Binding Studies of AICAR and Human Serum Albumin by Spectroscopic, Theoretical, and Computational Methodologies

 and
and
Abstract
:
1. Introduction
2. Results and Discussion
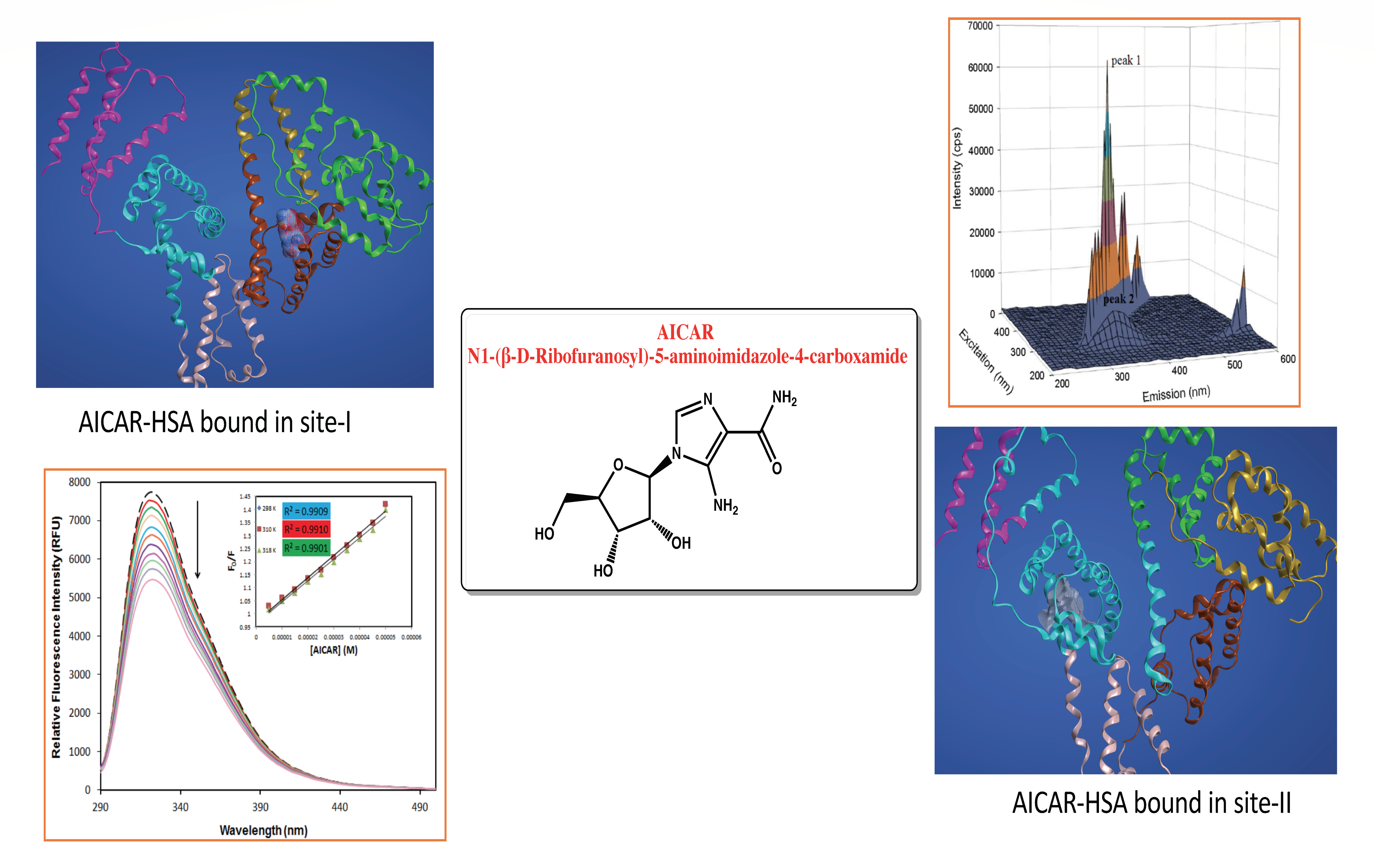
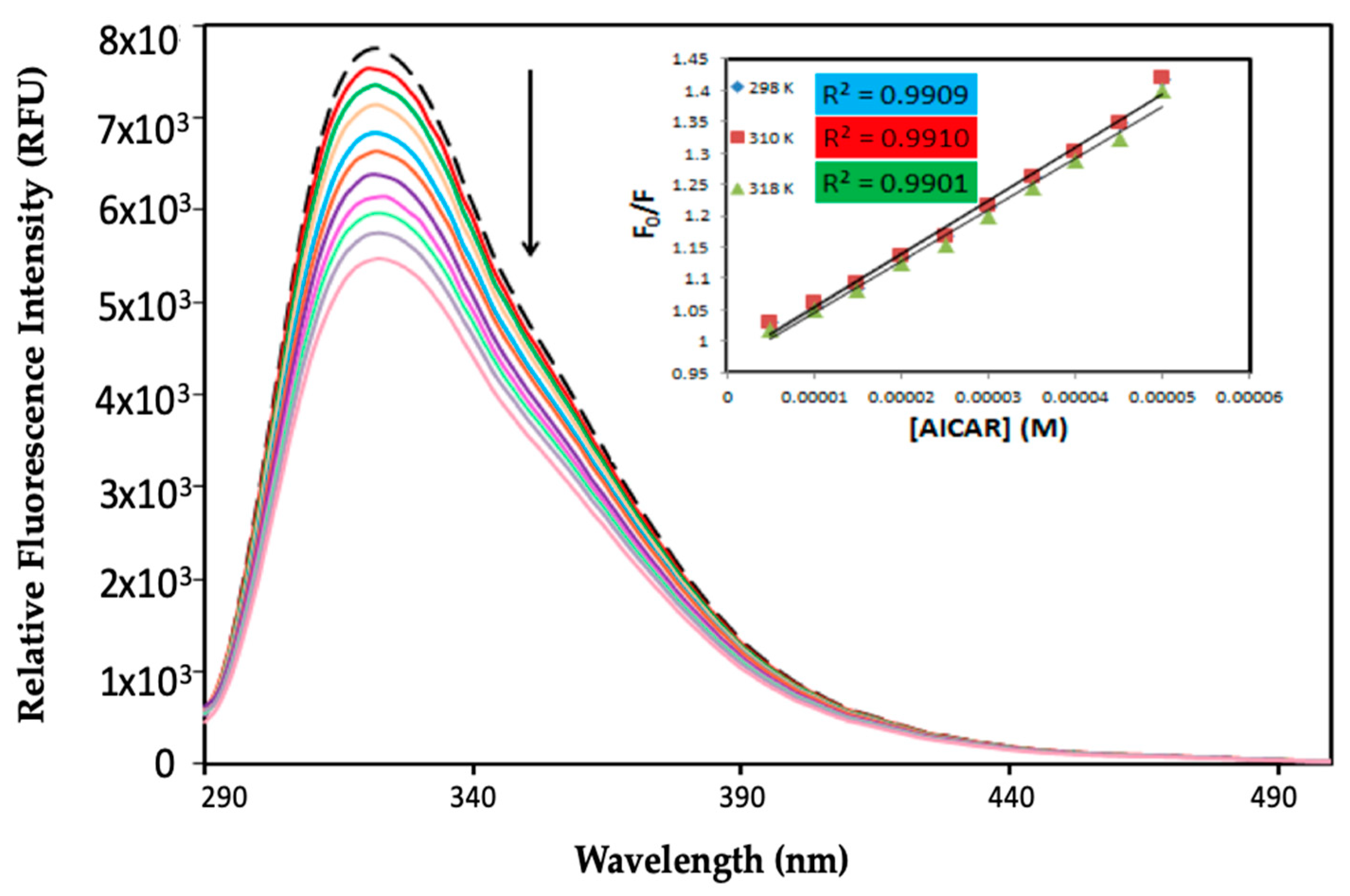
2.1. Protein Fluorescence Quenching of HSA by AICAR
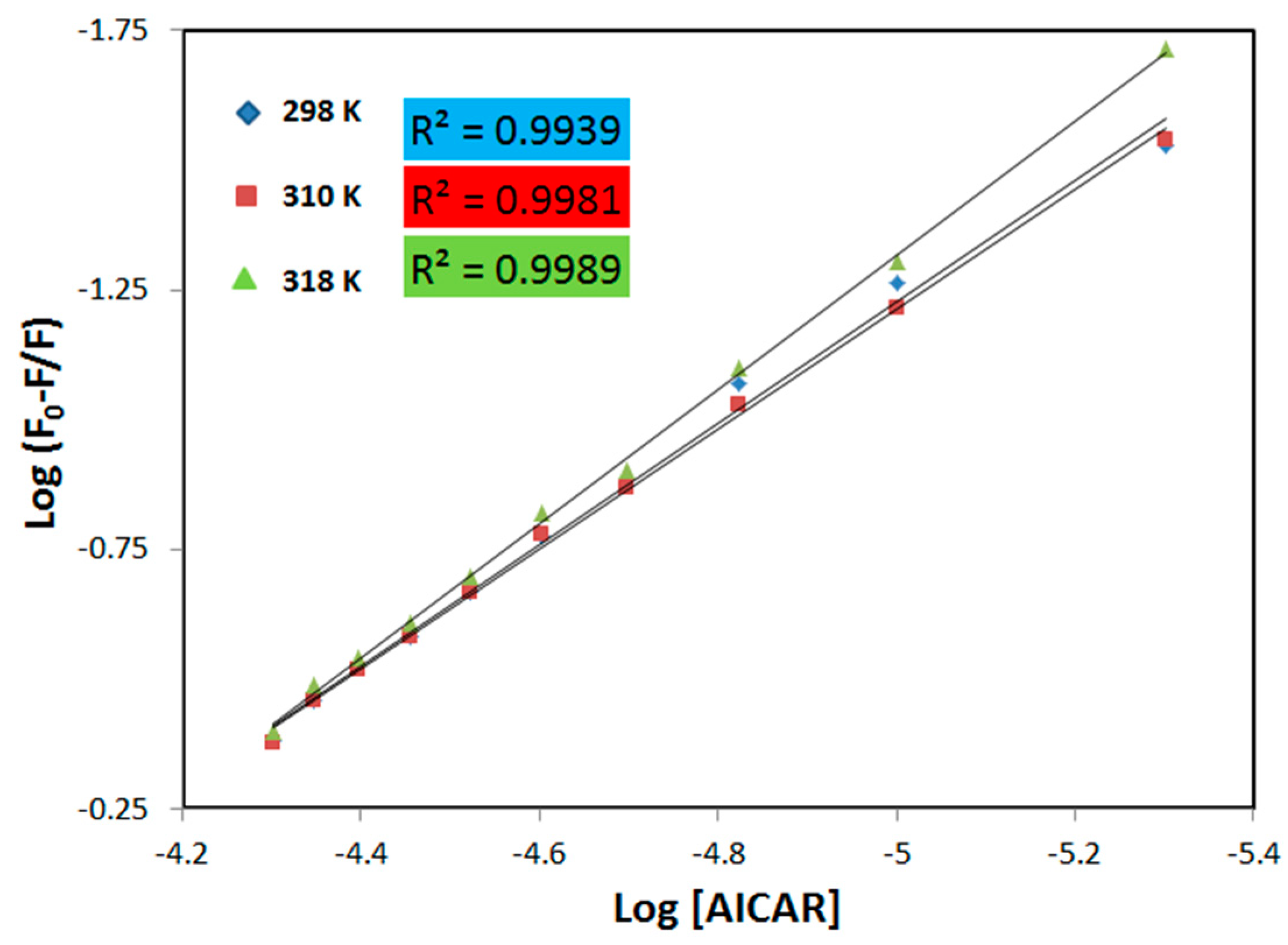
2.2. Fluorescence Quenching Mechanism
2.3. Thermodynamic Binding Constants and Interaction Sites between HSA and AICAR
2.3.1. Thermodynamic Binding Parameters
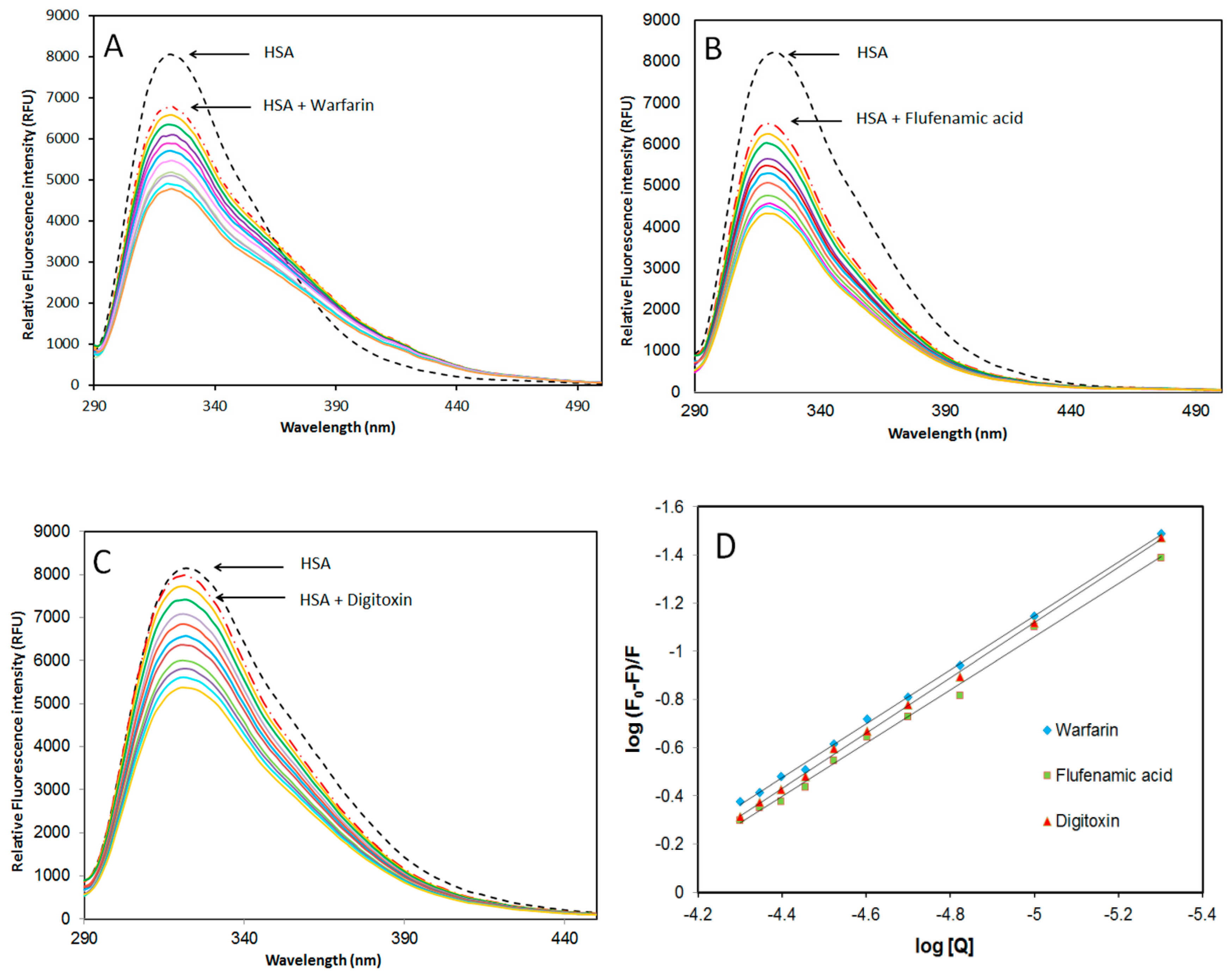
2.3.2. Site-Specific Ligand Competitive Displacement Experiments
2.4. Characterization of Conformational Change in HSA upon AICAR Binding
2.4.1. Synchronous Fluorescence Spectroscopy
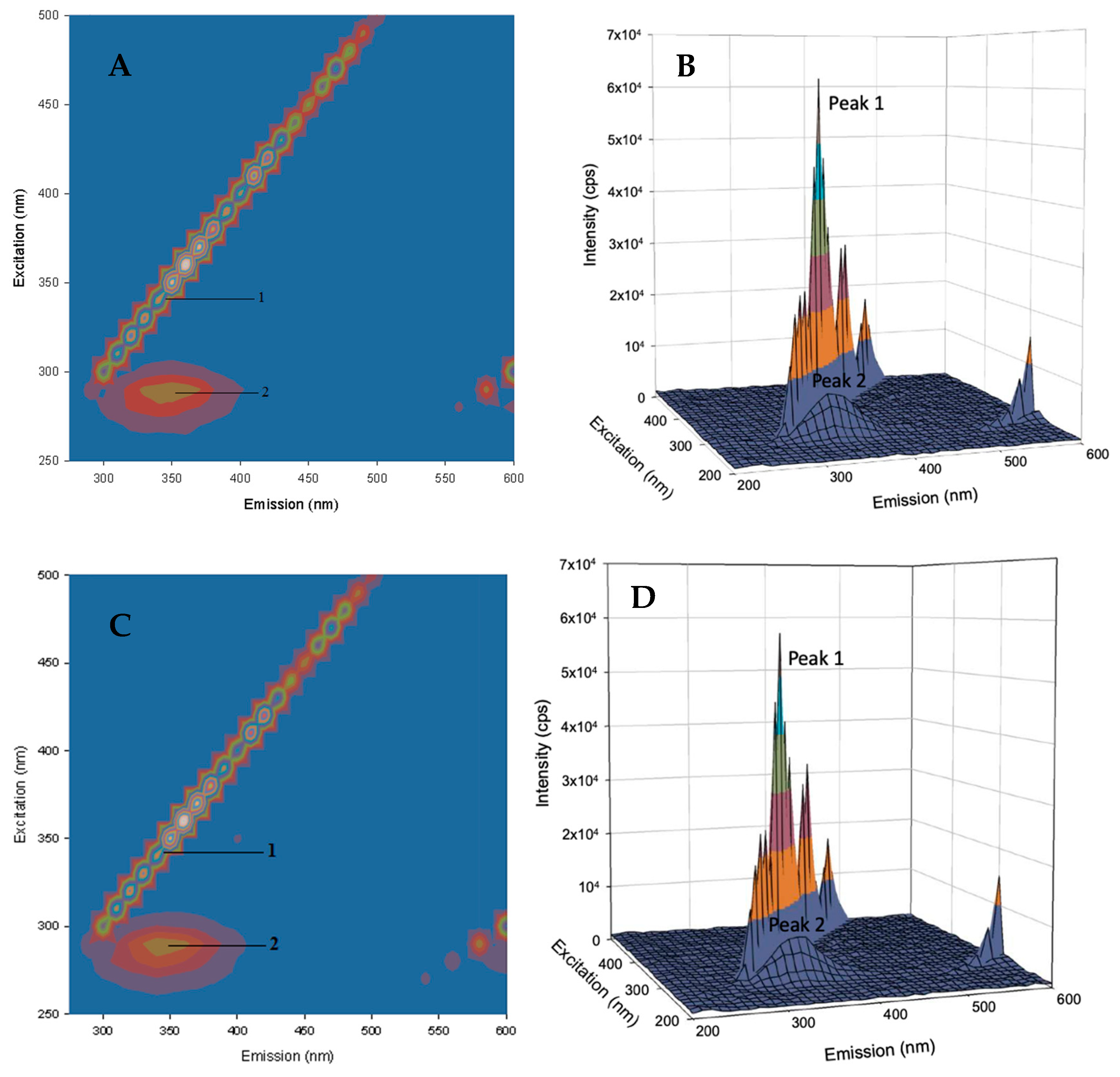
2.4.2. Three-Dimensional Fluorescence Spectroscopy
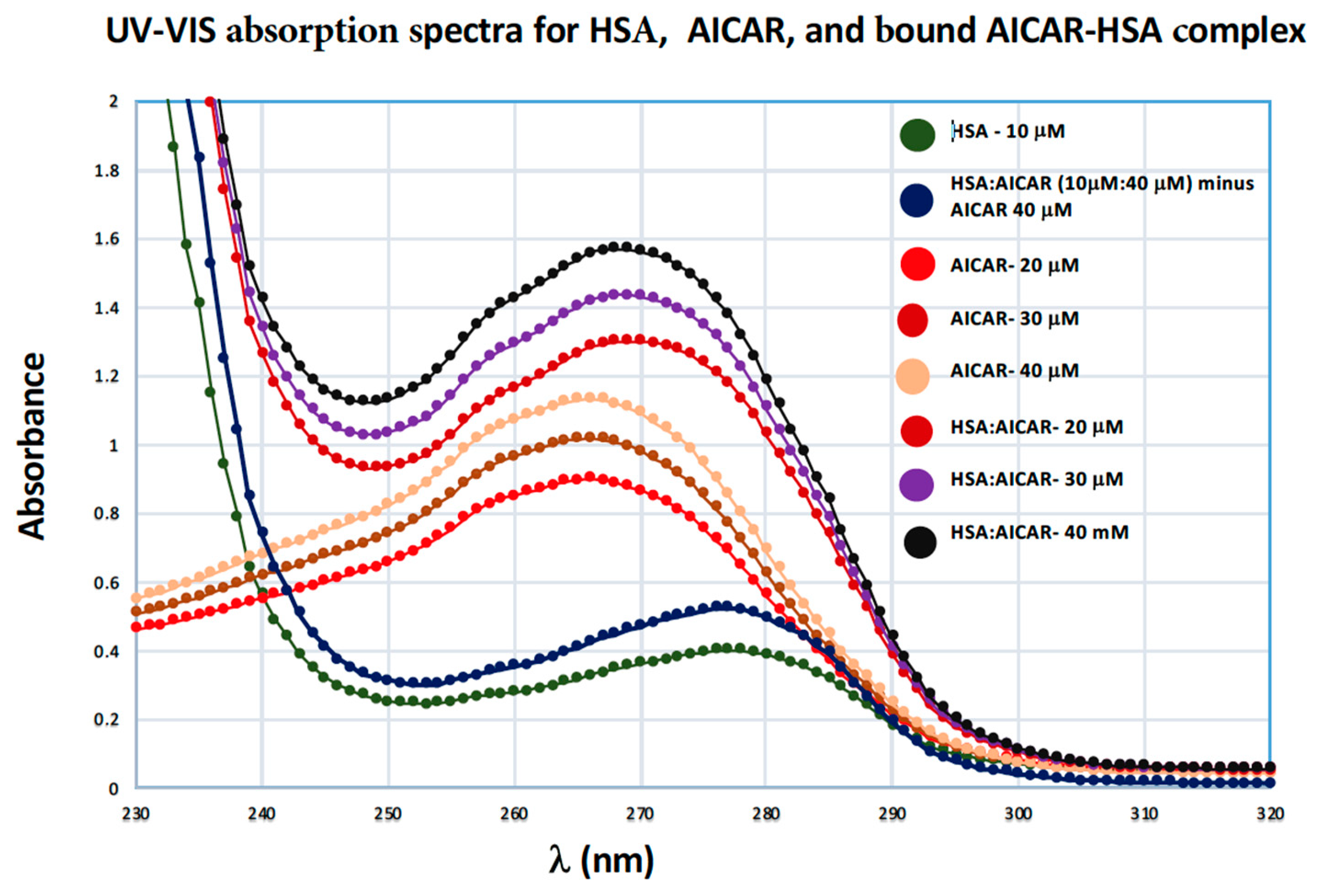
2.4.3. UV-VIS Absorption Spectroscopy
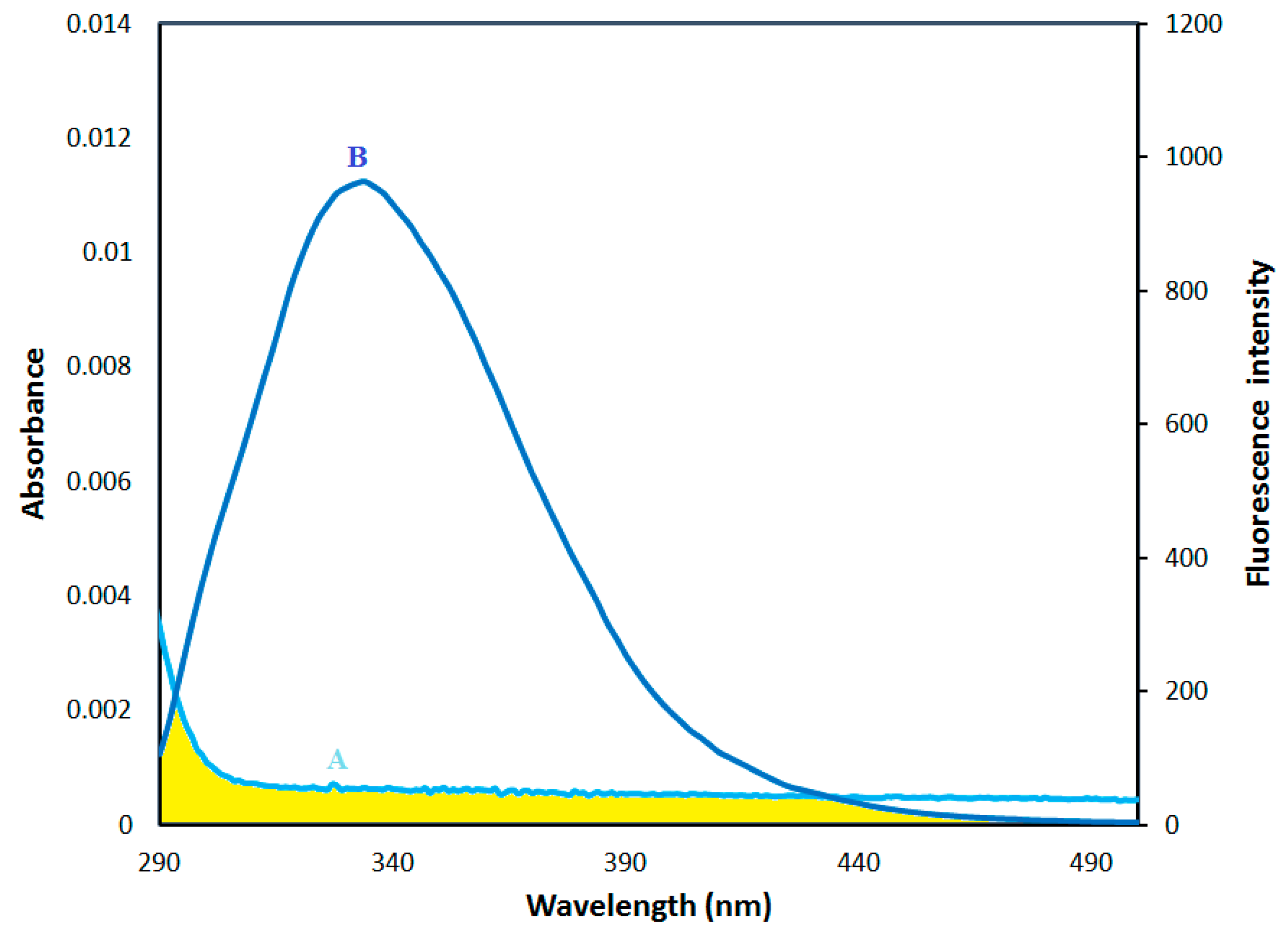
2.5. Fluorescence Energy Transfer between HSA and AICAR–HSA
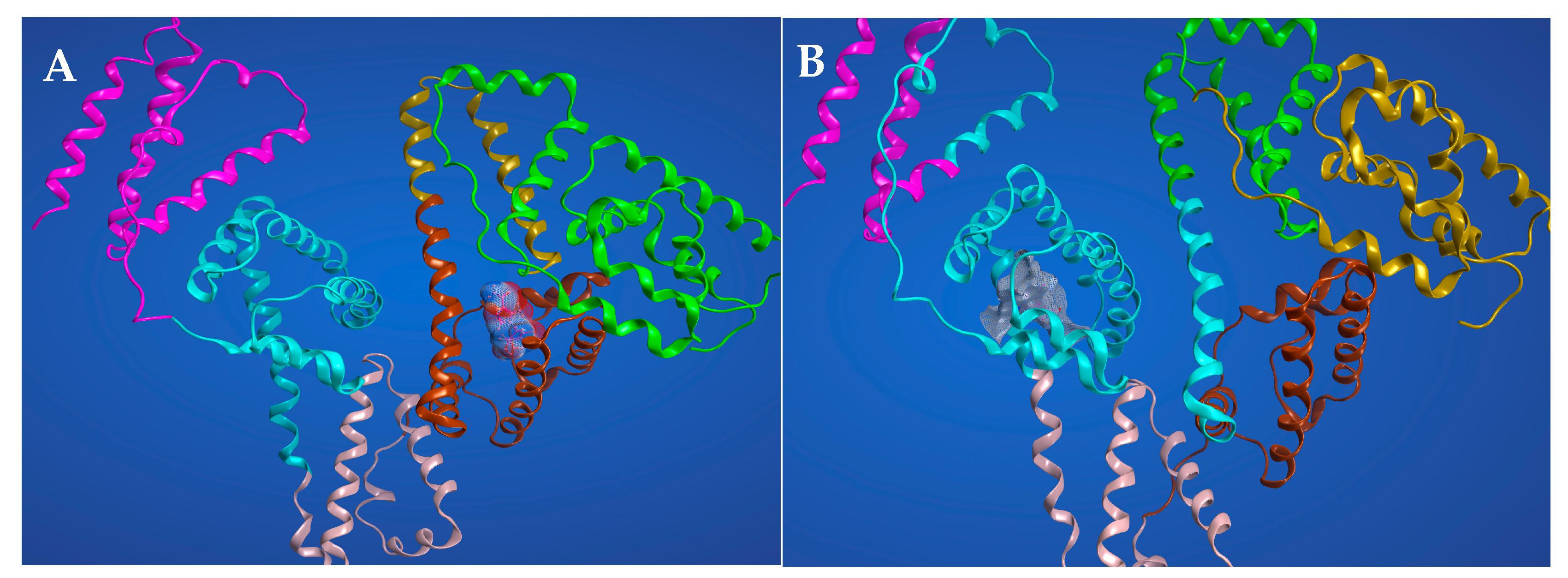
2.6. Molecular Docking Studies for the Interaction AICAR–HSA
3. Materials and Methods
3.1. Materials
Preparation of Stock Solutions
3.2. Major Equipment
UV-VIS Absorption Spectroscopy
3.3. Fluorescence Measurements
3.3.1. Fluorescence Spectroscopy Experiments
3.3.2. Synchronous Fluorescence Spectroscopy
3.3.3. Site Marker Competitive Fluorescence Experiments
3.3.4. 3D Fluorescence Spectra
3.4. Characterization of AICAR–HSA Binding Interactions by Molecular Docking
3.4.1. Protein Preparation
3.4.2. Ligand Preparation
