3. Materials and Methods
All used solvents were purified according to procedures described in [
34]. All starting compounds were commercially available reagents. The initial stages of the synthesis of the vector fragment
1–
5 (
Scheme 1) were made by methods previously developed by our scientific group [
21]. Spectral data of the compounds
7 and
8 (
Scheme 2) were described in [
35].
1H NMR was measured using a Bruker Avance spectrometer operating at 400 MHz for
1H using CDCl
3 and DMSO-d
6 as solvents. Chemical shifts were reported in δ units to 0.01 ppm precision with coupling constants reported to 0.1 Hz precision using residual solvent as an internal reference.
13C NMR was measured using a Bruker Avance spectrometer operating at 100 MHz using DMSO-d
6 as solvents. Chemical shifts were reported in δ units to 0.1 ppm precision using residual solvent as an internal reference. 2D NMR was measured using an Agilent 600 spectrometer operating at 600 MHz for
1H and 100 MHz for (
13C) using DMSO-d
6 as the solvent. As 2D NMR methods were used, such as heteronuclear single quantum coherence spectroscopy
1H-
13C (gHSQC) and heteronuclear multiple bond correlation
1H-
13C (gHMBC). NMR spectra were processed and analyzed using Mnova software (Mestrelab Research, Spain). High-resolution mass spectra were recorded on the Orbitrap Elite high-resolution mass spectrometer. Solutions of samples in acetonitrile with 1% formic acid were introduced into the ionization source by electrospray. For the HPLC analysis system with Shimadzu Prominence, an LC-20 column and a convection fraction collector connected with a single quadrupole mass spectrometer Shimadzu LCMS-2020 with dual ionization source DUIS-ESI-APCI were used. The analytical and preparative column was Phenomenex Luna 3u C18 100A. Preparative chromatographic separation of substances was carried out using the INTERCHIM puriFlash 430 chromatograph.
For better interpretation of the NMR spectra of target compound
19, the notation of structural fragments is shown in
Figure 4.
Cell Lines: LNCaP, 22Rv1, and PC-3 human prostate cancer cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA).
Cell Cultivation: Cells were maintained in RPMI-1640 medium (gibco), supplemented with 10% Fetal Bovine Serum (Sigma), 2 mM L-glutamine, and RPMI vitamin solution (Sigma). Cells were cultured at 37 °C in a humidified incubator (Sanyo) supplied with 5% CO2. Cells were seeded on glass coverslips or in 96-well plates (Corning) at concentrations of 120,000 cells per mL for LNCaP, 200,000 cells per mL for 22Rv1, and 90,000 cells per mL for PC-3 in experiments. The counting of cells was carried out using the automatic cell counter EVE.
Cell Incubation with Conjugates: A day after seeding the cells on glass coverslips, PSMA-Cy5 or fluorescently labeled compound 19 were added in culture medium at a concentration of 30 nM for 2 h. Later, cells were washed with PBS (pH 7.2–7.4) and fixed with 4% formaldehyde (Sigma) (on PBS) for 15 min. Cell nuclei were stained with DAPI (Sigma) for 10 min. Obtained preparations were imaged using an inverted fluorescence microscope EVOS (life technologies, objective PlanFluor 20×/0.45). Further processing of the photos was carried out by ImageJ software.
Cytotoxicity Assay: A day after cell seeding in 96-well plates, serial dilutions of conjugates and Docetaxel in culture medium were added to cells. Cells incubated in culture medium were used as control. DMSO diluted in the cell medium (20%) was used as a positive control. Cells were incubated for 72 h at 37 °C and 5% CO2. Later, the culture medium from each well was removed and 20 μL of MTS reagent (CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay, Promega) was added to each well with 100 μL of new culture medium. After 4 h of incubation at 37 °C in darkness, the absorbance of the obtained solution was measured at 490 nm wavelength using the Thermo Scientific Multiskan GO spectrometer. Cell viability was calculated as percent compared to cells incubated in culture medium. MTS assay revealed 100% cell death after incubation with 20% DMSO (data not shown). The absorbance of MTS reagent in culture medium without cells was taken as zero. Experiments were performed in triplicate.
Compound 6. To a solution of compound 5 (1 eq; 725 mg; 1.0 mmol) in 20 mL of DCM, DIPEA (1.4 eq; 244 μL; 1.4 mmol) and succinic anhydride (1.02 eq; 102 mg; 1.02 mmol) were added. The mixture was stirred for 12 h. After that, MeOH (2 eq.) was added and the resulting mixture was stirred for 1 h. Then, the solvent was removed under reduced pressure, and residue was dissolved in DCM and extracted with (1) 0.1 M HCl (2 × 30 mL) and (2) brine (2 × 30 mL). Then, the organic fraction was dried over Na2SO4, and concentrated under reduced pressure to obtain the final compound 6 as a yellow oil (801 mg, yield 97%).
1H-NMR (400 MHz, DMSO-d6, δ): 12.06 (br.s., 1H, X4C(O)OH), 7.81 (t, J = 5.2 Hz, m) & 7.77 (t, J = 5.2 Hz, n) (1H, X3NHk, m + n, m/n = 3/2), 7.40 (t, J = 7.7 Hz, X8He, n), 7.37–7.27 (m, X8Hd + X8He(m)), 7.26–7.21 (m, 1H, X8Ht, m + n), 7.19–7.10 (m, 1H, X8Hg, m + n), 6.34–6.20 (m, 2H, K2NH + E1NH, m + n), 4.56 (s, n) & 4.48 (s, m) (2H, X8Ha, m + n, m/n = 3/2), 4.07–4.00 (m, 1H, E1Ha, m + n), 4.00–3.90 (m, 1H, K2Ha, m + n), 3.22 (t, J = 7.3 Hz, n) & 3.19 (t, J = 7.3 Hz, m) (2H, K2He, m + n, m/n = 3/2), 3.01 (q, J = 6.4, 12.7 Hz, m) & 2.96 (q, J = 6.4, 12.7 Hz, n) (2H, X3He, m + n, m/n = 3/2), 2.44–2.38 (m, 2H, X4Hb, m + n), 2.36 (t, J = 7.4 Hz, X3Ha, m), 2.31–2.25 (m, 2H, X4Ha, m + n), 2.25–2.15 (m, E1Hg + X3Ha(n)), 1.91–1.80 (m, 1H, E1Hb(a)), 1.72–1.63 (m, 1H, E1Hb(b)), 1.63–1.56 (m, 1H, K2Hb(a)), 1.40–1.35 (m, 27H, tBu), 1.56–1.15 (m, 11H, K2Hb(b) + X3Hb + X3Hd+ K2Hd + K2Hg + X3Hg, m + n).
