
Targeting Lipid Peroxidation for Cancer Treatment

 , and
, and 
Abstract
:1. Introduction
2. Lipid Peroxidation: Non-Enzymatic Reactions vs. Enzymatic Reactions
2.1. Non-Enzyme-Dependent Lipid Peroxidation
2.2. Radiation Inducing Lipid Peroxidation
2.3. Enzyme-Dependent Lipid Peroxidation

{kind=link}
{kind=link}
| Tissue Location and Type of Cancer | Correlation with Other Biomarkers |
|---|---|
| Colon cancer [66], primary tumors and metastatic lymph nodes resections for colorectal adenocarcinoma [93], stage II and III colorectal cancer patients [94] | High levels of COX-2 correlates with high levels of MMP-2 and VEGF expression and shorter survival time [93,94]. |
| Cervical cancer [67] | Multivariate analysis of COX-2 levels in tumor/stromal compartments. The proportion of CD3+, CD4+, and CD25+ cells was lower in tumors with high tumor/stroma ratios, but in these tumors, mast cells were increased [67]. |
| Ovarian cancer [95,96,97] | No correlation between COX-2 expression and EGFR, and HER-2/neu status [96]. |
| Human breast cancer cell lines and tumors [26,98,99,100] | Elevated COX-2 expression associated with a large tumor size, a high histological grade, a negative hormone receptor status, a high proliferation rate, high p53 expression, and the presence of HER-2 oncogene amplification along with axillary node metastases and a ductal type of histology [98]. COX-2 inhibition may potentially prevent the development of ER-positive and ER-negative breast cancers [98]. Expression of PGE2 and IL-8 [101]. COX-2 over-expression induces an oncogenic microRNA (miR655) in human breast cancer cells by activation of EP4 [102]. |
| Ductal carcinoma in situ (DCIS) [103,104,105] | COX-2 expression stabilizes survivin, an inhibitor of apoptosis (IAP) [103]. CacyBP expression was significantly negatively associated with the COX expression [104]. |
| Non-small cell lung cancer [68,69] | Correlation between HER-2, EGFR, and COX-2 expression in patients of non-small cell lung cancer at different degrees [69] |
| Laryngeal cancer [71] | Cox-2 overexpression was significantly associated with radioresistant tumors [71]. |
| Papillary thyroid cancer [106] | The expression of COX-2 is increased with age in papillary thyroid cancer [106]. Immunohistochemically, expression of COX-2 and VEGF-C correlated strongly, and both were induced by the tumor promoter phorbol 12-myristate 13-acetate [107]. |
| Endometrial hyperplasia and carcinoma [108,109,110] | No correlation between COX-2 expression with estrogen (ER) or progesterone receptor (PR), p53, and neu [110]. Correlation between COX-2 (59%) and aromatase (65%) expression but not estrogen and progesterone receptor [111]. |
| Invasive gallbladder cancer [112] | COX-2, c-Met, β-catenin, c-erbB2 and EGFR were over-expressed in 80%, 74%, 71%, 62%, and 11% of invasive gallbladder cancers, respectively [112]. |
| Prostate cancer Metastatic primary prostate carcinoma compared to non-metastatic cancers [113,114,115,116] | COX-2 and Ki-67 antigen co-expression in 42.9% and 67% of the prostate cancer patients [113]. Patients with PSA > 7 ng/mL and high COX-2 expression had the highest probability of recurrence [114]. The expressions of COX-2 and E-cadherin are very firmly and inversely correlated as prognostic indicators. [115]. High expression of COX-2, TGF-beta, and Ki67 in metastatic primary prostate carcinoma was associated with death from prostate carcinoma [116]. |
| Gastric cancer [117,118] | A positive correlation between COX-2 and K-ras expression with the depth of invasion and lymph node metastasis in gastric cancer [117]. Epithelial MMP-2 expression in gastric cancer is associated with aggressive forms, COX-2 expression, and poor survival [118]. |
| Cervical cancer [119] | DNA hypermethylation of the COX-2 gene may be a potential prognostic marker in the early stages of cervical cancer [119]. |
| Pancreatic cancer [120,121] Anaplastic pancreatic cancer [122] | Tumor COX-2 expression portends a poor prognosis for patients with resected adenocarcinoma of the pancreas, particularly in tumors > or = 3 cm [121]. Expression of L1CAM, COX-2, and EGFR in the majority of undifferentiated pancreatic carcinomas [122]. |
2.4. Lipid Peroxidation Derived Products and Biological Targets
2.5. Antioxidants against Lipid Radical Reactions and Peroxidases
3. Lipid Hydroperoxides Generated by Stimulated Lipoxygenases (LOXs), Cyclooxygenases (COXs), and the Role of Their Metabolites in Cancer
3.1. COXs
3.2. The Use of COX Inhibitors to Induce Cancer Cell Death
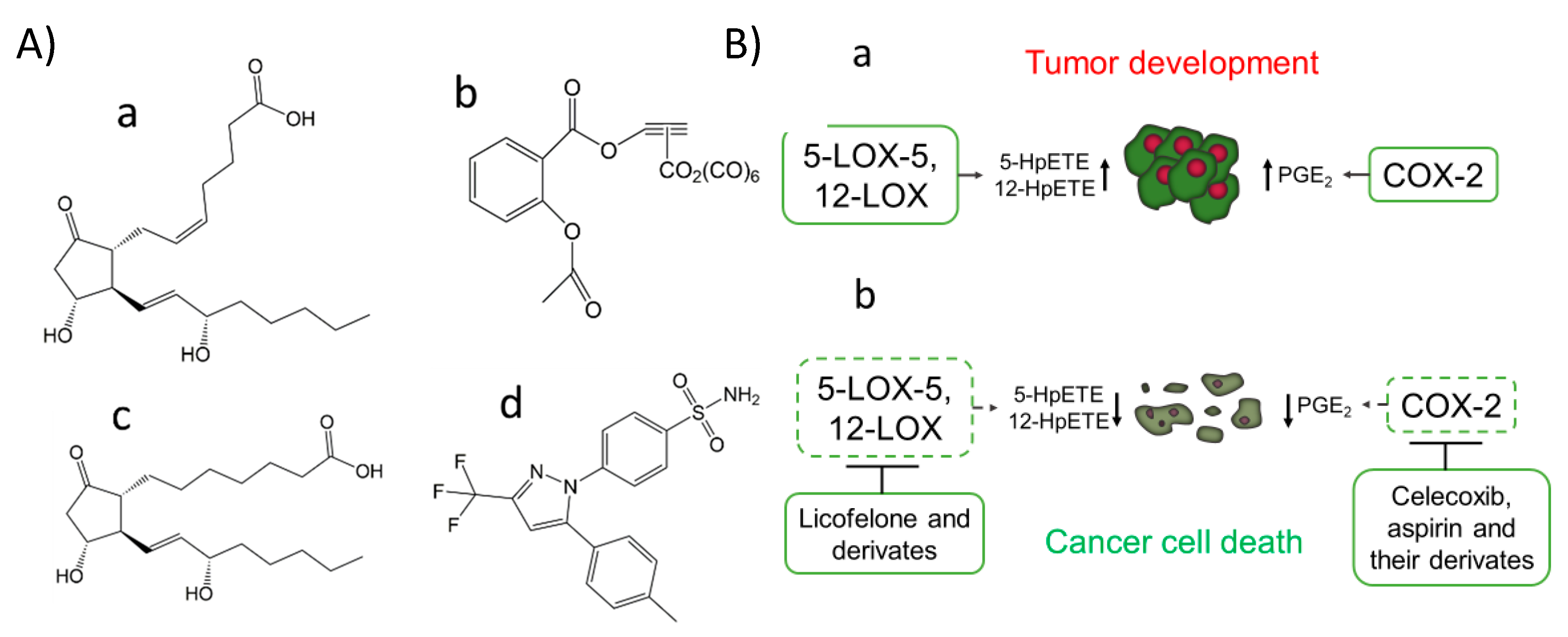
3.3. LOXs
3.4. The Use of LOX Inhibitors to Induce Cancer Cell Death
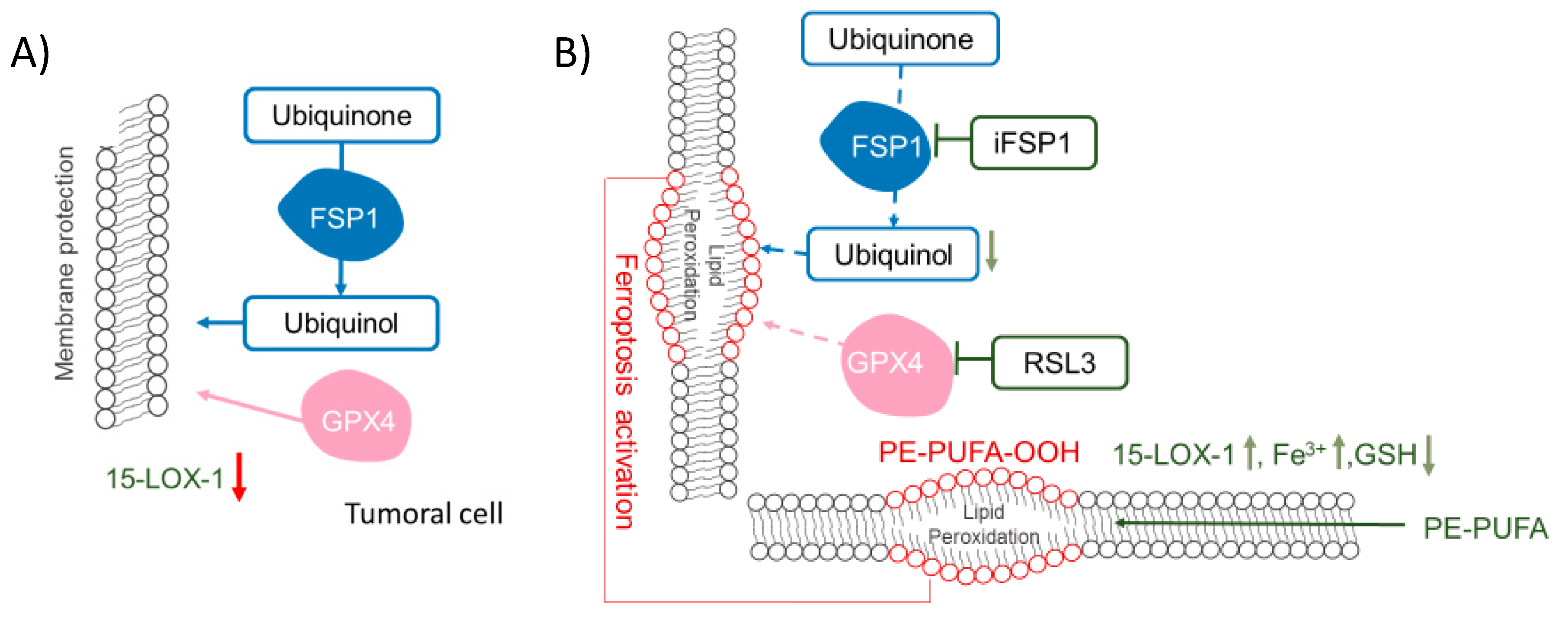
4. Stimulation of Peroxidases to Induce Cancer Cell Death: The Case of 15-LOX-1 Activity in Ferroptosis
5. Blockage of Antioxidant Enzymes to Increase Lipid Peroxidation in Tumoral Cells
5.1. GPX4
5.2. Quinone Reductases (QRs)
6. The Future of Nanoparticles (NP) for Cancer Targeting and Tissue Specificity
Author Contributions
Funding
Conflicts of Interest
References
- Bartsch, H.; Nair, J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: Role of lipid peroxidation, DNA damage, and repair. Langenbeck’s Arch. Surg. 2006, 391, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E.; Kamendulis, L.M. The Role of Oxidative Stress in Carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 239–267. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759–771. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaiswing, L.; St Clair, W.H.; St Clair, D.K. Redox Paradox: A Novel Approach to Therapeutics-Resistant Cancer. Antioxid. Redox Signal. 2018, 29, 1237–1272. [Google Scholar] [CrossRef] [PubMed]
- Sznarkowska, A.; Kostecka, A.; Meller, K.; Bielawski, K.P. Inhibition of cancer antioxidant defense by natural compounds. Oncotarget 2016, 8, 15996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Colombo, S.; Melo, T.; Martínez-López, M.; Carrasco, M.J.; Domingues, M.R.; Pérez-Sala, D.; Domingues, P. Phospholipidome of endothelial cells shows a different adaptation response upon oxidative, glycative and lipoxidative stress. Sci. Rep. 2018, 8, 12365. [Google Scholar] [CrossRef] [Green Version]
- Hazen, S.L. Oxidized phospholipids as endogenous pattern recognition ligands in innate immunity. J. Biol. Chem. 2008, 283, 15527–15531. [Google Scholar] [CrossRef] [Green Version]
- Dennis, E.A. Liberating Chiral Lipid Mediators, Inflammatory Enzymes, and LIPID MAPS from Biological Grease. J. Biol. Chem. 2016, 291, 24431–24448. [Google Scholar] [CrossRef] [Green Version]
- Freigang, S. The regulation of inflammation by oxidized phospholipids. Eur. J. Immunol. 2016, 46, 1818–1825. [Google Scholar] [CrossRef] [Green Version]
- Miller, Y.I.; Shyy, J.Y.J. Context-Dependent Role of Oxidized Lipids and Lipoproteins in Inflammation. Trends Endocrinol. Metab. 2017, 28, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Dias, I.H.K.; Milic, I.; Heiss, C.; Ademowo, O.S.; Polidori, M.C.; Devitt, A.; Griffiths, H.R. Inflammation, Lipid (Per)oxidation, and Redox Regulation. Antioxid. Redox Signal. 2020, 33, 166–190. [Google Scholar] [CrossRef]
- Van der Paal, J.; Neyts, E.C.; Verlackt, C.C.W.; Bogaerts, A. Effect of lipid peroxidation on membrane permeability of cancer and normal cells subjected to oxidative stress. Chem. Sci. 2016, 7, 489–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalá, A. Lipid peroxidation modifies the picture of membranes from the “Fluid Mosaic Model” to the “Lipid Whisker Model”. Biochimie 2012, 94, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Mouchlis, V.D.; Dennis, E.A. Phospholipase A(2) catalysis and lipid mediator lipidomics. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2019, 1864, 766–771. [Google Scholar] [CrossRef]
