
Synthesis, Kinetic and Conformational Studies of 2-Substituted-5-(β-d-glucopyranosyl)-pyrimidin-4-ones as Potential Inhibitors of Glycogen Phosphorylase

 , ,
, ,  ,
, 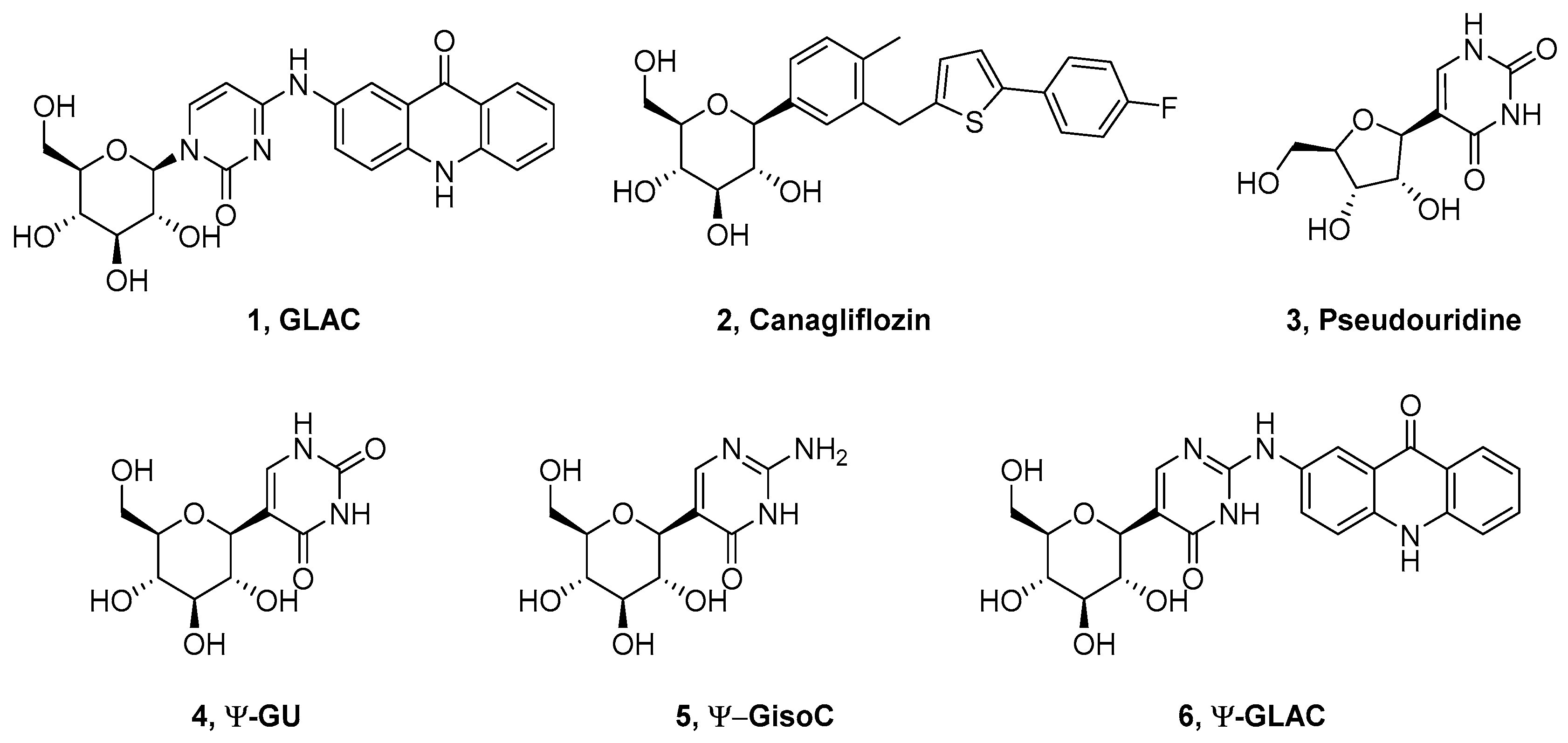{kind=link}
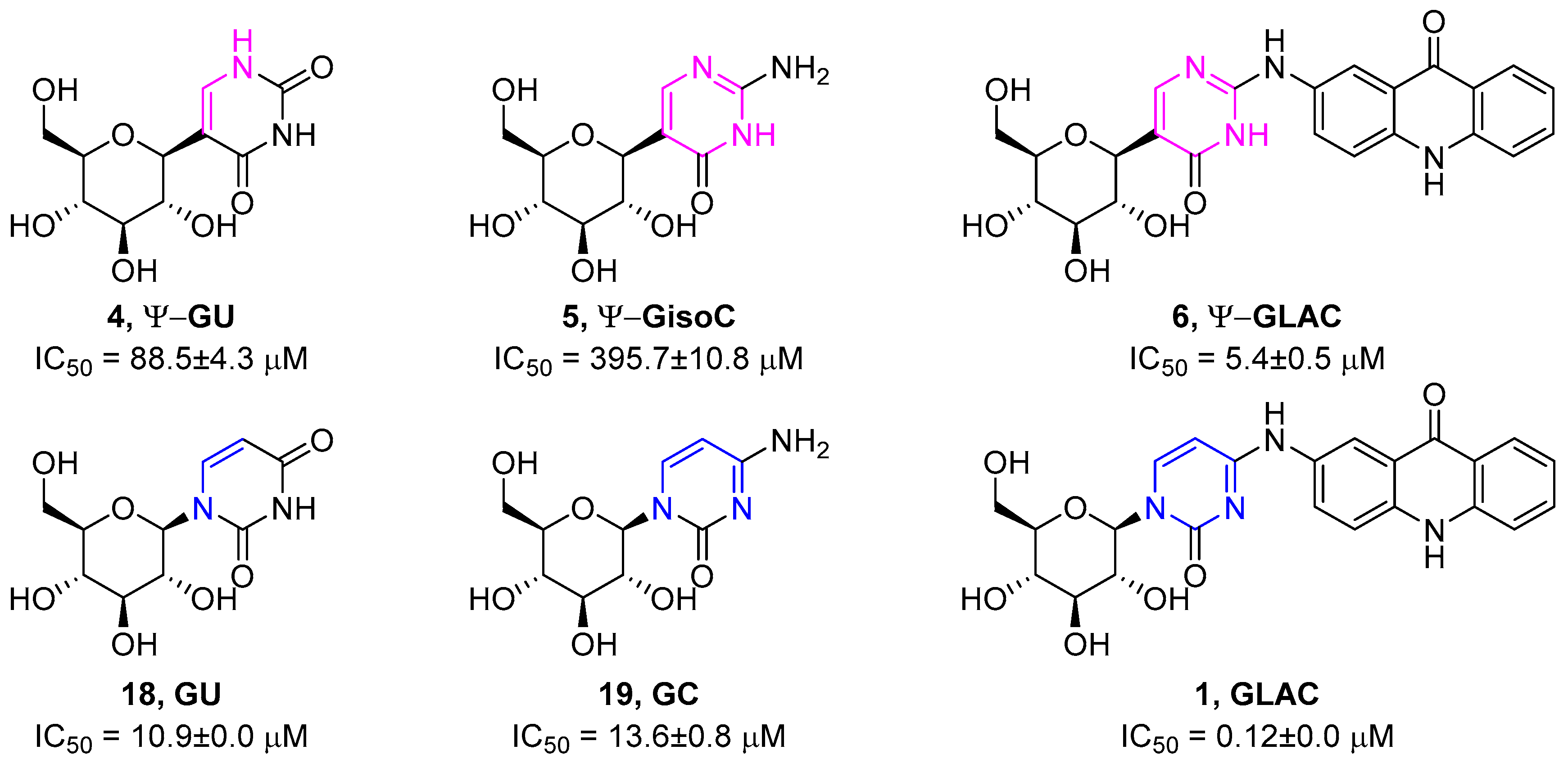{kind=link}
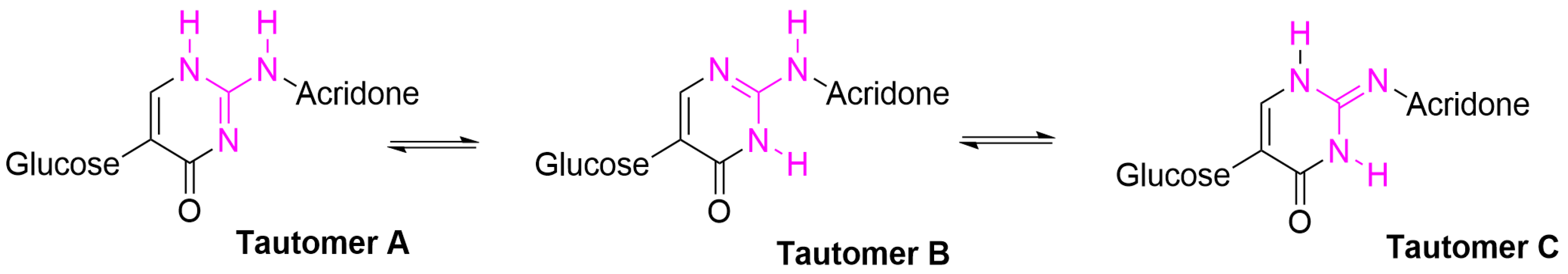{kind=link}
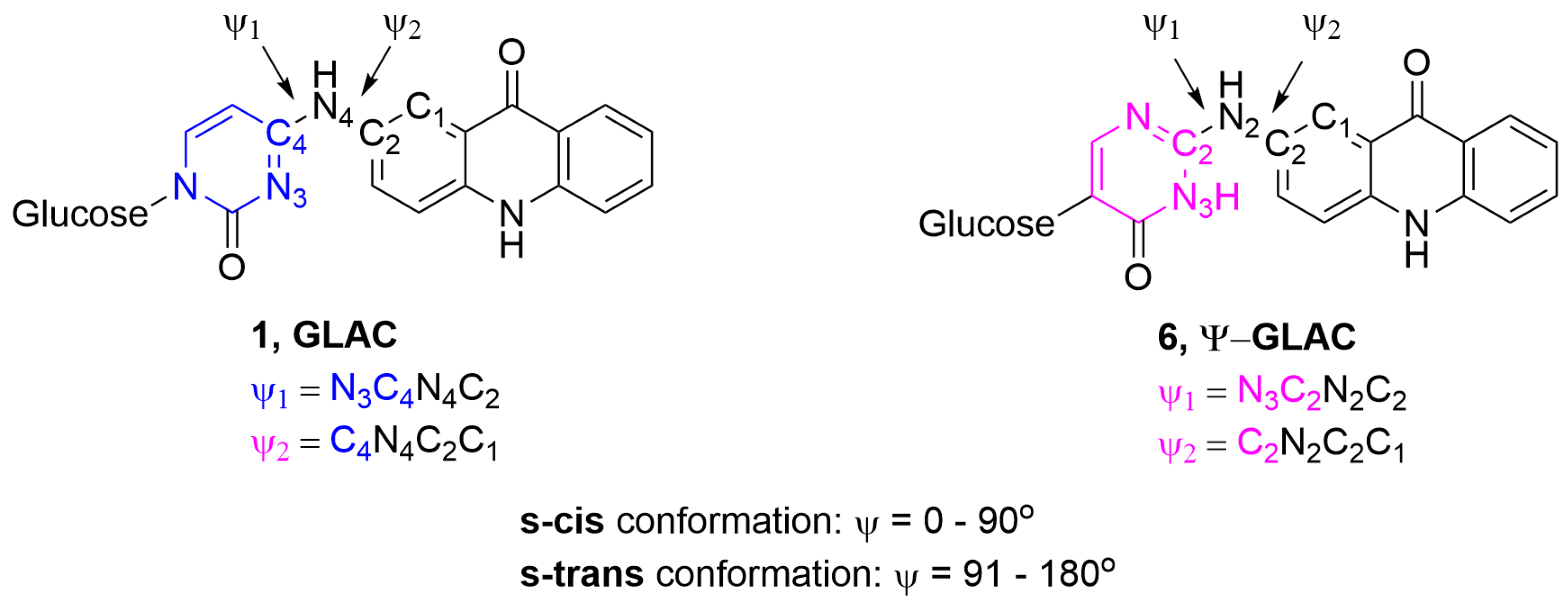{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
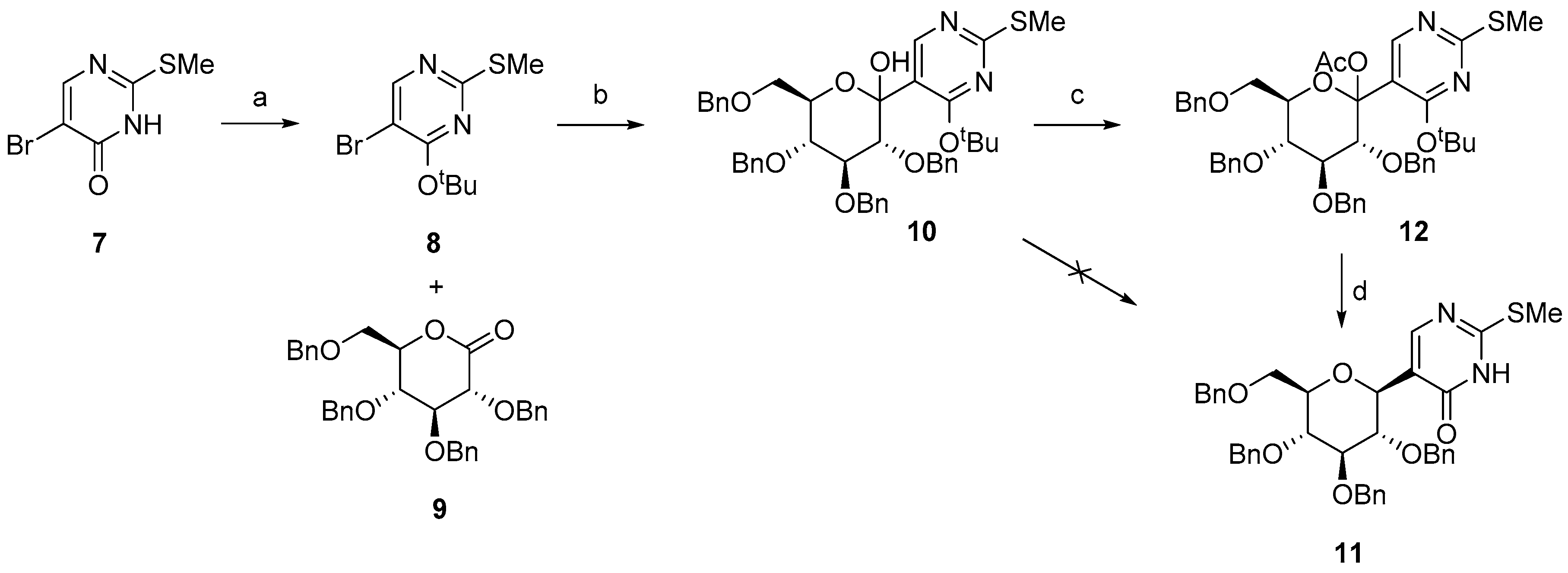
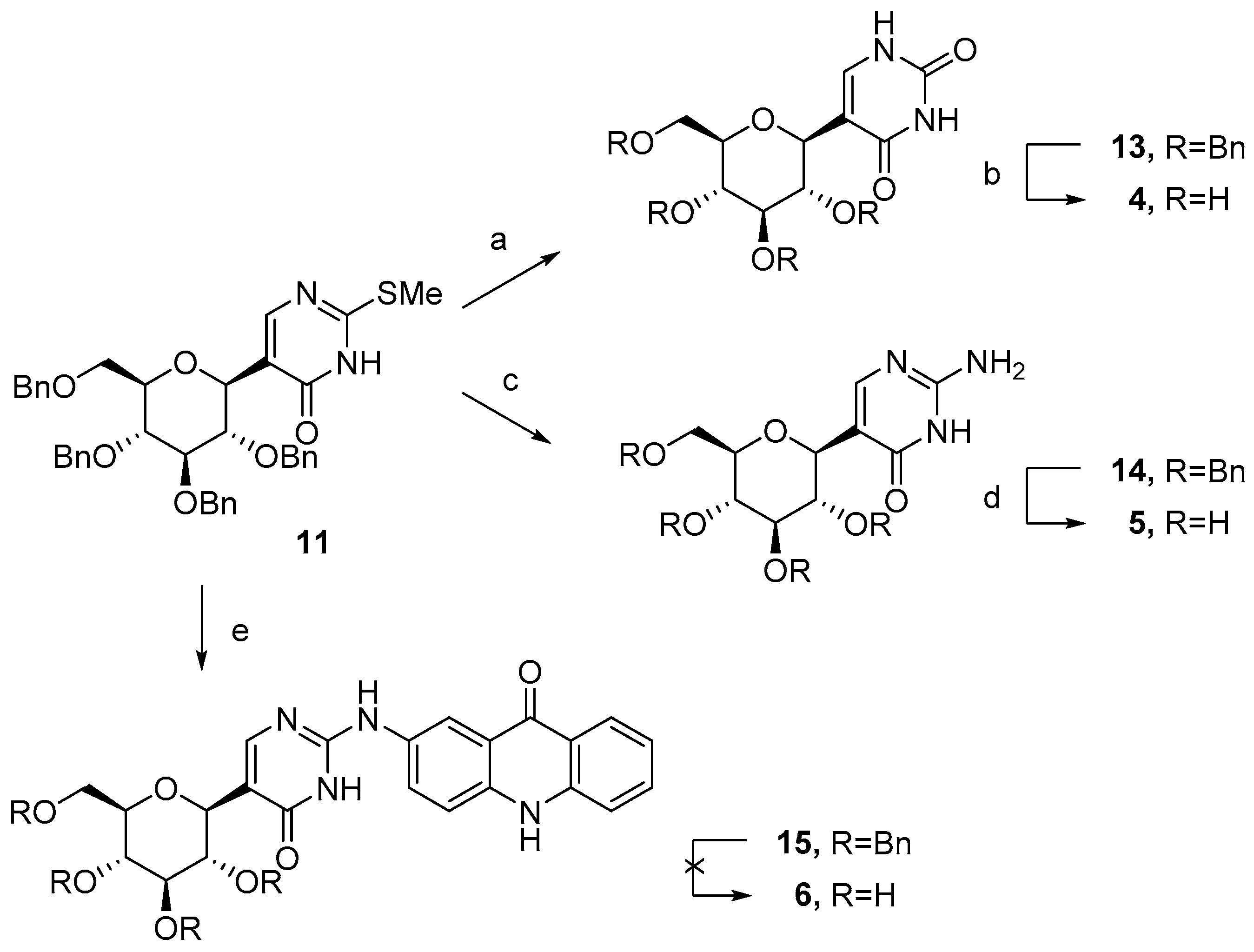
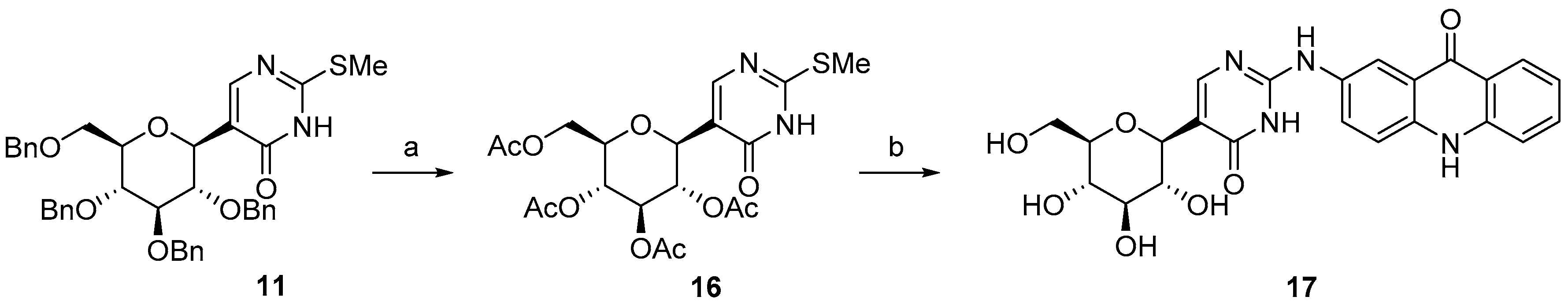
2.1. Synthesis
2.2. Enzyme Kinetics
3. Discussion
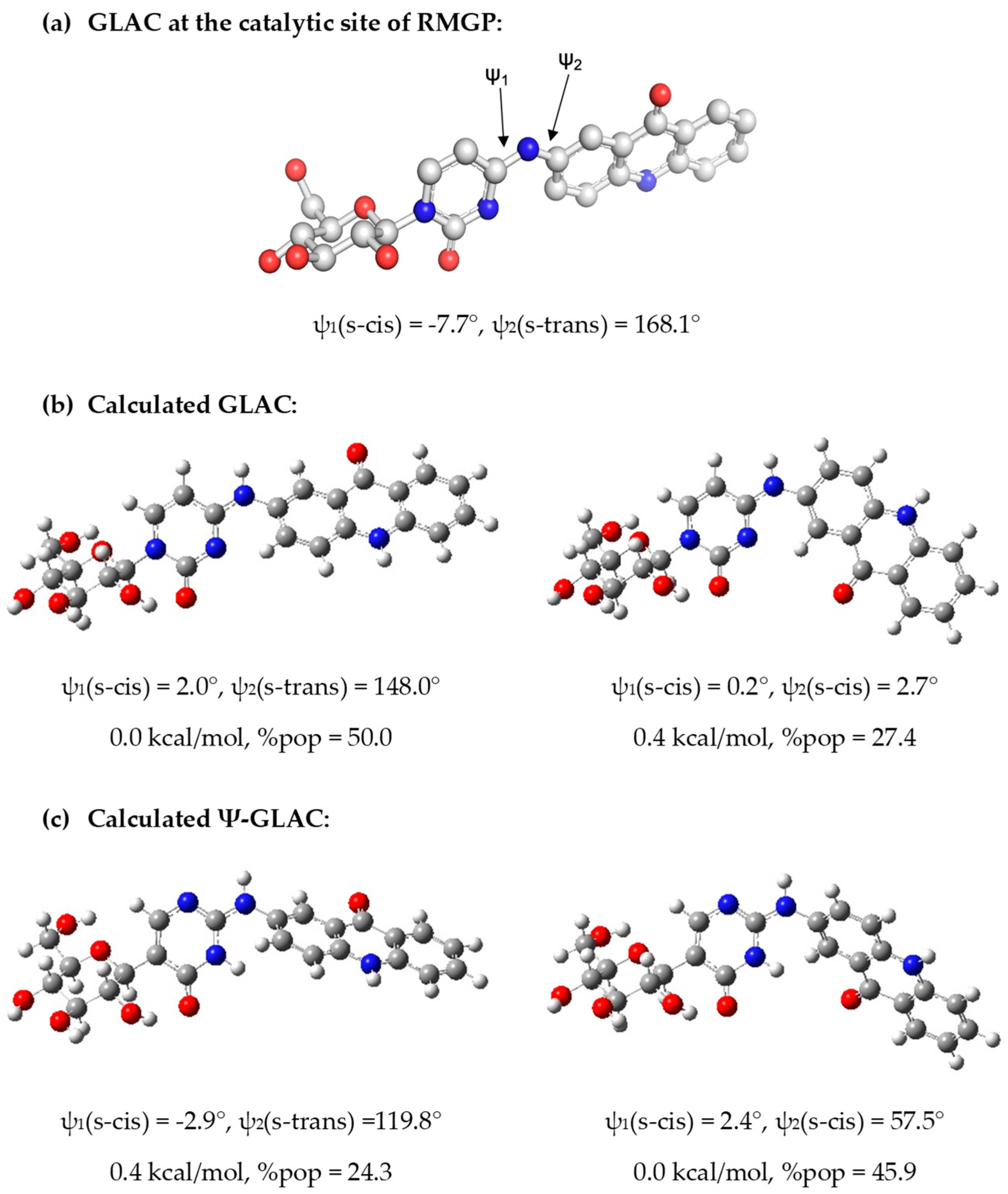
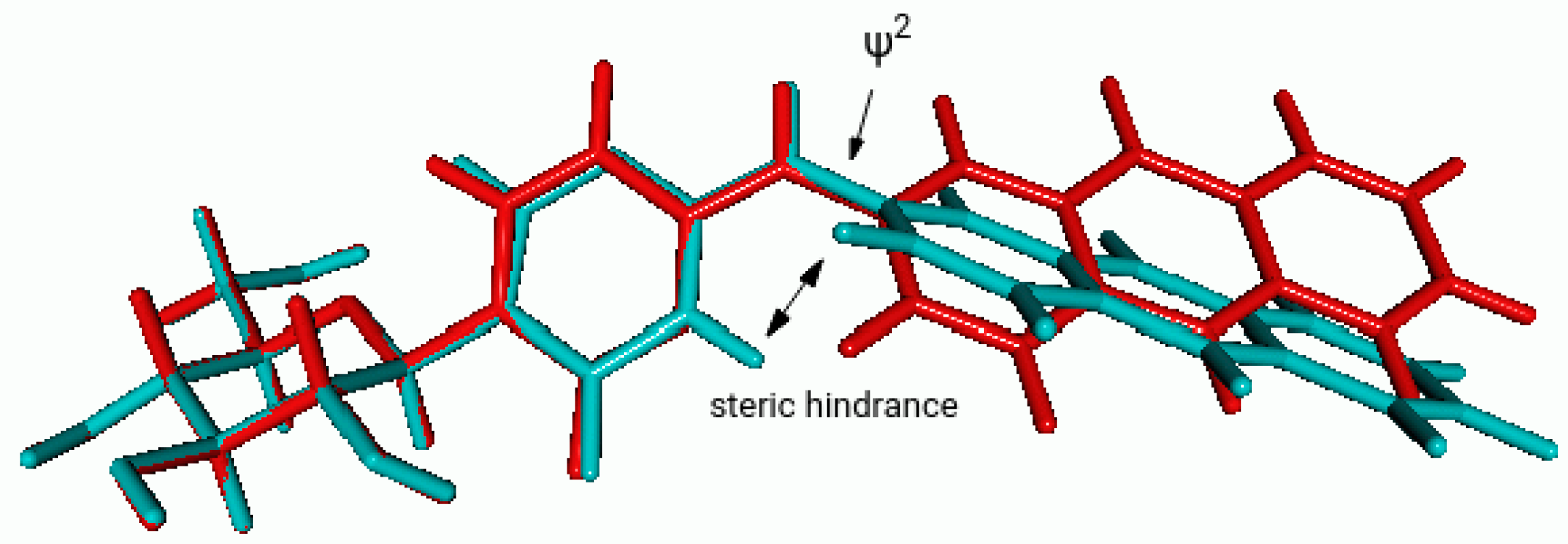
Computational Studies
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Belete, T.M. A recent achievement in the discovery and development of novel targets for the treatment of type-2 diabetes mellitus. J. Exp. Pharmacol. 2020, 12, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Praly, J.-P.; Vidal, S. Inhibition of glycogen phosphorylase in the context of type 2 diabetes, with focus on recent inhibitors bound at the active site. Mini Rev. Med. Chem. 2010, 10, 1102–1126. [Google Scholar] [CrossRef] [PubMed]
- Treadway, J.L.; Mendys, P.; Hoover, D.J. Expert opinion on investigational drugs glycogen phosphorylase inhibitors for treatment of type 2 diabetes mellitus. Expert Opin. Investig. Drugs 2001, 10, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Rousset, M.; Chevalier, G.; Rousset, J.P.; Dussaulx, E.; Zweibaum, A. Presence and cell growth-related variations of glycogen in human colorectal adenocarcinoma cell lines in culture. Cancer Res. 1979, 39, 531–534. [Google Scholar]
- Rousset, M.; Zweibaum, A.; Fogh, J. Presence of glycogen and growth-related variations in 58 cultured human tumor cell lines of various tissue origins. Cancer Res. 1981, 41, 1165–1170. [Google Scholar]