13C-NMR (100 MHz, DMSO-d6, δ): 173.93 (X4Cg), 172.26 (K2C(n)), 172.23 (K2C(m)), 172.22 (X3C(n)), 172.19 (X3C(m)), 171.95 (E1C), 171.47 (E1Cg), 170.76 (X4C(m)), 170.73 (X4C(n)), 157.18 (U(m)), 157.16 (U(n)), 141.20 (X9Cb(m)), 140.80 (X9Cb(n)), 133.45 (X9Ce(n)), 133.10 (X9Ce(m)), 130.63 (X9Cd(n)), 130.26 (X9Cd(m)), 127.24 (X9Ct(m)), 127.17 (X9Ck(n)), 126.88 (X9Ck(m)), 126.34 (X9Ct(n)), 126.08 (X9Cg(m)), 124.99 (X9Cg(n)), 80.59 (E1tBu), 80.42 (K2tBu(m)), 80.33 (K2tBu(n)), 79.77 (E1dtBu), 53.01 (K2Ca(n)), 52.88 (K2Ca(m)), 52.20 (E1Ca(m)), 52.18 (E1Ca(n)) 49.63 (X9Ca(n)), 47.11 (X9Ca(m)), 46.83 (K2Ce(m)), 45.20 (K2Ce(n)),38.49 (X3Ce(m)), 38.43 (X3Ce(n)), 32.34 (X3Ca(n)), 31.95 (X3Ca(m)), 31.83 (K2Cb), 30.93 (E1Cg), 30.06 (X4Ca), 29.25 (X4Cb), 29.13 (X3Cd(m)), 29.04 (X3Cd(n)), 27.75 (tBuE1), 27.69 (K2Cd(m)), 27.66 (tBuK2), 27.64 (tBuE1g + E1Cb), 26.72 (K2Cd(n)), 26.23 (X3Cg(m)), 26.15 (X3Cg(n)), 24.76 (X3Cb(m)), 24.63 (X3Cb(n)), 22.45 (K2Cg(n)), 22.27 (K2Cg(m)).
ESI-MS C41H65ClN4O11: m/z calcd. for [M + H+]+: 825.44, found: 825.45.
Compound 7. To a solution of FmocLys(L)(NHBoc)-OH (1 eq.; 1000 mg; 2.134 mmol) in DMF (20 mL), DIPEA (1.5 eq.; 556 μL; 3.2 mmol), HOBt (1.2 eq.; 344 mg; 2.56 mmol), and HBTU (1.2 eq.; 971 mg; 2.56 mmol), were added, then the resulting mixture was purged with Ar and stirred for 60 min. Then, NH2-(CH2)3-N3 (2 eq.; 38 mg; 0.38 mmol) was added and the mixture was stirred for 24 h under Ar atmosphere. At the next step, the solvent was removed under reduced pressure and re-evaporated with DCM twice. The residue was dissolved in DCM (50 mL) and extracted with 1) H2O (2 × 50 mL) and 2) brine (2 × 50 mL). Then, the organic fraction was dried over Na2SO4. After the solvent was removed under reduced pressure, the residue was purified by column chromatography (Puriflash 15μ 40g, eluent: Hex(100%)/EtOAc(0%) => Hex(0%)/EtOAc(100%) for 30 min. The eluent for TLC was EtOAc/Hex = 1:1. Compound 7 was obtained as a yellow oil (950 mg, 81% yield).
1H-NMR (400 MHz, CDCl3, δ): 7.76 (d, J = 7.5 Hz, 2H, Fmoc), 7.58 (d, J = 7.5 Hz, 2H, Fmoc), 7.40 (t, J = 7.4 Hz, 2H, Fmoc), 7.31 (t, J = 7.4 Hz, 2H, Fmoc), 6.43 (br.s., 1H, X8NH), 5.54 (br.d., 1H, K7NH), 4.63 (br.s., 1H, K7NHk), 4.40 (d, J = 6.8 Hz, 2H, Fmoc), 4.20 (t, J = 6.8 Hz, 1H, Fmoc), 4.15–4.01 (m, 1H, K7Ha), 3.40–3.25 (m, 4H, X8Hg + X8Ha), 3.18–3.01 (m, 2H, K7He), 1.92–1.81 (m, 1H, K7Hb(a)), 1.80–1.71 (m, 2H, X8Hb), 1.70–1.57 (m,1H, K7Hb(b)), 1.56–1.46 (m, 2H, K7Hd), 1.43 (s, 9H, tBu), 1.39–1.29 (m, 2H, K7Hg).
Compound 8. To a solution of 7 (1 eq.; 792 mg; 1.44 mmol) in DMF (10 mL), Et2NH (10 eq.; 1053 μL; 14.4 mmol) was added, then the resulting mixture was purged with Ar and stirred for 1 h. The control of the reaction was performed with TLC. The eluent for TLC was EtOAc/Hex = 1:1. At the next step, the solvent was removed under reduced pressure and re-evaporated with DCM twice. The residue was purified by column chromatography (Puriflash 15μ 25g, eluent: DCM(100%)/MeOH(0%) => DCM(85%)/MeOH(15%) for 30 min, after MeOH (100%) for 5 min. Compound 8 was obtained as a yellow oil (469 mg, 99% yield).
1H-NMR (400 MHz, CDCl3, δ): 4.59 (br.s., 1H, K7NHk), 3.40–3.25 (m, 5H, X8Hg + K7Ha + X8Ha), 3.19–3.02 (m, 2H, K7He), 1.91–1. 1.70 (m, 4H, K7Hb(a) + X8Hb + K7Hb(b)), 1.56–1.46 (m, 2H, K7Hd), 1.44 (s, 9H, tBu), 1.39–1.32 (m, 2H, K7Hg).
Compound 9. To a solution of FmocFF (1 eq.; 770 mg; 1.44 mmol) in DMF (20 mL), DIPEA (1.2 eq.; 301 μL; 1.73 mmol), HOBt (1.2 eq.; 233 mg; 1.73 mmol), HBTU (1.2 eq.; 655 mg; 1.73 mmol), and 8 (1 eq.; 469 mg; 1.43 mmol) were added, then the resulting mixture was purged with Ar and stirred for 16 h. The control of the reaction was performed with TLC. The eluent for TLC was DCM/MeOH = 19:1. At the next step, the solvent was removed under reduced pressure and re-evaporated with DCM twice. The residue was dissolved in DCM (50 mL), and extracted with 1) H2O (2 × 50 mL), 2) brine (2 × 50 mL). Then, the organic fraction was dried over Na2SO4. After the solvent was removed under reduced pressure, the residue was purified by column chromatography (Puriflash 15μ 40g, eluent: DCM(100%)/MeOH(0%) => DCM(90%)/MeOH(10%) for 30 min, after MeOH (100%) for 5 min. Compound 9 was obtained as a yellow oil (853 mg, 70% yield).
