
A Critical Overview of Current Theoretical Methods of Estimating the Energy of Intramolecular Interactions


Abstract
:1. Introduction
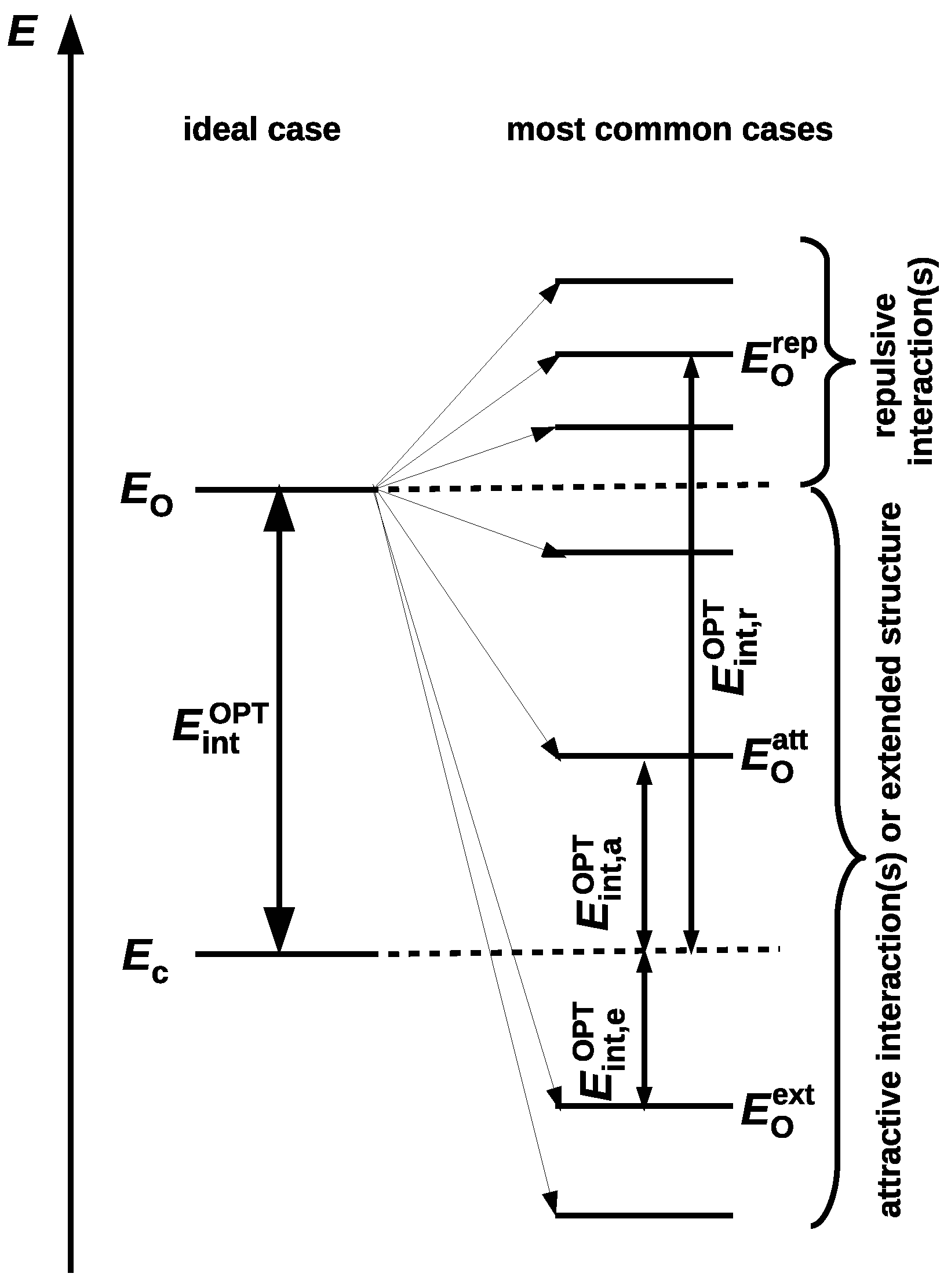
2. Theoretical Methods of Estimating the Energy of Intramolecular Interactions
2.1. Conformational Methods
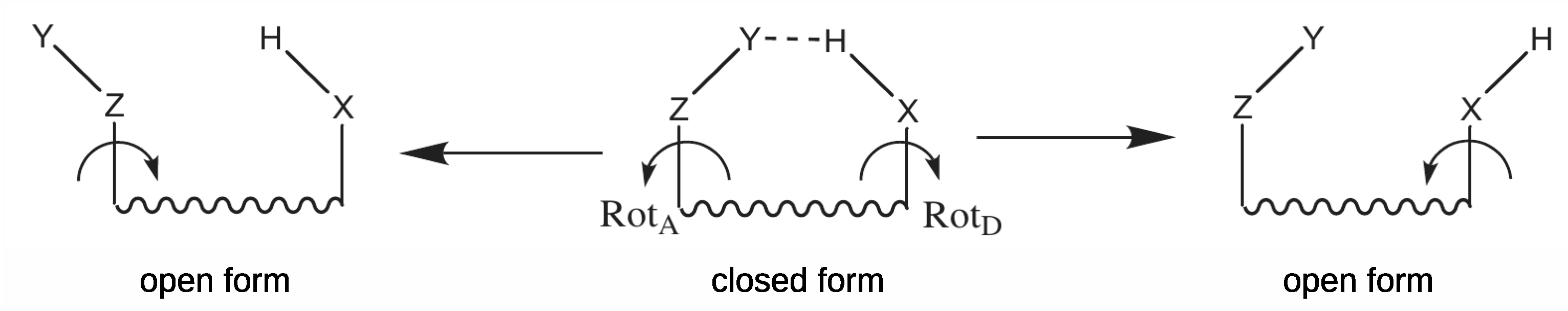
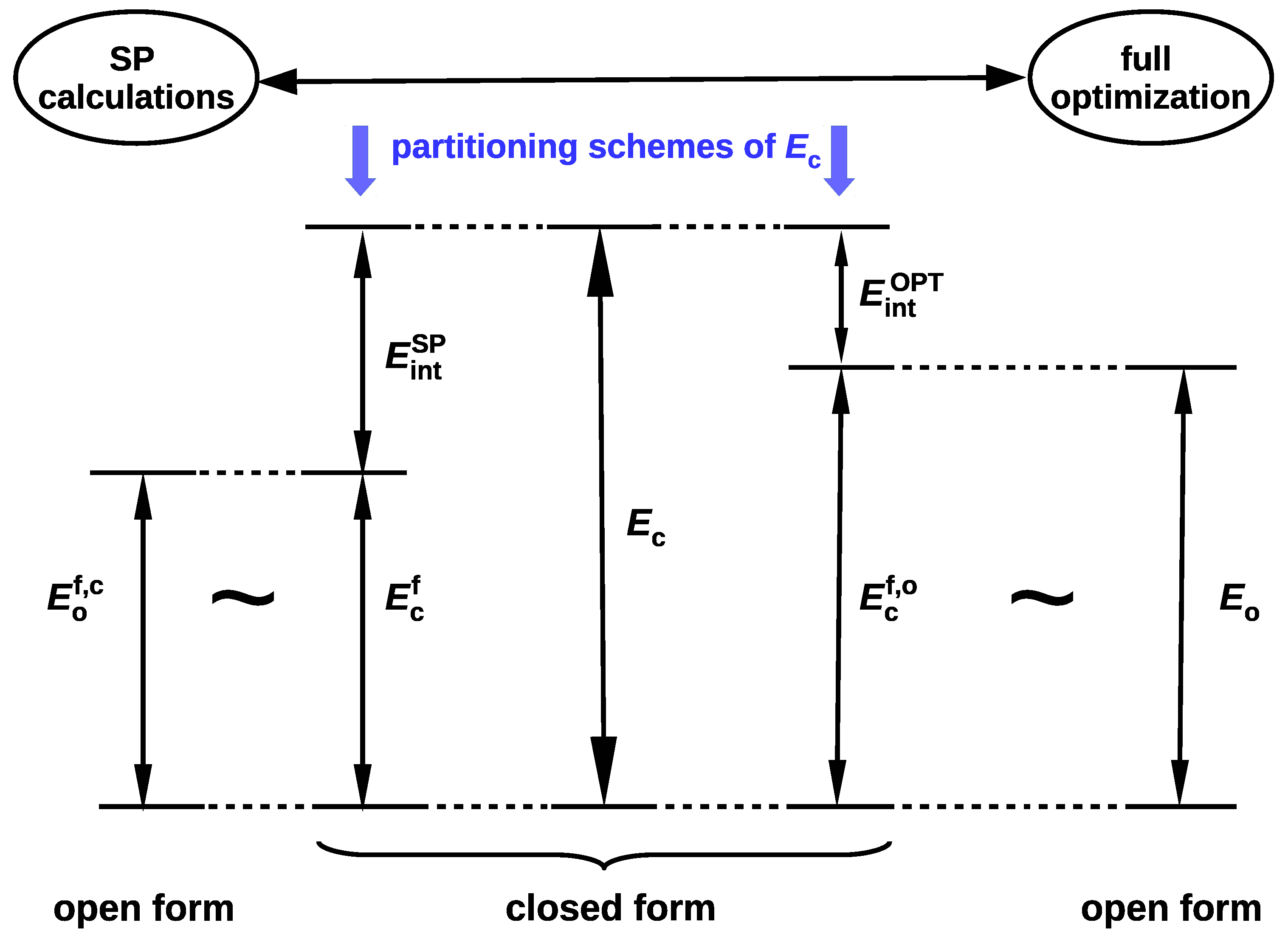
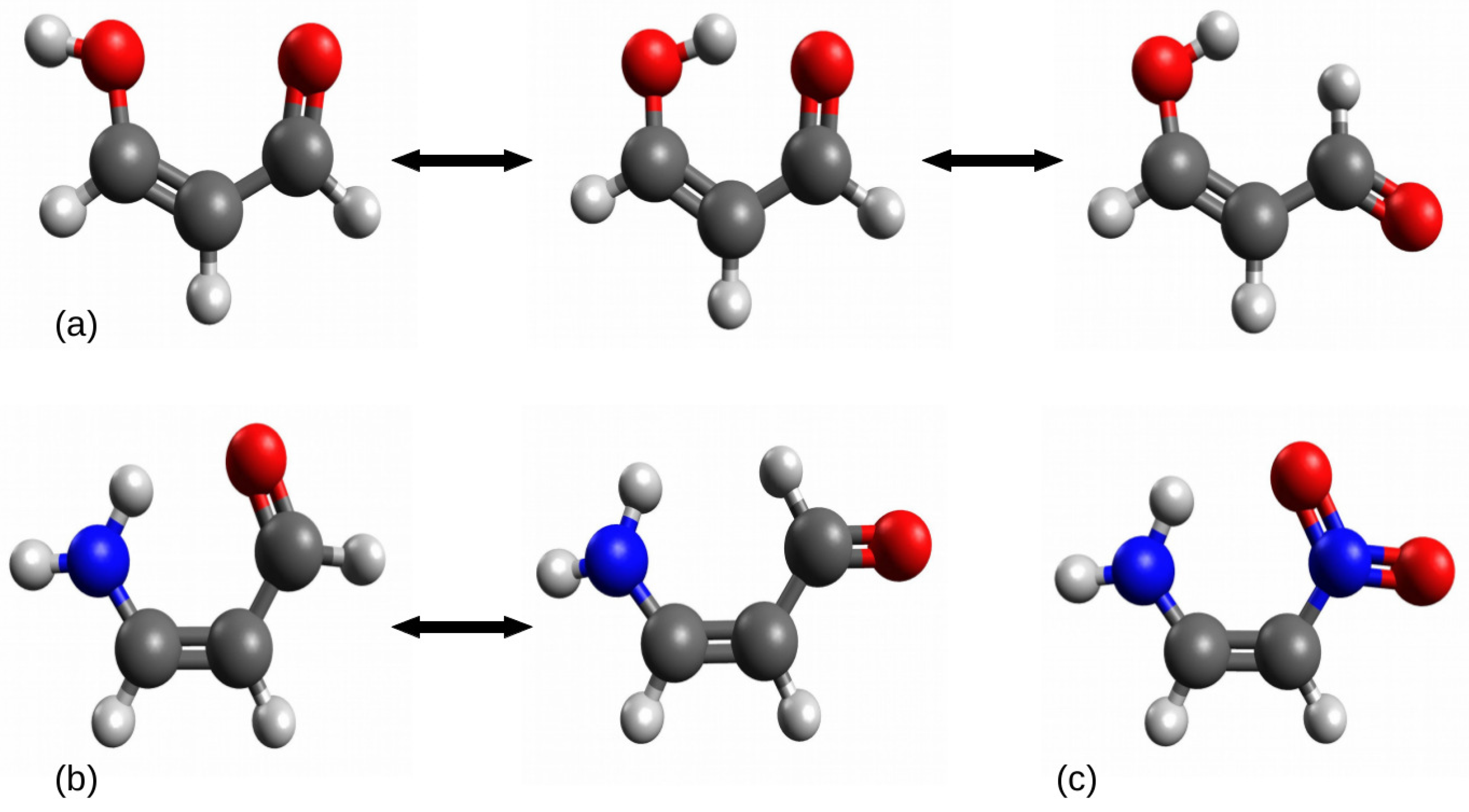
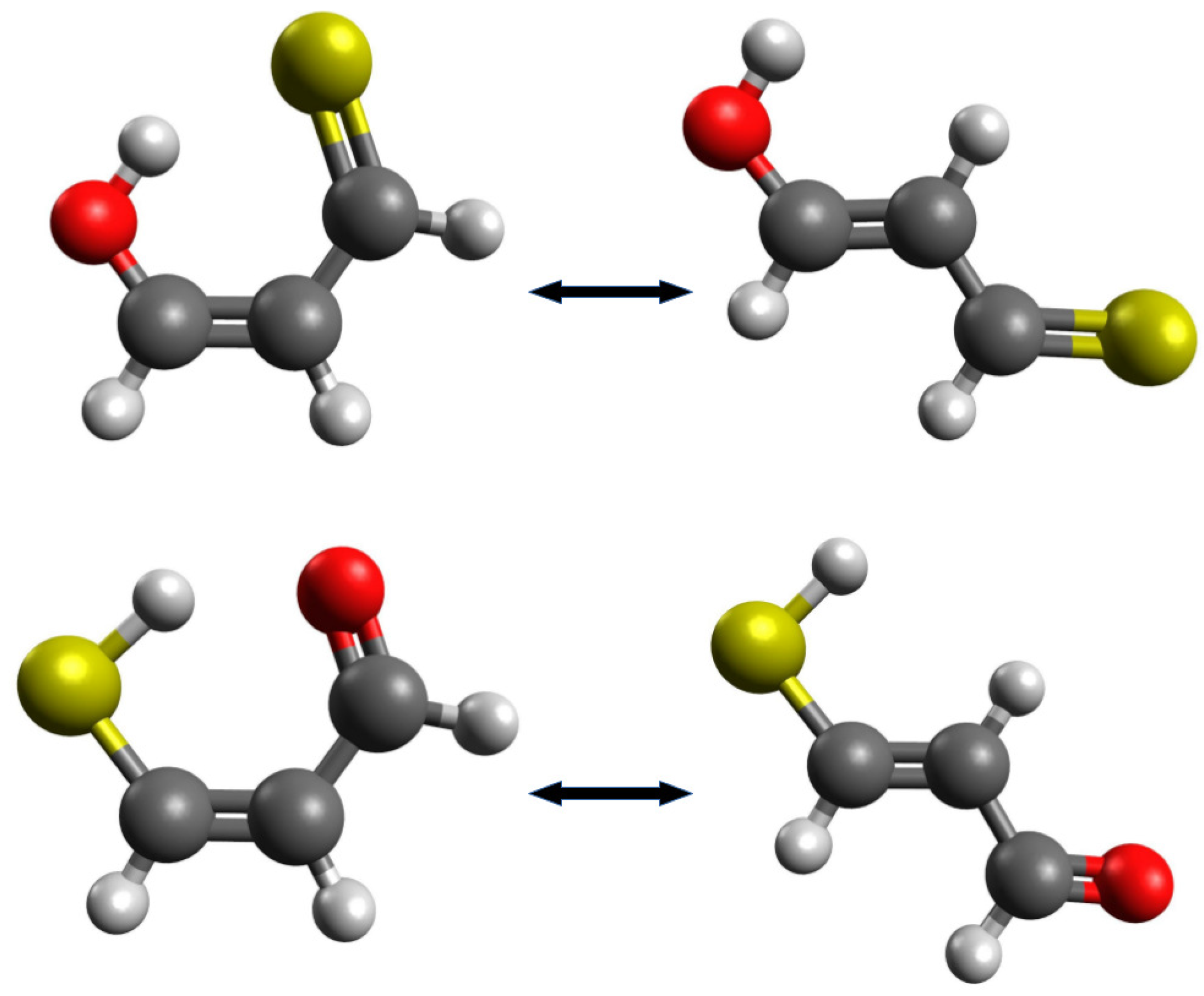
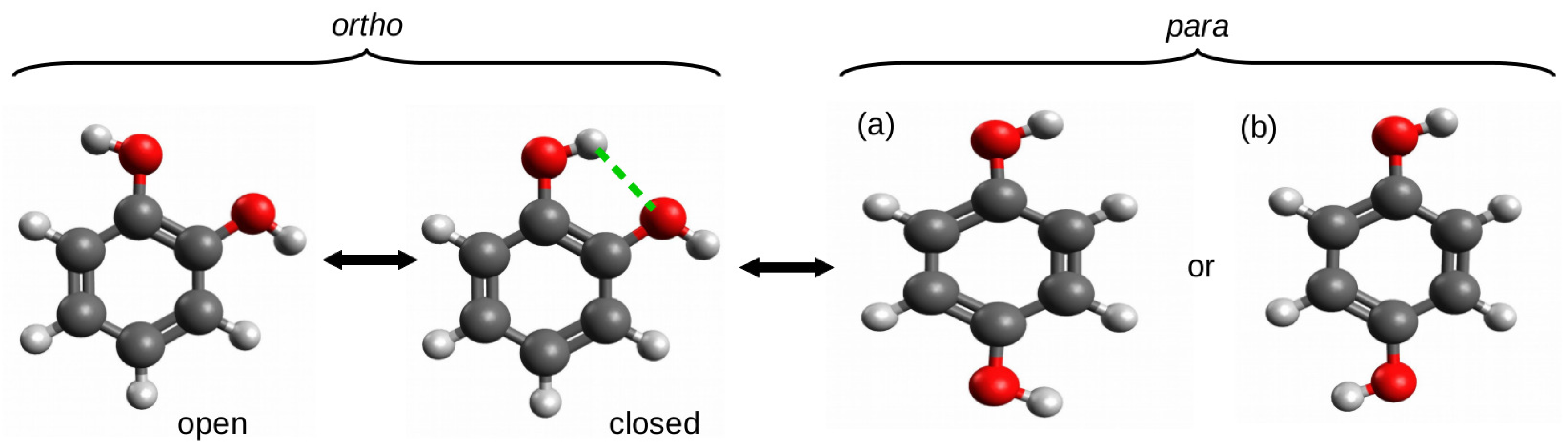
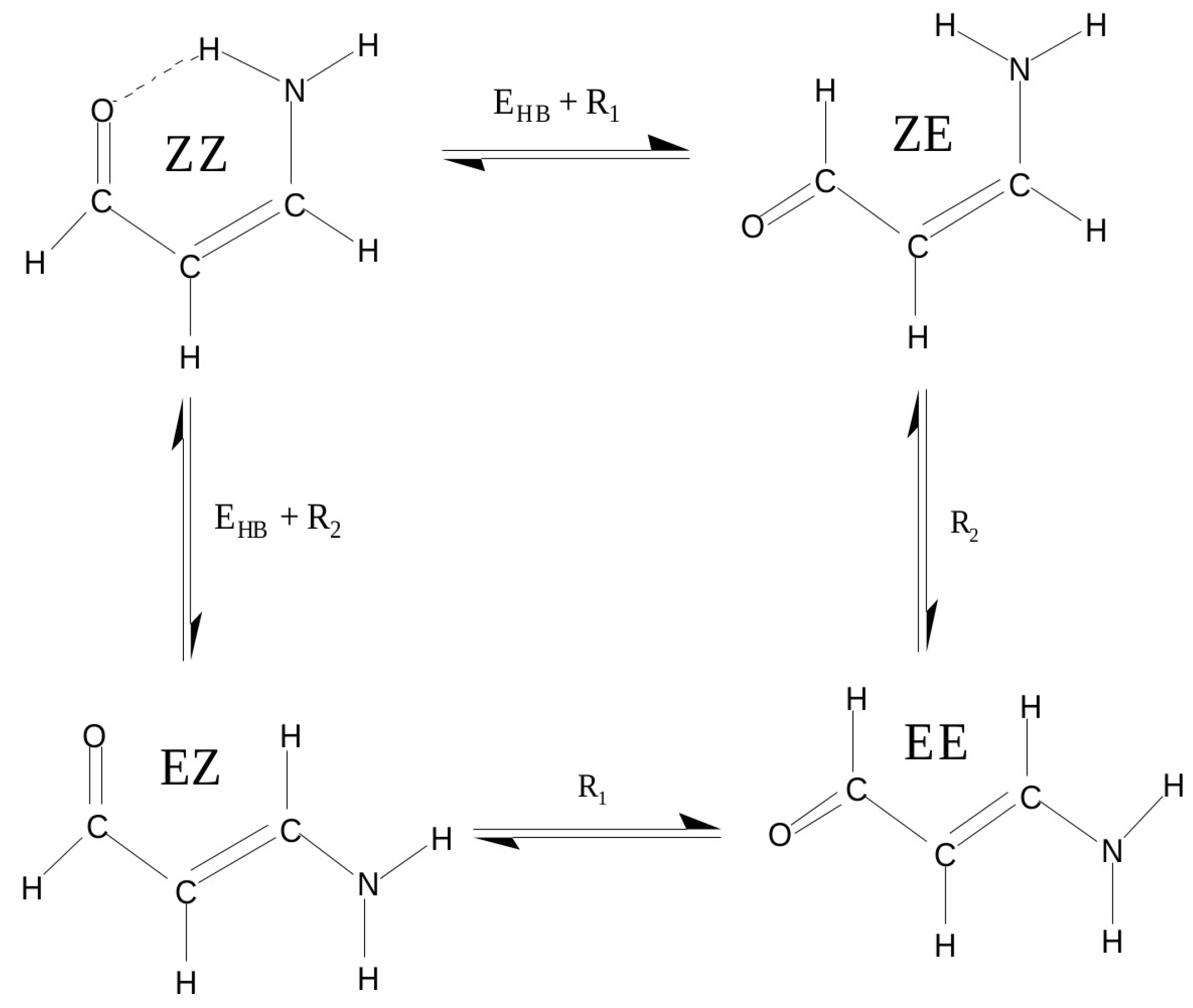
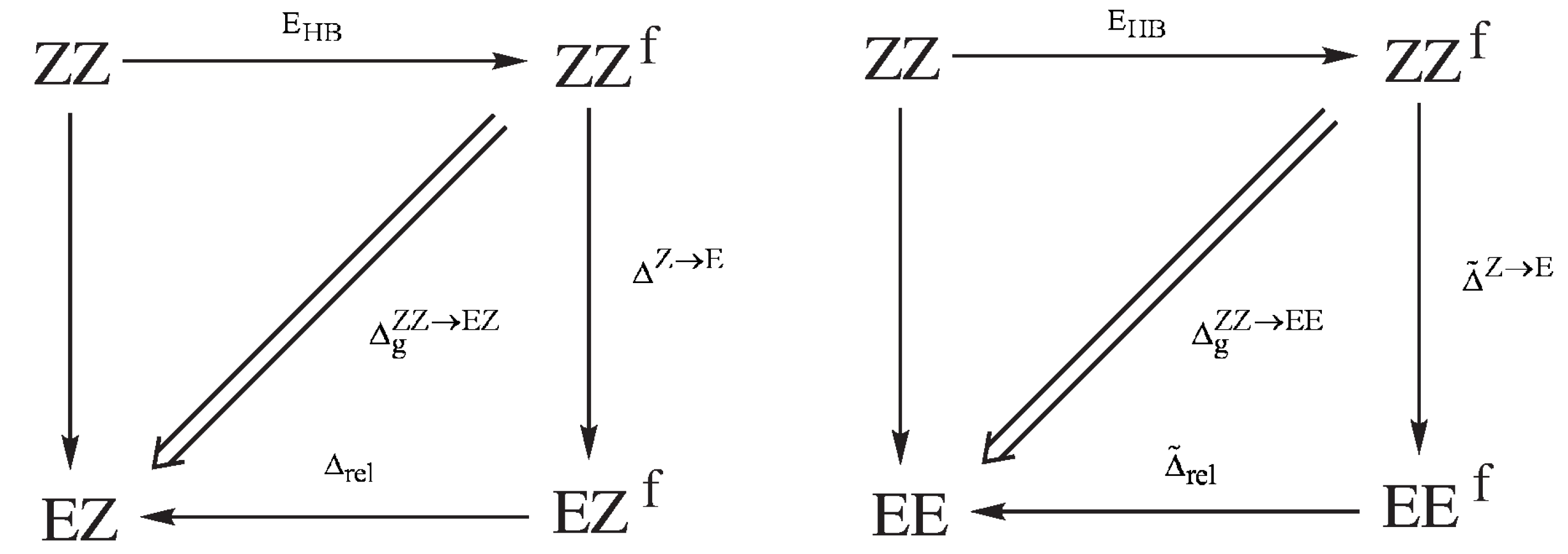
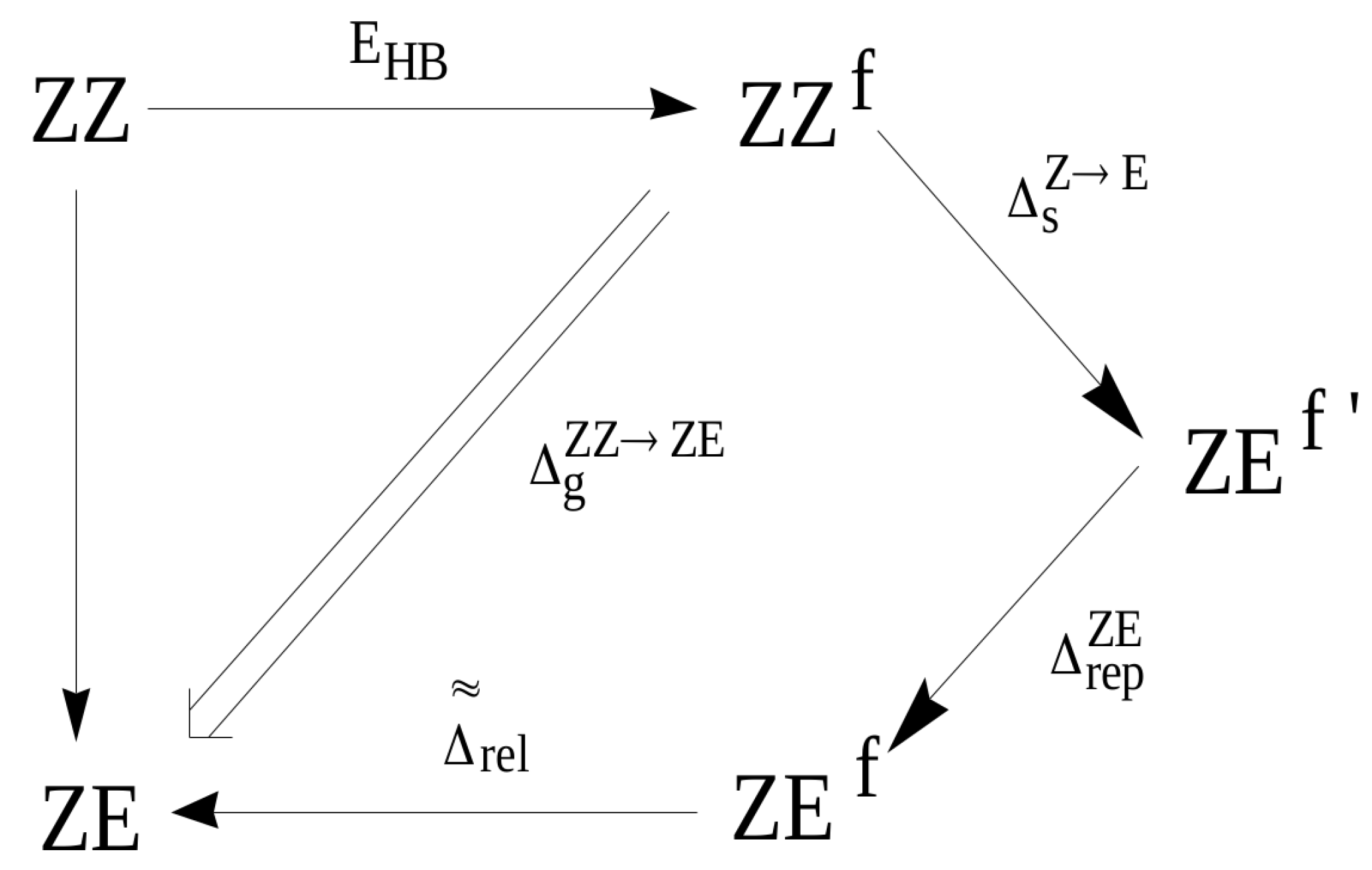
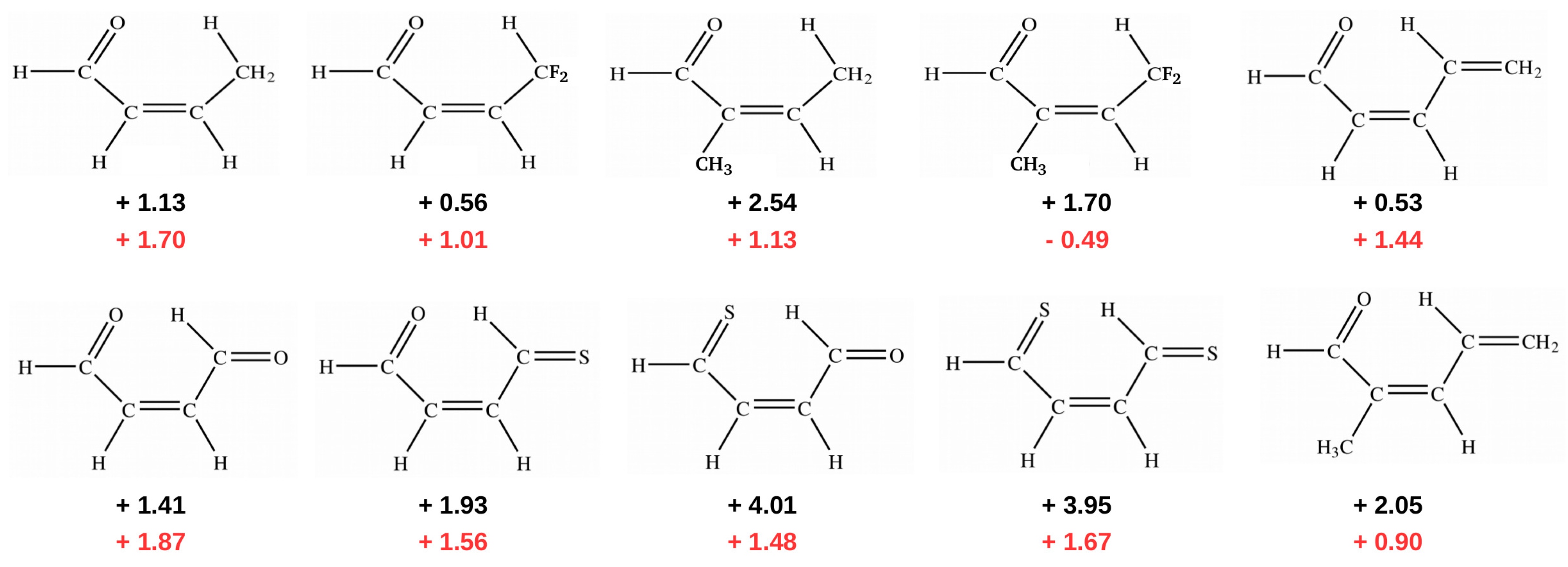
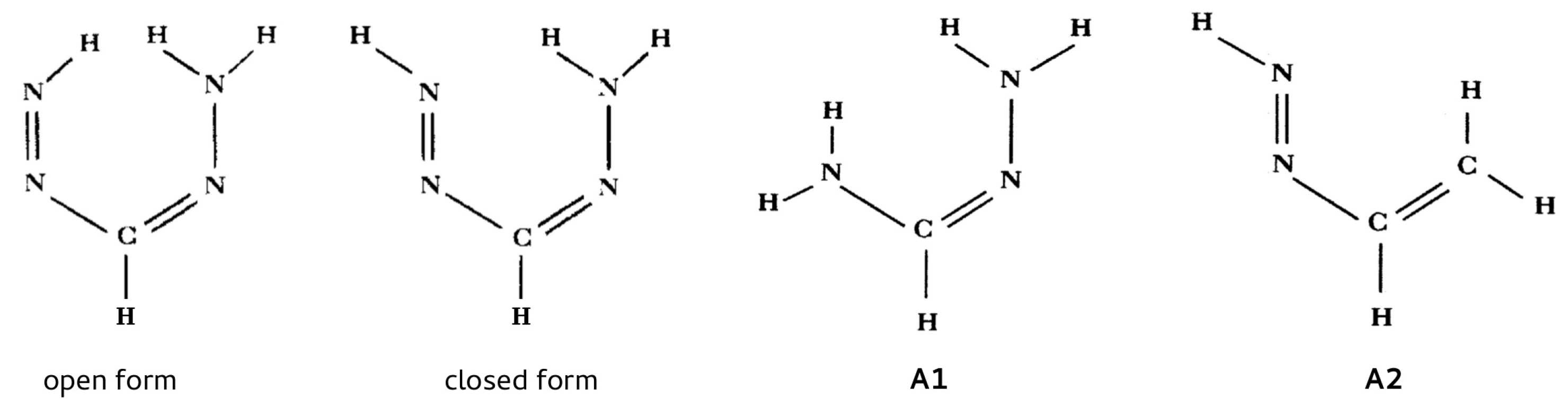
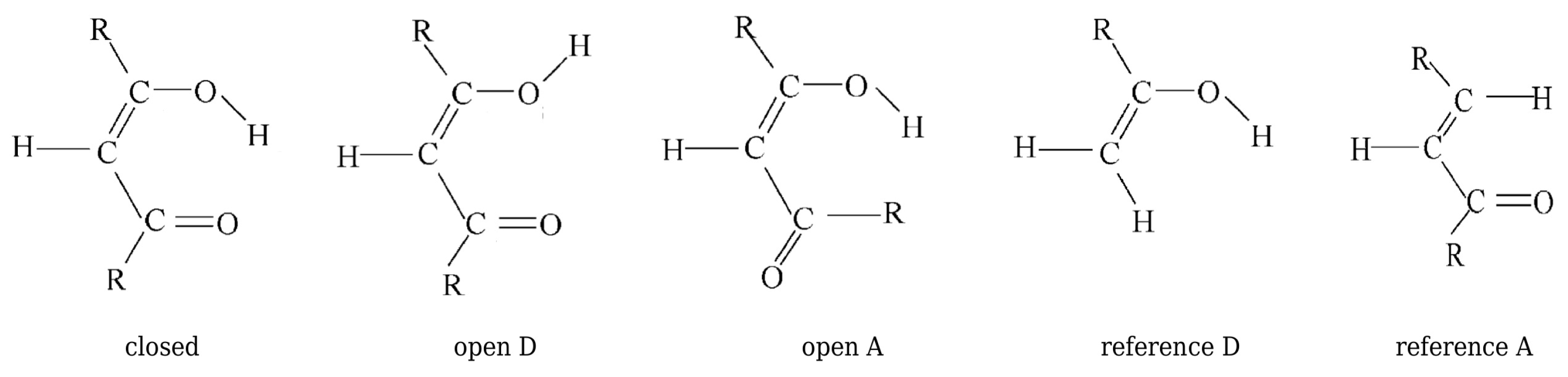
2.1.1. The Open-Closed Method (OCM)
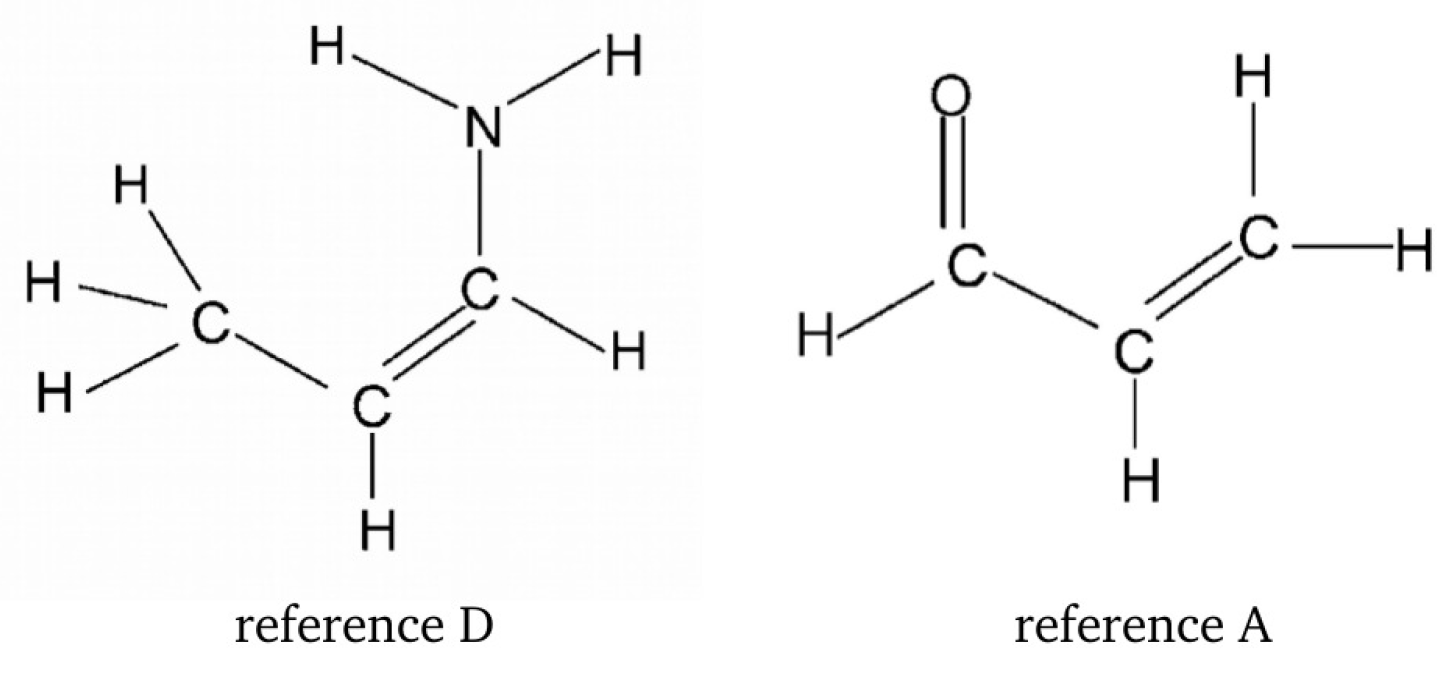
2.1.2. Ortho-Para Method (opM)
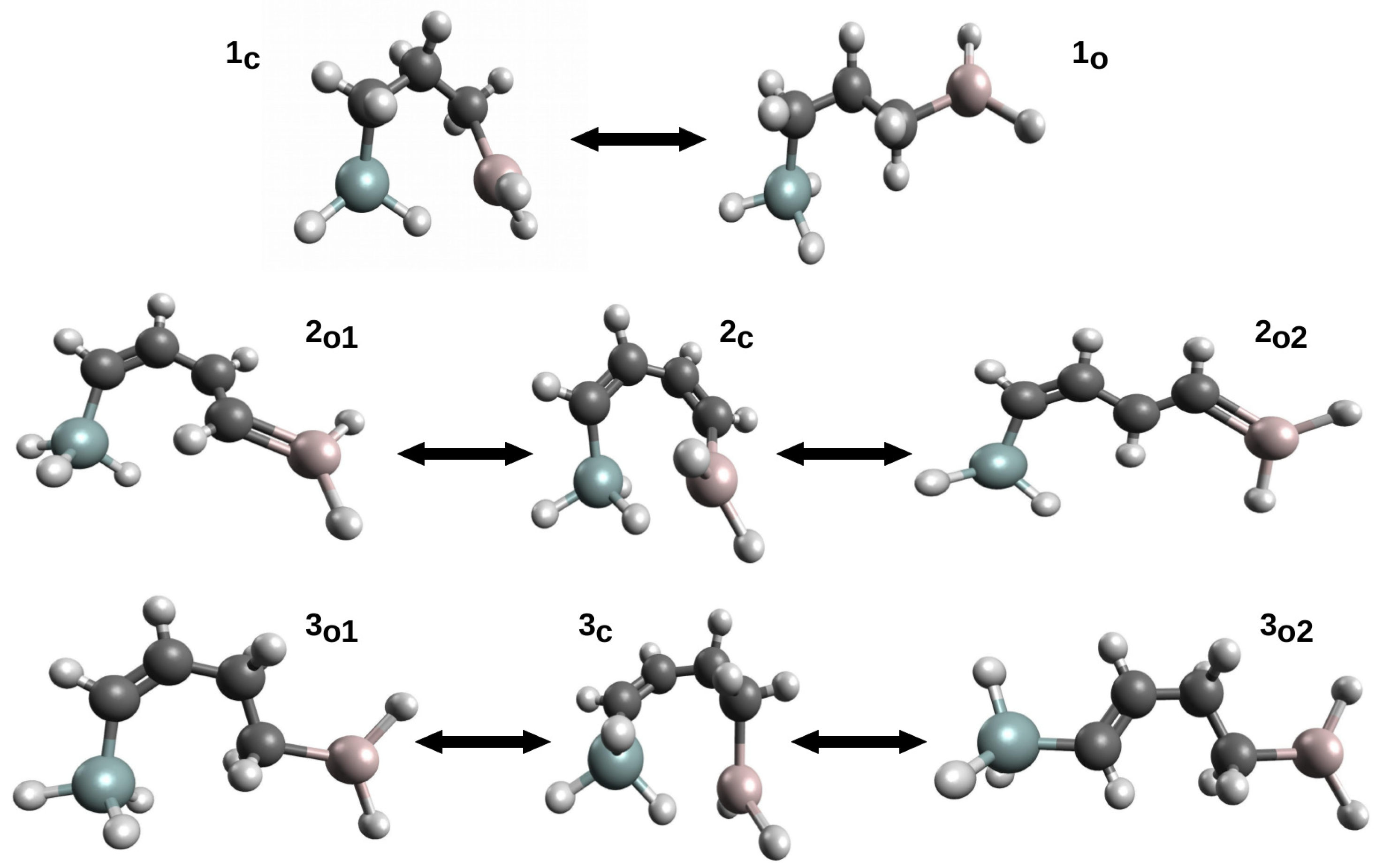
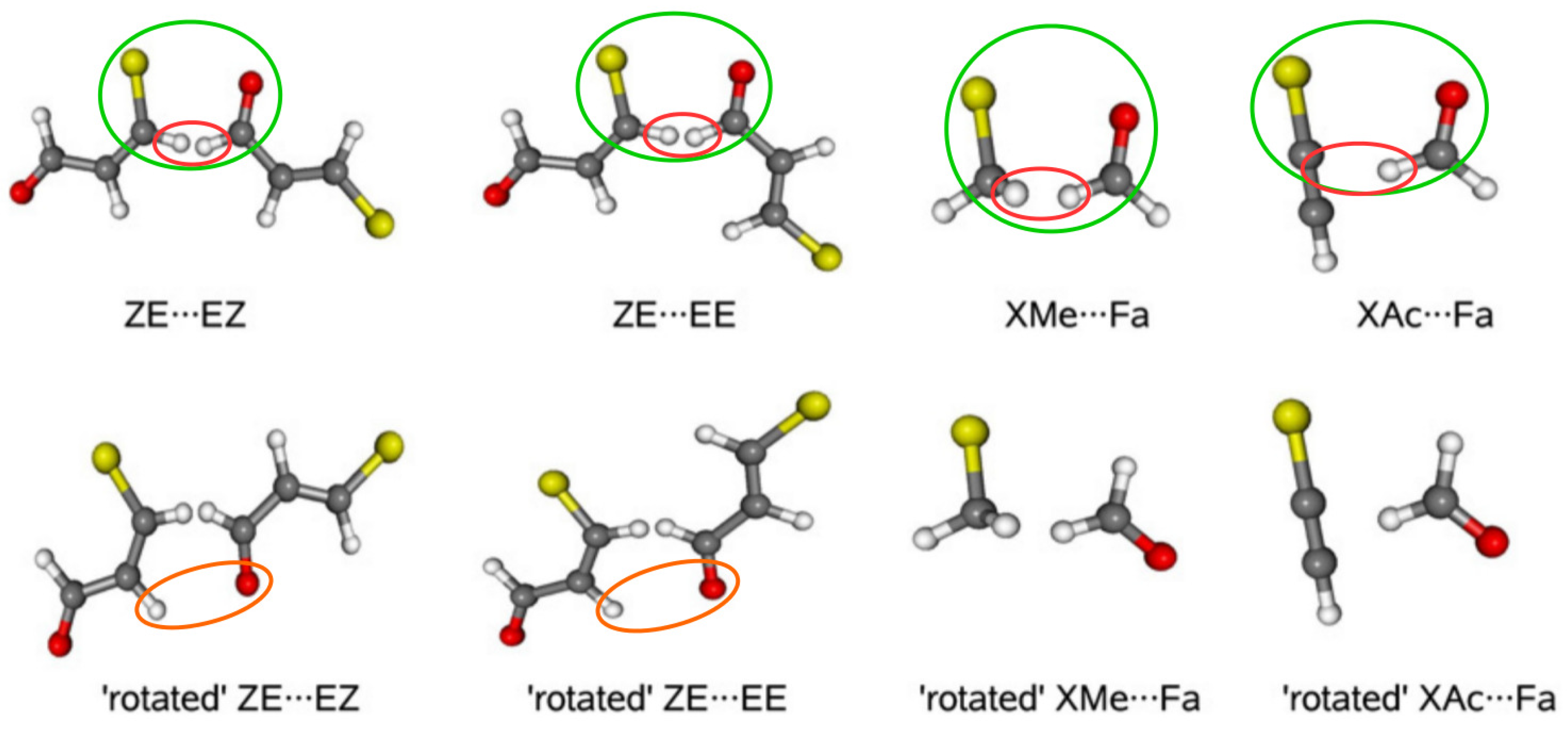
2.1.3. Related Rotamers Method (RRM)
2.1.4. Geometry-Corrected Method (GCM)
2.1.5. Geometry-Corrected Related Rotamer Method (GCRRM)
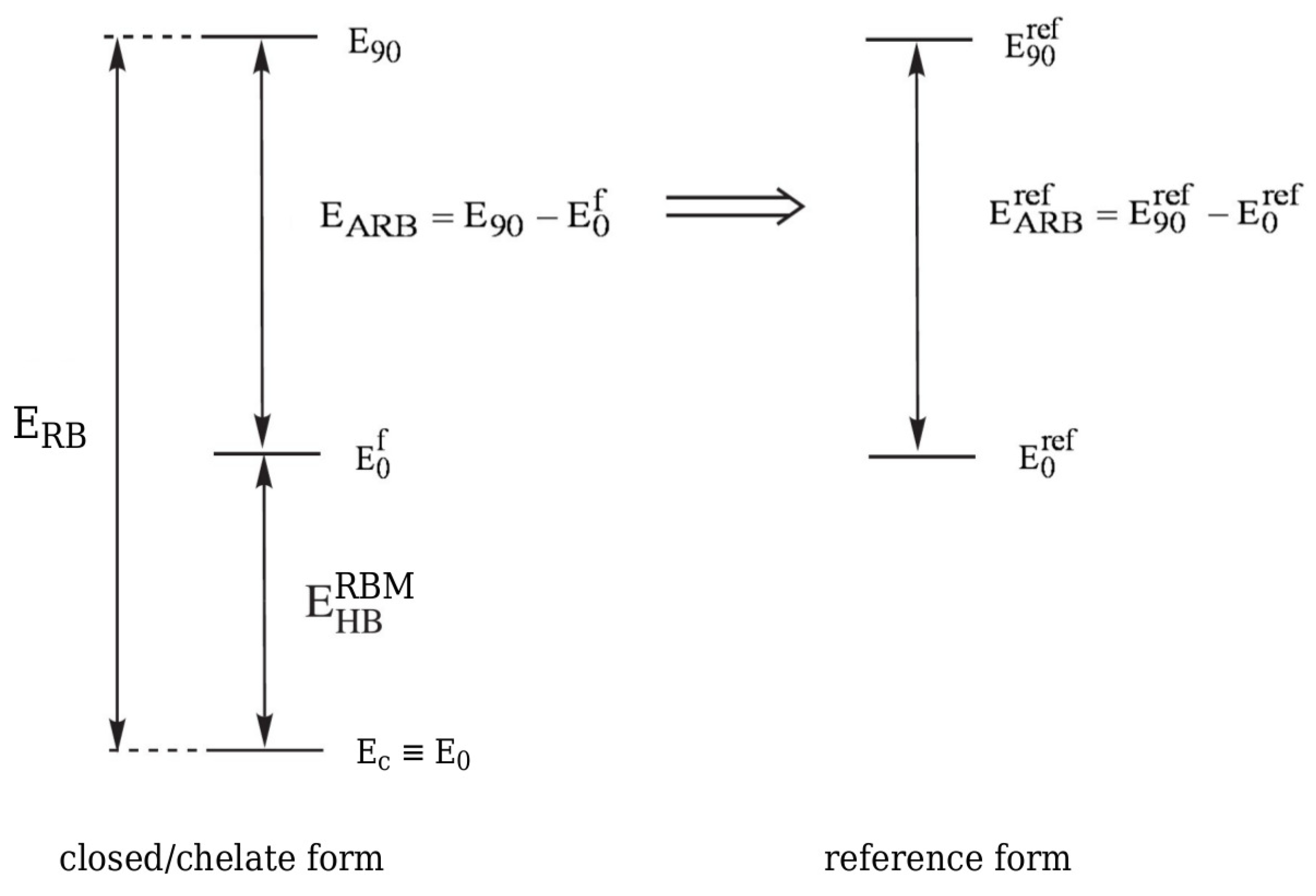
2.2. Rotation Barriers Method (RBM)
2.3. Dimer Model (DM)
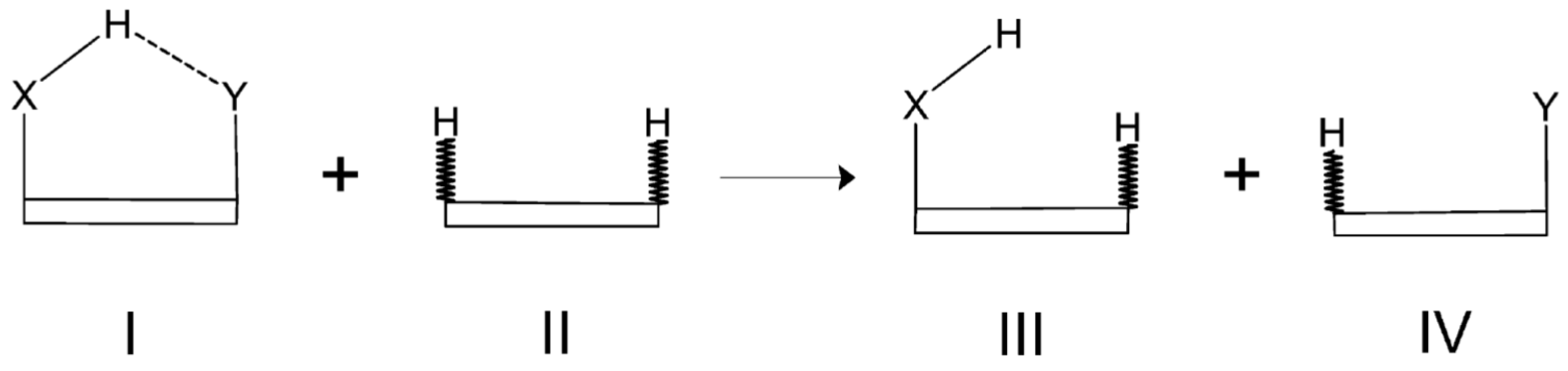
2.4. Isodesmic Reactions Method (IRM)
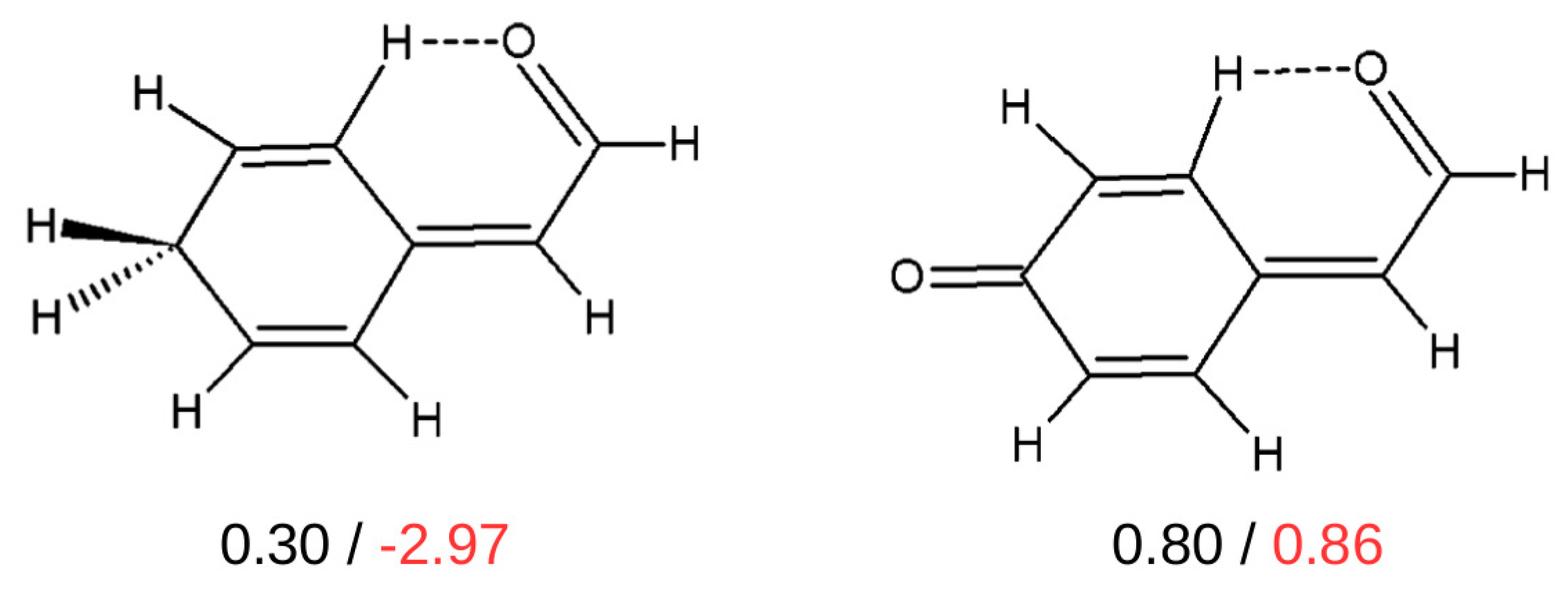
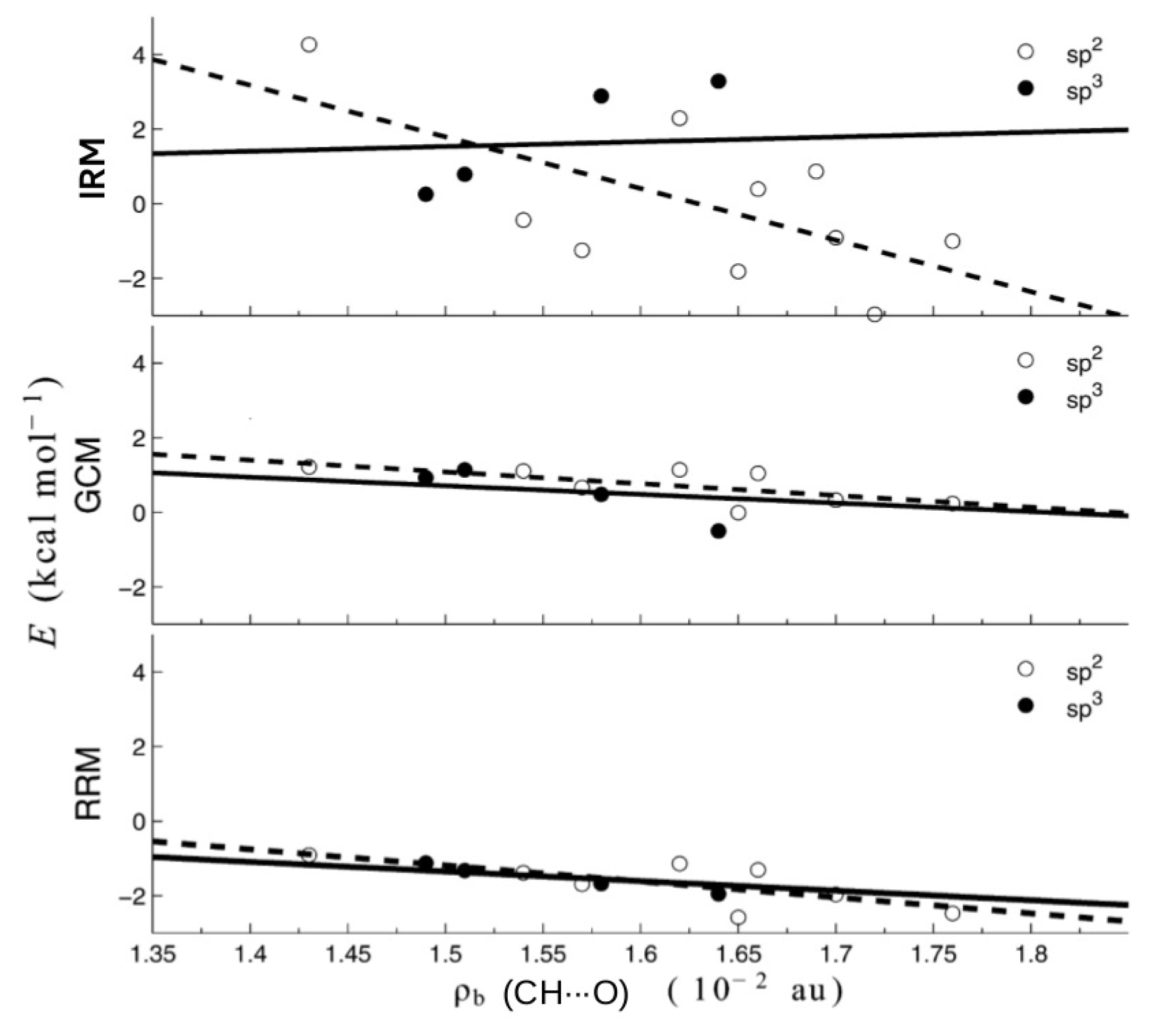
2.5. QTAIM-Based Methods
2.5.1. Espinosa’s Method (EM)
2.5.2. Interacting Quantum Atoms (IQA)
2.6. Empirically-Based Methods
2.6.1. Iogansen’s Relationship
2.6.2. Chemical Shift—Based Method
3. Summary
Funding
Conflicts of Interest
Abbreviations
| OCM | Open-Closed Method |
| opM | Ortho-Para Method |
| RRM | Related Rotamers Method |
| GCM | Geometry-Corrected Method |
| GCRRM | Geometry-Corrected Related Rotamers Method |
| RBM | Rotation Barriers Method |
| DM | Dimer Model |
| IRM | Isodesmic Reactions Method |
| EM | Espinosa’s Method |
| QTAIM | Quantum Theory of Atoms in Molecules |
| IQA | Interacting Quantum Atoms |
| BP | bond path |
| BCP | bond critical point |
References
- Pauling, L. The Nature of the Chemical Bond; Cornell University Press: New York, NY, USA, 1960. [Google Scholar]
- Pimentel, G.C.; McClellan, A.L. The Hydrogen Bond; W.H. Freeman & Co.: San Francisco, CA, USA, 1960. [Google Scholar]
- Hamilton, W.C.; Ibers, J.A. Hydrogen Bonding in Solids; W. A. Benjamin: New York, NY, USA, 1968. [Google Scholar]
- Vinogradov, S.N.; Linnell, R.H. Hydrogen Bonding; Van Nostrand-Reinhold: Princeton, NJ, USA, 1971. [Google Scholar]
- Schuster, P.; Zundel, G.; Sandorfy, C. (Eds.) The Hydrogen Bond. Recent Developments in Theory and Experiments; North Holland: Amsterdam, The Netherlands, 1976; Volumes I–III. [Google Scholar]
- Schuster, P. Intermolecular Interactions: From Diatomics to Biopolymers; Pullman, B., Ed.; Wiley-Interscience: New York, NY, USA, 1978. [Google Scholar]
- Hobza, P.; Zahradník, R. Weak Intermolecular Interactions in Chemistry and Biology; Academia: Prague, Czech Republic, 1980. [Google Scholar]
- Jeffrey, G.A.; Saenger, W. Hydrogen Bonding in Biological Structures; Springer: Berlin, Germany, 1991. [Google Scholar]
- Hadži, D. (Ed.) Theoretical Treatments of Hydrogen Bonding; Wiley: Chichester, UK, 1997. [Google Scholar]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Scheiner, S. (Ed.) Molecular Interactions. From van der Waals to Strongly Bound Complexes; Wiley: Chichester, UK, 1997. [Google Scholar]
- Scheiner, S. Hydrogen Bonding: A Theoretical Perspective; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford University Press: New York, NY, USA, 1999. [Google Scholar]
- Grabowski, S.J. Hydrogen Bonding—New Insights. Challenges and Advances in Computational Chemistry and Physics; Springer: Dordrecht, The Netherlands, 2006. [Google Scholar]
- Maréchal, Y. The Hydrogen Bond and the Water Molecule; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Gilli, G.; Gilli, P. The Nature of the Hydrogen Bond. Outline of a Comprehensive Hydrogen Bond Theory; Oxford University Press: Oxford, UK, 2009. [Google Scholar]
- Frenking, G.; Shaik, S. (Eds.) The Chemical Bond; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2014. [Google Scholar]
- Piela, L. Ideas of Quantum Chemistry; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Schuster, P. LCAO-MO-Beschreibung intramolekularer Wasserstoffbrücken. Monatshefte Chem. 1969, 100, 2084–2095. [Google Scholar] [CrossRef]
- Dietrich, S.W.; Jorgensen, E.C.; Kollman, P.A.; Rothenberg, S. A Theoretical Study of Intramolecular Hydrogen Bonding in Ortho-Substituted Phenols and Thiophenols. J. Am. Chem. Soc. 1976, 98, 8310–8324. [Google Scholar] [CrossRef]