3.4.3. Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Paolo Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005, 57, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Amiri, M.; Jankeje, K.; Albani, J.R. Characterization of Human Serum Albumin Forms with pH. Fluorescence Lifetime Studies. J. Pharm. Biomed. Anal. 2010, 51, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Burke, T.G.; Mi, Z. The structural basis of camptothecin interactions with human serum albumin: Impact on drug stability. J. Med. Chem. 1994, 37, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Wosikowski, K.; Biedermann, E.; Rattel, B. In vitro and in vivo antitumor activity of methotrexate conjugated to human serum albumin in human cancer cells. Clin. Cancer Res. 2003, 9, 1917–1926. [Google Scholar] [PubMed]
- Curry, S.; Mandelkow, H.; Brick, P.; Franks, N. Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat. Struct. Biol. 1998, 5, 827–835. [Google Scholar] [CrossRef]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. The Characterization of Two Specific Drug Binding Sites on Human Serum Albumin. Mol. Pharmacol. 1975, 11, 824–832. [Google Scholar]
- Möller, M.; Denicola, A. Protein tryptophan accessibility studied by fluorescence quenching. Biochem. Mol. Biol. Educ. 2002, 30, 175–178. [Google Scholar] [CrossRef]
- Scudiero, O.; Nigro, E.; Monaco, M.L.; Oliviero, G.; Polito, R.; Borbone, N.; D’Errico, S.; Mayol, L.; Daniele, A.; Piccialli, G. New synthetic AICAR derivatives with enhanced AMPK and ACC activation. J. Enzyme Inhib. Med. Chem. 2016, 31, 748–753. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.J.; Kim, T.Y.; Chung, N.; Yim, J.H.; Kim, W.G.; Kim, J.A.; Kim, W.B.; Shong, Y.K. The influence of the BRAF V600E mutation in thyroid cancer cell lines on the anticancer effects of 5-aminoimidazole-4-carboxamide-ribonucleoside. J. Endocrinol 2011, 211, 79–85. [Google Scholar] [CrossRef]
- Su, C.C.; Hsieh, K.L.; Liu, P.L.; Yeh, H.C.; Huang, S.P.; Fang, S.H.; Li, C.Y. AICAR Induces Apoptosis and Inhibits Migration and Invasion in Prostate Cancer Cells Through an AMPK/mTOR-Dependent Pathway. Int. J. Mol. Sci. 2019, 20, 1647. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.T.; Kuhlmann, M.; Hvam, M.L.; Howard, K.A. Albumin-based drug delivery: Harnessing nature to cure disease. Mol. Cell. Ther. 2016, 4, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.C.; Wang, H.M.; Niu, Q.X.; Ye, D.Y.; Liang, G.W. Binding between Saikosaponin C and Human Serum Albumin by Fluorescence Spectroscopy and Molecular Docking. Molecules 2016, 21, 153. [Google Scholar] [CrossRef] [PubMed]
- Gowda, J.I.; Nandibewoor, S.T. Binding and conformational changes of human serum albumin upon interaction with 4-aminoantipyrine studied by spectroscopic methods and cyclic voltammetry. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2014, 124, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Liu, Y.; Xie, M.-X.; Li, S.; Jiang, M.; Wang, Y.-D. Interactions of human serum albumin with chlorogenic acid and ferulic acid. Biochim. Biophysica Acta BBA 2004, 1674, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Meng-Xia, X.; Dong, Z.; Yuan, L.; Xiao-Yu, L.; Xing, C. Spectroscopic studies on the interaction of cinnamic acid and its hydroxyl derivatives with human serum albumin. J. Mol. Struct. 2004, 692, 71–80. [Google Scholar] [CrossRef]
