Synthesis and Conformational Analysis of Fluorinated Uridine Analogues Provide Insight into a Neighbouring-Group Participation Mechanism

 ,
,
Abstract
:
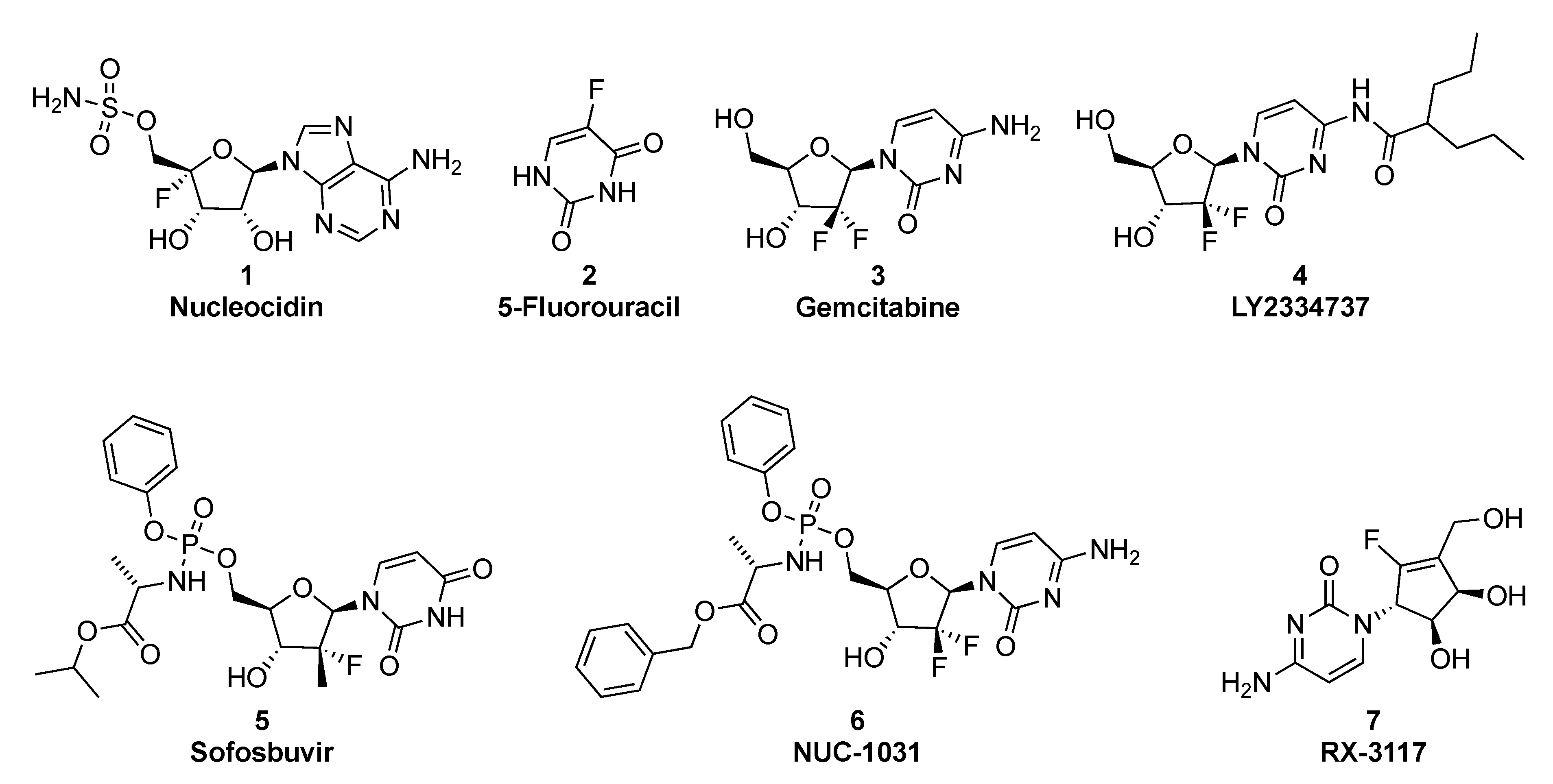
1. Introduction
2. Results
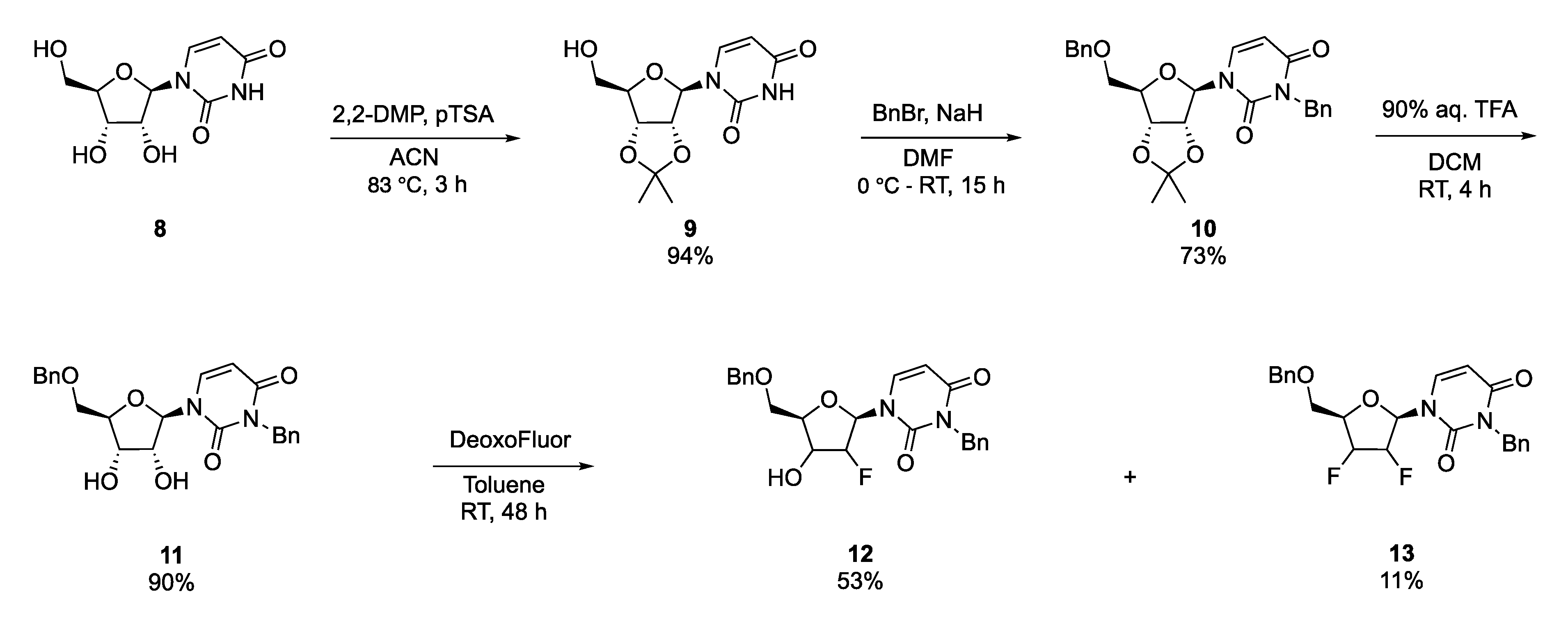
2.1. Synthesis of Fluorinated Nucleoside Analogues
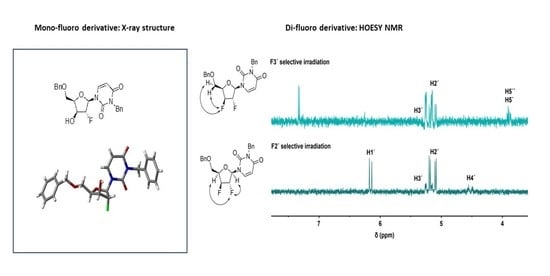
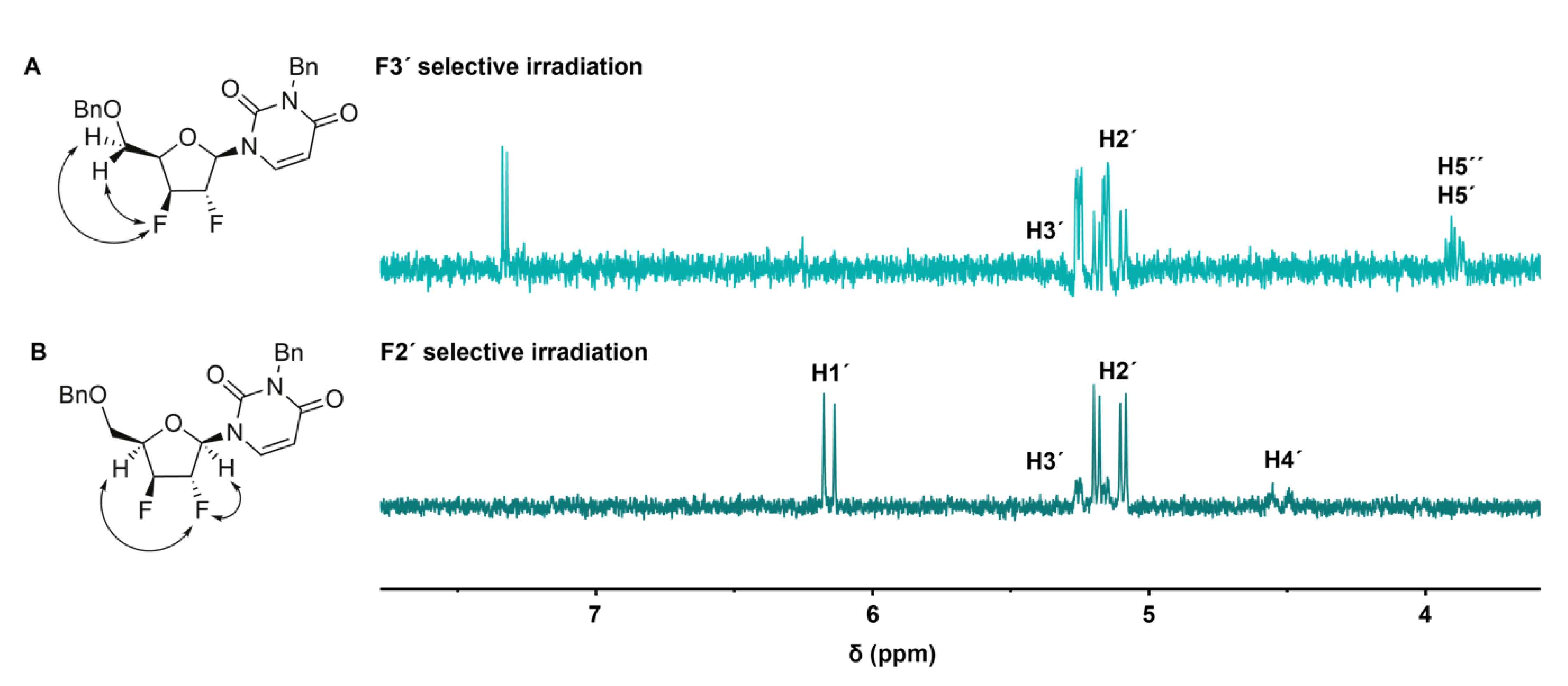
2.2. Conformational Analysis of the Synthesized Analogues
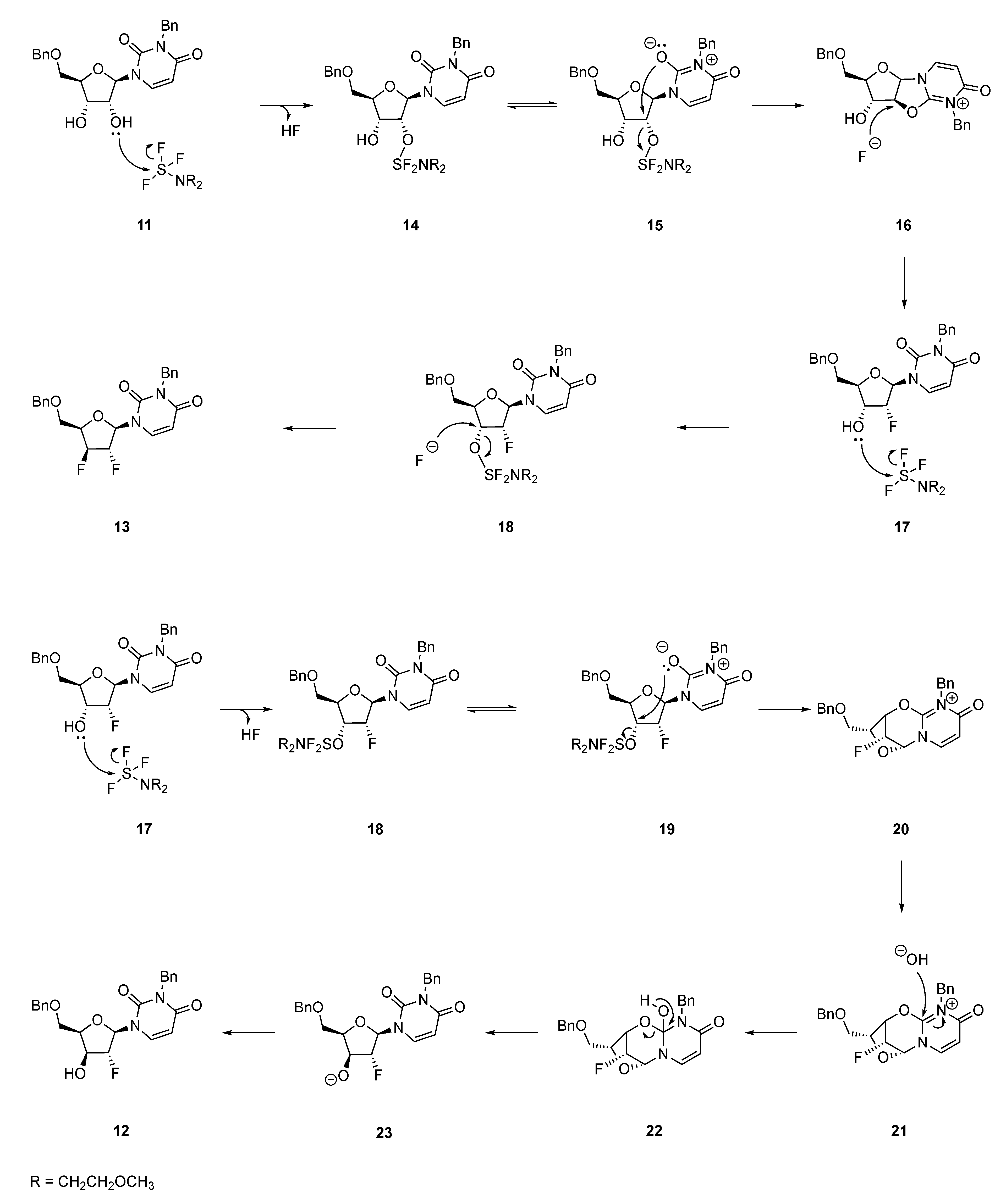
2.3. Mechanistic Proposal: Neighbouring-Group Participation
3. Discussion
4. Materials and Methods
4.1. General Considerations
4.2. Preparation of 2′,3′-O-Isopropylidene Uridine (9)
4.3. Preparation of 3,5′-dibenzyl-2′,3′-O–Isopropylidene Uridine (10)
4.4. Preparation of 3,5′-Dibenzyl–uridine (11)
4.5. General Procedure for the Fluorination Reactions