- Emsley, J. The Composition, Structure and Hydrogen Bonding of the β-Diketones. Struct. Bond. 1984, 57, 147–191. [Google Scholar]
- Buemi, G.; Gandolfo, C. Malondialdehyde and Acetylacetone: An AM1 Study of their Molecular Structures and Keto-Enol Tautomerism. J. Chem. Soc. Faraday Trans. 1989, 85, 215–227. [Google Scholar] [CrossRef]
- Millefiori, S.; Di Bella, S. Hydrogen Bonding and Tautomerism in 3-Substituted β-Thioxoketones: An Ab Initio Molecular Orbital Study. J. Chem. Soc. Faraday Trans. 1991, 87, 1297–1302. [Google Scholar] [CrossRef]
- Craw, J.S.; Bacskay, G.B. Quantum-chemical Studies of Hydrogen Bonding involving Thioxoketones, Thienols, Thioformaldehyde and Hydrogen Sulfide with Specific Reference to the Strength of Intramolecular Hydrogen Bonds. J. Chem. Soc. Faraday Trans. 1992, 88, 2315–2321. [Google Scholar] [CrossRef]
- Luth, K.; Scheiner, S. Excited-State Energetics and Proton-Transfer Barriers in Malonaldehyde. J. Phys. Chem. 1994, 98, 3582–3587. [Google Scholar] [CrossRef]
- Lampert, H.; Mikenda, W.; Karpfen, A. Intramolecular Hydrogen Bonding in 2-Hydroxybenzoyl Compounds: Infrared Spectra and Quantum Chemical Calculations. J. Phys. Chem. 1996, 100, 7418–7425. [Google Scholar] [CrossRef]
- Scheiner, S.; Kar, T.; Čuma, M. Excited State Intramolecular Proton Transfer in Anionic Analogues of Malonaldehyde. J. Phys. Chem. A 1997, 101, 5901–5909. [Google Scholar] [CrossRef]
- Chung, G.; Kwon, O.; Kwon, Y. Theoretical Study on 1,2-Dihydroxybenzene and 2-Hydroxythiophenol: Intramolecular Hydrogen Bonding. J. Phys. Chem. A 1997, 101, 9415–9420. [Google Scholar] [CrossRef]
- Cuma, M.; Scheiner, S.; Kar, T. Effect of adjoining aromatic ring upon excited state proton transfer, o-hydroxybenzaldehyde. J. Mol. Struct. 1999, 467, 37–49. [Google Scholar] [CrossRef]
- Forés, M.; Scheiner, S. Effects of chemical substitution upon excited state proton transfer. Fluoroderivatives of salicylaldimine. Chem. Phys. 1999, 246, 65–74. [Google Scholar] [CrossRef]
- Koll, A.; Rospenk, M.; Jagodzińska, E.; Dziembowska, T. Dipole moments and conformation of Schiff bases with intramolecular hydrogen bonds. J. Mol. Struct. 2000, 552, 193–204. [Google Scholar] [CrossRef]
- Rozas, I.; Alkorta, I.; Elguero, J. Intramolecular Hydrogen Bonds in ortho-Substituted Hydroxybenzenes and in 8-Susbtituted 1-Hydroxynaphthalenes: Can a Methyl Group Be an Acceptor of Hydrogen Bonds? J. Phys. Chem. A 2001, 105, 10462–10467. [Google Scholar] [CrossRef]
- Buemi, G.; Zuccarello, F. Is the intramolecular hydrogen bond energy valuable from internal rotation barriers? J. Mol. Struct. 2002, 581, 71–85. [Google Scholar] [CrossRef]
- Kovács, A.; Szabó, A.; Hargittai, I. Structural Characteristics of Intramolecular Hydrogen Bonding in Benzene Derivatives. Acc. Chem. Res. 2002, 35, 887–894. [Google Scholar] [CrossRef]
- Korth, H.-G.; de Heer, M.I.; Mulder, P. A DFT Study on Intramolecular Hydrogen Bonding in 2-Substituted Phenols: Conformations, Enthalpies, and Correlation with Solute Parameters. J. Phys. Chem. A 2002, 106, 8779–8789. [Google Scholar] [CrossRef]
- Grabowski, S.J. π-Electron delocalisation for intramolecular resonance assisted hydrogen bonds. J. Phys. Org. Chem. 2003, 16, 797–802. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen bonding strength–measures based on geometric and topological parameters. J. Phys. Org. Chem. 2004, 17, 18–31. [Google Scholar] [CrossRef]
- Estácio, S.G.; do Couto, P.C.; Costa Cabral, B.J.; Minas da Piedade, M.E.; Martinho Simões, J.A. Energetics of Intramolecular Hydrogen Bonding in Di-substituted Benzenes by the ortho–para Method. J. Phys. Chem. A 2004, 108, 10834–10843. [Google Scholar] [CrossRef]
- Jabłoński, M.; Kaczmarek, A.; Sadlej, A.J. Estimates of the Energy of Intramolecular Hydrogen Bonds. J. Phys. Chem. A 2006, 110, 10890–10898. [Google Scholar] [CrossRef] [PubMed]
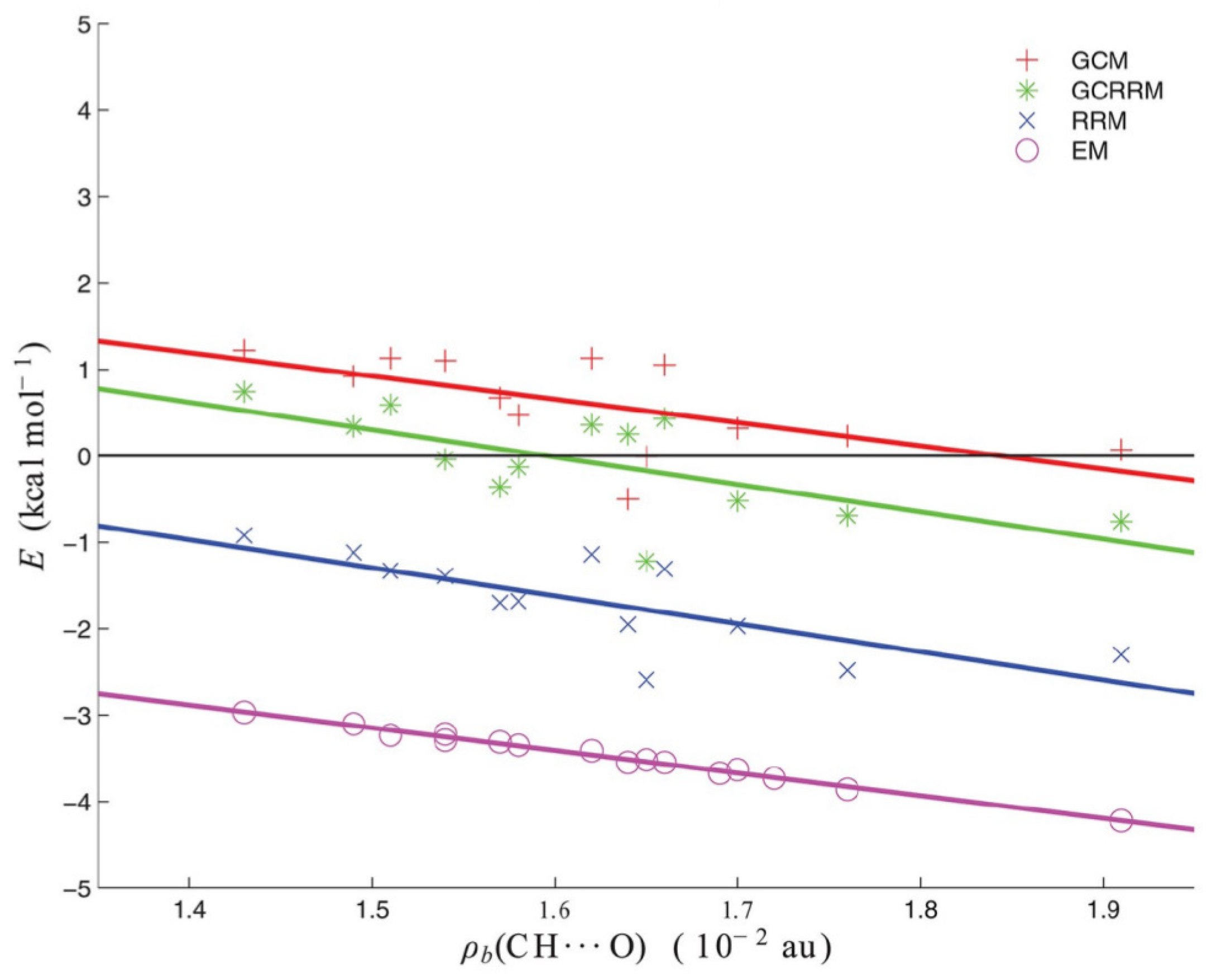
- Jabłoński, M.; Sadlej, A.J. Blue-Shifting Intramolecular C–H⋯O Interactions. J. Phys. Chem. A 2007, 111, 3423–3431. [Google Scholar] [CrossRef] [PubMed]
- Jabłoński, M. Blue-shifting intramolecular C–H⋯O(S) contacts in sterically strained systems. J. Mol. Struct. 2007, 820, 118–127. [Google Scholar]
- Jabłoński, M. Full vs. constrain geometry optimization in the open–closed method in estimating the energy of intramolecular charge-inverted hydrogen bonds. Chem. Phys. 2010, 376, 76–83. [Google Scholar] [CrossRef]