- Boaz, H.; Rollefson, G. The quenching of fluorescence. Deviations from the Stern-Volmer law. J. Am. Chem. Soc. 1950, 72, 3435–3443. [Google Scholar] [CrossRef]
- Eftink, M.R.; Hagaman, K.A. Fluorescence lifetime and anisotropy studies with liver alcohol dehydrogenase and its complexes. Biochemistry 1986, 25, 6631–6637. [Google Scholar] [CrossRef]
- Meti, M.D.; Nandibewoor, S.T.; Joshi, S.D.; More, U.A.; Chimatadar, S.A. Multi-spectroscopic investigation of the binding interaction of fosfomycin with bovine serum albumin. J. Pharm. Anal. 2015, 5, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Sudlow, G.; Birkett, D.; Wade, D. Further characterization of specific drug binding sites on human serum albumin. Mol. Pharmacol. 1976, 12, 1052–1061. [Google Scholar]
- Zunszain, P.A.; Ghuman, J.; Komatsu, T.; Tsuchida, E.; Curry, S. Crystal structural analysis of human serum albumin complexed with hemin and fatty acid. BMC Struct. Biol. 2003, 3, 3–6. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Zhao, N.; Wang, L. Fluorescence spectrometric studies on the binding of puerarin to human serum albumin using warfarin, ibuprofen and digitoxin as site markers with the aid of chemometrics. J. Lumin. 2011, 131, 2716–2724. [Google Scholar] [CrossRef]
- Meneghini, C.; Leboffe, L.; Bionducci, M.; Fanali, G.; Meli, M.; Colombo, G.; Fasano, M.; Ascenzi, P.; Mobilio, S. The five-to-six-coordination transition of ferric human serum heme-albumin is allosterically-modulated by ibuprofen and warfarin: A combined XAS and MD study. PLoS ONE 2015, 10, e0123144. [Google Scholar] [CrossRef]