- Schnier, J.B.; Nishi, K.; Monks, A.; Gorin, F.A.; Bradbury, E.M. Inhibition of glycogen phosphorylase (GP) by CP-91,149 induces growth inhibition correlating with brain GP expression. Biochem. Biophys. Res. Commun. 2003, 309, 126–134. [Google Scholar] [CrossRef]
- Rousset, M.; Paris, H.; Chevalier, G.; Terrain, B.; Murat, J.C.; Zweibaum, A. Growth-related enzymatic control of glycogen metabolism in cultured human tumor cells. Cancer Res. 1984, 44, 154–160. [Google Scholar] [CrossRef] [Green Version]
- Uhlén, M.; Björling, E.; Agaton, C.; Szigyarto, C.A.K.; Amini, B.; Andersen, E.; Andersson, A.C.; Angelidou, P.; Asplund, A.; Asplund, C.; et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteomics 2005, 4, 1920–1932. [Google Scholar] [CrossRef] [Green Version]
- Favaro, E.; Bensaad, K.; Chong, M.G.; Tennant, D.A.; Ferguson, D.J.P.; Snell, C.; Steers, G.; Turley, H.; Li, J.-L.L.; Günther, U.L.; et al. Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab. 2012, 16, 751–764. [Google Scholar] [CrossRef] [Green Version]
- Shen, G.-M.; Zhang, F.-L.; Liu, X.-L.; Zhang, J.-W. Hypoxia-inducible factor 1-mediated regulation of PPP1R3C promotes glycogen accumulation in human MCF-7 cells under hypoxia. FEBS Lett. 2010, 584, 4366–4372. [Google Scholar] [CrossRef] [Green Version]
- Ros, S.; Schulze, A. Linking glycogen and senescence in cancer cells. Cell Metab. 2012, 16, 687–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zois, C.E.; Favaro, E.; Harris, A.L. Glycogen metabolism in cancer. Biochem. Pharmacol. 2014, 92, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Zois, C.E.; Harris, A.L. Glycogen metabolism has a key role in the cancer microenvironment and provides new targets for cancer therapy. J. Mol. Med. 2016, 94, 137–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bak, L.K.; Walls, A.B. Astrocytic glycogen metabolism in the healthy and diseased brain. J. Biol. Chem. 2018, 293, 7108–7116. [Google Scholar] [CrossRef] [Green Version]