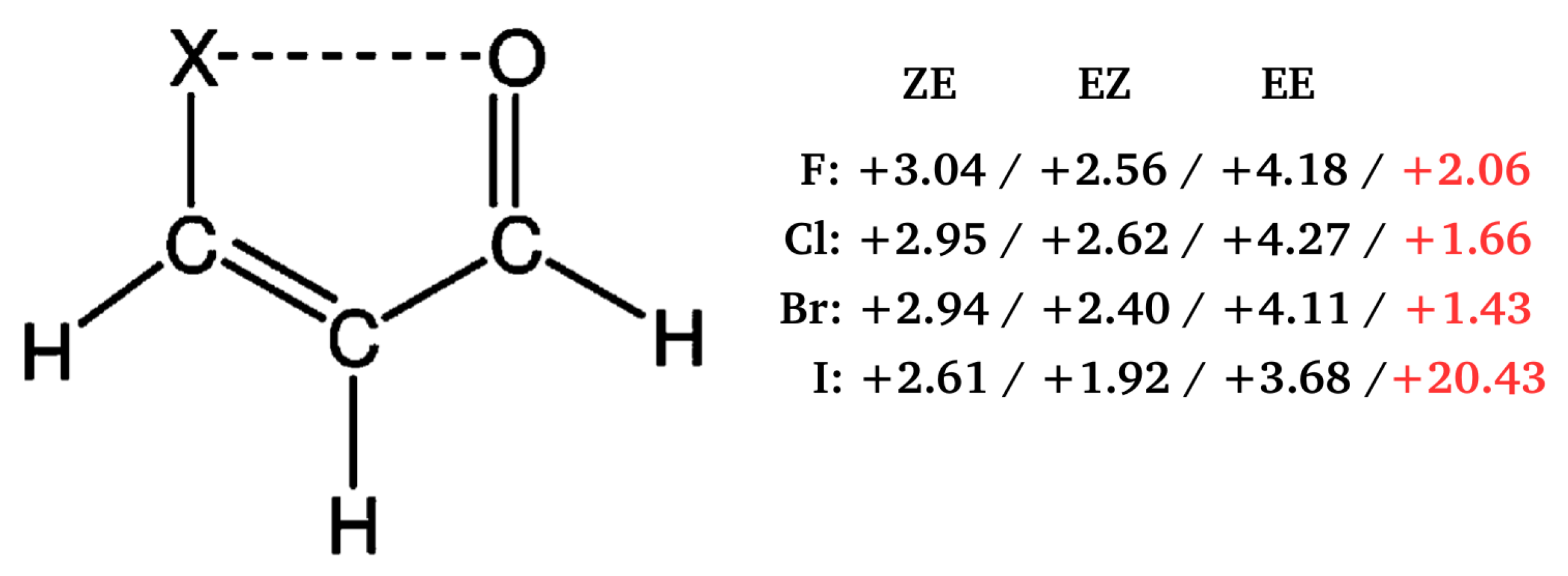
- Jabłoński, M. Energetic and Geometrical Evidence of Nonbonding Character of Some Intramolecular Halogen⋯Oxygen and Other Y⋯Y Interactions. J. Phys. Chem. A 2012, 116, 3753–3764. [Google Scholar] [CrossRef]
- Jabłoński, M.; Monaco, G. Different Zeroes of Interaction Energies As the Cause of Opposite Results on the Stabilizing Nature of C–H⋯O Intramolecular Interactions. J. Chem. Inf. Model. 2013, 53, 1661–1675. [Google Scholar] [CrossRef]
- Jabłoński, M. Strength of Si–H⋯B charge-inverted hydrogen bonds in 1-silacyclopent-2-enes and 1-silacyclohex-2-enes. Struct. Chem. 2017, 28, 1697–1706. [Google Scholar] [CrossRef] [Green Version]
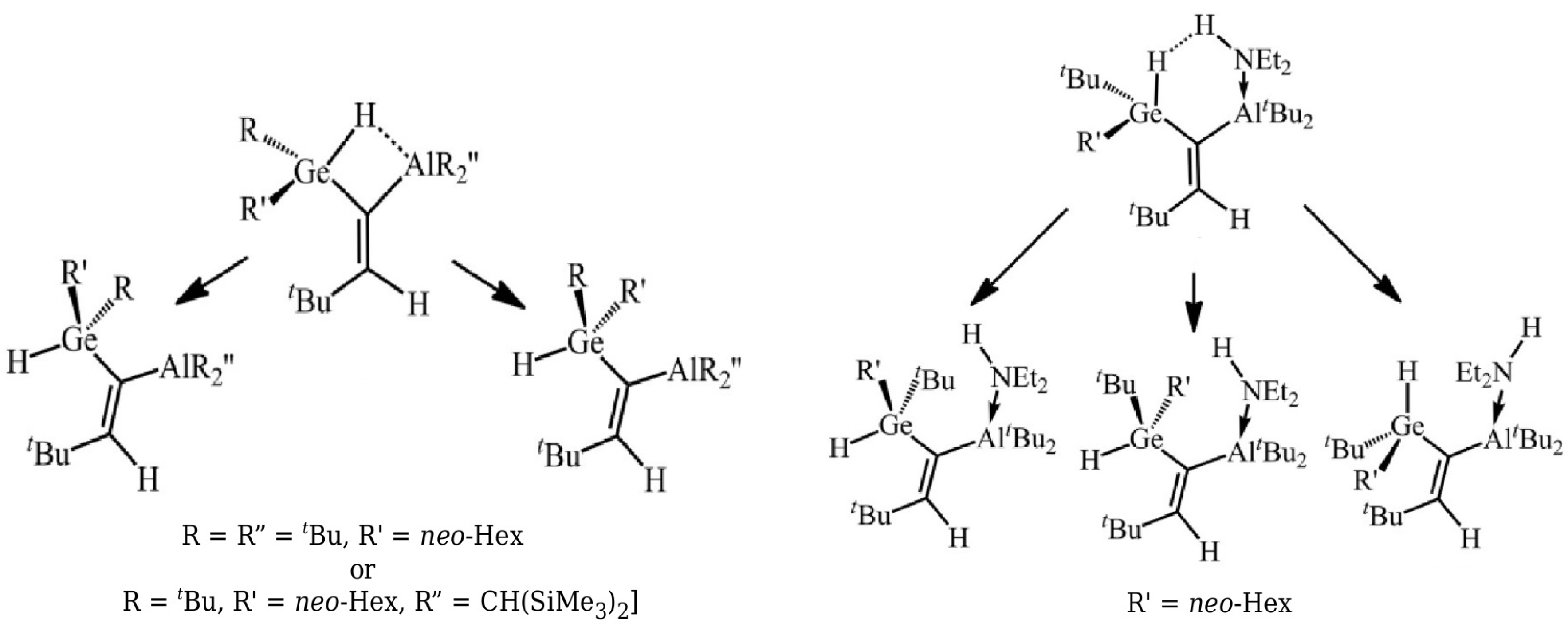
- Honacker, C.; Kappelt, B.; Jabłoński, M.; Hepp, A.; Layh, M.; Rogel, F.; Uhl, W. Aluminium Functionalized Germanes: Intramolecular Activation of Ge-H Bonds, Formation of a Dihydrogen Bond and Facile Hydrogermylation of Unsaturated Substrates. Eur. J. Inorg. Chem. 2019, 3287–3300. [Google Scholar] [CrossRef]
- Jabłoński, M. Enaminoketony: Teoretyczne Badania Dynamiki Wewnątrzmolekularnej w Układzie Modelowym 3-Aminoakroleiny. Master’s Thesis, Nicolaus Copernicus University, Toruń, Poland, 2000. [Google Scholar]
- Buemi, G. AM1 Study of Tautomerism and Intramolecular Hydrogen Bonding in Thiomalondialdehyde and Thioacetylacetone. J. Chem. Soc. Faraday Trans. 1990, 86, 2813–2818. [Google Scholar] [CrossRef]
- Buemi, G. Intramolecular Hydrogen Bonding of Malondialdehyde and its Monothio and Dithio Analogues Studied by the PM3 Method. J. Mol. Struct. 1990, 208, 253–260. [Google Scholar] [CrossRef]
- Gonzales, L.; Mó, O.; Yáñez, M. High-Level ab Initio Calculations on the Intramolecular Hydrogen Bond in Thiomalonaldehyde. J. Phys. Chem. A 1997, 101, 9710–9719. [Google Scholar] [CrossRef]
- Jabłoński, M. Binding of X-H to the lone-pair vacancy: Charge-inverted hydrogen bond. Chem. Phys. Lett. 2009, 477, 374–376. [Google Scholar] [CrossRef]
- Jabłoński, M. Ten years of charge-inverted hydrogen bonds. Struct. Chem. 2020, 31, 61–80. [Google Scholar] [CrossRef]
- Wrackmeyer, B.; Tok, O.L.; Milius, W.; Khan, A.; Badshah, A. 1-Silacyclopent-2-enes and 1-silacyclohex-2-enes bearing functionally substituted silyl groups in 2-positions. Novel electron-deficient Si–H–B bridges. Appl. Organomet. Chem. 2006, 20, 99–105. [Google Scholar] [CrossRef]
- Pinto, S.S.; Diogo, H.P.; Guedes, R.C.; Costa Cabral, B.J.; Minas da Piedade, M.E.; Martinho Simões, J.A. Energetics of Hydroxybenzoic Acids and of the Corresponding Carboxyphenoxyl Radicals. Intramolecular Hydrogen Bonding in 2-Hydroxybenzoic Acid. J. Phys. Chem. A 2005, 109, 9700–9708. [Google Scholar] [CrossRef]
- Roy, D.; Sunoj, R.B. Quantification of Intramolecular Nonbonding Interactions in Organochalcogens. J. Phys. Chem. A 2006, 110, 5942–5947. [Google Scholar] [CrossRef]
- Hammett, L.P. The Effect of Structure upon the Reactions of Organic Compounds. Benzene Derivatives. J. Am. Chem. Soc. 1937, 59, 96–103. [Google Scholar] [CrossRef]
- Hammett, L.P. Physical Organic Chemistry; McGraw-Hill: New York, NY, USA, 1940. [Google Scholar]
- Hansch, C.; Leo, A.; Taft, R.W. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Stępień, B.T. Sigma- and Pi-Electron Delocalization: Focus on Substituent Effects. Chem. Rev. 2005, 105, 3482–3512. [Google Scholar] [CrossRef]
- Lipkowski, P.; Koll, A.; Karpfen, A.; Wolschann, P. An approach to estimate the energy of the intramolecular hydrogen bond. Chem. Phys. Lett. 2002, 360, 256–263. [Google Scholar] [CrossRef]
- Nowroozi, A.; Raissi, H.; Farzad, F. The presentation of an approach for estimating the intramolecular hydrogen bond strength in conformational study of β-Aminoacrolein. J. Mol. Struct. 2005, 730, 161–169. [Google Scholar] [CrossRef]
- Nowroozi, A.; Raissi, H.; Hajiabadi, H.; Mohammadzadeh Jahani, P. Reinvestigation of Intramolecular Hydrogen Bond in Malonaldehyde Derivatives: An Ab initio, AIM and NBO Study. Int. J. Quant. Chem. 2011, 111, 3040–3047. [Google Scholar] [CrossRef]
- Nowroozi, A.; Masumian, E. Comparative study of NH⋯O and NH⋯S intramolecular hydrogen bonds in β-aminoacrolein, β-thioaminoacrolein and their halogenated derivatives by some usual methods. Struct. Chem. 2017, 28, 587–596. [Google Scholar] [CrossRef]
- Tayyari, S.F.; Fazli, M.; Milani-nejad, F. Molecular conformation and intramolecular hydrogen bonding in 4-amino-3-penten-2-one. J. Mol. Struct. 2001, 541, 11–15. [Google Scholar] [CrossRef]