- Xu, Y.; Qian, S.Y. Anti-cancer activities of ω-6 polyunsaturated fatty acids. Biomed. J. 2014, 37, 112–119. [Google Scholar] [CrossRef]
- Anderson, D.W.; Crowle, A.J. Regression and differentiation of neuroblastoma tumors in mice treated with differentiating agents-prostaglandin E1 and a phosphodiesterase inhibitor, RO 20-1724. Cancer Lett. 1982, 16, 287–295. [Google Scholar] [CrossRef]
- Hanazaki, K.; Kajikawa, S.; Fujimori, Y.; Nakata, S.; Shimozawa, N.; Koide, N.; Adachi, W.; Amano, J. Effects of prostaglandin E1 administration during hepatectomy for cirrhotic hepatocellular carcinoma. Hepatogastroenterology 2000, 47, 461–464. [Google Scholar] [PubMed]
- Wang, X.; Lin, H.; Gu, Y. Multiple roles of dihomo-γ-linolenic acid against proliferation diseases. Lipids Health Dis. 2012, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Davies, P.; Bailey, P.J.; Goldenberg, M.M.; Ford-Hutchinson, A.W. The Role of Arachidonic Acid Oxygenation Products in Pain and Inflammation. Annu. Rev. Immunol. 1984, 2, 335–357. [Google Scholar] [CrossRef] [PubMed]
- Castellone, M.D.; Teramoto, H.; Williams, B.O.; Druey, K.M.; Gutkind, J.S. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science 2005, 310, 1504–1510. [Google Scholar] [CrossRef]
- Pai, R.; Soreghan, B.; Szabo, I.L.; Pavelka, M.; Baatar, D.; Tarnawski, A.S. Prostaglandin E2 transactivates EGF receptor: A novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat. Med. 2002, 8, 289–293. [Google Scholar] [CrossRef]
- Schrey, M.P.; Patel, K.V. Prostaglandin E2 production and metabolism in human breast cancer cells and breast fibroblasts. Regulation by inflammatory mediators. Br. J. Cancer 1995, 72, 1412–1419. [Google Scholar] [CrossRef] [Green Version]
- Uotila, P. Inhibition of prostaglandin E2 formation and histamine action in cancer immunotherapy. Cancer Immunol. Immunother. 1993, 37, 251–254. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef]
- Tachtsis, B.; Whitfield, J.; Hawley, J.A.; Hoffman, N.J. Omega-3 Polyunsaturated Fatty Acids Mitigate Palmitate-Induced Impairments in Skeletal Muscle Cell Viability and Differentiation. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef]
- Magtanong, L.; Ko, P.J.; Dixon, S.J. Emerging roles for lipids in non-apoptotic cell death. Cell Death Differ. 2016, 23, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Khadge, S.; Sharp, J.G.; McGuire, T.R.; Thiele, G.M.; Talmadge, J.E. Lipid Inflammatory Mediators in Cancer Progression and Therapy. In Tumor Immune Microenvironment in Cancer Progression and Cancer Therapy; Kalinski, P., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 145–156. [Google Scholar]
- Zhang, Q.; Zhu, B.; Li, Y. Resolution of Cancer-Promoting Inflammation: A New Approach for Anticancer Therapy. Front. Immunol. 2017, 8, 71. [Google Scholar] [CrossRef] [Green Version]
- Ungaro, F.; D’Alessio, S.; Danese, S. The Role of Pro-Resolving Lipid Mediators in Colorectal Cancer-Associated Inflammation: Implications for Therapeutic Strategies. Cancers 2020, 12, 2060. [Google Scholar] [CrossRef]
- Rigas, B.; Goldman, I.S.; Levine, L. Altered eicosanoid levels in human colon cancer. J. Lab. Clin. Med. 1993, 122, 518–523. [Google Scholar]
- Wang, D.; DuBois, R.N. Cyclooxygenase-2: A potential target in breast cancer. Semin. Oncol. 2004, 31, 64–73. [Google Scholar] [CrossRef]
- Koppenol, W.H. The centennial of the Fenton reaction. Free Radic. Biol. Med. 1993, 15, 645–651. [Google Scholar] [CrossRef]
- Koppenol, W.H. The Haber-Weiss cycle—70 years later. Redox Rep. 2001, 6, 229–234. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 4th ed.; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Johnson, D.R.; Decker, E.A. The Role of Oxygen in Lipid Oxidation Reactions: A Review. Annu. Rev. Food Sci. Technol. 2015, 6, 171–190. [Google Scholar] [CrossRef] [PubMed]
- Schafer, F.Q.; Qian, S.Y.; Buettner, G.R. Iron and free radical oxidations in cell membranes. Cell Mol. Biol. 2000, 46, 657–662. [Google Scholar]
- Qian, S.Y.; Buettner, G.R. Iron and dioxygen chemistry is an important route to initiation of biological free radical oxidations: An electron paramagnetic resonance spin trapping study. Free Radic. Biol. Med. 1999, 26, 1447–1456. [Google Scholar] [CrossRef]
- Minotti, G.; Aust, S.D. The role of iron in the initiation of lipid peroxidation. Chem. Phys. Lipids 1987, 44, 191–208. [Google Scholar] [CrossRef]
- Reis, A.; Spickett, C.M. Chemistry of phospholipid oxidation. Biochim. Biophys. Acta (BBA)-Biomembr. 2012, 1818, 2374–2387. [Google Scholar] [CrossRef] [Green Version]
- Pratt, D.A.; Tallman, K.A.; Porter, N.A. Free radical oxidation of polyunsaturated lipids: New mechanistic insights and the development of peroxyl radical clocks. Acc Chem. Res. 2011, 44, 458–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repetto, M.G.; Ferrarotti, N.F.; Boveris, A. The involvement of transition metal ions on iron-dependent lipid peroxidation. Arch. Toxicol. 2010, 84, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Repetto, M.; Boveris, A. Transition Metals: Bioinorganic and Redox Reactions in Biological Systems; Transition Metals: Characteristics, Properties and Uses; Nova Science Publishers Inc.: New York, NY, USA, 2011; pp. 349–370. [Google Scholar]
- Pike, L.J.; Han, X.; Chung, K.-N.; Gross, R.W. Lipid Rafts Are Enriched in Arachidonic Acid and Plasmenylethanolamine and Their Composition Is Independent of Caveolin-1 Expression: A Quantitative Electrospray Ionization/Mass Spectrometric Analysis. Biochemistry 2002, 41, 2075–2088. [Google Scholar] [CrossRef]
- Gutierrez-Merino, C.; Marques-da-Silva, D.; Fortalezas, S.; Samhan-Arias, A.K. The critical role of lipid rafts nanodomains in the cross-talk between calcium and reactive oxygen and nitrogen species in cerebellar granule neurons apoptosis by extracellular potassium deprivation. Aims. Mol. Sci. 2016, 3, 12–29. [Google Scholar] [CrossRef]
- Yaqoob, P. Fatty acids as gatekeepers of immune cell regulation. Trends Immunol. 2003, 24, 639–645. [Google Scholar] [CrossRef]
- Tyurina, Y.Y.; Tyurin, V.A.; Kapralova, V.I.; Amoscato, A.A.; Epperly, M.W.; Greenberger, J.S.; Kagan, V.E. Mass-Spectrometric Characterization of Phospholipids and Their Hydroperoxide Derivatives In Vivo: Effects of Total Body Irradiation. In Lipidomics: Volume 2: Methods and Protocols; Armstrong, D., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 153–183. [Google Scholar]
- Stark, G. The effect of ionizing radiation on lipid membranes. Biochim. Biophys. Acta (BBA)-Rev. Biomembr. 1991, 1071, 103–122. [Google Scholar] [CrossRef]
- Corre, I.; Niaudet, C.; Paris, F. Plasma membrane signaling induced by ionizing radiation. Mutat. Res. Rev. Mutat. Res. 2010, 704, 61–67. [Google Scholar] [CrossRef]
- Emerit, J.; Klein, J.M.; Coutellier, A.; Congy, F. [Free radicals and lipid peroxidation in cell biology: Physiopathologic prospects]. Pathol. Biol. (Paris) 1991, 39, 316–327. [Google Scholar] [PubMed]
- Vlasova, I.I. Peroxidase Activity of Human Hemoproteins: Keeping the Fire under Control. Molecules 2018, 23, 2561. [Google Scholar] [CrossRef] [Green Version]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef]