- Swanson, R.A. Brain glycogen–Vestigial no more. Metab. Brain Dis. 2015, 30, 251–253. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Sun, H. Pharmacological manipulation of brain glycogenolysis as a therapeutic approach to cerebral ischemia. Mini Rev. Med. Chem. 2010, 10, 1188–1193. [Google Scholar] [CrossRef]
- Donnier-Maréchal, M.; Vidal, S. Glycogen phosphorylase inhibitors: A patent review (2013–2015). Expert Opin. Ther. Pat. 2016, 26, 199–212. [Google Scholar] [CrossRef]
- D Chrysina, E.; Chajistamatiou, A.; Chegkazi, M. From structure–based to knowledge-based drug design through X-ray protein crystallography: Sketching glycogen phosphorylase binding sites. Curr. Med. Chem. 2011, 18, 2620–2629. [Google Scholar] [CrossRef]
- Gimisis, T. Synthesis of N-glucopyranosidic derivatives as potential inhibitors that bind at the catalytic site of glycogen phosphorylase. Mini Rev. Med. Chem. 2010, 10, 1127–1138. [Google Scholar] [CrossRef]
- Somsak, L.; Nagy, V.; Hadady, Z.; Docsa, T.; Gergely, P. Glucose analog inhibitors of glycogen phosphorylases as potential antidiabetic agents: Recent developments. Curr. Pharm. Des. 2003, 9, 1177–1189. [Google Scholar] [CrossRef]
- Somsak, L.; Czifrak, K.; Toth, M.; Bokor, E.; Chrysina, E.; Alexacou, K.-M.; Hayes, J.; Tiraidis, C.; Lazoura, E.; Leonidas, D.; et al. New inhibitors of glycogen phosphorylase as potential antidiabetic agents. Curr. Med. Chem. 2008, 15, 2933–2983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikonomakos, N.G.; Somsák, L. Advances in glycogen phosphorylase inhibitor design. Curr. Opin. Investig. Drugs 2008, 9, 379–395. [Google Scholar] [PubMed]
- Somsák, L. Glucose derived inhibitors of glycogen phosphorylase. Comptes Rendus Chim. 2011, 14, 211–223. [Google Scholar] [CrossRef] [Green Version]
- Bokor, É. N- and C-Glycopyranosyl heterocycles as glycogen phosphorylase inhibitors. In Recent Trends in Carbohydrate Chemistry; Rauter, A.P., Christensen, B.E., Somsák, L., Kosma, P., Adamo, R., Eds.; Elsevier: New York, NY, USA, 2020; pp. 253–300. [Google Scholar] [CrossRef]