- Sandhya, B.; Hegde, A.H.; Kalanur, S.S.; Katrahalli, U.; Seetharamappa, J. Interaction of triprolidine hydrochloride with serum albumins: Thermodynamic and binding characteristics, and influence of site probes. J. Pharm. Biomed. Anal. 2011, 54, 1180–1186. [Google Scholar] [CrossRef]
- Byadagi, K.; Meti, M.; Nandibewoor, S.; Chimatadar, S. Investigation of binding behaviour of procainamide hydrochloride with human serum albumin using synchronous, 3D fluorescence and circular dichroism. J. Pharm. Anal. 2017, 7, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; He, J.; Yang, H.; Tang, P.; Tang, B.; Sun, Q.; Li, H. Investigation on the interaction of antibacterial drug moxifloxacin hydrochloride with human serum albumin using multi-spectroscopic approaches, molecular docking and dynamical simulation. RSC Adv. 2017, 7, 48942–48951. [Google Scholar] [CrossRef] [Green Version]
- Mei, P.; Zhang, Y.-Z.; Zhang, X.-P.; Yan, C.-X.; Zhang, H.; Liu, Y. Spectroscopic investigation of the interaction between copper (II) 2-oxo-propionic acid salicyloyl hydrazone complex and bovine serum albumin. Biol. Trace Elem. Res. 2008, 124, 269. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhu, M.; Xu, C.; Ji, B. Characterization of the baicalein–bovine serum albumin complex without or with Cu2+ or Fe3+ by spectroscopic approaches. Eur. J. Med. Chem. 2011, 46, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Abdulrahman, A.; Haitham, A.H.; Eman, H.; Rashed, N. Spectroscopic and molecular docking studies reveal binding characteristics of nazartinib (EGF816) to human serum albumin. R. Soc. Open Sci. 2020, 7, 191595. [Google Scholar]
