Chem. 2010, 53, 1397–1401. [Google Scholar] [CrossRef] [PubMed]
- Boyd, M.R.; Cardellina, J.H., II; Gustafson, K.R.; McMahon, J.B.; Fuller, R.W.; Cragg, G.M.; Kashman, Y.; Soejarto, D. Calanolide and related antiviral compounds, compositions, and uses thereof. U.S. Patent 5,859,049, 12 January 1999. [Google Scholar]
- Uckun, F.M.; Sudbeck, E. Calanolides for Inhibiting BTK. Official Gazette of the United States Patents and Trademark Office Patents. U.S. Patent 6,306,897, 23 October 2001. [Google Scholar]
- Aalipour, A.; Advani, R.H. Bruton’s tyrosine kinase inhibitors and their clinical potential in the treatment of B-cell malignancies: Focus on ibrutinib. Ther. Adv. Hematol. 2014, 5, 121–133. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Calanolides | Sources | Physical State | Mol. Formula | Mol. Weight | Optical Rotation [a]D | UV λmax (MeOH) nm | References |
|---|---|---|---|---|---|---|---|
| Calanolide A (1) | Calophyllum lanigerum var. austrocoriaceum | Oil | C22H26O5 | 370.44 | [a]D +60° (c, 0.7 in CHCl3) | 228, 284 and 325 | [1,9,11,16] |
| Calophyllum brasiliense | [4,17,18] | ||||||
| Calophyllum inophyllum | [6,11] | ||||||
| Calophyllum teysmannii | [19] | ||||||
| Clausena excavata | [10] | ||||||
| 12-O-Acetylcalanolide A (2) | Calophyllum lanigerum var. austrocoriaceum | Oil | C24H28O6 | 412.48 | [a]D +20° (c, 0.5 in CHCl3) | 228, 284 and 325 | [1] |
| 12-O-Methylcalanolide A (3) | Calophyllum lanigerum var. austrocoriaceum | Oil | C23H28O5 | 384.47 | [a]D +32° (c, 0.8 in CHCl3) | 228, 284 and 325 | [1] |
| Calanolide B (4) | Calophyllum lanigerum var. austrocoriaceum | Oil | C22H26O5 | 370.44 | [a]D +8° (c, 1.0 in acetone) | 228, 284 and 325 | [1] |
| Calophyllum brasiliense | [5,17] | ||||||
| Calophyllum teysmannii var. inophylloide | [9] | ||||||
| 12-O-Methyl-calanolide B (5) | Calophyllum lanigerum var. austrocoriaceum | Oil | C23H28O5 | 384.47 | [a]D +34° (c, 0.5 in CHCl3) | 228, 284 and 325 | [1] |
| Calanolide C (6) | Calophyllum brasiliense | Oil | C22H26O5 | 370.44 | - | - | [4,5] |
| Calanolide D (7) | Calophyllum brasiliense | Amorphous solid | C22H24O5 | 368.42 | - | - | [6] |
| Calanolide E (10) | Calophyllum lanigerum var. austrocoriaceum | Amorphous powder | C22H28O6 | 388.50 | [a]D +30° (c, 0.7 in acetone) | - | [1,9,20] |
| Calophyllum membranaceum | [21] | ||||||
| Calophyllum molle | [9] | ||||||
| Calophyllum polyanthum | [22] | ||||||
| Calophyllum teysmannii var. inophylloide | [20] | ||||||
| Calophyllum wallichianum | [15] | ||||||
| Calanolide E1 (11) | Calophyllum lanigerum var. austrocoriaceum | Amorphous powder | C22H28O6 | 388.50 | - | - | [9,20] |
| Calophyllum brasiliense | [9,14] | ||||||
| Calophyllum molle | [9] | ||||||
| Calanolide E2 (12) | Calophyllum lanigerum var. austrocoriaceum | Amorphous powder | C22H28O6 | 388.50 | - | - | [9,20] |
| Calophyllum brasiliense Cambess. | [14] | ||||||
| Calophyllum membranaceum | [21] | ||||||
| Calophyllum molle | [9] | ||||||
| Calophyllum polyanthum | [22,23] | ||||||
| Calophyllum teysmannii var. inophylloide | [9,20] | ||||||
| Calanolide F (13) | Calophyllum lanigerum var. austrocoriaceum | Amorphous powder | C22H26O5 | 370.44 | [a]D −51.5 (c, 0.3 in CHCl3) | 227, 283, 322 | [9,20] |
| Calophyllum teysmannii var. inophylloide | [9,20] | ||||||
| Costatolide (14) [(−)-Calanolide B] | Calophyllum brasiliense | Crystals (M.p. 181–183°) | C22H26O5 | 370.44 | [a]D −19.9 (c, 0.42 in CHCl3) | 228, 284 and 325 | [4] |
| Calophyllum costatum | [24] | ||||||
| Calophyllum inophyllum L. | [24] | ||||||
| Calophyllum teysmannii var. inophylloide | [9,19,25] | ||||||
| 7,8-Dihydrocalanolide A (15) | Calophyllum lanigerum var. austrocoriaceum | Amorphous solid | C22H28O5 | 372.46 | Negative optical rotation | - | [25] |
| Dihydrocostatolide (16) | Calophyllum costatum | Amorphous solid | C22H28O5 | 372.46 | - | - | [26] |
| Pseudocalanolide C (8) [incorrectly named as calanolide C (6)] | Calophyllum lanigerum var. austrocoriaceum | Amorphous solid | C22H26O5 | 370.44 | [a]D +68° (c, 0.7 in CHCl3) | - | [1,3,9] |
| Pseudocalanolide D (9) [incorrectly named as calanolide D (7)] | Calophyllum lanigerum var. austrocoriaceum | Amorphous solid | C22H24O5 | 368.43 | [a]D +60° (c, 0.5 in CHCl3) | - | [1,3] |
| Tomentolide B (17) | Calophyllum tomentosa | Amorphous solid (M.p. 158–160o) | C22H24O5 | 368.43 | Racemic mixture | - | [1,3,9] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nahar, L.; Talukdar, A.D.; Nath, D.; Nath, S.; Mehan, A.; Ismail, F.M.D.; Sarker, S.D. Naturally Occurring Calanolides: Occurrence, Biosynthesis, and Pharmacological Properties Including Therapeutic Potential. Molecules 2020, 25, 4983. https://doi.org/10.3390/molecules25214983
Nahar L, Talukdar AD, Nath D, Nath S, Mehan A, Ismail FMD, Sarker SD. Naturally Occurring Calanolides: Occurrence, Biosynthesis, and Pharmacological Properties Including Therapeutic Potential. Molecules. 2020; 25(21):4983. https://doi.org/10.3390/molecules25214983
Chicago/Turabian StyleNahar, Lutfun, Anupam Das Talukdar, Deepa Nath, Sushmita Nath, Aman Mehan, Fyaz M. D. Ismail, and Satyajit D. Sarker. 2020. "Naturally Occurring Calanolides: Occurrence, Biosynthesis, and Pharmacological Properties Including Therapeutic Potential" Molecules 25, no. 21: 4983. https://doi.org/10.3390/molecules25214983
APA StyleNahar, L., Talukdar, A. D., Nath, D., Nath, S., Mehan, A., Ismail, F. M. D., & Sarker, S. D. (2020). Naturally Occurring Calanolides: Occurrence, Biosynthesis, and Pharmacological Properties Including Therapeutic Potential. Molecules, 25(21), 4983. https://doi.org/10.3390/molecules25214983