4.5.1. 1-(5-O-Benzyl-3-O-2′-deoxy-2′-fluoro–β-d-arabinofuranosyl)-N3-benzyluracil (12)
4.5.2. 1-(5-O-Benzyl-3-O-2′,3′-dideoxy-2′,3′-difluoro-β-D-xylofuranosyl)-N3-benzyluracil (13)
4.6. Modelling of the Conformers of Compound 12
4.7. Heteronuclear NMR Spectroscopy
4.8. X-Ray Crystallography
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cavaliere, A.; Probst, C.K.; Westwell, D.A.; Slusarczyk, M. Fluorinated nucleosides as an important class of anticancer and antiviral agents. Future Med. Chem. 2017, 9, 809–1833. [Google Scholar] [CrossRef]
- Shelton, J.; Lu, X.; Hollenbaugh, J.A.; Cho, J.H.; Amblard, F.; Schinazi, R.F. Metabolism, Biochemical Actions, and Chemical Synthesis of Anticancer Nucleosides, Nucleotides, and Base Analogs. Chem. Rev. 2016, 116, 14379–14455. [Google Scholar] [CrossRef] [PubMed]
- Pankiewicz, K.W. Fluorinated Nucleosides. Carbohydr. Res. 2000, 327, 87–105. [Google Scholar] [CrossRef]
- Ferraboschi, P.; Ciceri, S.; Grisenti, P. Synthesis of Antitumor Fluorinated Pyrimidine Nucleosides. Org. Prep. Proced. Int. 2017, 49, 69–154. [Google Scholar] [CrossRef]
- Muller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Remete, A.M.; Dobson, L.S.; Kiss, L.; Izawa, K.; Moriwaki, H.; Soloshonok, V.A.; O’Hagan, D. Next generation organofluorine containing blockbuster drugs. J. Fluor. Chem. 2020, 239, 109639. [Google Scholar] [CrossRef]
- Inoue, M.; Sumii, Y.; Shibata, N. Contribution of Organofluorine Compounds to Pharmaceuticals. Acs Omega 2020, 5, 10633–10640. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Dixey, M.; Weymouth-Wilson, A.; Linclau, B. The synthesis of gemcitabine. Carbohydr. Res. 2014, 387, 59–73. [Google Scholar] [CrossRef] [Green Version]