- Chatterjee, T.; Pal, A.; Dey, S.; Chatterjee, B.K.; Chakrabarti, P. Interaction of virstatin with human serum albumin: Spectroscopic analysis and molecular modeling. PLoS ONE 2012, 7, e37468. [Google Scholar] [CrossRef] [Green Version]
- Shahabadi, N.; Hadidi, S. Mechanistic and conformational studies on the interaction of a platinum (II) complex containing an antiepileptic drug, levetiracetam, with bovine serum albumin by optical spectroscopic techniques in aqueous solution. Appl. Biochem. Biotechnol. 2015, 175, 1843–1857. [Google Scholar] [CrossRef]
- Wu, C.-W.; Stryer, L. Proximity relationships in rhodopsin. Proc. Natl. Acad. Sci USA 1972, 69, 1104–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buddanavar, A.T.; Nandibewoor, S.T. Multi-spectroscopic characterization of bovine serum albumin upon interaction with atomoxetine. J. Pharm. Anal. 2017, 7, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Wardell, M.; Wang, Z.; Ho, J.X.; Robert, J.; Ruker, F.; Ruble, J.; Carter, D.C. The atomic structure of human methemalbumin at 1.9 Å. Biochem. Biophys. Res. Commun. 2002, 291, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Petitpas, I.; Bhattacharya, A.; Twine, S.; Curry, S. Crystal Structure Analysis of Warfarin Binding to Human Serum Albumin: Anatomy of Drug Site I. J. Biol. Chem. 2001, 276, 22804–22809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, F.; Liu, G.; Chen, T.; Fu, X.; Niu, M.M. Structure-Based Virtual Screening and Biological Evaluation of Peptide Inhibitors for Polo-Box Domain. Molecules 2020, 25, 107. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: All samples can be available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T (K) | Kq (M−1 s−1) × 1011 | Ksv (M−1) × 103 | R2 |
|---|---|---|---|
| 298 | 8.539 | 8.539 ± 0.293 | 0.991 |
| 310 | 8.653 | 8.653 ± 0.337 | 0.988 |
| 318 | 8.223 | 8.223 ± 0.292 | 0.990 |
| T (K) | Kb(M−1) × 103 | n | R2 | ΔG (kJ·M−1) |
|---|---|---|---|---|
| 298 | 38.301 ± 0.00146 | 1.161 ± 0.0356 | 0.9925 | −26.15 |
| 310 | 48.031 ± 0.00120 | 1.181 ± 0.0170 | 0.9983 | −27.78 |
| 318 | 139.064 ± 0.00119 | 1.292 ± 0.0161 | 0.9988 | −31.31 |
| T (K) | System | Kb(M−1) × 103 | n | R2 | ΔG (kJ·M−1) |
|---|---|---|---|---|---|
| 298 | AICAR–HSA | 38.30 ± 0.001 | 1.16 ± 0.04 | 0.9925 | −26.15 |
| 298 | AICAR–HSA–Warfarin | 28.38 ± 0.001 | 1.12 ± 0.01 | 0.9987 | −25.40 |
| 298 | AICAR–HSA–Digitoxin | 40.18 ± 0.001 | 1.14 ± 0.01 | 0.9988 | −26.27 |
| 298 | AICAR–HSA–Flufenamic Acid | 24.55 ± 0.001 | 1.08 ± 0.03 | 0.9953 | −25.04 |
| (A) | Intensity (cps) ± SD | Wavelength (nm) |
| HSA | 4303 ± 100 | 302 |
| AICAR–HSA | 3910 ± 55 | 302 |
| % Change | −9.14 | |
| Shift | 0 | |
| (B) | Intensity (cps) ± SD | Wavelength (nm) |
| HSA | 12,166 ± 219 | 346 |
| AICAR–HSA | 11,430 ± 255 | 348 |
| % Change | −6.05 | |
| Shift | 2 |
| System | Peak Number | Peak Position λex/λem (nm/nm) | Intensity (cps) ± SD | % Relative Intensity Change | Shift λem (nm) |
|---|---|---|---|---|---|