Compound 10. To a solution of 9 (1 eq.; 840 mg; 0.995 mmol) in DMF (7 mL), Et2NH (10 eq.; 1029 μL; 9.95 mmol) was added, then the resulting mixture was purged with Ar and stirred for 1 h. The control of the reaction was performed with TLC. The eluent for TLC was DCM/MeOH = 19:1. At the next step, the solvent was removed under reduced pressure and re-evaporated with DCM twice. The residue was purified by column chromatography (Puriflash 15μ 25g, eluent: DCM(100%)/MeOH(0%) => DCM(90%)/MeOH(10%) for 30 min, after MeOH (100%) for 5 min. Compound 10 was obtained as a yellow oil (490 mg, 79% yield).
1H-NMR (400 MHz, DMSO-d6, δ): 8.14 (d, J = 7.9 Hz, 1H, F6NH), 8.06 (d, J = 7.7 Hz, 1H, K7NH), 7.90 (t, J = 5.7 Hz, 1H, X8NH), 7.29–7.09 (m, 10H, Ph + Ph), 6.77 (t, J = 5.5 Hz, 1H, K7NHk), 4.64–4.54 (m, 1H, F6Ha), 4.21–4.09 (m, 1H, K7Ha), 3.40–3.29 (m, 4H, F5Ha + F6Hb(a) + X8Hg), 3.11 (q, J = 6.0, 6.5 Hz, 2H, X8Ha), 2.97 (dd, J = 13.8, 4.9 Hz, 1H, F6Hb(b)), 2.91–2.78 (m, 4H, F5Hb(ab) + K7He), 1.73 (br.s., 2H, F5NH2), 1.69–1.56 (m, 3H, X8Hb + K7Hb(a)), 1.56–1.44 (m, 1H, K7Hb(b)), 1.36 (s, 9H, tBu), 1.35–1.29 (m, 2H, K7Hd), 1.28–1.11 (m, 2H, K7Hg).
Compound 15. Activation of 2-CTC. The mixture of 2-CTC (1 eq.; 1 g; 1.2–1.4 mmol/g; 100–200 mesh) in DCM (10 mL) was stirred for 10 min, then the mixture was purged with Ar, then SOCl2 (3 eq.; 305 µL; 4.2 mmol) was added dropwise, and then DMF (16 µL; 5% V/V to SOCl2) was added and stirred at 40 °C for 4 h. After that, the resin was filtered and transferred to a polypropylene reactor and washed with DMF (3 × 10 mL, 1 min) and DCM (3 × 10 mL, 1 min).
The addition of FmocLys(L)(NHBoc)-OH. To the mixture of CTC-2 (1 equiv; 1 g; 1.2–1.4 mmol/g; 100–200 mesh) in DMF (10 mL), FmocLys(NHBoc)-OH (2 eq.; 1.312 g; 2.8 mmol) and DIPEA (10 eq.; 2.44 mL; 14 mmol) were added, and the mixture was stirred for 2 h. Then, the resin was filtered off and washed with MeOH (3 × 10 mL, 5 min), DCM (3 × 0 mL, 1 min), DMF (3 × 10 mL, 1 min), and DCM (3 × 10 mL, 1 min).
Deprotection of Fmoc. FmocK(NHBoc) on a 2-CTC resin (1 eq.) was washed with DMF (2 × 15 mL, 1 min), then 4-methylpiperidine in DMF (20%/80% V/V, 15 mL) was added and stirred for 15 min, then the resin was filtered off and washed with DMF (3 × 15 mL, 1 min), then 4-methylpiperidine in DMF (20%/80% V/V,15 mL) was added and stirred for 15 min. After the resin was filtered off, the resulting solution was washed with DMF (3 × 15 mL, 1 min) and DCM (3 × 15 mL, 1 min).
The addition of FmocPhe(L)-OH. To the mixture of NH2-K(NHBoc) on a CTC-2 resin (1 eq.) in DMF (15 mL), FmocPhe(L)-OH (2 eq.; 1.085 g; 2.8 mmol), HOBt (0.5 eq.; 95 mg; 0.7 mmol), HBTU (2 eq.; 1.062 g; 2.8 mmol), and DIPEA (3 eq.; 0.73 mL; 4.2 mmol) were added and stirred for 2 h. Then the resin was filtered off and washed with DMF (3 × 15 mL, 1 min) and DCM (3 × 15 mL, 1 min).
Deprotection of Fmoc. FmocFK(NHBoc) on a CTC-2 resin (1 eq.) was washed with DMF (2 × 15 mL, 1 min), then 4-methylpiperidineine in DMF (20%/80% V/V, 15 mL) was added and stirred for 15 min, then the resin was filtered off and washed with DMF (3 × 15 mL, 1 min), then 4-methylpiperidine in DMF (20%/80% V/V, 15 mL) was added and stirred for 15 min. After the resin was filtered off, DMF (3 × 15 mL, 1 min) and DCM (3 × 15 mL, 1 min) wash was carried out.
Addition of FmocPhe(L)-OH. To the mixture of NH2-FK(NHBoc) on a CTC-2 resin (1 eq.) in DMF (15 mL), FmocPhe(L)-OH (2 eq.; 1.085 g; 2.8 mmol), HOBt (0.5 eq.; 95 mg; 0.7 mmol), HBTU (2 eq.; 1.062 g; 2.8 mmol), and DIPEA (3 eq.; 0.73 mL; 4.2 mmol) were added and stirred for 2 h. Then, the resin was filtered off and washed with DMF (3 × 15 mL, 1 min) and DCM (3 × 15 mL, 1 min).
Deprotection of Fmoc. FmocFFK(NHBoc) on a CTC-2 resin (1 eq.) was washed with DMF (2 × 15 mL, 1 min), then 4-methylpiperidineine in DMF (20%/80% V/V, 15 mL) was added and stirred for 15 min. Then, the resin was filtered off, washed with DMF (3 × 15 mL, 1 min), then 4-methylpiperidine in DMF (20%/80% V/V, 15 mL) was added and stirred for 15 min. After the resin was filtered off, DMF (3 × 15 mL, 1 min) and DCM (3 × 15 mL, 1 min) wash was carried out. Thus, the NH2-FFK(NHBoc) tripeptide was obtained on 2-CTC resin (1.95 g, ~1.4 mmol).
Compound 16. To the mixture of tripeptide 15 NH2-F5F6K7(NHBoc) on 2-CTC resin (1 eq.; 463 mg; 0,33 mmol) in DMF (5 mL) in a polypropylene reactor, compound 6 (1.2 eq.; 327 mg; 0,396 mmol), HOBt (0.5 eq.; 22 mg; 0.165 mmol), HBTU (2 eq.; 250 mg; 0.66 mmol), and DIPEA (3 eq.; 172 μL; 0.99 mmol) were added. The mixture was stirred for 2 h. Then, the solvent was removed by filtration on a porous reactor filter and the resin was washed with DMF (3 × 5 mL), DCM (3 × 5 mL), and then dried from residue of solvents.