- Buemi, G.; Zuccarello, F.; Venuvanalingam, P.; Ramalingam, M. Ab initio study of tautomerism and hydrogen bonding of β-carbonylamine in the gas phase and in water solution. Theor. Chem. Acc. 2000, 104, 226–234. [Google Scholar] [CrossRef]
- Buemi, G.; Zuccarello, F. Molecular conformations and intramolecular hydrogen bonding of 3-formylmalondialdehyde and 3-formylacetylacetone. An ab initio study. Electr. J. Theor. Chem. 1997, 2, 118–129. [Google Scholar] [CrossRef]
- Buemi, G.; Zuccarello, F.; Venuvanalingam, P.; Ramalingam, M.; Ammal, S.S.C. Ab initio study of formazan and 3-nitroformazan. J. Chem. Soc. Faraday Trans. 1998, 94, 3313–3319. [Google Scholar] [CrossRef] [Green Version]
- Buemi, G. Ab initio DFT study of the hydrogen bridges in hexafluoro-acetylacetone, trifluoro-acetylacetone and some 3-substituted derivatives. J. Mol. Struct. 2000, 499, 21–34. [Google Scholar] [CrossRef]
- Buemi, G. Ab initio study of 2,4-dihalosubstituted malonaldehyde and 2-halo-phenols in gas phase and solution. Chem. Phys. 2002, 277, 241–256. [Google Scholar] [CrossRef]
- Buemi, G. Ab initio DFT study of the rotation barriers and competitive hydrogen bond energies (in gas phase and water solution) of 2-nitroresorcinol, 4,6-dinitroresorcinol and 2-nitrophenol in their neutral and deprotonated conformations. Chem. Phys. 2002, 282, 181–195. [Google Scholar] [CrossRef]
- Buemi, G.; Zuccarello, F. DFT study of the intramolecular hydrogen bonds in the amino and nitro-derivatives of malonaldehyde. Chem. Phys. 2004, 306, 115–129. [Google Scholar] [CrossRef]
- Ramalingam, M.; Venuvanalingam, P.; Swaminathan, J.; Buemi, G. Ab initio and DFT studies on conformations, hydrogen bonding and electronic structures of glyoxalmonoxime and its methyl derivatives. J. Mol. Struct. 2004, 712, 175–185. [Google Scholar] [CrossRef]
- Palusiak, M.; Krygowski, T.M. Unusual electron density topology and intramolecular steric π–π interaction in 1,3,5,7-cyclooctatetraene. Chem. Phys. Lett. 2009, 481, 34–38. [Google Scholar] [CrossRef]
- Jabłoński, M.; Palusiak, M. The halogen⋯oxygen interaction in 3-halogenopropenal revisited—The dimer model vs. QTAIM indications. Chem. Phys. 2013, 415, 207–213. [Google Scholar] [CrossRef]
- Palusiak, M.; Grabowski, S.J. Do intramolecular halogen bonds exist? Ab initio calculations and crystal structures’ evidences. Struct. Chem. 2007, 18, 859–865. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Radom, L.; Pople, J.A. Molecular Orbital Theory of the Electronic Structure of Organic Compounds. V. Molecular Theory of Bond Separation. J. Am. Chem. Soc. 1970, 92, 4796–4801. [Google Scholar] [CrossRef]
- Radom, L.; Hehre, W.J.; Pople, J.A. Molecular Orbital Theory of the Electronic Structure of Organic Compounds. VII. A Systematic Study of Energies, Conformations, and Bond Interactions. J. Am. Chem. Soc. 1971, 93, 289–300. [Google Scholar] [CrossRef]
- Radom, L. Ab Initio Molecular Orbital Calculations on Anions. Determination of Gas Phase Acidities. J. Chem. Soc. Chem. Commun. 1974, 403–404. [Google Scholar] [CrossRef]
- George, P.; Trachtman, M.; Bock, C.W.; Brett, A.M. An Alternative Approach to the Problem of Assessing Stabilization Energies in Cyclic Conjugated Hydrocarbons. Theor. Chim. Acta 1975, 38, 121–129. [Google Scholar] [CrossRef]
- George, P.; Trachtman, M.; Bock, C.W.; Brett, A.M. Homodesmotic Reactions for the Assessment of Stabilization Energies in Benzenoid and Other Conjugated Cyclic Hydrocarbons. J. Chem. Soc. Perkin Trans. 1976, 2, 1222–1227. [Google Scholar] [CrossRef]
- George, P.; Trachtman, M.; Bock, C.W.; Brett, A.M. An Alternative Approach to the Problem of Assessing Destabilization Energies (Strain Energies) in Cyclic Hydrocarbons. Tetrahedron 1976, 32, 317–323. [Google Scholar] [CrossRef]
- George, P.; Trachtman, M.; Brett, A.M.; Bock, C.W. Comparison of Various lsodesmic and Homodesmotic Reaction Heats with Values derived from Published Ab initio Molecular Orbital Calculations. J. Chem. Soc. Perkin Trans. 1977, 2, 1036–1047. [Google Scholar] [CrossRef]
- Hehre, W.J.; Radom, L.; von Ragué Schleyer, P.; Pople, J.A. Ab Initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Varnali, T.; Hargittai, I. Geometrical consequences of resonance-assisted hydrogen bonding in 2-nitrovinyl alcohol and indication of a slight attractive O⋯H interaction in 2-nitroethanol. An ab initio molecular orbital investigation. J. Mol. Struct. 1996, 388, 315–319. [Google Scholar]