- Ji, J.; Kline, A.E.; Amoscato, A.; Samhan-Arias, A.K.; Sparvero, L.J.; Tyurin, V.A.; Tyurina, Y.Y.; Fink, B.; Manole, M.D.; Puccio, A.M.; et al. Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nat. Neurosci. 2012, 15, 1407–1413. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, J.; Kapralov, A.A.; Yanamala, N.; Tyurina, Y.Y.; Amoscato, A.A.; Pearce, L.; Peterson, J.; Huang, Z.; Jiang, J.; Samhan-Arias, A.K.; et al. A mitochondria-targeted inhibitor of cytochrome c peroxidase mitigates radiation-induced death. Nat. Commun. 2011, 2, 497. [Google Scholar] [CrossRef] [Green Version]
- Samhan-Arias, A.K.; Tyurina, Y.Y.; Kagan, V.E. Lipid antioxidants: Free radical scavenging versus regulation of enzymatic lipid peroxidation. J. Clin. Biochem. Nutr. 2011, 48, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Belikova, N.A.; Tyurina, Y.Y.; Borisenko, G.; Tyurin, V.; Samhan Arias, A.K.; Yanamala, N.; Furtmuller, P.G.; Klein-Seetharaman, J.; Obinger, C.; Kagan, V.E. Heterolytic reduction of fatty acid hydroperoxides by cytochrome c/cardiolipin complexes: Antioxidant function in mitochondria. J. Am. Chem. Soc. 2009, 131, 11288–11289. [Google Scholar] [CrossRef]
- Samhan-Arias, A.K.; Cordas, C.M.; Carepo, M.S.; Maia, L.B.; Gutierrez-Merino, C.; Moura, I.; Moura, J.J.G. Ligand accessibility to heme cytochrome b5 coordinating sphere and enzymatic activity enhancement upon tyrosine ionization. J. Biol. Inorg. Chem. A Publ. Soc. Biol. Inorg. Chem. 2019, 24, 317–330. [Google Scholar] [CrossRef]
- Samhan-Arias, A.K.; Maia, L.B.; Cordas, C.M.; Moura, I.; Gutierrez-Merino, C.; Moura, J.J.G. Peroxidase-like activity of cytochrome b5 is triggered upon hemichrome formation in alkaline pH. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 373–378. [Google Scholar] [CrossRef]
- Ascenzi, P.; Coletta, M.; Wilson, M.T.; Fiorucci, L.; Marino, M.; Polticelli, F.; Sinibaldi, F.; Santucci, R. Cardiolipin–cytochrome c complex: Switching cytochrome c from an electron-transfer shuttle to a myoglobin- and a peroxidase-like heme-protein. Iubmb Life 2015, 67, 98–109. [Google Scholar] [CrossRef]
- Beckerson, P.; Svistunenko, D.; Reeder, B. Effect of the distal histidine on the peroxidatic activity of monomeric cytoglobin. F1000Res 2015, 4, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulos, T.L. Heme enzyme structure and function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Eberhart, C.E.; Coffey, R.J.; Radhika, A.; Giardiello, F.M.; Ferrenbach, S.; Dubois, R.N. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 1994, 107, 1183–1188. [Google Scholar] [CrossRef]
- Ferrandina, G.; Lauriola, L.; Zannoni, G.F.; Distefano, M.G.; Legge, F.; Salutari, V.; Gessi, M.; Maggiano, N.; Scambia, G.; Ranelletti, F.O. Expression of cyclooxygenase-2 (COX-2) in tumour and stroma compartments in cervical cancer: Clinical implications. Br. J. Cancer 2002, 87, 1145–1152. [Google Scholar] [CrossRef] [Green Version]
- Tian, F.; Wang, T.Y.; Gong, M.; Lu, X.M.; Hu, J.; Wang, J.; Zhang, C.H. [Overexpression of COX-2 and its clinical significance in non-small cell lung cancer]. Zhonghua Wai Ke Za Zhi 2003, 41, 407–410. [Google Scholar]
- Brattstrom, D.; Wester, K.; Bergqvist, M.; Hesselius, P.; Malmstrom, P.U.; Nordgren, H.; Wagenius, G.; Brodin, O. HER-2, EGFR, COX-2 expression status correlated to microvessel density and survival in resected non-small cell lung cancer. Acta Oncol. 2004, 43, 80–86. [Google Scholar] [CrossRef]
- Liao, Z.; Milas, L. COX-2 and its inhibition as a molecular target in the prevention and treatment of lung cancer. Expert Rev. Anticancer Ther. 2004, 4, 543–560. [Google Scholar] [CrossRef]
- Nix, P.; Lind, M.; Greenman, J.; Stafford, N.; Cawkwell, L. Expression of Cox-2 protein in radioresistant laryngeal cancer. Ann. Oncol. 2004, 15, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, V.; Leach, D.A.; Zinonos, I.; Ponomarev, V.; Licari, G.; Liapis, V.; Ingman, W.V.; Anderson, P.; DeNichilo, M.O.; Evdokiou, A. Inflammatory peroxidases promote breast cancer progression in mice via regulation of the tumour microenvironment. Int. J. Oncol. 2017, 50, 1191–1200. [Google Scholar] [CrossRef] [Green Version]
- Tabariès, S.; Ouellet, V.; Hsu, B.E.; Annis, M.G.; Rose, A.A.N.; Meunier, L.; Carmona, E.; Tam, C.E.; Mes-Masson, A.-M.; Siegel, P.M. Granulocytic immune infiltrates are essential for the efficient formation of breast cancer liver metastases. Breast Cancer Res. 2015, 17, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samoszuk, M.; Lin, F.; Rim, P.; Strathearn, G. New marker for blood vessels in human ovarian and endometrial cancers. Clin. Cancer Res. 1996, 2, 1867–1871. [Google Scholar]
- Cormier, S.A.; Taranova, A.G.; Bedient, C.; Nguyen, T.; Protheroe, C.; Pero, R.; Dimina, D.; Ochkur, S.I.; O’Neill, K.; Colbert, D.; et al. Pivotal Advance: Eosinophil infiltration of solid tumors is an early and persistent inflammatory host response. J. Leukoc. Biol. 2006, 79, 1131–1139. [Google Scholar] [CrossRef] [Green Version]
- Hou, Z.; Falcone, D.J.; Subbaramaiah, K.; Dannenberg, A.J. Macrophages induce COX-2 expression in breast cancer cells: Role of IL-1beta autoamplification. Carcinogenesis 2011, 32, 695–702. [Google Scholar] [CrossRef] [Green Version]
- Mustea, A.; Heinze, G.; Sehouli, J.; Koensgen, D.; Wolf, A.; Gutu, L.; Sofroni, D.; Pirvulescu, C.; Braicu, E.I.; Schuster, E.; et al. The -463G/A polymorphism in myeloperoxidase gene and cervical cancer. Anticancer Res. 2007, 27, 1531–1535. [Google Scholar]
- Zhu, H.; Yang, L.; Zhou, B.; Yu, R.; Tang, N.; Wang, B. Myeloperoxidase G-463A polymorphism and the risk of gastric cancer: A case-control study. Carcinogenesis 2006, 27, 2491–2496. [Google Scholar] [CrossRef] [Green Version]
- London, S.J.; Lehman, T.A.; Taylor, J.A. Myeloperoxidase genetic polymorphism and lung cancer risk. Cancer Res. 1997, 57, 5001–5003. [Google Scholar]
- Limami, Y.; Pinon, A.; Leger, D.Y.; Pinault, E.; Delage, C.; Beneytout, J.-L.; Simon, A.; Liagre, B. The P2Y2/Src/p38/COX-2 pathway is involved in the resistance to ursolic acid-induced apoptosis in colorectal and prostate cancer cells. Biochimie 2012, 94, 1754–1763. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.; Weichert, H.; Porzel, A.; Wasternack, C.; Kuhn, H.; Feussner, I. Enzymatic and non-enzymatic lipid peroxidation in leaf development. Biochim. Biophys. Acta 2001, 1533, 266–276. [Google Scholar] [CrossRef]
- Stoyanovsky, D.A.; Tyurina, Y.Y.; Shrivastava, I.; Bahar, I.; Tyurin, V.A.; Protchenko, O.; Jadhav, S.; Bolevich, S.B.; Kozlov, A.V.; Vladimirov, Y.A.; et al. Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? Free Radic. Biol. Med. 2019, 133, 153–161. [Google Scholar] [CrossRef]
- Noguchi, N.; Yamashita, H.; Hamahara, J.; Nakamura, A.; Kuhn, H.; Niki, E. The specificity of lipoxygenase-catalyzed lipid peroxidation and the effects of radical-scavenging antioxidants. Biol. Chem. 2002, 383, 619–626. [Google Scholar] [CrossRef]
- Drew, D.A.; Cao, Y.; Chan, A.T. Aspirin and colorectal cancer: The promise of precision chemoprevention. Nat. Rev. Cancer 2016, 16, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Elwood, P.C.; Morgan, G.; Pickering, J.E.; Galante, J.; Weightman, A.L.; Morris, D.; Kelson, M.; Dolwani, S. Aspirin in the Treatment of Cancer: Reductions in Metastatic Spread and in Mortality: A Systematic Review and Meta-Analyses of Published Studies. PLoS ONE 2016, 11, e0152402. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Anderson, A.J.; Malkowski, M.G. Arg-513 and Leu-531 Are Key Residues Governing Time-Dependent Inhibition of Cyclooxygenase-2 by Aspirin and Celebrex. Biochemistry 2019, 58, 3990–4002. [Google Scholar] [CrossRef]