| HSA | 1 | 360/360 | 252,756 ± 23,400 | ||
| 2 | 280/340 | 9080 ± 537 | |||
| AICAR–HAS (1:1) | 1 | 360/360 | 235,360 ± 17,600 | −6.9 | 0 |
| 2 | 280/340 | 8393 ± 575 | −7.6 | 0 | |
| AICAR–HSA (2:1) | 1 | 360/360 | 220,564 ± 9960 | −12.7 | 0 |
| 2 | 280/350 | 8293 ± 269 | −8.7 | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hashempour, S.; Shahabadi, N.; Adewoye, A.; Murphy, B.; Rouse, C.; Salvatore, B.A.; Stratton, C.; Mahdavian, E. Binding Studies of AICAR and Human Serum Albumin by Spectroscopic, Theoretical, and Computational Methodologies. Molecules 2020, 25, 5410. https://doi.org/10.3390/molecules25225410
Hashempour S, Shahabadi N, Adewoye A, Murphy B, Rouse C, Salvatore BA, Stratton C, Mahdavian E. Binding Studies of AICAR and Human Serum Albumin by Spectroscopic, Theoretical, and Computational Methodologies. Molecules. 2020; 25(22):5410. https://doi.org/10.3390/molecules25225410
Chicago/Turabian StyleHashempour, Shokoufeh, Nahid Shahabadi, Aishat Adewoye, Brennen Murphy, Camaray Rouse, Brian A. Salvatore, Christopher Stratton, and Elahe Mahdavian. 2020. "Binding Studies of AICAR and Human Serum Albumin by Spectroscopic, Theoretical, and Computational Methodologies" Molecules 25, no. 22: 5410. https://doi.org/10.3390/molecules25225410
APA StyleHashempour, S., Shahabadi, N., Adewoye, A., Murphy, B., Rouse, C., Salvatore, B. A., Stratton, C., & Mahdavian, E. (2020). Binding Studies of AICAR and Human Serum Albumin by Spectroscopic, Theoretical, and Computational Methodologies. Molecules, 25(22), 5410. https://doi.org/10.3390/molecules25225410