- Wiberg, K.B.; Ochterski, J.W. Comparison of Different Ab Initio Theoretical Models for Calculating Isodesmic Reaction Energies for Small Ring and Related Compounds. J. Comput. Chem. 1997, 18, 108–114. [Google Scholar] [CrossRef]
- Howard, S.T. Relationship between Basicity, Strain, and Intramolecular Hydrogen-Bond Energy in Proton Sponges. J. Am. Chem. Soc. 2000, 122, 8238–8244. [Google Scholar] [CrossRef]
- Sanz, P.; Mó, O.; Yáñez, M. Characterization of intramolecular hydrogen bonds and competitive chalcogen–chalcogen interactions on the basis of the topology of the charge density. Phys. Chem. Chem. Phys. 2003, 5, 2942–2947. [Google Scholar] [CrossRef]
- Cyrański, M.K. Energetic Aspects of Cyclic Pi-Electron Delocalization: Evaluation of the Methods of Estimating Aromatic Stabilization Energies. Chem. Rev. 2005, 105, 3773–3811. [Google Scholar] [CrossRef]
- Raab, V.; Gauchenova, E.; Merkoulov, A.; Harms, K.; Sundermeyer, J.; Kovačević, B.; Maksić, Z.B. 1,8-Bis(hexamethyltriaminophosphazenyl)naphthalene, HMPN: A Superbasic Bisphosphazene “Proton Sponge”. J. Am. Chem. Soc. 2005, 127, 15738–15743. [Google Scholar] [CrossRef]
- Emel’yanenko, V.N.; Strutynska, A.; Verevkin, S.P. Enthalpies of Formation and Strain of Chlorobenzoic Acids from Thermochemical Measurements and from ab Initio Calculations. J. Phys. Chem. A 2005, 109, 4375–4380. [Google Scholar] [CrossRef]
- Deshmukh, M.M.; Suresh, C.H.; Gadre, S.R. Intramolecular Hydrogen Bond Energy in Polyhydroxy Systems: A Critical Comparison of Molecular Tailoring and Isodesmic Approaches. J. Phys. Chem. A 2007, 111, 6472–6480. [Google Scholar] [CrossRef]
- Wheeler, S.E.; Houk, K.N.; Schleyer, P.; Allen, W.D. A Hierarchy of Homodesmotic Reactions for Thermochemistry. J. Am. Chem. Soc. 2009, 131, 2547–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ukomska, M.; Rybarczyk-Pirek, A.J.; Jabłoński, M.; Palusiak, M. The nature of NO-bonding in N-oxide group. Phys. Chem. Chem. Phys. 2015, 17, 16375–16387. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Popelier, P.L.A. Atoms in Molecules. An Introduction; Longman: Singapore, 2000. [Google Scholar]
- Matta, C.F.; Boyd, R.J. The Quantum Theory of Atoms in Molecules; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Bader, R.F.W. A Quantum Theory of Molecular Structure and Its Appllcatlons. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Bader, R.F.W. A Bond Path: A Universal Indicator of Bonded Interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Cioslowski, J.; Mixon, S.T.; Edwards, W.D. Weak Bonds in the Topological Theory of Atoms in Molecules. J. Am. Chem. Soc. 1991, 113, 1083–1085. [Google Scholar] [CrossRef]
- Cioslowski, J.; Mixon, S.T. Topological Properties of Electron Density in Search of Steric Interactions in Molecules: Electronic Structure Calculations on Ortho-Substituted Biphenyls. J. Am. Chem. Soc. 1992, 114, 4382–4387. [Google Scholar] [CrossRef]
- Haaland, A.; Shorokhov, D.J.; Tverdova, N.V. Topological Analysis of Electron Densities: Is the Presence of an Atomic Interaction Line in an Equilibrium Geometry a Sufficient Condition for the Existence of a Chemical Bond? Chem. Eur. J. 2004, 10, 4416–4421. [Google Scholar] [CrossRef]
- Cerpa, E.; Krapp, A.; Vela, A.; Merino, G. The Implications of Symmetry of the External Potential on Bond Paths. Chem. Eur. J. 2008, 14, 10232–10234. [Google Scholar] [CrossRef]
- Poater, J.; Solà, M.; Bickelhaupt, F.M. Hydrogen–Hydrogen Bonding in Planar Biphenyl, Predicted by Atoms-in-Molecules Theory, Does Not Exist. Chem. Eur. J. 2006, 12, 2889–2895. [Google Scholar] [CrossRef]
- Poater, J.; Solà, M.; Bickelhaupt, F.M. A Model of the Chemical Bond Must Be Rooted in Quantum Mechanics, Provide Insight, and Possess Predictive Power. Chem. Eur. J. 2006, 12, 2902–2905. [Google Scholar] [CrossRef]
- Dem’yanov, P.; Polestshuk, P. A Bond Path and an Attractive Ehrenfest Force Do Not Necessarily Indicate Bonding Interactions: Case Study on M2X2 (M = Li, Na, K; X = H, OH, F, Cl). Chem. Eur. J. 2012, 18, 4982–4993. [Google Scholar] [CrossRef] [PubMed]
- Demy’anov, P.I.; Polestshuk, P.M. Forced Bonding and QTAIM Deficiencies: A Case Study of the Nature of Interactions in He@Adamantane and the Origin of the High Metastability. Chem. Eur. J. 2013, 19, 10945–10957. [Google Scholar] [CrossRef] [PubMed]
- Tognetti, V.; Joubert, L. On the physical role of exchange in the formation of an intramolecular bond path between two electronegative atoms. J. Chem. Phys. 2013, 138, 024102. [Google Scholar] [CrossRef] [PubMed]