- Bartholome, A.; Janso, J.E.; Reilly, U.; O‘Hagan, D. Fluorometabolite biosynthesis: Isotopically labelled glycerol incorporations into the antibiotic nucleocidin in Streptomyces calvus. Org. Biomol. Chem. 2016, 15, 61–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.H.; Yao, G.M.; Li, Y.M.; Lu, J.H.; Lin, C.J.; Wang, X.; Kong, C.H. 5-Fluorouracil Derivatives from the Sponge Phakellia fusca. J. Nat. Prod. 2003, 66, 285–288. [Google Scholar] [CrossRef]
- Williams, R. Discontinued in 2013: Oncology drugs. Expert Opin. Investig. Drugs 2014, 24, 1–16. [Google Scholar] [CrossRef]
- Infante, J.R.; Benhadji, K.A.; Dy, G.K.; Fetterly, G.; Ma, W.W.; Bendell, J.; Callies, S.; Adjei, A.A. Phase 1b study of the oral gemcitabine ‘Pro-drug‘ LY2334737 in combination with capecitabine in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Blagden, S.P.; Rizzuto, I.; Suppiah, P.; O‘Shea, D.; Patel, M.; Spiers, L.; Sukumaran, A.; Bharwani, N.; Rockall, A.; Gabra, H.; et al. Anti-tumour activity of a first-in-class agent NUC-1031 in patients with advanced cancer: Results of a phase I study. Br. J. Cancer 2018, 119, 815–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slusarczyk, M.; Lopez, M.H.; Balzarini, J.; Mason, M.; Jiang, W.G.; Blagden, S.; Thompson, E.; Ghazaly, E.; McGuigan, C. Application of ProTide technology to gemcitabine: A successful approach to overcome the key cancer resistance mechanisms leads to a new agent (NUC-1031) in clinical development. J. Med. Chem. 2014, 57, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- A Targeted Anti-Cancer Therapy. Available online: https://www.rexahn.com/product-pipeline/rx-3117 (accessed on 16 November 2020).