- Mamais, M.; Degli Esposti, A.; Kouloumoundra, V.; Gustavsson, T.; Monti, F.; Venturini, A.; Chrysina, E.D.; Markovitsi, D.; Gimisis, T. A new potent inhibitor of glycogen phosphorylase reveals the basicity of the catalytic site. Chem. A Eur. J. 2017, 23, 8800–8805. [Google Scholar] [CrossRef]
- Kantsadi, A.L.; Bokor, É.; Kun, S.; Stravodimos, G.A.; Chatzileontiadou, D.S.M.; Leonidas, D.D.; Juhász-Tóth, É.; Szakács, A.; Batta, G.; Docsa, T.; et al. Synthetic, enzyme kinetic, and protein crystallographic studies of C-β-d-glucopyranosyl pyrroles and imidazoles reveal and explain low nanomolar inhibition of human liver glycogen phosphorylase. Eur. J. Med. Chem. 2016, 123, 737–745. [Google Scholar] [CrossRef] [Green Version]
- Bokor, É.; Kun, S.; Goyard, D.; Tóth, M.; Praly, J.-P.; Vidal, S.; Somsák, L. C-glycopyranosyl arenes and hetarenes: Synthetic methods and bioactivity focused on antidiabetic potential. Chem. Rev. 2017, 117, 1687–1764. [Google Scholar] [CrossRef]
- Deeks, E.D.; Scheen, A.J. Canagliflozin: A review in type 2 diabetes. Drugs 2017, 77, 1577–1592. [Google Scholar] [CrossRef]
- Somsák, L.; Bokor, É.; Juhász, L.; Kun, S.; Lázár, L.; Juhász-Tóth, É.; Tóth, M. New syntheses towards C-glycosyl type glycomimetics. Pure Appl. Chem. 2019, 91, 1159–1175. [Google Scholar] [CrossRef]
- Bokor, É.; Kun, S.; Docsa, T.; Gergely, P.; Somsák, L. 4(5)-Aryl-2-C-glucopyranosyl-imidazoles as new nanomolar glucose analogue inhibitors of glycogen phosphorylase. ACS Med. Chem. Lett. 2015, 6, 1215–1219. [Google Scholar] [CrossRef] [Green Version]