- Lucido, M.J.; Orlando, B.J.; Vecchio, A.J.; Malkowski, M.G. Crystal Structure of Aspirin-Acetylated Human Cyclooxygenase-2: Insight into the Formation of Products with Reversed Stereochemistry. Biochemistry 2016, 55, 1226–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giménez-Bastida, J.A.; Boeglin, W.E.; Boutaud, O.; Malkowski, M.G.; Schneider, C. Residual cyclooxygenase activity of aspirin-acetylated COX-2 forms 15 R-prostaglandins that inhibit platelet aggregation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 1033–1041. [Google Scholar] [CrossRef]
- Jeske, L.; Placzek, S.; Schomburg, I.; Chang, A.; Schomburg, D. BRENDA in 2019: A European ELIXIR core data resource. Nucleic Acids Res. 2018, 47, D542–D549. [Google Scholar] [CrossRef]
- Fan, X.M.; Zheng, F.S.; Liu, H.Y.; Ma, Y.H.; Wong, B.C. [Mechanism of apoptosis induced by specific COX-2 inhibitor SC236 in gastric cancer cells]. Zhonghua Zhong Liu Za Zhi 2005, 27, 145–147. [Google Scholar]
- Ott, I.; Schmidt, K.; Kircher, B.; Schumacher, P.; Wiglenda, T.; Gust, R. Antitumor-Active Cobalt−Alkyne Complexes Derived from Acetylsalicylic Acid: Studies on the Mode of Drug Action. J. Med. Chem. 2005, 48, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Zanellato, I.; Bonarrigo, I.; Ravera, M.; Gabano, E.; Gust, R.; Osella, D. The hexacarbonyldicobalt derivative of aspirin acts as a CO-releasing NSAID on malignant mesothelioma cells. Met. Integr. Biometal Sci. 2013, 5, 1604–1613. [Google Scholar] [CrossRef]
- Soumaoro, L.T.; Uetake, H.; Takagi, Y.; Iida, S.; Higuchi, T.; Yasuno, M.; Enomoto, M.; Sugihara, K. Coexpression of VEGF-C and Cox-2 in human colorectal cancer and its association with lymph node metastasis. Dis. Colon Rectum 2006, 49, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.H.; Wan, D.S.; Li, L.R.; Chen, G.; Lu, Z.H.; Wu, X.J.; Kong, L.H.; Pan, Z.Z. Expression of COX-2, MMP-2 and VEGF in stage II and III colorectal cancer and the clinical significance. Hepatogastroenterology 2011, 58, 369–376. [Google Scholar]
- Ferrandina, G.; Lauriola, L.; Zannoni, G.F.; Fagotti, A.; Fanfani, F.; Legge, F.; Maggiano, N.; Gessi, M.; Mancuso, S.; Ranelletti, F.O.; et al. Increased cyclooxygenase-2 (COX-2) expression is associated with chemotherapy resistance and outcome in ovarian cancer patients. Ann. Oncol. 2002, 13, 1205–1211. [Google Scholar] [CrossRef]
- Ferrandina, G.; Ranelletti, F.O.; Lauriola, L.; Fanfani, F.; Legge, F.; Mottolese, M.; Nicotra, M.R.; Natali, P.G.; Zakut, V.H.; Scambia, G. Cyclooxygenase-2 (COX-2), epidermal growth factor receptor (EGFR), and Her-2/neu expression in ovarian cancer. Gynecol. Oncol. 2002, 85, 305–310. [Google Scholar] [CrossRef]
- Denkert, C.; Köbel, M.; Pest, S.; Koch, I.; Berger, S.; Schwabe, M.; Siegert, A.; Reles, A.; Klosterhalfen, B.; Hauptmann, S. Expression of Cyclooxygenase 2 Is an Independent Prognostic Factor in Human Ovarian Carcinoma. Am. J. Pathol. 2002, 160, 893–903. [Google Scholar] [CrossRef] [Green Version]
- Ristimäki, A.; Sivula, A.; Lundin, J.; Lundin, M.; Salminen, T.; Haglund, C.; Joensuu, H.; Isola, J. Prognostic Significance of Elevated Cyclooxygenase-2 Expression in Breast Cancer. Cancer Res. 2002, 62, 632–635. [Google Scholar]
- Mazhar, D.; Ang, R.; Waxman, J. COX inhibitors and breast cancer. Br. J. Cancer 2006, 94, 346–350. [Google Scholar] [CrossRef] [Green Version]
- Fulton, A.M.; Heppner, G.H. Relationships of Prostaglandin E and Natural Killer Sensitivity to Metastatic Potential in Murine Mammary Adenocarcinomas. Cancer Res. 1985, 45, 4779–4784. [Google Scholar]
- Singh, B.; Berry, J.A.; Vincent, L.E.; Lucci, A. Involvement of IL-8 in COX-2-mediated bone metastases from breast cancer. J. Surg. Res. 2006, 134, 44–51. [Google Scholar] [CrossRef]
- Majumder, M.; Dunn, L.; Liu, L.; Hasan, A.; Vincent, K.; Brackstone, M.; Hess, D.; Lala, P.K. COX-2 induces oncogenic micro RNA miR655 in human breast cancer. Sci. Rep. 2018, 8, 327. [Google Scholar] [CrossRef] [Green Version]
- Barnes, N.; Haywood, P.; Flint, P.; Knox, W.F.; Bundred, N.J. Survivin expression in in situ and invasive breast cancer relates to COX-2 expression and DCIS recurrence. Br. J. Cancer 2006, 94, 253–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, F.; Yu, X.L.; Wang, X.G.; Tang, Y.F.; Wang, L.L.; Ma, L. Down-regulation of CacyBP is associated with poor prognosis and the effects on COX-2 expression in breast cancer. Int. J. Oncol. 2010, 37, 1261–1269. [Google Scholar] [CrossRef]
- Li, E.X.; Wu, Y.Y.; Shi, F.; Wu, Y.; Guo, J.J.; Dong, D.F. [Relationship between serum VEGF level and VEGF, COX-2 and MVD expression in breast cancer tissues]. Zhonghua Zhong Liu Za Zhi 2007, 29, 522–525. [Google Scholar]
- Siironen, P.; Ristimäki, A.; Nordling, S.; Louhimo, J.; Haapiainen, R.; Haglund, C. Expression of COX-2 is increased with age in papillary thyroid cancer. Histopathology 2004, 44, 490–497. [Google Scholar] [CrossRef]
- Siironen, P.; Ristimaki, A.; Narko, K.; Nordling, S.; Louhimo, J.; Andersson, S.; Haapiainen, R.; Haglund, C. VEGF-C and COX-2 expression in papillary thyroid cancer. Endocr. Relat. Cancer 2006, 13, 465–473. [Google Scholar] [CrossRef]
- Fujiwaki, R.; Iida, K.; Kanasaki, H.; Ozaki, T.; Hata, K.; Miyazaki, K. Cyclooxygenase-2 expression in endometrial cancer: Correlation with microvessel count and expression of vascular endothelial growth factor and thymidine phosphorylase. Hum. Pathol. 2002, 33, 213–219. [Google Scholar] [CrossRef]
- Landen, C.N.; Mathur, S.P.; Richardson, M.S.; Creasman, W.T. Expression of cyclooxygenase-2 in cervical, endometrial, and ovarian malignancies. Am. J. Obstet. Gynecol. 2003, 188, 1174–1176. [Google Scholar] [CrossRef]
- Ferrandina, G.; Ranelletti, F.O.; Gallotta, V.; Martinelli, E.; Zannoni, G.F.; Gessi, M.; Scambia, G. Expression of cyclooxygenase-2 (COX-2), receptors for estrogen (ER), and progesterone (PR), p53, ki67, and neu protein in endometrial cancer. Gynecol. Oncol. 2005, 98, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.M.; Ramirez, N.; Cohn, D.E.; Kelbick, N.; Pavelka, J.; Ben-Shachar, I.; Morrison, C. Correlation of cyclooxygenase-2 (COX-2) and aromatase expression in human endometrial cancer: Tissue microarray analysis. Am. J. Obstet. Gynecol. 2005, 192, 1262–1271. [Google Scholar] [CrossRef]
- Moon, W.S.; Park, H.S.; Lee, H.; Pai, R.; Tarnawski, A.S.; Kim, K.R.; Jang, K.Y. Co-expression of cox-2, C-met and beta-catenin in cells forming invasive front of gallbladder cancer. Cancer Res. Treat. 2005, 37, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Rubio, J.; Ramos, D.; Lopez-Guerrero, J.A.; Iborra, I.; Collado, A.; Solsona, E.; Almenar, S.; Llombart-Bosch, A. Immunohistochemical expression of Ki-67 antigen, cox-2 and Bax/Bcl-2 in prostate cancer; prognostic value in biopsies and radical prostatectomy specimens. Eur. Urol. 2005, 48, 745–751. [Google Scholar] [CrossRef]
- Cohen, B.L.; Gomez, P.; Omori, Y.; Duncan, R.C.; Civantos, F.; Soloway, M.S.; Lokeshwar, V.B.; Lokeshwar, B.L. Cyclooxygenase-2 (COX-2) expression is an independent predictor of prostate cancer recurrence. Int. J. Cancer 2006, 119, 1082–1087. [Google Scholar] [CrossRef]
- Rao, D.S.; Gui, D.; Koski, M.E.; Popoviciu, L.M.; Wang, H.; Reiter, R.E.; Said, J.W. An Inverse Relation Between COX-2 and E-cadherin Expression Correlates With Aggressive Histologic Features in Prostate Cancer. Appl. Immunohistochem. Mol. Morphol. 2006, 14, 375–383. [Google Scholar] [CrossRef]
- Richardsen, E.; Uglehus, R.D.; Due, J.; Busch, C.; Busund, L.T. COX-2 is overexpressed in primary prostate cancer with metastatic potential and may predict survival. A comparison study between COX-2, TGF-beta, IL-10 and Ki67. Cancer Epidemiol. 2010, 34, 316–322. [Google Scholar] [CrossRef]