After that, a mixture of DCM/TFA (99.25%/0.75%, 6.5 mL) was added to the resin and stirred for 15 min, then the solution was filtered off from the resin. The solvent was removed under reduced pressure and the residue was re-evaporated three times with DCM. The product was purified by column chromatography (Puriflash, column of PF-15C18AQ-F0025 (15μ 40g), eluent: H2O(80%)/MeCN(20%) => H2O(0%)/MeCN (100%) for 15 min after MeCN (100%) for 5 min. Compound 16 was obtained as a colorless oil (338 mg, 76% yield).
1H-NMR (400 MHz, DMSO-d6, δ): 12.53 (br.s., 1H, K7COOH), 8.18 (d, J = 7.5 Hz, 2H, F5NH + F6NH), 7.99–7.90 (m, 1H, K7NH), 7.89 (t, J = 5.2 Hz, m) & 7.86 (t, J = 5.2 Hz, n) (1H, X3NHk, m + n, m/n = 3/2), 7.42–7.08 (m, 14H, Ph + Ph + X9H), 6.79 (t, J = 5.1 Hz, 1H,K7NHk), 6.35–6.22 (m, 2H, K2NH + E1NH, m + n), 4.60–4.48 (m, F6Ha + X9Ha(n)), 4.48 (s, X9Ha(m), m + n, m/n = 3/2), 4.40–4.30 (m, 1H, F5Ha), 4.20–4.09 (m, 1H, K7Ha), 4.08–4.00 (m, 1H, E1Ha), 4.00–3.90 (m, 1H, K2Ha), 3.22 (t, J = 7.3 Hz, n) & 3.17 (t, J = 7.3 Hz, m) (2H, K2He, m + n, m/n = 3/2), 3.14–3.06 (m, 1H, F6Hb(a)), 3.05–2.82 (m, 6H, F6Hb(b) + X3He + K7He + F5Hb(a)), 2.70–2.57 (m, 1H, F5Hb(b)), 2.37–2.11 (m, 8H, X4Hb + E1Hg + X4Ha + X3Ha), 1.91–1.80 (m, 1H, E1Hb(a)), 1.77–1.12 (m, 19H, E1Hb(b) + K7Hb(a) + K2Hb(a) + K7Hb(b) + K2Hb(b) + X3Hb + X3Hd + K2Hd + K2Hg + X3Hg, m + n), 1.41–1.32 (m, 36H, tBu).
13C-NMR (100 MHz, DMSO-d6, δ): 173.35 (K7C), 172.24 (K2C(n)), 172.20 (K2C(m)), 172.14 (X3C(n)+ F6C(m)), 172.12 (X3C(m)), 172.08 (F6C(n)), 171.92 (E1C), 171.45 (E1Cd), 171.39 (X4Cg(mn)), 171.07 (F5C(mn)), 171.00 (X4C(mn)), 157.14 (U(m)), 157.12 (U(n)), 155.60 (K7Boc), 141.17 (X9Cb(m)), 140.77 (X9Cb(n)),138.13 (F6Cg), 137.99 (F5Cg), 133.43 (X9Ce(n)), 133.07 (X9Ce(m)), 130.60 (X9Cd(n)), 130.27 (X9Cd(m)), 129.18 (F6Cd), 129.07 (F5Cd), 128.09 (F6Ce), 128.03 (F5Ce), 127.21 (X9Ct(m)), 127.15 (X9Ck(n)), 126.86 (X9Ck(m)), 126.31 (F6Ck), 126.25 (F5Ck), 126.19 (X9Ct(n)),126.06 (X9Cg(m)), 124.95 (X9Cg(n)), 80.58 (E1tBu), 80.41 (K2tBu(m)), 80.32 (K2tBu(n)), 79.77 (E1dtBu), 77.37 (K7BoctBu), 54.43 (F5Ca), 53.79 (F6Ca(m)), 52.99 (K2Ca(n)), 52.86 (K2Ca(m)), 52.18 (E1Ca), 52.00 (K7Ca), 49.60 (X9Ca(n)), 47.09 (X9Ca(m)), 46.79 (K2Ce(m)), 45.20 (K2Ce(n)), 39.10 (K7Ce(mn), 38.61 (X3Ce(m)), 38.55 (X3Ce(n)), 37.07 (F5Cb), 36.95 (F6Cb), 32.32 (X3Ca(n)), 31.95 (X3Ca(m)), 31.82 (K2Cb), 30.91 (E1Cg), 30.77 (X4Ca + K7Cb), 30.63 (X4Cb), 29.19 (K7Cd), 29.09 (X3Cd(m)), 28.99 (X3Cd(n)), 28.29 (tBuK7), 27.75 (tBuE1), 27.66 (tBuK2+ K2Cd(m)), 27.63 (tBuE1d+ E1Cb), 26.71 (K2Cd(n)), 26.31 (X3Cg(m)), 26.22 (X3Cg(n)), 24.75 (X3Cb(m)), 24.60 (X3Cb(n)), 22.73 (K7Cg), 22.44 (K2Cg(n)) 22.26 (K2Cg(m)).
ESI-MS C70H103ClN8O16: m/z calcd. for [M + H+]+:1347.72, found:1347.55.
HRMS (m/z, ESI): calcd for C70H103ClN8O16-[M + H]+1347.7253, found: 1347.7236, 1369.7073 [M + Na]+, 1385.6801 [M + K]+.
Compound 11. Scheme 2. Method 2. To a solution of compound 6 (1 eq.; 245 mg; 0.257 mmol) in DMF (15 mL), DIPEA (1.5 eq.; 66 μL; 0.385 mmol), HOBt(Cl) (1.2 eq.; 44 mg; 0.308 mmol), and HBTU (1.2 eq.; 97 mg; 0.308 mmol) were added, then the resulting mixture was purged with Ar and stirred for 120 min, then compound 10 (1 equiv; 160 mg; 0.257 mmol) was added and the mixture was stirred for 24 h under Ar atmosphere. Then, the solvent was removed under reduced pressure and the residue was dissolved in DCM (25 mL), then extraction was carried out: (1) H2O (2 × 30 mL), (2) brine (2 × 30 mL). Then, the organic fraction was dried over Na2SO4. After the solvent was removed under reduced pressure, the residue was purified by column chromatography (Puriflash 15μ 25g, eluent: DCM(98%)/MeOH(2%) => DCM(92%)/MeOH(8%) for 40 min, where the eluent for TLC was DCM/MeOH = 19:1. As a result, several fractions were obtained with the content of the claimed substance from 21% to 57%. Re-purification was performed using column chromatography (Puriflash on the column PF-15C18HP-F0035 (15μ 35g); eluent: H2O(70%)/MeCN(30%) => H2O(0%)/MeCN(100%) for 15 min, after MeCN (100%) for 5 min. Compound 12 was obtained as a colorless oil (244 mg, 66% yield).