- Mamais, M.; Kouloumoundra, V.; Smyrli, E.; Grammatopoulos, P.; Chrysina, E.D.; Gimisis, T. Synthesis of N4-aryl-β-d-glucopyranosylcytosines: A methodology study. Tetrahedron Lett. 2015, 56, 5549–5552. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yu, B. Recent advances in the chemical synthesis of C-glycosides. Chem. Rev. 2017, 117, 12281–12356. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.D.; Cha, J.K.; Kishi, Y. Highly stereoselective approaches to α- and β-C-glycopyranosides. J. Am. Chem. Soc. 1982, 104, 4976–4978. [Google Scholar] [CrossRef]
- Kraus, G.A.; Molina, M.T.; Teresa Molina, M. A direct synthesis of C-glycosyl compounds. J. Org. Chem. 1988, 53, 752–753. [Google Scholar] [CrossRef] [Green Version]
- Horton, D.; Priebe, W. Synthetic routes to higher-carbon sugars. Reaction of lactones with 2-lithio-,3-dithiane. Carbohydr. Res. 1981, 94, 27–41. [Google Scholar] [CrossRef]
- RajanBabu, T.V.; Reddy, G.S. 1-methylene sugars as C-glycoside precursors. J. Org. Chem. 1986, 51, 5458–5461. [Google Scholar] [CrossRef]
- Tibiletti, F.; Simonetti, M.; Nicholas, K.M.; Palmisano, G.; Parravicini, M.; Imbesi, F.; Tollari, S.; Penoni, A. One-pot synthesis of meridianins and meridianin analogues via indolization of nitrosoarenes. Tetrahedron 2010, 66, 1280–1288. [Google Scholar] [CrossRef]
- Barrett, H.W.; Goodman, I.; Dittmer, K. The synthesis of 5-halogeno-2-thiouracil and 6-methyl-5-halogeno-2-thiouracil derivatives 1. J. Am. Chem. Soc. 1948, 70, 1753–1756. [Google Scholar] [CrossRef]
- Hanessian, S.; Machaalani, R. A highly stereocontrolled and efficient synthesis of α- and β-pseudouridines. Tetrahedron Lett. 2003, 44, 8321–8323. [Google Scholar] [CrossRef]
- Ulbricht, T.L.V.L.V. Syntheses with pyrimidine-lithium compounds. Tetrahedron 1959, 6, 225–231. [Google Scholar] [CrossRef]
- Boudet, N.; Knochel, P. Chemo- and regioselective functionalization of uracil derivatives. Applications to the synthesis of oxypurinol and emivirine. Org. Lett. 2006, 8, 3737–3740. [Google Scholar] [CrossRef]
- Kofink, C.C.; Knochel, P. Synthesis of functionalized diarylmethanes via a copper-catalyzed cross-coupling of arylmagnesium reagents with benzylic phosphates. Org. Lett. 2006, 8, 4121–4124. [Google Scholar] [CrossRef] [PubMed]