- Li, M.; Liu, W.; Zhu, Y.F.; Chen, Y.L.; Zhang, B.Z.; Wang, R. Correlation of COX-2 and K-ras expression to clinical outcome in gastric cancer. Acta Oncol. 2006, 45, 1115–1119. [Google Scholar] [CrossRef]
- Mrena, J.; Wiksten, J.P.; Nordling, S.; Kokkola, A.; Ristimäki, A.; Haglund, C. MMP-2 but not MMP-9 associated with COX-2 and survival in gastric cancer. J. Clin. Pathol. 2006, 59, 618–623. [Google Scholar] [CrossRef] [Green Version]
- Jo, H.; Kang, S.; Kim, J.W.; Kang, G.H.; Park, N.H.; Song, Y.S.; Park, S.Y.; Kang, S.B.; Lee, H.P. Hypermethylation of the COX-2 gene is a potential prognostic marker for cervical cancer. J. Obs. Gynaecol. Res. 2007, 33, 236–241. [Google Scholar] [CrossRef]
- Matsumoto, G.; Muta, M.; Tsuruta, K.; Horiguchi, S.; Karasawa, K.; Okamoto, A. Tumor size significantly correlates with postoperative liver metastases and COX-2 expression in patients with resectable pancreatic cancer. Pancreatology 2007, 7, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Matsubayashi, H.; Infante, J.R.; Winter, J.; Klein, A.P.; Schulick, R.; Hruban, R.; Visvanathan, K.; Goggins, M. Tumor COX-2 expression and prognosis of patients with resectable pancreatic cancer. Cancer Biol. 2007, 6, 1569–1575. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, F.; Moldenhauer, G.; Herpel, E.; Gaida, M.M.; Strobel, O.; Werner, J.; Esposito, I.; Muerkoster, S.S.; Schirmacher, P.; Kern, M.A. Expression of L1CAM, COX-2, EGFR, c-KIT and Her2/neu in anaplastic pancreatic cancer: Putative therapeutic targets? Histopathology 2010, 56, 440–448. [Google Scholar] [CrossRef]
- Yin, H.; Porter, N.A. New Insights Regarding the Autoxidation of Polyunsaturated Fatty Acids. Antioxid. Redox Signal. 2004, 7, 170–184. [Google Scholar] [CrossRef]
- Russell, G.A. Deuterium-isotope Effects in the Autoxidation of Aralkyl Hydrocarbons. Mechanism of the Interaction of PEroxy Radicals1. J. Am. Chem. Soc. 1957, 79, 3871–3877. [Google Scholar] [CrossRef]
- Pizzimenti, S.; Ciamporcero, E.; Daga, M.; Pettazzoni, P.; Arcaro, A.; Cetrangolo, G.; Minelli, R.; Dianzani, C.; Lepore, A.; Gentile, F.; et al. Interaction of aldehydes derived from lipid peroxidation and membrane proteins. Front. Physiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.J.; Yaffe, M.B. Protein Regulation in Signal Transduction. Cold Spring Harb. Perspect. Biol. 2016, 8, a005918. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, N.; Costello, C.E. Emerging roles of post-translational modifications in signal transduction and angiogenesis. Proteomics 2015, 15, 300–309. [Google Scholar] [CrossRef]
- Tomko, N.; Kluever, M.; Wu, C.; Zhu, J.; Wang, Y.; Salomon, R.G. 4-Hydroxy-7-oxo-5-heptenoic acid lactone is a potent inducer of brain cancer cell invasiveness that may contribute to the failure of anti-angiogenic therapies. Free Radic. Biol. Med. 2020, 146, 234–256. [Google Scholar] [CrossRef]
- Sousa, B.C.; Ahmed, T.; Dann, W.L.; Ashman, J.; Guy, A.; Durand, T.; Pitt, A.R.; Spickett, C.M. Short-chain lipid peroxidation products form covalent adducts with pyruvate kinase and inhibit its activity in vitro and in breast cancer cells. Free Radic. Biol. Med. 2019, 144, 223–233. [Google Scholar] [CrossRef]
- Sakuma, S.; Sumi, H.; Kohda, T.; Arakawa, Y.; Fujimoto, Y. Effects of Lipid Peroxidation-Derived Products on the Growth of Human Colorectal Cancer Cell Line HT-29. J. Clin. Biochem. Nutr. 2009, 45, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Gasparovic, A.C.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. Cancer growth regulation by 4-hydroxynonenal. Free Radic. Biol. Med. 2017, 111, 226–234. [Google Scholar] [CrossRef]
- Rossin, D.; Calfapietra, S.; Sottero, B.; Poli, G.; Biasi, F. HNE and cholesterol oxidation products in colorectal inflammation and carcinogenesis. Free Radic. Biol. Med. 2017, 111, 186–195. [Google Scholar] [CrossRef]
- Tappel, A.L. Vitamin E as the Biological Lipid Antioxidant. In Vitamins & Hormones; Harris, R.S., Wool, I.G., Marrian, G.F., Thimann, K.V., Eds.; Academic Press: New York, NY, USA, 1962; Volume 20, pp. 493–510. [Google Scholar]
- Buettner, G.R. The pecking order of free radicals and antioxidants: Lipid peroxidation, alpha-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef]
- Samhan-Arias, A.K.; Duarte, R.O.; Martin-Romero, F.J.; Moura, J.J.; Gutierrez-Merino, C. Reduction of ascorbate free radical by the plasma membrane of synaptic terminals from rat brain. Arch. Biochem. Biophys. 2008, 469, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, M.; Rigo, A.; Maiorino, M.; Ursini, F.; Gregolin, C. Formation of alpha-tocopherol radical and recycling of alpha-tocopherol by ascorbate during peroxidation of phosphatidylcholine liposomes. An electron paramagnetic resonance study. Biochim. Biophys. Acta 1984, 801, 215–219. [Google Scholar] [CrossRef]
- Buettner, G.R.; Jurkiewicz, B.A. Catalytic metals, ascorbate and free radicals: Combinations to avoid. Radiat. Res. 1996, 145, 532–541. [Google Scholar] [CrossRef] [Green Version]
- Mendiratta, S.; Qu, Z.C.; May, J.M. Enzyme-dependent ascorbate recycling in human erythrocytes: Role of thioredoxin reductase. Free Radic. Biol. Med. 1998, 25, 221–228. [Google Scholar] [CrossRef]
- Schneider, M.; Wortmann, M.; Mandal, P.K.; Arpornchayanon, W.; Jannasch, K.; Alves, F.; Strieth, S.; Conrad, M.; Beck, H. Absence of glutathione peroxidase 4 affects tumor angiogenesis through increased 12/15-lipoxygenase activity. Neoplasia 2010, 12, 254–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [Green Version]
- Bakhle, Y.S. COX-2 and cancer: A new approach to an old problem. Br. J. Pharm. 2001, 134, 1137–1150. [Google Scholar] [CrossRef]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arter. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Murakami, M.; Naraba, H.; Tanioka, T.; Semmyo, N.; Nakatani, Y.; Kojima, F.; Ikeda, T.; Fueki, M.; Ueno, A.; Oh-ishi, S.; et al. Regulation of Prostaglandin E2 Biosynthesis by Inducible Membrane-associated Prostaglandin E2 Synthase That Acts in Concert with Cyclooxygenase-2. J. Biol. Chem. 2000, 275, 32783–32792. [Google Scholar] [CrossRef] [Green Version]
- Jakobsson, P.J.; Thorén, S.; Morgenstern, R.; Samuelsson, B. Identification of human prostaglandin E synthase: A microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc. Natl. Acad. Sci. USA 1999, 96, 7220–7225. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.G.; Douglas-Jones, A.; Mansel, R.E. Levels of expression of lipoxygenases and cyclooxygenase-2 in human breast cancer. Prostaglandinsleukotrienes Essent. Fat. Acids 2003, 69, 275–281. [Google Scholar] [CrossRef]
- Nassar, A.; Radhakrishnan, A.; Cabrero, I.A.; Cotsonis, G.; Cohen, C. COX-2 expression in invasive breast cancer: Correlation with prognostic parameters and outcome. Appl. Immunohistochem. Mol. Morphol. 2007, 15, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wu, S.; Ahmad, M.; Jiang, J.; Liu, H.; Nagayama, T.; Rose, M.E.; Tyurin, V.A.; Tyurina, Y.Y.; Borisenko, G.G.; et al. The cyclooxygenase site, but not the peroxidase site of cyclooxygenase-2 is required for neurotoxicity in hypoxic and ischemic injury. J. Neurochem. 2010, 113, 965–977. [Google Scholar] [CrossRef] [Green Version]