- Tognetti, V.; Joubert, L. On critical points and exchange-related properties of intramolecular bonds between two electronegative atoms. Chem. Phys. Lett. 2013, 579, 122–126. [Google Scholar] [CrossRef]
- Shahbazian, S. Why Bond Critical Points Are Not “Bond” Critical Points. Chem. Eur. J. 2018, 24, 5401–5405. [Google Scholar] [CrossRef]
- Wick, C.R.; Clark, T. On bond-critical points in QTAIM and weak interactions. J. Mol. Model. 2018, 24, 142. [Google Scholar] [CrossRef]
- Jabłoński, M. Hydride-Triel Bonds. J. Comput. Chem. 2018, 39, 1177–1191. [Google Scholar] [CrossRef]
- Jabłoński, M. Bond Paths Between Distant Atoms Do Not Necessarily Indicate Dominant Interactions. J. Comput. Chem. 2018, 39, 2183–2195. [Google Scholar] [CrossRef]
- Jabłoński, M. On the Uselessness of Bond Paths Linking Distant Atoms and on the Violation of the Concept of Privileged Exchange Channels. ChemistryOpen 2019, 8, 497–507. [Google Scholar] [CrossRef]
- Jabłoński, M. Counterintuitive bond paths: An intriguing case of the C(NO2)3- ion. Chem. Phys. Lett. 2020, 759, 137946. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strength revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Blanco, M.A.; Pendás, A.M.; Francisco, E. Interacting Quantum Atoms: A Correlated Energy Decomposition Scheme Based on the Quantum Theory of Atoms in Molecules. J. Chem. Theory Comput. 2005, 1, 1096–1109. [Google Scholar] [CrossRef]
- Guevara-Vela, J.M.; Francisco, F.; Rocha-Rinza, T.; Pendás, A.M. Interacting Quantum Atoms—Review. Molecules 2020, 25, 4028. [Google Scholar] [CrossRef] [PubMed]
- Gatti, C.; May, E.; Destro, R.; Cargnoni, F. Fundamental Properties and Nature of CH··O Interactions in Crystals on the Basis of Experimental and Theoretical Charge Densities. The Case of 3,4-Bis(dimethylamino)-3-cyclobutene-1,2-dione (DMACB) Crystal. J. Phys. Chem. A 2002, 106, 2707–2720. [Google Scholar] [CrossRef]
- Nikolaienko, T.Y.; Bulavin, L.A.; Hovorun, D.M. Bridging QTAIM with vibrational spectroscopy: The energy of intramolecular hydrogen bonds in DNA-related biomolecules. Phys. Chem. Chem. Phys. 2012, 14, 7441–7447. [Google Scholar] [CrossRef]
- Mata, I.; Alkorta, I.; Espinosa, E.; Molins, E. Relationships between interaction energy, intermolecular distance and electron density properties in hydrogen bonded complexes under external electric fields. Chem. Phys. Lett. 2011, 507, 185–189. [Google Scholar] [CrossRef]
- Afonin, A.V.; Vashchenko, A.V.; Sigalov, M.V. Estimating the energy of intramolecular hydrogen bonds from 1H NMR and QTAIM calculations. Org. Biomol. Chem. 2016, 14, 11199–11211. [Google Scholar] [CrossRef] [PubMed]
- Pendás, A.M.; Blanco, M.A.; Francisco, E. The nature of the hydrogen bond: A synthesis from the interacting quantum atoms picture. J. Chem. Phys. 2006, 125, 184112. [Google Scholar] [CrossRef]
- García-Revilla, M.; Francisco, E.; Popelier, P.L.A.; Pendás, A.M. Domain-Averaged Exchange-Correlation Energies as a Physical Underpinning for Chemical Graphs. ChemPhysChem 2013, 14, 1211–1218. [Google Scholar] [CrossRef]
- Guevara-Vela, J.M.; Chávez-Calvillo, R.; García-Revilla, M.; Hernández-Trujillo, J.; Christiansen, O.; Francisco, E.; Pendás, A.M.; Rocha-Rinza, T. Hydrogen-Bond Cooperative Effects in Small Cyclic Water Clusters as Revealed by the Interacting Quantum Atoms Approach. Chem. Eur. J. 2013, 19, 14304–14315. [Google Scholar] [CrossRef]
- Guevara-Vela, J.M.; Romero-Montalvo, E.; Gómez, V.A.M.; Chávez-Calvillo, R.; García-Revilla, M.; Francisco, E.; Pendás, A.M.; Rocha-Rinza, T. Hydrogen bond cooperativity and anticooperativity within the water hexamer. Phys. Chem. Chem. Phys. 2016, 18, 19557–19566. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Mata, I.; Molins, E.; Espinosa, E. Charged versus Neutral Hydrogen-Bonded Complexes: Is There a Difference in the Nature of the Hydrogen Bonds? Chem. Eur. J. 2016, 22, 9226–9234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guevara-Vela, J.M.; Romero-Montalvo, E.; Costales, A.; Pendás, A.M.; Rocha-Rinza, T. The nature of resonance-assisted hydrogen bonds: A quantum chemical topology perspective. Phys. Chem. Chem. Phys. 2016, 18, 26383–26390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guevara-Vela, J.M.; Romero-Montalvo, E.; del Río Lima, A.; Pendás, A.M.; Hernández-Rodríguez, M.; Rocha Rinza, T. Hydrogen-Bond Weakening through π Systems: Resonance-Impaired Hydrogen Bonds (RIHB). Chem. Eur. J. 2017, 23, 16605–16611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Montalvo, E.; Guevara-Vela, J.M.; Costales, A.; Pendás, A.M.; Rocha-Rinza, T. Cooperative and anticooperative effects in resonance assisted hydrogen bonds in merged structures of malondialdehyde. Phys. Chem. Chem. Phys. 2017, 19, 97–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimi, S.; Dabbagh, H.A.; Eskandari, K. Nature of intramolecular interactions of vitamin C in view of interacting quantum atoms: the role of hydrogen bond cooperativity on geometry. Phys. Chem. Chem. Phys. 2016, 18, 18278–18288. [Google Scholar] [CrossRef]