- Ellsworth, B.A.; Doyle, A.G.; Patel, M.; Caceres-Cortes, J.; Meng, W.; Deshpande, P.P.; Pullockaran, A.; Washburn, W.N. C-Arylglucoside synthesis: Triisopropylsilane as a selective reagent for the reduction of an anomeric C-phenyl ketal. Tetrahedron Asymm. 2003, 14, 3243–3247. [Google Scholar] [CrossRef]
- Deshpande, P.P.; Singh, J.; Pullockaran, A.; Kissick, T.; Ellsworth, B.A.; Gougoutas, J.Z.; Dimarco, J.; Fakes, M.; Reyes, M.; Lai, C.; et al. A practical stereoselective synthesis and novel cocrystallizations of an amphiphatic SGLT-2 inhibitor. Org. Process Res. Dev. 2012, 16, 577–585. [Google Scholar] [CrossRef]
- Urabe, D.; Nagatomo, M.; Hagiwara, K.; Masuda, K.; Inoue, M. Symmetry-driven synthesis of 9-demethyl-10,15-dideoxyryanodol. Chem. Sci. 2013, 4, 1615. [Google Scholar] [CrossRef]
- Dondoni, A.; Scherrmann, M. Thiazole-based synthesis of formyl C-Glycosides. J. Org. Chem. 1994, 59, 6404–6412. [Google Scholar] [CrossRef]
- Xie, J.; Molina, A.; Czernecki, S. Alkylidenation of sugar lactones and further transformation to C-glycosides. J. Carbohydr. Chem. 1999, 18, 481–498. [Google Scholar] [CrossRef]
- Khalifa, N.M.; Al-Omar, M.A.; Amr, A.E. Synthesis and characterization of novel 5-allyl-6-{(benzo[d]thiazol-2-yl)methyl}-2-(alkylsulfanyl)oxopyrimidine derivatives. Russ. J. Gen. Chem. 2016, 86, 2752–2758. [Google Scholar] [CrossRef]
- Grosjean, S.; Triki, S.; Meslin, J.C.; Julienne, K.; Deniaud, D. Synthesis of nitrogen bicyclic scaffolds: Pyrimido[1,2-a]pyrimidine-2,6-diones. Tetrahedron 2010, 66, 9912–9924. [Google Scholar] [CrossRef]
- Cho, A.; Zhang, L.; Xu, J.; Babusis, D.; Butler, T.; Lee, R.; Saunders, O.L.; Wang, T.; Parrish, J.; Perry, J.; et al. Synthesis and characterization of 2′-C-Me branched C-nucleosides as HCV polymerase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 4127–4132. [Google Scholar] [CrossRef]