- Markovič, T.; Jakopin, Ž.; Dolenc, M.S.; Mlinarič-Raščan, I. Structural features of subtype-selective EP receptor modulators. Drug Discov. Today 2017, 22, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, Y.; Narumiya, S. Prostaglandin E receptors. J. Biol. Chem. 2007, 282, 11613–11617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reader, J.; Holt, D.; Fulton, A. Prostaglandin E2 EP receptors as therapeutic targets in breast cancer. Cancer Metastasis Rev. 2011, 30, 449–463. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Han, C.; Wu, T. 15-PGDH inhibits hepatocellular carcinoma growth through 15-keto-PGE2/PPARγ-mediated activation of p21WAF1/Cip1. Oncogene 2014, 33, 1101–1112. [Google Scholar] [CrossRef] [Green Version]
- Tai, H.H.; Cho, H.; Tong, M.; Ding, Y. NAD+-linked 15-hydroxyprostaglandin dehydrogenase: Structure and biological functions. Curr. Pharm. Des. 2006, 12, 955–962. [Google Scholar] [CrossRef]
- Tachibana, K.; Yamasaki, D.; Ishimoto, K.; Doi, T. The Role of PPARs in Cancer. Ppar. Res. 2008, 2008, 102737. [Google Scholar] [CrossRef] [Green Version]
- Savage, D.B. PPARγ as a metabolic regulator: Insights from genomics and pharmacology. Expert Rev. Mol. Med. 2005, 7, 1–16. [Google Scholar] [CrossRef]
- Robbins, G.T.; Nie, D. PPAR gamma, bioactive lipids, and cancer progression. Front. Biosci. 2012, 17, 1816–1834. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Xu, H.; Ji, J.; Shi, X.; Lyu, J.; Zhu, Y.; Yu, H.; Wang, F. Heterogeneity of PTEN and PPAR-gamma in cancer and their prognostic application to bladder cancer. Exp. Med. 2019, 18, 3177–3183. [Google Scholar] [CrossRef] [Green Version]
- Dana, N.; Vaseghi, G.; Haghjooy Javanmard, S. PPAR gamma agonist, pioglitazone, suppresses melanoma cancer in mice by inhibiting TLR4 signaling. J. Pharm. Pharm. Sci. 2019, 22, 418–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawayama, H.; Ishimoto, T.; Watanabe, M.; Yoshida, N.; Sugihara, H.; Kurashige, J.; Hirashima, K.; Iwatsuki, M.; Baba, Y.; Oki, E.; et al. Small molecule agonists of PPAR-gamma exert therapeutic effects in esophageal cancer. Cancer Res. 2014, 74, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Forman, B.M.; Tontonoz, P.; Chen, J.; Brun, R.P.; Spiegelman, B.M.; Evans, R.M. 15-Deoxy-Δ12,14-Prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ. Cell 1995, 83, 803–812. [Google Scholar] [CrossRef] [Green Version]
- De Jong, E.; Winkel, P.; Poelstra, K.; Prakash, J. Anticancer effects of 15d-prostaglandin-J2 in wild-type and doxorubicin-resistant ovarian cancer cells: Novel actions on SIRT1 and HDAC. PLoS ONE 2011, 6, e25192. [Google Scholar] [CrossRef] [Green Version]
- Pommery, N.; Taverne, T.; Telliez, A.; Goossens, L.; Charlier, C.; Pommery, J.; Goossens, J.F.; Houssin, R.; Durant, F.; Henichart, J.P. New COX-2/5-LOX inhibitors: Apoptosis-inducing agents potentially useful in prostate cancer chemotherapy. J. Med. Chem. 2004, 47, 6195–6206. [Google Scholar] [CrossRef]
- Ye, Y.N.; Wu, W.K.; Shin, V.Y.; Bruce, I.C.; Wong, B.C.; Cho, C.H. Dual inhibition of 5-LOX and COX-2 suppresses colon cancer formation promoted by cigarette smoke. Carcinogenesis 2005, 26, 827–834. [Google Scholar] [CrossRef] [Green Version]
- Goossens, L.; Pommery, N.; Henichart, J.P. COX-2/5-LOX dual acting anti-inflammatory drugs in cancer chemotherapy. Curr. Top. Med. Chem. 2007, 7, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Che, X.H.; Chen, C.L.; Ye, X.L.; Weng, G.B.; Guo, X.Z.; Yu, W.Y.; Tao, J.; Chen, Y.C.; Chen, X. Dual inhibition of COX-2/5-LOX blocks colon cancer proliferation, migration and invasion in vitro. Oncol. Rep. 2016, 35, 1680–1688. [Google Scholar] [CrossRef]
- Chang, J.; Tang, N.; Fang, Q.; Zhu, K.; Liu, L.; Xiong, X.; Zhu, Z.; Zhang, B.; Zhang, M.; Tao, J. Inhibition of COX-2 and 5-LOX regulates the progression of colorectal cancer by promoting PTEN and suppressing PI3K/AKT pathway. Biochem. Biophys. Res. Commun. 2019, 517, 1–7. [Google Scholar] [CrossRef]
- Sarkar, F.H.; Adsule, S.; Li, Y.; Padhye, S. Back to the future: COX-2 inhibitors for chemoprevention and cancer therapy. Mini Rev. Med. Chem. 2007, 7, 599–608. [Google Scholar] [CrossRef]
- Sun, L.; Zhao, Y.; Wang, L.; Song, M.; Song, J. [Suppression of COX-2 protein to cell apoptosis in non-small cell lung cancer]. Zhongguo Fei Ai Za Zhi 2007, 10, 188–191. [Google Scholar] [CrossRef]
- Bocca, C.; Bozzo, F.; Bassignana, A.; Miglietta, A. Antiproliferative effects of COX-2 inhibitor celecoxib on human breast cancer cell lines. Mol. Cell. Biochem. 2011, 350, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Lepage, C.; Léger, D.Y.; Bertrand, J.; Martin, F.; Beneytout, J.L.; Liagre, B. Diosgenin induces death receptor-5 through activation of p38 pathway and promotes TRAIL-induced apoptosis in colon cancer cells. Cancer Lett. 2011, 301, 193–202. [Google Scholar] [CrossRef]
- Holmes, M.D.; Chen, W.Y.; Schnitt, S.J.; Collins, L.; Colditz, G.A.; Hankinson, S.E.; Tamimi, R.M. COX-2 expression predicts worse breast cancer prognosis and does not modify the association with aspirin. Breast Cancer Res. Treat 2011, 130, 657–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisk, G.; Ekberg, S.; Lidbrink, E.; Eloranta, S.; Sund, M.; Fredriksson, I.; Lambe, M.; Smedby, K.E. No association between low-dose aspirin use and breast cancer outcomes overall: A Swedish population-based study. Breast Cancer Res. 2018, 20, 142. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Xia, H.H.; Tu, S.P.; Fan, D.M.; Lin, M.C.; Kung, H.F.; Lam, S.K.; Wong, B.C. 15-Lipoxygenase-1 mediates cyclooxygenase-2 inhibitor-induced apoptosis in gastric cancer. Carcinogenesis 2003, 24, 243–247. [Google Scholar] [CrossRef] [Green Version]
- Haeggström, J.Z.; Funk, C.D. Lipoxygenase and Leukotriene Pathways: Biochemistry, Biology, and Roles in Disease. Chem. Rev. 2011, 111, 5866–5898. [Google Scholar] [CrossRef]
- Krieg, P.; Fürstenberger, G. The role of lipoxygenases in epidermis. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2014, 1841, 390–400. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. MCP 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Nie, D.; Krishnamoorthy, S.; Jin, R.; Tang, K.; Chen, Y.; Qiao, Y.; Zacharek, A.; Guo, Y.; Milanini, J.; Pages, G.; et al. Mechanisms regulating tumor angiogenesis by 12-lipoxygenase in prostate cancer cells. J. Biol. Chem. 2006, 281, 18601–18609. [Google Scholar] [CrossRef] [Green Version]
- Pidgeon, G.P.; Kandouz, M.; Meram, A.; Honn, K.V. Mechanisms Controlling Cell Cycle Arrest and Induction of Apoptosis after 12-Lipoxygenase Inhibition in Prostate Cancer Cells. Cancer Res. 2002, 62, 2721–2727. [Google Scholar] [PubMed]
- Pidgeon, G.P.; Tang, K.; Cai, Y.L.; Piasentin, E.; Honn, K.V. Overexpression of Platelet-type 12-Lipoxygenase Promotes Tumor Cell Survival by Enhancing αvβ3 and αvβ5 Integrin Expression. Cancer Res. 2003, 63, 4258–4267. [Google Scholar] [PubMed]
- Nie, D.; Hillman, G.G.; Geddes, T.; Tang, K.; Pierson, C.; Grignon, D.J.; Honn, K.V. Platelet-Type 12-Lipoxygenase in a Human Prostate Carcinoma Stimulates Angiogenesis and Tumor Growth. Cancer Res. 1998, 58, 4047–4051. [Google Scholar]
- Ghosh, J.; Myers, C.E. Arachidonic Acid Stimulates Prostate Cancer Cell Growth: Critical Role of 5-Lipoxygenase. Biochem. Biophys. Res. Commun. 1997, 235, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.J.; Kim, B.R.; Kim, J.L.; Kang, S.; Jeong, Y.A.; Park, S.H.; Jo, M.J.; Kim, J.Y.; Kim, H.J.; Oh, S.C.; et al. Deficiency of 15-LOX-1 Induces Radioresistance through Downregulation of MacroH2A2 in Colorectal Cancer. Cancers (Basel) 2019, 11, 1776. [Google Scholar] [CrossRef] [Green Version]
- Shureiqi, I.; Zuo, X.; Broaddus, R.; Wu, Y.; Guan, B.; Morris, J.S.; Lippman, S.M. The transcription factor GATA-6 is overexpressed in vivo and contributes to silencing 15-LOX-1 in vitro in human colon cancer. Faseb J. 2007, 21, 743–753. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Mao, F.; Zuo, X.; Moussalli, M.J.; Elias, E.; Xu, W.; Shureiqi, I. 15-LOX-1 suppression of hypoxia-induced metastatic phenotype and HIF-1alpha expression in human colon cancer cells. Cancer Med. 2014, 3, 472–484. [Google Scholar] [CrossRef]