1H-NMR (400 MHz, CDCl3, δ): 8.02–7.93 (m, 1H, F5NH), 7.87–7.72 (m, 1H, X3NH(mn)), 7.43–6.98 (m, 15H, X9H(mn) + F6NH + Ph + Ph), 6.98–6.88 (m, 1H, X8NH(mn), 6.30–6.14 (m, 1H, K7NH(m + n), 5.52–5.27 (m, 2H, K2NH(m + n)+ E1NH(m + n), 5.05–4.90 (m, 1H, K7NHk(m + n)), 4.60–4.18 (m, 7H, X9Ha(n) + F6Ha + X9Ha(m) + F5Ha + K7Ha + E1Ha + K2Ha), 3.41–3.00 (m, 10H, X8Hg + K2He + X8Ha + X3He + K7He), 2.97–2.85 (m, 1H, F6Hb(a)), 2.84–2.72 (m, 1H, F6Hb(b)), 2.71–2.59 (m, 1H, F5Hb(a)), 2.47–2.38 (m, 1H, F5Hb(b)), 2.36–2.11 (m, 8H, X4Hb(mn) + X4Ha + X3Ha(mn) + E1Hg), 2.12–1.97 (m, 1H, E1Hb(a)), 1.91–1.77 (m, 3H, X8Hb + E1Hb(b)), 1.77–1.68 (m, 2H, K7Hb(a) + K2Hb(a)), 1.68–1.10 (m, 16H, K7Hb(b) + K2Hb(b) + X3Hb + X3Hd + K7Hd + K2Hd + K7Hg + K2Hg + X3Hg, m + n), 1.46–1.38 (m, 36H, tBu).
ESI-MS C73H109ClN12O15: m/z calcd. for [M + H+]+: 1429.79, found: 1430.60.
Scheme 3. Method 1. To a solution of compound 16 (1 eq.; 30 mg; 0.022 mmol) in DMF (3 mL) NH2-(CH2)3-N3 (2 eq.; 4 mg; 0.044 mmol), HOBt (1.2 eq.; 4 mg; 0.0264 mmol), HBTU (1.2 eq.; 10 mg; 0.0264 mmol), and DIPEA (1.5 eq.; 6 μL; 0.033 mmol) were added. The mixture was stirred for 24 h in an inert atmosphere. Then, the solvent was removed under reduced pressure and was twice reevaporated with DCM. The residue was purified by column chromatography (Puriflash on column PF-15C18AQ-F0004 (15μ 4g)); eluent: system H2O(80%)/MeCN(20%) => H2O(0%)/MeCN (100%) for 10 min, after MeCN (100%) for 5 min. Compound 11 was obtained as a colorless oil (21 mg, yield 67%).
1H-NMR (400 MHz, DMSO-d6, δ): 8.34 (d, J = 7.2 Hz, 1H, F5NH), 8.19 (d, J = 7.5 Hz, 1H, F6NH), 8.01–7.89 (m, 1H, X3NHk(mn), m/n = 3/2), 7.77–7.67 (m, 1H, K7NH(mn)), 7.65–7.56 (m, 1H, X8NH(mn)), 7.42–7.09 (m, 14H, Ph + Ph + X9H(mn)), 6.79 (t, J = 5.1 Hz, 1H,K7NHk), 6.34–6.21 (m, K2NH(mn) + E1NH(mn)), 4.59–4.39 (m, 3H, X9Ha(n) + X9Ha(m) + F6Ha), 4.37–4.25 (m, 1H, F5Ha), 4.17–4.07 (m, 1H, K7Ha), 4.05–4.00 (m, 1H, E1Ha), 4.00–3.91 (m, 1H, K2Ha), 3.33 (t, J = 6.9 Hz, 2H, X8Hg), 3.21 (t, J = 7.3 Hz, n) & 3.16 (t, J = 7.3 Hz, m) (2H, K2He, m + n, m/n = 3/2), 3.14–2.81 (m, 9H, X8Ha + F6Hb(a) + F6Hb(b) + X3He(mn) + K7He + F5Hb(a)), 2.71–2.60 (m, 1H, F5Hb(b)), 2.38–2.12 (m, 8H, X4Hb + E1Hg + X4Ha + X3Ha), 1.93–1.80 (m, 1H, E1Hb(a)), 1.72–1.60 (m, 4H, E1Hb(b) + X8Hb + K7Hb(a)), 1.60–1.10 (m, 17H, K2Hb(a) + K7Hb(b) + K2Hb(b) + X3Hb + X3Hd + K2Hd + K2Hg + X3Hg, m + n), 1.41–1.32 (m, 36H, tBu).
ESI-MS C73H109ClN12O15: m/z calcd. for[M − H+]−: 1429.79, found: 1430.70.
HRMS (m/z, ESI): calcd. for C73H109ClN12O15-[M + Na]+: 1451.7748, found: 1451.7716.
Compound 12. Scheme 2. Compound 11 (1 eq.; 243 mg; 0.17 mmol) was dissolved in mixture of DCM/TFA (9 mL of DCM, 1 mL of TFA). The mixture was stirred for 12 h, then the solvent was removed under reduced pressure and re-evaporated with DCM three times. The product was precipitated with Et2O and washed twice with Et2O (10 mL). After, the residue was purified by column chromatography (Puriflash on a column of PF-15C18AQ-F0025 (15μ 25g), eluent: H2O(80%)/MeCN(20%) => H2O(0%)/MeCN(100%) for 15 min after MeCN (100%) for 5 min. Compound 12 was obtained as a colorless oil (166 mg, yield 84%).
1H-NMR (400 MHz, DMSO-d6, δ): 8.76–8.68 (m, F5NH(m)), 8.60–8.53 (m, F5NH(n)), 8.54–8.43 (m, F6NH(m) + X3NHk(m)), 8.42–8.37 (m, F6NH(n)), 8.36–8.27 (m, X3NHk(n), m/n = 3/2), 7.76–7.62 (m, 1H, K7NH(mn)), 7.59–7.46 (m, 1H, X8NH(n) + X8NH(m)), 7.43–7.07 (m, 14H, Ph + Ph + X9H(mn)) 6.43–6.23 (m, K2NH(mn) + E1NH(mn)), 4.59–4.44 (m, 2H, X9Ha(n) + X9Ha(m)), 4.43–4.33 (m, 1H, F6Ha), 4.26–4.16 (m, 1H, F5Ha), 4.16–4.05 (m, 1H, K7Ha), 4.04–3.91 (m, 2H, E1Ha + K2Ha), 3.33 (t, J = 6,9 Hz, 2H, X8Hg), 3.25–2.95 (m, 8H, K2He(mn) + X8Ha + F6Hb(a) + F6Hb(b) + X3He(mn), 2.94–2.82 (m, 1H, F5H(a)), 2.73 (t, J = 7.5 Hz, 2H, K7He), 2.70–2.61 (m, 1H, F5Hb(b)), 2.44–2.26 (m, X4Hb(mn) + X4Ha(a) + X3Ha(m)), 2.25–2.11 (m, E1Hg + X3Ha(n) + X4Ha(b)), 1.84-1.69 (m, 3H, E1Hb(a) + E1Hb(b) + K7Hb(a)), 1.69–1.60 (m, 3H, X8Hb + K2Hb(a)), 1.60–1.31 (m, 10H, K7Hb(b) + K2Hb(b) + K7Hd + X3Hb + X3Hd + K2Hd), 1.30–1.12 (m, 6H, K2Hg + K7Hg + X3Hg).