- Li, Y.; Manickam, G.; Ghoshal, A.; Subramaniam, P. More efficient palladium catalyst for hydrogenolysis of benzyl groups. Synth. Commun. 2006, 36, 925–928. [Google Scholar] [CrossRef]
- Maddess, M.L.; Carter, R. SNAr Reactions of 2-Methylthio-4-pyrimidinones in Pivalic Acid: Access to Functionalized Pyrimidinones and Pyrimidines. Synthesis. 2012, 44, 1109–1118. [Google Scholar] [CrossRef]
- Alzeer, J.; Vasella, A. Oligosaccharide analogues of polysaccharides. Part 2. Regioselective deprotection of monosaccharide-derived monomers and dimers. Helv. Chim. Acta 1995, 78, 177–193. [Google Scholar] [CrossRef]
- Oikonomakos, N.G.; Kontou, M.; Zographos, S.E.; Watson, K.A.; Johnson, L.N.; Bichard, C.J.F.; Fleet, G.W.J.; Acharya, K.R. N-acetyl-β-d-glucopyranosylamine: A potent T-state inhibitor of glycogen phosphorylase. A comparison with α-d-glucose. Protein Sci. 1995, 4, 2469–2477. [Google Scholar] [CrossRef] [PubMed]
- Maffeis, V.; Mavreas, K.; Monti, F.; Mamais, M.; Gustavsson, T.; Chrysina, E.D.; Markovitsi, D.; Gimisis, T.; Venturini, A. Multiscale time-resolved fluorescence study of a glycogen phosphorylase inhibitor combined with quantum chemistry calculations. Phys. Chem. Chem. Phys. 2019, 21, 7685–7696. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. G16_c01; Gaussian, Inc.: Oxfordshire, UK, 2016. [Google Scholar]
- Szennyes, E.; Gyémánt, G.; Somsák, L.; Bokor, É. Synthesis of new series of 2-C-(β-d-glucopyranosyl)-pyrimidines and their evaluation as inhibitors of some glycoenzymes. Molecules 2020, 25, 701. [Google Scholar] [CrossRef] [Green Version]
- Szennyes, E.; Bokor, É.; Langer, P.; Gyémánt, G.; Docsa, T.; Sipos, Á.; Somsák, L. The first general synthesis of 2-C-(β-d-glycopyranosyl)pyrimidines and their evaluation as inhibitors of some glycoenzymes. New J. Chem. 2018, 42, 17439–17446. [Google Scholar] [CrossRef]