- Pendás, A.M.; Francisco, E.; Blanco, M.A.; Gatti, C. Bond Paths as Privileged Exchange Channels. Chem. Eur. J. 2007, 13, 9362–9371. [Google Scholar] [CrossRef]
- Badger, R.M.; Bauer, S.H. Spectroscopic Studies of the Hydrogen Bond. II. The Shift of the O–H Vibrational Frequency in the Formation of the Hydrogen Bond. J. Chem. Soc. 1937, 5, 839–851. [Google Scholar] [CrossRef]
- Joesten, M.D.; Drago, R.S. The Validity of Frequency Shift-Enthalpy Correlations. I. Adducts of Phenol with Nitrogen and Oxygen Donors. J. Am. Chem. Soc. 1962, 84, 3817–3821. [Google Scholar] [CrossRef]
- Epley, T.D.; Drago, R.S. Calorimetric Studies on Some Hydrogen-Bonded Adducts. J. Am. Chem. Soc. 1967, 89, 5770–5773. [Google Scholar] [CrossRef]
- Odinokov, S.E.; Glazunov, V.P.; Nabiullin, A.A. Infrared Spectroscopic Studies of Hydrogen Bonding in Triethylammonium Salts. Part 3.—Strong Hydrogen Bonding. J. Chem. Soc. Faraday Trans. 1984, 80, 899–908. [Google Scholar] [CrossRef]
- Ratajczak, H.; Orville-Thomas, W.J.; Rao, C.N.R. Charge transfer theory of hydrogen bonds: Relations between vibrational spectra and energy of hydrogen bonds. Chem. Phys. 1976, 17, 197–216. [Google Scholar] [CrossRef]
- Jabłoński, M.; Sadlej, A.J. Infrared and Raman Intensities in Proper and Improper Hydrogen-Bonded Systems. Pol. J. Chem. 2007, 81, 767–782. [Google Scholar]
- Pecul, M.; Leszczynski, J.; Sadlej, J. Comprehensive ab initio studies of nuclear magnetic resonance shielding and coupling constants in XH⋯O hydrogen-bonded complexes of simple organic molecules. J. Chem. Phys. 2000, 112, 7930–7938. [Google Scholar] [CrossRef]
- McDowell, S.A.C.; Buckingham, A.D. Shift of proton magnetic shielding in ClH⋯Y dimers (Y = N2, CO, BF). Mol. Phys. 2006, 104, 2527–2531. [Google Scholar] [CrossRef]
- Scheiner, S. Interpretation of Spectroscopic Markers of Hydrogen Bonds. ChemPhysChem 2016, 17, 2263–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheiner, S. Assessment of the Presence and Strength of H-Bonds by Means of Corrected NMR. Molecules 2016, 21, 1426. [Google Scholar] [CrossRef] [Green Version]
- Fliegl, H.; Lehtonen, O.; Sundholm, D. Kailaz, V.R.I. Hydrogen-bond strengths by magnetically induced currents. Phys. Chem. Chem. Phys. 2011, 13, 434–437. [Google Scholar] [CrossRef]
- Monaco, G.; Della Porta, P.; Jabłoński, M.; Zanasi, R. Topology of the magnetically induced current density and proton magnetic shielding in hydrogen bonded systems. Phys. Chem. Chem. Phys. 2015, 17, 5966–5972. [Google Scholar] [CrossRef]
- Busfield, W.K.; Ennis, M.P.; McEwen, I.J. An infrared study of intramolecular hydrogen bonding in α, ω diols. Spectrochim. Acta A 1973, 29, 1259–1264. [Google Scholar] [CrossRef]
- Schaefer, T. A Relationship between Hydroxyl Proton Chemical Shifts and Torsional Frequencies in Some Ortho-Substituted Phenol Derivatives. J. Phys. Chem. 1975, 79, 1888–1890. [Google Scholar] [CrossRef]
- Takasuka, M.; Matsui, Y. Experimental Observations and CNDO/2 Calculations for Hydroxy Stretching Frequency Shifts, Intensities, and Hydrogen Bond Energies of Intramolecular Hydrogen Bonds in ortho-Substituted Phenols. J. Chem. Soc. Perkin Trans. 2 1979, 1743–1750. [Google Scholar] [CrossRef]
- Shagidullin, R.R.; Chernova, A.V.; Shagidullin, R.R. Intramolecular hydrogen bonds and conformations of the 1,4-butanediol molecule. Russ. Chem. Bull. 1993, 42, 1505–1510. [Google Scholar] [CrossRef]
- Dziembowska, T.; Szczodrowska, B.; Krygowski, T.M.; Grabowski, S.J. Estimation of the OH⋯O interaction energy in intramolecular hydrogen bonds: A comparative study. J. Phys. Org. Chem. 1994, 7, 142–146. [Google Scholar] [CrossRef]
- Vashchenko, A.V.; Afonin, A.V. Comparative estimation of the energies of intramolecular C-H⋯O, N-H⋯O, and O-H⋯O hydrogen bonds according to the QTAIM analysis and NMR spectroscopy data. J. Struct. Chem. 2014, 55, 636–643. [Google Scholar] [CrossRef]