- Balboni, B.; El Hassouni, B.; Honeywell, R.J.; Sarkisjan, D.; Giovannetti, E.; Poore, J.; Heaton, C.; Peterson, C.; Benaim, E.; Lee, Y.B.; et al. RX-3117 (fluorocyclopentenyl cytosine): A novel specific antimetabolite for selective cancer treatment. Expert Opin. Investig. Drugs 2019, 28, 311–322. [Google Scholar] [CrossRef] [PubMed]
- O‘Hagan, D. Understanding organofluorine chemistry. An introduction to the C-F bond. Chem. Soc. Rev. 2008, 37, 308–319. [Google Scholar] [CrossRef]
- Withers, S.G.; Street, I.P.; Percival, M.D. Fluorinated Carbohydrates as Probes of Enzyme Specificity and Mechanism. In Fluorinated Carbohydrates; ACS Publications: Washington, DC, USA, 1988; pp. 59–77. [Google Scholar] [CrossRef]
- Walsh, C. Fluorinated substrate analogs: Routes of metabolism and selective toxicity. Adv. Enzym. Relat. Areas Mol. Biol. 1983, 55, 197–289. [Google Scholar]
- Ferraris, D.; Duvall, B.; Delahanty, G.; Mistry, B.; Alt, J.; Rojas, C.; Rowbottom, C.; Sanders, K.; Schuck, E.; Huang, K.C.; et al. Design, synthesis, and pharmacological evaluation of fluorinated tetrahydrouridine derivatives as inhibitors of cytidine deaminase. J. Med. Chem. 2014, 57, 2582–2588. [Google Scholar] [CrossRef]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef]
- Bohm, H.J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Muller, K.; Obst-Sander, U.; Stahl, M. Fluorine in medicinal chemistry. Chembiochem 2004, 5, 637–643. [Google Scholar] [CrossRef]
- Qiu, X.-L.; Xu, X.-H.; Qing, F.-L. Recent advances in the synthesis of fluorinated nucleosides. Tetrahedron 2010, 66, 789–843. [Google Scholar] [CrossRef]
- De Clercq, E. Highlights in antiviral drug research: Antivirals at the horizon. Med. Res. Rev. 2013, 33, 1215–1248. [Google Scholar] [CrossRef] [PubMed]
- Yates, M.K.; Seley-Radtke, K.L. The evolution of antiviral nucleoside analogues: A review for chemists and non-chemists. Part II: Complex modifications to the nucleoside scaffold. Antivir. Res 2019, 162, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Galmarini, C.M.; Mackey, J.R.; Dumontet, C. Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol. 2002, 3, 415–424. [Google Scholar] [CrossRef]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, M.; Silverman, S.M.; Lehmann, J.; Adluri, B.; Wang, Y.; Cohen, R.; Campeau, L.C.; Britton, R. A short de novo synthesis of nucleoside analogs. Science 2020, 369, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Pankiewicz, K.W.; Watanabe, K.A. Synthesis of 2′-β-fluoro-substituted nucleosides by a direct approach. J. Fluor. Chem. 1993, 64, 15–36. [Google Scholar] [CrossRef]
- Takamatsu, S.; Maruyama, T.; Katayama, S.; Hirose, N.; Naito, M.; Izawa, K.J. Synthesis of 9-(2,3-Dideoxy-2-fluoro-β-D-threo-pentofuranosyl)adenine (FddA) via a Purine 3’-Deoxynucleoside. J. Org. Chem. 2001, 66, 7469–7477. [Google Scholar] [CrossRef]
- Srivastav, N.C.; Shakya, N.; Mak, M.; Liang, C.; Tyrrell, D.L.; Agrawal, B.; Kumar, R. Synthesis and in vitro antiviral activities of 3’-fluoro (or chloro) and 2’,3’-difluoro 2’,3’-dideoxynucleoside analogs against hepatitis B and C viruses. Bioorg. Med. Chem. 2010, 18, 7542–7547. [Google Scholar] [CrossRef]