- Waschke, D.; Thimm, J.; Thiem, J. Highly efficient synthesis of ketoheptoses. Org. Lett. 2011, 13, 3628–3631. [Google Scholar] [CrossRef]
- Damager, I.; Erik Olsen, C.; Lindberg Møller, B.; Saddik Motawia, M. Chemical synthesis of 6’’’-α-maltotriosyl-maltohexaose as substrate for enzymes in starch biosynthesis and degradation. Carbohydr. Res. 1999, 320, 19–30. [Google Scholar] [CrossRef]
- Saheki, S.; Takeda, A.; Shimazu, T. Assay of inorganic phosphate in the mild pH range, suitable for measurement of glycogen phosphorylase activity. Anal. Biochem. 1985, 148, 277–281. [Google Scholar] [CrossRef]









Sample Availability: Samples of the compounds are not available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mavreas, K.F.; Neofytos, D.D.; Chrysina, E.D.; Venturini, A.; Gimisis, T. Synthesis, Kinetic and Conformational Studies of 2-Substituted-5-(β-d-glucopyranosyl)-pyrimidin-4-ones as Potential Inhibitors of Glycogen Phosphorylase. Molecules 2020, 25, 5463. https://doi.org/10.3390/molecules25225463
Mavreas KF, Neofytos DD, Chrysina ED, Venturini A, Gimisis T. Synthesis, Kinetic and Conformational Studies of 2-Substituted-5-(β-d-glucopyranosyl)-pyrimidin-4-ones as Potential Inhibitors of Glycogen Phosphorylase. Molecules. 2020; 25(22):5463. https://doi.org/10.3390/molecules25225463
Chicago/Turabian StyleMavreas, Konstantinos F., Dionysios D. Neofytos, Evangelia D. Chrysina, Alessandro Venturini, and Thanasis Gimisis. 2020. "Synthesis, Kinetic and Conformational Studies of 2-Substituted-5-(β-d-glucopyranosyl)-pyrimidin-4-ones as Potential Inhibitors of Glycogen Phosphorylase" Molecules 25, no. 22: 5463. https://doi.org/10.3390/molecules25225463
APA StyleMavreas, K. F., Neofytos, D. D., Chrysina, E. D., Venturini, A., & Gimisis, T. (2020). Synthesis, Kinetic and Conformational Studies of 2-Substituted-5-(β-d-glucopyranosyl)-pyrimidin-4-ones as Potential Inhibitors of Glycogen Phosphorylase. Molecules, 25(22), 5463. https://doi.org/10.3390/molecules25225463