- Iogansen, A.V.; Rassadin, B.V. Dependence of Intensity and Shift in the Infrared Bands of the Hydroxyl Group on Hydrogen Bonding Energy. J. Appl. Spectrosc. 1969, 11, 1318–1325. [Google Scholar] [CrossRef]
- Iogansen, A.V. Direct proportionality of the hydrogen bonding energy and the intensification of the stretching ν(XH) vibration in infrared spectra. Spectrochim. Acta Part A 1999, 55, 1585–1612. [Google Scholar] [CrossRef]
- Yurenko, Y.P.; Zhurakivsky, R.O.; Ghomi, M.; Samijlenko, S.P.; Hovorun, D.M. Comprehensive Conformational Analysis of the Nucleoside Analogue 2’-β-Deoxy-6-azacytidine by DFT and MP2 Calculations. J. Phys. Chem. B 2007, 111, 6263–6271. [Google Scholar] [CrossRef]
- Yurenko, Y.P.; Zhurakivsky, R.O.; Ghomi, M.; Samijlenko, S.P.; Hovorun, D.M. How Many Conformers Determine the Thymidine Low-Temperature Matrix Infrared Spectrum? DFT and MP2 Quantum Chemical Study. J. Phys. Chem. B 2007, 111, 9655–9663. [Google Scholar] [CrossRef]
- Yurenko, Y.P.; Zhurakivsky, R.O.; Ghomi, M.; Samijlenko, S.P.; Hovorun, D.M. Ab Initio Comprehensive Conformational Analysis of 2’-Deoxyuridine, the Biologically Significant DNA Minor Nucleoside, and Reconstruction of Its Low-Temperature Matrix Infrared Spectrum. J. Phys. Chem. B 2008, 112, 1240–1250. [Google Scholar] [CrossRef]
- Gränacher, I. Einfluss der Wasserstoffbrückenbindung auf das Kernresonanzspektrum von Phenolen. Helv. Phys. Acta 1961, 34, 272–304. [Google Scholar]
- Hobza, P.; Halvas, Z. Blue-Shifting Hydrogen Bonds. Chem. Rev. 2000, 100, 4253–4264. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.; Jemmis, E.D. Red-, Blue-, or No-Shift in Hydrogen Bonds: A Unified Explanation. J. Am. Chem. Soc. 2007, 129, 4620–4632. [Google Scholar] [CrossRef] [PubMed]
- Lippincott, E.R.; Schroeder, R. One-Dimensional Model of the Hydrogen Bond. J. Chem. Phys. 1955, 23, 1099–1106. [Google Scholar] [CrossRef]
- Schroeder, R.; Lippincott, E.R. Potential Function Model of Hydrogen Bonds. II. J. Phys. Chem. 1957, 61, 921–928. [Google Scholar] [CrossRef]
- Musin, R.N.; Mariam, Y.H. An integrated approach to the study of intramolecular hydrogen bonds in malonaldehyde enol derivatives and naphthazarin: Trend in energetic versus geometrical consequences. J. Phys. Org. Chem. 2006, 19, 425–444. [Google Scholar] [CrossRef]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Rotated Group | SP | P1 | P2 | P3 | P4 | P5 | OPT |
|---|---|---|---|---|---|---|---|---|
| 1 | −SiH2 | −5.31 | −4.88 | −4.41 | −3.36 | −1.50 | −0.99 | −1.08 |
| −AlH2 | −7.01 | −6.61 | −5.79 | −4.41 | −3.24 | −1.75 | ||
| 2 | −SiH2 | −5.23 | −4.75 | −3.93 | −2.46 | −2.42 | −2.42 | Al |
| −AlH2 | −8.09 | −7.37 | −6.27 | −4.85 | −4.73 | −4.72 | ||
| 3 | −SiH2 | −6.04 | −5.03 | −3.47 | −1.41 | −1.40 | −1.40 | −4.97/−0.75 |
| −AlH2 | −10.61 | −10.05 | −8.44 | −6.23 | −6.19 | −6.19 |
| Y | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| O | 4.77 | 3.77 | 6.50 | −6.50 | −5.28 | 1.840 | 2.141 | −0.301 | −1.87 |
| S | 6.07 | 4.09 | 6.02 | −6.02 | −6.96 | 1.968 | 2.127 | −0.159 | −1.86 |
| Molecule | ||||||
|---|---|---|---|---|---|---|
| malondialdehyde | −14.0 | −10.7 | −14.1 | −14.0 | −12.4 | −12.9 |
| acetylacetone | −16.2 | −13.3 | −15.1 | −16.9 | −12.3 | −14.5 |
| X | ||||
|---|---|---|---|---|
| F | 1.82 | 1.97 | 0.52 | 0.44 |
| Cl | 2.98 | 3.34 | 1.06 | 0.38 |
| Br | 3.49 | 3.95 | 1.08 | 0.19 |
| Type | A | B | R | |
|---|---|---|---|---|
| O-H⋯O | −239 ± 2.2 | 3.09 ± 0.07 | 0.93 | 0.86 |
| O-H⋯N | −142 ± 2.1 | −1.72 ± 0.08 | 0.97 | 0.94 |
| N-H⋯O | −225 ± 12 | 2.03 ± 0.25 | 0.85 | 0.72 |
| O-H⋯C | −288 ± 19 | 0.29 ± 0.22 | 0.86 | 0.74 |
| together | −200 ± 2.2 | 1.70 ± 0.07 | 0.88 | 0.77 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jabłoński, M. A Critical Overview of Current Theoretical Methods of Estimating the Energy of Intramolecular Interactions. Molecules 2020, 25, 5512. https://doi.org/10.3390/molecules25235512
Jabłoński M. A Critical Overview of Current Theoretical Methods of Estimating the Energy of Intramolecular Interactions. Molecules. 2020; 25(23):5512. https://doi.org/10.3390/molecules25235512
Chicago/Turabian StyleJabłoński, Mirosław. 2020. "A Critical Overview of Current Theoretical Methods of Estimating the Energy of Intramolecular Interactions" Molecules 25, no. 23: 5512. https://doi.org/10.3390/molecules25235512
APA StyleJabłoński, M. (2020). A Critical Overview of Current Theoretical Methods of Estimating the Energy of Intramolecular Interactions. Molecules, 25(23), 5512. https://doi.org/10.3390/molecules25235512