13C-NMR (100 MHz, DMSO-d6, δ): 175.56 (K2C(m)), 175.32 (K2C(n)), 175.06 (E1C(m)), 174.99 (E1C(n)), 174.62 (E1Cd(mn)), 173.66 (X4Cg(m)), 173.40 (X4Cg(n)), 172.32 (F5C(mn)), 172.23 (X3C(m)), 172.13 (X3C(n)), 171.97 (X4C(m)), 171.84 (X4C(n)), 171.26 (K7C(mn)), 171.16 (F6C(m)), 171.08 (F6C(n)), 157.38 (U(mn)), 141.22 (X9Cb(m)), 140.79 (X9Cb(n)), 138.16 (F6Cg(m)), 138.08 (F6Cg(n)), 137.89 (F5Cg(mn)), 133.40 (X9Ce(n)), 133.04 (X9Ce(m)), 130.61 (X9Cd(n)), 130.24 (X9Cd(m)), 129.00 (F6Cd), 128.93 (F5Cd), 128.27 (F6Ce), 128.14 (F5Ce), 127.21 (X9Ct(m)), 127.13 (X9Ck(n)), 126.84 (X9Ck(m)), 126.38 (F6Ck), 126.34 (F5Ck), 126.28 (X9Ct(n)), 126.09 (X9Cg(m)), 125.00 (X9Cg(n)), 55.97 (F5Ca(m)), 55.65 (F5Ca(n)), 55.27 (F6Ca(m)), 55.07 (F6Ca(n)), 53.17 (K2Ca(m)), 52.88 (K2Ca(n) + E1Ca(mn) + K7Ca(mn)), 49.54 (X9Ca(n)), 48.20 (X8Cg), 47.05 (X9Ca(m)), 46.94 (K2Ce(m)), 44.89 (K2Ce(n)), 38.76 (X3Ce(m)), 38.68 (X3Ce(n)), 38.50 (K7Ce(mn), 36.62 (F5Cb(mn)), 36.25 (F6Cb(n)), 36.10 (F6Cb(m)), 35.87 (X8Ca), 32.51 (K2Cb(m)), 32.28 (K2Cb(n) + X3Ca(n)), 31.76 (X3Ca(m)), 31.17 (K7Cb + E1Cg), 30.72 (X4Ca), 30.46 (X4Cb), 28.93 (E1Cb + X3Cd(n)), 28.73 (X3Cd(m)), 28.20 (X8Cb), 27.93 (K2Cd(m)), 26.89 (K2Cd(n) + K7Cd(mn)), 26.13 (X3Cg(mn)), 24.63 (X3Cb(mn)), 22.66 (K7Cg(m)), 22.58 (K7Cg(n)), 22.40 (K2Cg(n)), 22.33 (K2Cg(m)).
ESI-MS C56H77ClN12O13: m/z calcd. for [M + H+]+:1161.55, found: 1161.55.
Scheme 3. 11 (1 eq.; 39 mg; 0.027 mmol) was dissolved in the system of DCM/TFA/TIPS/H2O (46.25%/46.25%/2.5%/5%; V/V respectively, 2 mL). The mixture was stirred for 3 h, then the solvent was removed under reduced pressure and re-evaporated with DCM three times. The product was precipitated with Et2O and washed twice with Et2O (1 mL). After, the compound was purified by column chromatography (Puriflash on the column PF-15C18AQ-F0004 (15μ 4g), eluent: H2O(80%)/MeCN (20%) => H2O(0%)/MeCN (100%) for 15 min, after MeCN (100%) for 5 min. Individual 12 was obtained as a colorless oil (28 mg, yield 88%).
1H-NMR (400 MHz, DMSO-d6, δ): 8.76–8.67 (m, F5NH(m)), 8.60–8.53 (m, F5NH(n)), 8.52–8.42 (m, F6NH(m) + X3NHk(m)), 8.42–8.35 (m, F6NH(n)), 8.35–8.26 (m, X3NHk(n), m/n = 3/2), 7.75–7.62 (m, 1H, K7NH(mn)), 7.59–7.41 (m, 1H, X8NH(n) + X8NH(m)), 7.43–7.07 (m, 14H, Ph + Ph + X9H(mn)) 6.43–6.23 (m, K2NH(mn) + E1NH(mn)), 4.54–4.44 (m, 2H, X9Ha(n) + X9Ha(m)), 4.43–4.33 (m, 1H, F6Ha), 4.27–4.17 (m, 1H, F5Ha), 4.17–4.07 (m, 1H, K7Ha), 4.04–3.91 (m, 2H, E1Ha + K2Ha), 3.33 (t, J = 6,9 Hz, 2H, X8Hg), 3.25–2.95 (m, 8H, K2He(mn) + X8Ha + F6Hb(a) + F6Hb(b) + X3He(mn), 2.94–2.85 (m, 1H, F5H(a)), 2.73 (t, J = 7.5 Hz, 2H, K7He), 2.70–2.61 (m, 1H, F5Hb(b)), 2.44–2.26 (m, X4Hb(mn) + X4Ha(a) + X3Ha(m)), 2.25–2.11 (m, E1Hg + X3Ha(n) + X4Ha(b)), 1.84–1.69 (m, 3H, E1Hb(a) + E1Hb(b) + K7Hb(a)), 1.69–1.60 (m, 3H, X8Hb + K2Hb(a)), 1.60–1.31 (m, 10H, K7Hd + K7Hb(b) + K2Hb(b) + X3Hb(m) + X3Hb(n) + X3Hd + K2Hd), 1.30–1.12 (m, 6H, K2Hg + K7Hg + X3Hg).