- Zuo, X.; Morris, J.S.; Broaddus, R.; Shureiqi, I. 15-LOX-1 transcription suppression through the NuRD complex in colon cancer cells. Oncogene 2009, 28, 1496–1505. [Google Scholar] [CrossRef] [Green Version]
- Wolff, C.; Zoschke, C.; Kalangi, S.K.; Reddanna, P.; Schäfer-Korting, M. Tumor microenvironment determines drug efficacy in vitro-apoptotic and anti-inflammatory effects of 15-lipoxygenase metabolite, 13-HpOTrE. Eur. J. Pharm. Biopharm. 2019, 142, 1–7. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; St. Croix, C.M.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641.e626. [Google Scholar] [CrossRef]
- Lu, X.; Huang, L.; Zhang, W.; Ning, X. Tepoxalin a dual 5-LOX-COX inhibitor and erlotinib an EGFR inhibitor halts progression of gastric cancer in tumor xenograft mice. Am. J. Transl. Res. 2018, 10, 3847–3856. [Google Scholar]
- Tavolari, S.; Bonafe, M.; Marini, M.; Ferreri, C.; Bartolini, G.; Brighenti, E.; Manara, S.; Tomasi, V.; Laufer, S.; Guarnieri, T. Licofelone, a dual COX/5-LOX inhibitor, induces apoptosis in HCA-7 colon cancer cells through the mitochondrial pathway independently from its ability to affect the arachidonic acid cascade. Carcinogenesis 2008, 29, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Cartwright, C.; Chan, D.; Vijjeswarapu, M.; Ding, J.; Newman, R.A. Zyflamend®-mediated inhibition of human prostate cancer PC3 cell proliferation: Effects on 12-LOX and Rb protein phosphorylation. Cancer Biol. Ther. 2007, 6, 228–236. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.K.; Singh, R.; Naz, F.; Chauhan, S.S.; Dinda, A.; Shukla, A.A.; Gill, K.; Kapoor, V.; Dey, S. Structure based design and synthesis of peptide inhibitor of human LOX-12: In vitro and in vivo analysis of a novel therapeutic agent for breast cancer. PLoS ONE 2012, 7, e32521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, B.C.Y.; Wang, W.P.; Cho, C.H.; Fan, X.M.; Lin, M.C.M.; Kung, H.F.; Lam, S.K. 12-Lipoxygenase inhibition induced apoptosis in human gastric cancer cells. Carcinogenesis 2001, 22, 1349–1354. [Google Scholar] [CrossRef] [Green Version]
- Lovey, J.; Nie, D.; Tovari, J.; Kenessey, I.; Kandouz, M.; Timar, J.; Kasler, M.; Honn, K.V. [Selective 12-lipoxygenase inhibition potentiates the effect of radiation on human prostate cancer cells in vitro and in vivo]. Magy. Onkol. 2014, 58, 211–218. [Google Scholar] [PubMed]
- Lovey, J.; Nie, D.; Tovari, J.; Kenessey, I.; Timar, J.; Kandouz, M.; Honn, K.V. Radiosensitivity of human prostate cancer cells can be modulated by inhibition of 12-lipoxygenase. Cancer Lett. 2013, 335, 495–501. [Google Scholar] [CrossRef]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations With Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cao, F.; Yin, H.-l.; Huang, Z.-j.; Lin, Z.-t.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Hayano, M.; Yang, W.S.; Corn, C.K.; Pagano, N.C.; Stockwell, B.R. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016, 23, 270–278. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Kagan, V.; Serbinova, E.; Packer, L. Antioxidant effects of ubiquinones in microsomes and mitochondria are mediated by tocopherol recycling. Biochem. Biophys. Res. Commun. 1990, 169, 851–857. [Google Scholar] [CrossRef]
- Forsmark, P.; Åberg, F.; Norling, B.; Nordenbrand, K.; Dallner, G.; Ernster, L. Inhibition of lipid peroxidation by ubiquinol in submitochondrial particles in the absence of vitamin E. Febs Lett. 1991, 285, 39–43. [Google Scholar] [CrossRef] [Green Version]
- Frei, B.; Kim, M.C.; Ames, B.N. Ubiquinol-10 is an effective lipid-soluble antioxidant at physiological concentrations. Proc. Natl. Acad. Sci. USA 1990, 87, 4879–4883. [Google Scholar] [CrossRef] [Green Version]
- Kagan, V.E.; Tiurin, V.A.; Kitanova, S.A.; Serbinova, E.A.; Quinn, P.J. [The action of a homologous series of ubiquinols on lipid peroxidation in brain mitochondrial and synaptosomal membranes]. Biull. Eksp. Biol. Med. 1989, 107, 420–422. [Google Scholar] [CrossRef]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS–RAF–MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 865–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. Acs Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef]
- Stadtman, E.R. Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal-catalyzed reactions. Annu. Rev. Biochem. 1993, 62, 797–821. [Google Scholar] [CrossRef]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell. Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Xia, L.; Nordman, T.; Olsson, J.M.; Damdimopoulos, A.; Björkhem-Bergman, L.; Nalvarte, I.; Eriksson, L.C.; Arnér, E.S.J.; Spyrou, G.; Björnstedt, M. The Mammalian Cytosolic Selenoenzyme Thioredoxin Reductase Reduces Ubiquinone: A novel mechanism for defense against oxidative stress. J. Biol. Chem. 2003, 278, 2141–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Shieh, B.; Reigan, P.; Zhang, Z.; Colucci, M.A.; Chilloux, A.; Newsome, J.J.; Siegel, D.; Chan, D.; Moody, C.J.; et al. Potent activity of indolequinones against human pancreatic cancer: Identification of thioredoxin reductase as a potential target. Mol. Pharmacol. 2009, 76, 163–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkinson, E.I.; Hergenrother, P.J. Deoxynyboquinones as NQO1-Activated Cancer Therapeutics. Acc. Chem. Res. 2015, 48, 2715–2723. [Google Scholar] [CrossRef] [PubMed]
- Navarro, F.; Villalba, J.M.; Crane, F.L.; Mackellar, W.C.; Navas, P. A phospholipid-dependent NADH-coenzyme Q reductase from liver plasma membrane. Biochem. Biophys. Res. Commun. 1995, 212, 138–143. [Google Scholar] [CrossRef]
- Nakashima, Y.; Shinzawa-Itoh, K.; Watanabe, K.; Naoki, K.; Hano, N.; Yoshikawa, S. Steady-state kinetics of NADH:coenzyme Q oxidoreductase isolated from bovine heart mitochondria. J. Bioenerg. Biomembr. 2002, 34, 11–19. [Google Scholar] [CrossRef]
- Reda, A.; Refaat, A.; Abd-Rabou, A.A.; Mahmoud, A.M.; Adel, M.; Sabet, S.; Ali, S.S. Role of mitochondria in rescuing glycolytically inhibited subpopulation of triple negative but not hormone-responsive breast cancer cells. Sci. Rep. 2019, 9, 13748. [Google Scholar] [CrossRef] [Green Version]
- Grivennikova, V.G.; Gavrikova, E.V.; Timoshin, A.A.; Vinogradov, A.D. Fumarate reductase activity of bovine heart succinate-ubiquinone reductase. New assay system and overall properties of the reaction. Biochim. Biophys. Acta (BBA)-Bioenerg. 1993, 1140, 282–292. [Google Scholar] [CrossRef]
- Ralph, S.J.; Moreno-Sánchez, R.; Neuzil, J.; Rodríguez-Enríquez, S. Inhibitors of Succinate: Quinone Reductase/Complex II Regulate Production of Mitochondrial Reactive Oxygen Species and Protect Normal Cells from Ischemic Damage but Induce Specific Cancer Cell Death. Pharm. Res. 2011, 28, 2695. [Google Scholar] [CrossRef]
- Wang, A.; Lim, H.; Cheng, S.-Y.; Xie, L. ANTENNA, a Multi-Rank, Multi-Layered Recommender System for Inferring Reliable Drug-Gene-Disease Associations: Repurposing Diazoxide as a Targeted Anti-Cancer Therapy. bioRxiv 2017, 192385. [Google Scholar] [CrossRef]
- Olsson, J.M.; Xia, L.; Eriksson, L.C.; Björnstedt, M. Ubiquinone is reduced by lipoamide dehydrogenase and this reaction is potently stimulated by zinc. Febs Lett. 1999, 448, 190–192. [Google Scholar] [CrossRef] [Green Version]