- Kim, K.R.; Moon, H.R.; Park, A.Y.; Chun, M.W.; Jeong, L.S. Design, synthesis, and biological evaluation of novel iso-D-2’,3’-dideoxy-3’-fluorothianucleoside derivatives. Bioorg. Med. Chem. 2007, 15, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Snyder, N.L.; Seeberger, P.H.; Mukosera, G.T.; Held, E.M.K. 9.05 Technology-Enabled Synthesis of Carbohydrates. In Comprehensive Organic Synthesis II; Elsevier: Amsterdam, The Netherlands, 2014; pp. 111–142. [Google Scholar] [CrossRef]
- Ragab, A.E.; Gruschow, S.; Tromans, D.R.; Goss, R.J. Biogenesis of the unique 4’,5’-dehydronucleoside of the uridyl peptide antibiotic pacidamycin. J. Am. Chem. Soc. 2011, 133, 15288–15291. [Google Scholar] [CrossRef] [PubMed]
- Erande, N.; Gunjal, A.D.; Fernandes, M.; Gonnade, R.; Kumar, V.A. Synthesis and structural studies of S-type/N-type-locked/frozen nucleoside analogues and their incorporation in RNA-selective, nuclease resistant 2’-5’ linked oligonucleotides. Org. Biomol. Chem. 2013, 11, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Plavec, J.; Tong, W.; Chattopadhyaya, J. How Do the Gauche and Anomeric Effects Drive the Pseudorotational Equilibrium of the Pentofuranose Moiety of Nucleosides? J. Am. Chem. Soc. 1993, 115, 9734–9746. [Google Scholar] [CrossRef]
- Michalik, M.; Hein, M.; Frank, M. NMR spectra of fluorinated carbohydrates. Carbohydr. Res. 2000, 327, 185–218. [Google Scholar] [CrossRef]
- Barchi, J.J.; Karki, R.G.; Nicklaus, M.C.; Siddiqui, M.A.; George, C.; Mikhailopulo, I.A.E. Marquez, V.E. Comprehensive Structural Studies of 2’,3’-Difluorinated Nucleosides: Comparison of Theory, Solution, and Solid State. J. Am. Chem. Soc. 2008, 130, 9048–9057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Montero, S.; Deleavey, G.F.; Kulkarni, A.; Martin-Pintado, N.; Lindovska, P.; Thomson, M.; Gonzalez, C.; Gotte, M.; Damha, M.J. Rigid 2’,4’-difluororibonucleosides: Synthesis, conformational analysis, and incorporation into nascent RNA by HCV polymerase. J. Org. Chem. 2014, 79, 5627–5635. [Google Scholar] [CrossRef]
- Martinez-Montero, S.; Deleavey, G.F.; Dierker-Viik, A.; Lindovska, P.; Ilina, T.; Portella, G.; Orozco, M.; Parniak, M.A.; Gonzalez, C.; Damha, M.J. Synthesis and properties of 2’-deoxy-2’,4’-difluoroarabinose-modified nucleic acids. J. Org. Chem. 2015, 80, 3083–3091. [Google Scholar] [CrossRef] [Green Version]
- Gosselin, G.; Puech, F.; Génu-Dellac, C.; Imbach, J.L. 1,2-Di-O-acetyl-5-O-benzoyl-3-deoxy-%fluoro-D- xylofuranose. A versatile precursor for the synthesis of %deoxy-3-fluoro-P-mxylofuranosyl nucleosides as potential antiviral agents. Carbohydr. Res. 1993, 249, 1–17. [Google Scholar] [CrossRef]
- Haasnoot, C.; De Leeuw, F.; Altona, C. The relationship between proton-proton NMR coupling constants and substituent electronegativities—I: An empirical generalization of the Karplus equation. Tetrahedron 1980, 36, 2783–2792. [Google Scholar] [CrossRef]
- Thibaudeau, C.; Plavec, J.; Chattopadhyaya, J. A New Generalized Karplus-type Equation Relating Vicinal Proton-Fluorine Coupling Constants to H-C-C-F Torsion Angles. J. Org. Chem. 1998, 63, 4967–4984. [Google Scholar] [CrossRef]
- Szlenkier, M.; Boryski, J. Application of Sugar-Base Anhydro Bridge for Modification of Nucleosides in the 2′- and/or 3′-Positions—Revisited. Curr. Org. Chem. 2019, 23, 409–438. [Google Scholar] [CrossRef]
- Jeonga, L.S.; Nicklausa, M.C.; George, C.; Marquez, V.E. Facile fluorination of deoxy-4’-thiopyrimidine nucleosides with “down“ hydroxyl groups. Retention of configuration after fluoride opening of the quaternized N3-MEM anhydronucleosides. Tetrahedron Lett. 1994, 35, 7573–7576. [Google Scholar] [CrossRef]