13C-NMR (100 MHz, DMSO-d6, δ): 175.62 (K2C(m)), 175.46 (K2C(n)), 175.22 (E1C(m)), 175.15 (E1C(n)), 174.75 (E1Cd(m)), 174.71 (E1Cd(n)), 173.54 (X4Cg(m)), 173.33 (X4Cg(n)), 172.30 (F5C(mn) + X3C(m)), 172.17 (X3C(n)), 171.99 (X4C(m)), 171.89 (X4C(n)), 171.39 (K7C(mn)), 171.23 (F6C(m)), 171.18 (F6C(n)), 157.51 (U(mn)), 141.26 (X9Cb(m)), 140.84 (X9Cb(n)), 138.18 (F6Cg(m)), 138.12 (F6Cg(n)), 137.96 (F5Cg(mn)), 133.50 (X9Ce(n)), 133.14 (X9Ce(m)), 130.65 (X9Cd(n)), 130.29 (X9Cd(m)), 129.11 (F6Cd), 129.03 (F5Cd), 128.31 (F6Ce), 128.20 (F5Ce), 127.26 (X9Ct(m)), 127.20 (X9Ck(n)), 126.90 (X9Ck(m)), 126.40 (F6Ck+ F5Ck), 126.32 (X9Ct(n)), 126.14 (X9Cg(m)), 125.03 (X9Cg(n)), 55.83 (F5Ca(m)), 55.60 (F5Ca(n)), 55.24 (F6Ca(m)), 55.10 (F6Ca(n)), 53.20 (K2Ca(m)), 52.95 (K2Ca(n) + E1Ca(mn) + K7Ca(mn)), 49.72 (X9Ca(n)), 48.29 (X8Cg), 47.19 (X9Ca(m)), 47.07 (K2Ce(m)), 45.26 (K2Ce(n)), 38.79 (X3Ce(m)), 38.69 (X3Ce(n)), 38.62 (K7Ce(mn), 36.78 (F5Cb(mn)), 36.42 (F6Cb(n)), 36.31 (F6Cb(m)), 35.95 (X8Ca), 32.55 (K2Cb(m)), 32.38 (K2Cb(n) + X3Ca(n)), 31.90 (X3Ca(m)), 31.23 (K7Cb), 31.05 (E1Cg), 30.81 (X4Ca), 30.58 (X4Cb), 28.96 (E1Cb), 28.90 (X3Cd), 28.29 (X8Cb), 28.05 (K2Cd(m)), 26.91 (K2Cd(n) + K7Cd(mn)), 26.28 (X3Cg(mn)), 24.76 (X3Cb(m)), 24.68 (X3Cb(n)), 22.68 (K7Cg(m)), 22.58 (K7Cg(n)), 22.53 (K2Cg(n)) 22.45 (K2Cg(m)).
ESI-MS C56H77ClN12O13: m/z calcd. for [M − H+]−:1161.55, found:1161.55.
HRMS (m/z, ESI): calcd. for C56H77ClN12O13-[M + H]+ 1161.5494, found: 1161,5505.
Compound 17. A solution of docetaxel (1 eq.; 500 mg; 0.619 mmol), hex-5-ynoic acid (1.1 eq.; 76 mg; 0.68 mmol), and DMAP (0.1 eq.; 7 mg; 0.062 mmol) in DCM was cooled to 0 °C. DIC (1.5 eq.; 117 mg; 0.928 mmol)) was then added dropwise. The reaction mixture was stirred for 4 h at 0 °C and then stirred at room temperature overnight. The solvent was evaporated under reduced pressure. The crude product was purified by chromatography ((Puriflash on column PF-15C18HP-F0040 (15μ 40g), eluent: Hex(95%)/EtOAc(5%) => Hex(0%)/EtOAc(100%) for 25 min after EtOAc (100%) for 5 min.). Compound 17 was obtained as a white crystalline powder (335 mg, yield 60%).
1H-NMR (400 MHz, DMSO-d6, δ): 7.99 (d, J = 7.2 Hz, 2H, 25 + 29), 7.89 (d, J = 8.9 Hz, 1H, NHBoc), 7.73 (t, J = 7.3 Hz, 1H, 27), 7.65 (t, J = 7.5 Hz, 2H, 26 + 28), 7.42 (t, J = 7.5 Hz, 2H, 35 + 37), 7.39–7.32 (m, 2H, 34 + 38), 7.17 (t, J = 7.2 Hz, 1H, 36), 5.83–5.70 (m, 1H, 13), 5.40 (d, J = 6.5 Hz, 1H, 2), 5.14–5.04 (m, 3H, 31 + 10 + 32), 5.02 (d, J = 7.2 Hz, 1H, 7OH), 4.93 (d, J = 2.5 Hz, 1H, 10OH), 4.92–4.87 (m, 1H, 5), 4.45(br.s, 1H, 1OH), 4.10–3.98 (m, 3H, 7 + 20a + 20b), 3.63 (d, J = 6.2 Hz, 1H, 3), 2.83 (t, J = 2.6 Hz, 1H, X10Hk), 2.55–2.50 (m, 2H, X10Hb), 2.32–2.25 (m, 1H, 6Hb), 2.24 (s, 3H, 22), 2.23–2.16 (m, 2H, X10Hd), 1.87–1.78 (m, 1H, 14Hb), 1.77–1.71 (m, 2H, X10Hg), 1.69 (s, 3H, 18), 1.67–1.59 (m, 1H, 6Ha), 1.55–1.46 (m, 4H, 19 + 14Ha), 1.38 (s, 9H, tBu), 0.98 (s, 6H, 16 + 17).
13C-NMR (100 MHz, DMSO-d6, δ): 209.35 (9), 171.81 (X10Ca), 169.61 (21), 169.11 (30), 165.32 (23), 155.21 (C(O)Boc), 137.49 (33), 136.93 (11), 135.93 (12), 133.46 (27), 130.06 (24), 129.60 (25/29), 128.71 (26/28), 128.61 (35/37), 128.10 (36), 127.46 (34/38), 83.80 (5), 83.51 (X10Ce), 80.30 (4), 78.52 (CBoc), 76.81 (1), 75.43 (20), 75.11 (31), 74.81 (2), 73.76 (10), 71.91 (X10Ck), 71.21 (13), 70.77 (7), 57.00 (8), 55.14 (32), 45.98 (3), 42.91 (15), 36.50 (6), 34.71 (14), 32.10 (X10Cb), 28.16 (tBu), 26.46 (16), 23.47 (X10Cg), 22.53 (22), 20.79 (17), 17.03 (X10Cd), 13.68 (18), 9.83 (19).
HRMS (m/z, ESI): calcd. for C49H59N15O13-[M + H]+ 902.3957, found: 902.3981.
Compound 18. Compounds 12 (1 eq.; 162 mg; 0.127 mmol) and 17 (1 eq.; 115 mg; 0.127 mmol), CuSO4·5H2O (0.4 eq.; 13 mg; 0.05 mmol) were dissolved in DMF/H2O (6 mL/1 mL). After, the system was purged with argon. To the mixture, sodium ascorbate (1.2 eq.; 30 mg; 0.152 mmol) was added in H2O (1 mL) with a syringe. The resulting solution was stirred for 24 h in an inert atmosphere. After, EDTA (0.8 eq.; 30 mg; 0.1 mmol) was added. The mixture was stirred for 3 h. After the reaction, the mixture was filtered from the precipitate and the solvent was removed under reduced pressure. The product was precipitated with MeCN and washed twice with MeCN (2 mL). After, the residue was purified by column chromatography (Puriflash on a column of PF-15C18AQ-F0025 (15μ 25g), eluent: H2O(90%)/MeCN(10%) => H2O(0%)/MeCN(100%) for 20 min after MeCN (100%) for 5 min. Compound 18 was obtained as a pink powder (99 mg, yield 38%).