- Kwak, C.H.; Lee, J.H.; Kim, E.Y.; Han, C.W.; Kim, K.J.; Lee, H.; Cho, M.; Jang, S.B.; Kim, C.H.; Chung, T.W.; et al. Huzhangoside A Suppresses Tumor Growth through Inhibition of Pyruvate Dehydrogenase Kinase Activity. Cancers (Basel) 2019, 11, 712. [Google Scholar] [CrossRef] [Green Version]
- Ullrich, A.; Knecht, W.; Fries, M.; Loffler, M. Recombinant expression of N-terminal truncated mutants of the membrane bound mouse, rat and human flavoenzyme dihydroorotate dehydrogenase. A versatile tool to rate inhibitor effects? Eur. J. Biochem. 2001, 268, 1861–1868. [Google Scholar] [CrossRef]
- Madak, J.T.; Bankhead, A.; Cuthbertson, C.R.; Showalter, H.D.; Neamati, N. Revisiting the role of dihydroorotate dehydrogenase as a therapeutic target for cancer. Pharmacol. Ther. 2019, 195, 111–131. [Google Scholar] [CrossRef]
- Lucas-Hourani, M.; Munier-Lehmann, H.; El Mazouni, F.; Malmquist, N.A.; Harpon, J.; Coutant, E.P.; Guillou, S.; Helynck, O.; Noel, A.; Scherf, A.; et al. Original 2-(3-Alkoxy-1H-pyrazol-1-yl)azines Inhibitors of Human Dihydroorotate Dehydrogenase (DHODH). J. Med. Chem. 2015, 58, 5579–5598. [Google Scholar] [CrossRef] [Green Version]
- Bahrami, B.; Mohammadnia-Afrouzi, M.; Bakhshaei, P.; Yazdani, Y.; Ghalamfarsa, G.; Yousefi, M.; Sadreddini, S.; Jadidi-Niaragh, F.; Hojjat-Farsangi, M. Folate-conjugated nanoparticles as a potent therapeutic approach in targeted cancer therapy. Tumor Biol. 2015, 36, 5727–5742. [Google Scholar] [CrossRef] [PubMed]
- Bretin, L.; Pinon, A.; Bouramtane, S.; Ouk, C.; Richard, L.; Perrin, M.L.; Chaunavel, A.; Carrion, C.; Bregier, F.; Sol, V.; et al. Photodynamic Therapy Activity of New Porphyrin-Xylan-Coated Silica Nanoparticles in Human Colorectal Cancer. Cancers (Basel) 2019, 11, 1474. [Google Scholar] [CrossRef] [Green Version]
- Jasim, K.A.; Gesquiere, A.J. Ultrastable and Biofunctionalizable Conjugated Polymer Nanoparticles with Encapsulated Iron for Ferroptosis Assisted Chemodynamic Therapy. Mol. Pharm. 2019, 16, 4852–4866. [Google Scholar] [CrossRef]
- Thomas, R.G.; Moon, M.J.; Lee, H.; Sasikala, A.R.; Kim, C.S.; Park, I.K.; Jeong, Y.Y. Hyaluronic acid conjugated superparamagnetic iron oxide nanoparticle for cancer diagnosis and hyperthermia therapy. Carbohydr. Polym. 2015, 131, 439–446. [Google Scholar] [CrossRef]
- Emami, M.; Shamsipur, M.; Saber, R.; Irajirad, R. An electrochemical immunosensor for detection of a breast cancer biomarker based on antiHER2-iron oxide nanoparticle bioconjugates. Analyst 2014, 139, 2858–2866. [Google Scholar] [CrossRef]
- Kim, S.E.; Zhang, L.; Ma, K.; Riegman, M.; Chen, F.; Ingold, I.; Conrad, M.; Turker, M.Z.; Gao, M.; Jiang, X.; et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat. Nanotechnol. 2016, 11, 977–985. [Google Scholar] [CrossRef] [Green Version]
- Alarifi, S.; Ali, D.; Alkahtani, S.; Alhader, M.S. Iron Oxide Nanoparticles Induce Oxidative Stress, DNA Damage, and Caspase Activation in the Human Breast Cancer Cell Line. Biol. Trace Elem. Res. 2014, 159, 416–424. [Google Scholar] [CrossRef]
- Jaganjac, M.; Borovic Sunjic, S.; Zarkovic, N. Utilizing Iron for Targeted Lipid Peroxidation as Anticancer Option of Integrative Biomedicine: A Short Review of Nanosystems Containing Iron. Antioxidants 2020, 9, 191. [Google Scholar] [CrossRef] [Green Version]
- Sang, M.; Luo, R.; Bai, Y.; Dou, J.; Zhang, Z.; Liu, F.; Feng, F.; Xu, J.; Liu, W. Mitochondrial membrane anchored photosensitive nano-device for lipid hydroperoxides burst and inducing ferroptosis to surmount therapy-resistant cancer. Theranostics 2019, 9, 6209–6223. [Google Scholar] [CrossRef]
- Abrams, R.P.; Carroll, W.L.; Woerpel, K.A. Five-Membered Ring Peroxide Selectively Initiates Ferroptosis in Cancer Cells. Acs Chem. Biol. 2016, 11, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.; Song, J.; Yung, B.C.; Zhou, Z.; Wu, A.; Chen, X. Emerging Strategies of Cancer Therapy Based on Ferroptosis. Adv. Mater. 2018, 30, 1704007. [Google Scholar] [CrossRef]
- Said-Elbahr, R.; Nasr, M.; Alhnan, M.A.; Taha, I.; Sammour, O. Nebulizable colloidal nanoparticles co-encapsulating a COX-2 inhibitor and a herbal compound for treatment of lung cancer. Eur. J. Pharm. Biopharm. 2016, 103, 1–12. [Google Scholar] [CrossRef]
- Kim, T.-H.; Jeong, Y.-I.; Jin, S.-G.; Pei, J.; Jung, T.-Y.; Moon, K.-S.; Kim, I.-Y.; Kang, S.-S.; Jung, S. Preparation of polylactide-co-glycolide nanoparticles incorporating celecoxib and their antitumor activity against brain tumor cells. Int. J. Nanomed. 2011, 6, 2621–2631. [Google Scholar] [CrossRef] [Green Version]


| ALOX Gene | Name | Cell and Tissue Location |
|---|---|---|
| ALOX5 PubMed Gene ID: 240 | arachidonate 5-lipoxygenase or 5-lipoxygenase (5-LOX) | Monocytes, macrophages, B lymphocytes cells [175] and appendix, bone marrow, gall bladder, lung, lymph node, spleen, and urinary bladder [176]. |
| ALOX12 PubMed Gene ID: 239 | arachidonate 12-lipoxygenase, 12S type or 12-lipoxygenase (12-LOX) | Esophagus and skin [176]. |
| ALOX15 PubMed Gene ID: 246 | arachidonate 15-lipoxygenase or platelet type platelet lipoxygenase or 15-lipoxygenase-1 (15-LOX-1) | Reticulocytes, eosinophils [175] and lung, small intestine, testis urinary bladder [176]. |
| ALOX15B PubMed Gene ID: 247 | arachidonate 15-lipoxygenase type B or 15-lipoxygenase-2 (15-LOX-2) | Human skin [175] and prostate, lung, and esophagus [176]. |
| ALOX12B PubMed Gene ID: 242 | arachidonate 12-lipoxygenase, 12R type or 12R-lipoxygenase (12R-LOX) | Skin and esophagus [176]. |
| ALOXE3 PubMed Gene ID: 59344 | arachidonate lipoxygenase 3, lipoxygenase, epidermis type (eLOX3) | Skin, tongue, prostate, tonsils [175,176]. |
| Name | Km (CoQ) (µM) | Specific Inhibitor with Anti-Cancer Properties | IC50 (µM) and Cancer Cell Line | Structure |
|---|---|---|---|---|
| FSP1 (AFM2) | 12 [217] | 1-amino-3-(4-methylphenyl)-pyrido [1,2-a]benzimidazole-2,4-dicarbonitrile (iFSP1) [217] | ≈1 variety of human cancer cell lines co-treatment with RSL3 [217] |  |
| TrxR | 22 [218] | 5-methoxy-1-methyl-3-[(2,4,6-trifluorophenoxy)methyl]indole-4,7-dione [219] | 0.034 (MIA PaCa-2) Human pancreatic cancer [219] |  |
| NQO1 | 0.79 [217] | 1-isobutyl-4,6-dimethylpyrido[3,2-g]quinoline-2,5,8,10(1H,9H)-tetraone (IB-DNQ) [220] | 0.08 (A549) Human Lung Cancer [220] |  |
| Cb5R | 625 [221] | N.D. | N.D. | |
| NADH:ubiquinone reductase Complex I | 10 [222] | Rotenone [223] | 0.5 > (MBA-MD-231) Triple negative breast cancer [223] |  |
| Succinate-quinone oxidoreductase Complex II | 0.3 [224] | 7-chloro-3-methyl-4H-1,2,4-benzothiadiazine 1,1-dioxide (Idra-21) [225] | 0.87 [226] |  |
| DLD | 5 [227] | Huzhangoside A [228] | 1.5 (A549) Human Lung Cancer [228] |  |
| DHODH | 14 [229] | 2-(4-(2,6-difluorophenoxy)-3-isopropoxy-5-methyl-1H-pyrazol-1-yl)-5-ethylpyrimidine (BDBM50070908) [230] | 0.02 (Jurkat cells) Human lymphocytes [231] |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clemente, S.M.; Martínez-Costa, O.H.; Monsalve, M.; Samhan-Arias, A.K. Targeting Lipid Peroxidation for Cancer Treatment. Molecules 2020, 25, 5144. https://doi.org/10.3390/molecules25215144
Clemente SM, Martínez-Costa OH, Monsalve M, Samhan-Arias AK. Targeting Lipid Peroxidation for Cancer Treatment. Molecules. 2020; 25(21):5144. https://doi.org/10.3390/molecules25215144
Chicago/Turabian StyleClemente, Sofia M., Oscar H. Martínez-Costa, Maria Monsalve, and Alejandro K. Samhan-Arias. 2020. "Targeting Lipid Peroxidation for Cancer Treatment" Molecules 25, no. 21: 5144. https://doi.org/10.3390/molecules25215144
APA StyleClemente, S. M., Martínez-Costa, O. H., Monsalve, M., & Samhan-Arias, A. K. (2020). Targeting Lipid Peroxidation for Cancer Treatment. Molecules, 25(21), 5144. https://doi.org/10.3390/molecules25215144