- Agyei-aye, K.; Yan, S.; Hebbler, A.K.; Baker, D.C. Preparation of 2,3’-Anhydropyrimidine Nucleosides using N,N-Diethylaminosulfur Trifluoride (DAST). Nucleosides Nucleotides 1989, 8, 327–337. [Google Scholar] [CrossRef]
- Jones, G.H.; Taniguchi, M.; Tegg, D.; Moffatt, J.G. 4’-Substituted nucleosides. 5. Hydroxymethylation of nucleoside 5’-aldehydes. J. Org. Chem. 1979, 44, 1309–1317. [Google Scholar] [CrossRef]
- Combettes, L.E.; Clausen-Thue, P.; King, M.A.; Odell, B.; Thompson, A.L.; Gouverneur, V.; Claridge, T.D. Conformational analysis of fluorinated pyrrolidines using 19F-1H scalar couplings and heteronuclear NOEs. Chemistry 2012, 18, 13133–13141. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| C1 (RS-north) | |||||||
| Relative E (kJ/mol) | JF-H1′ (Hz) | JF-H3′ (Hz) | JH1′-H2′ (Hz) | JH2′-H3′ (Hz) | JH3′-H4′ (Hz) | Avg. Deviation | |
| A1 (SR-north) | 0 | 3.7 | 18.0 | 7.2 | 6.3 | 8.6 | 4.48 |
| A2 (SR-south) | 2.58 | 32.9 | 18.7 | 1.9 | 0.9 | 1.9 | 3.00 |
| B1 (RR-north) | 0 | 20.0 | 39.9 | 0.8 | 3.4 | 8.6 | 6.21 |
| B2 (RR-south) | 9.47 | 16.9 | 3.2 | 6.1 | 5.8 | 1.0 | 2.31 |
| C1 (RS-north) | 0 | 22.8 | 17.7 | 0.8 | 1.2 | 3.2 | 1.68 |
| C2 (RS-south) | 31.44 | 20.0 | 24.5 | 4.2 | 6.2 | 7.3 | 3.46 |
| D1 (SS-north) | 9.83 | 6.2 | 3.5 | 7.2 | 6.4 | 2.7 | 3.77 |
| D2 (SS-south) | 0 | 24.1 | 39.2 | 3.6 | 3.8 | 7.5 | 6.11 |
| Experimental | 21.2 | 9.6 | 0 | 0 | 2.8 | ||
| d(F-H2′) (Å) | d(F-H4′) (Å) | d(F-H1′) (Å) | d(F-H3′) (Å) | Avg. Deviation | |
|---|---|---|---|---|---|
| A1 (SR-north) | 2.017 | 4.150 | 3.025 | 2.511 | 0.541 |
| A2 (SR-south) | 2.038 | 4.006 | 3.300 | 2.496 | 0.534 |
| B1 (RR-north) | 2.028 | 2.757 | 2.452 | 3.292 | 0.289 |
| B2 (RR-south) | 2.025 | 4.292 | 2.497 | 2.917 | 0.554 |
| C1 (RS-north) | 2.036 | 2.630 | 2.419 | 2.511 | 0.048 |
| C2 (RS-south) | 2.025 | 4.313 | 2.462 | 2.417 | 0.536 |
| D1 (SS-north) | 2.014 | 4.076 | 3.108 | 2.951 | 0.552 |
| D2 (SS-south) | 2.017 | 4.046 | 3.285 | 3.288 | 0.618 |
| NOE | 1.000 | 0.174 | 0.437 | 0.330 | |
| Distance | 2.027 | 2.713 | 2.327 | 2.438 |
Sample Availability:Not available. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michailidou, F.; Lebl, T.; Slawin, A.M.Z.; Sharma, S.V.; Brown, M.J.B.; Goss, R.J.M. Synthesis and Conformational Analysis of Fluorinated Uridine Analogues Provide Insight into a Neighbouring-Group Participation Mechanism. Molecules 2020, 25, 5513. https://doi.org/10.3390/molecules25235513
Michailidou F, Lebl T, Slawin AMZ, Sharma SV, Brown MJB, Goss RJM. Synthesis and Conformational Analysis of Fluorinated Uridine Analogues Provide Insight into a Neighbouring-Group Participation Mechanism. Molecules. 2020; 25(23):5513. https://doi.org/10.3390/molecules25235513
Chicago/Turabian StyleMichailidou, Freideriki, Tomas Lebl, Alexandra M. Z. Slawin, Sunil Vishnuprasadji Sharma, Murray J. B. Brown, and Rebecca Jane Miriam Goss. 2020. "Synthesis and Conformational Analysis of Fluorinated Uridine Analogues Provide Insight into a Neighbouring-Group Participation Mechanism" Molecules 25, no. 23: 5513. https://doi.org/10.3390/molecules25235513
APA StyleMichailidou, F., Lebl, T., Slawin, A. M. Z., Sharma, S. V., Brown, M. J. B., & Goss, R. J. M. (2020). Synthesis and Conformational Analysis of Fluorinated Uridine Analogues Provide Insight into a Neighbouring-Group Participation Mechanism. Molecules, 25(23), 5513. https://doi.org/10.3390/molecules25235513