1H-NMR (600 MHz, DMSO-d6, δ): 8.70-8.62 (m, F5NH(m)), 8.55-8.48 (m, F5NH(n)), 8.48–8.43 (m, F6NH(m)), 8.43–8.37 (X3NHk(m)), 8.37–8.31 (m, F6NH(n)), 8.28–8.20 (m, X3NHk(n)), 7.98 (d, J = 7.2 Hz, 2H, 25 + 29), 7.87 (d, J = 8.9 Hz, 1H, NHBoc), 7.83 (s, 1H, X10Hk), 7.76–7.68 (m, 2H, 27 + K7NH(mn)), 7.68–7.57 (m, 3H, 26 + 28 + X8NH(n) + X8NH(m)), 7.40 (t, J = 7.5 Hz, 2H, 35 + 37), 7.38–7.34 (m, 2H, 34 + 38), 7.35–7.06 (m, 15H, Ph + Ph + X9H(mn) + 36), 6.43–6.20 (m, 2H, K2NH(mn) + E1NH(mn)), 5.82–5.72 (m, 1H, 13), 5.39 (d, J = 6.5 Hz, 1H, 2), 5.13–5.00 (m, 3H, 31 + 10 + 32), 4.94 (br.s., 1H, OH), 4.90 (d, J = 9.4 Hz, 1H, 5), 4.57–4.47 (m, 2H, X9Ha(n) + X9Ha(m)), 4.45-4.37 (m, 2H, OH + F6Ha), 4.30 (t, J = 6.9 Hz,2H, X8Hg), 4.27-4.18 (m, 1H, F5Ha), 4.18–4.10 (m, 1H, K7Ha), 4.08–3.94 (m, 5H, 7 + 20a + E1Ha + 20b + K2Ha), 3.63 (d, J = 6.2 Hz, 1H, 3), 3.22–2.95 (m, 8H, K2He(mn) + F6Hb(a) + X8Ha +F6Hb(b) + X3He(mn)), 2.93–2.84 (m, 1H, F5Hb(a)), 2.74 (t, J = 7.5 Hz, 2H, K7He), 2.71–2.65 (m, 1H, F5Hb(b)), 2.62 (t, J = 7.0 Hz, 2H, X10Hd), 2.45 (t, J = 7.0 Hz, 2H, X10Hb), 2.42–2.11 (m, 9H, X4Hb(mn) + X4Ha(a) + X3Ha(m) + 6Hb + E1Hg + X3Ha(n) + X4Ha(b)), 2.23 (s. 3H, 22), 1.99–1.90 (m, 2H, X8Hb), 1.90–1.84 (m, 2H, X10Hg), 1.84–1.69 (m, 4H, 14Hb + E1Hb(a) + E1Hb(b) + K7Hb(a)), 1.70 (s. 3H, 18), 1.68–1.58 (m, 3H, 14Ha + 6Ha + K2Hb(a)), 1.58–1.31 (m, 10H, K7Hd + K7Hb(b) + K2Hb(b) + X3Hb(m) + X3Hb(n) +X3Hd(mn) + K2Hd(mn)), 1.50 (s. 3H, 19), 1.33 (s. 9H, tBu), 1.30–1.16 (m, 6H, K2Hg + K7Hg + X3Hg), 0.97 (s. 6H, 16 + 17).
13C-NMR (100 MHz, DMSO-d6, δ): 209.31 (9), 175.25 (K2C(m)), 175.21 (K2C(n)), 174.81 (E1C(m)), 174.76 (E1C(n)), 174.34 (E1Cd(m)), 174.25 (E1Cd(n)), 173.59 (X4Cg(mn)), 172.30 (F5C(mn)), 172.21 X3C(mn)), 172.07 (X10Ca + X3C(n)), 171.92 (X4C(m)), 171.87 (X4C(n)), 171.33 (K7C(mn)), 171.15 (F6C(m)), 171.10 (F6C(n)), 169.57 (21), 169.11 (30), 165.29 (23), 157.30 (U(mn)), 155.19 (C(O)Boc), 145.87 (X10Ce), 141.19 (X9Cb(m)), 140.76 (X9Cb(n)), 138.10 (F6Cg(m)), 138.02 (F6Cg(n)), 137.78 (F5Cg(mn)), 137.50 (33), 136.89 (11), 135.93 (12), 133.43 (27), 133.37 (X9Ce(n)), 133.01 (X9Ce(m)), 130.59 (X9Cd(n)), 130.22 (X9Cd(m)), 130.03 (24), 129.56 (25/29), 128.97 (F6Cd), 128.89 (F5Cd), 128.68 (26/28), 128.57 (35/37), 128.23 (F6Ce), 128.11 (F5Ce), 128.06 (36), 127.41 (34/38), 127.20 (X9Ct(m)), 127.15 (X9Ck(n)), 126.82 (X9Ck(m)), 126.36 (F6Ck), 126.30 (X9Ct(n) + F5Ck), 126.08 (X9Cg(m)), 124.99 (X9Cg(n)), 122.03 (X10Ck), 83.76 (5), 80.27 (4), 78.46 (CBoc), 76.80 (1), 75.40 (20), 75.01 (31), 74.79 (2), 73.72 (10), 71.16 (13), 70.74 (7), 56.97 (8), 55.89 (F5Ca(mn)), 55.20 F6Ca(mn)), 55.11 (32), 52.85 (K2Ca(mn) + E1Ca(mn)), 52.65 (K7Ca(mn)), 49.60 (X9Ca(n)), 47.00 (X9Ca(m)), 46.85 (X8Cg+ K2Ce(m)), 45.96 (3), 45.31 (K2Ce(n)), 42.88 (15), 38.71 (X3Ce(m)), 38.61 (X3Ce(n)), 38.50 (K7Ce(mn), 36.66 (F5Cb(mn)), 36.47 (6 + F6Cb(mn)), 35.82 (X8Ca), 34.69 (14), 32.65 (X10Cb), 32.30 (K2Cb(m)), 32.25 (K2Cb(n) + X3Ca(n)), 31.74 (X3Ca(m)), 31.09 (K7Cb), 30.92 (E1Cg), 30.70 (X4Ca), 30.48 (X4Cb), 29.81 (X8Cb), 28.66(E1Cb + X3Cd), 28.08 (tBu), 27.83 (K2Cd(m)), 26.85 (K2Cd(n) + K7Cd(mn)), 26.44 (16), 26.08 (X3Cg(mn)), 24.59 (X3Cb(m)), 24.56 (X3Cb(n)), 24.29 (X10Cg), 24.18 (X10Cd), 22.60 (K7Cg(mn)), 22.50 (22), 22.27 (K2Cg(mn)), 20.77 (17), 13.65 (18), 9.79 (19).
ESI-MS C105H136ClN13O28: m/z calcd. for [M + 2H+]2+ 1031.97, found:1032.60.
HRMS (m/z, ESI): calcd. for C105H136ClN13O28- [M + 2H]2+ 1031.9726, found: 1031,9761.
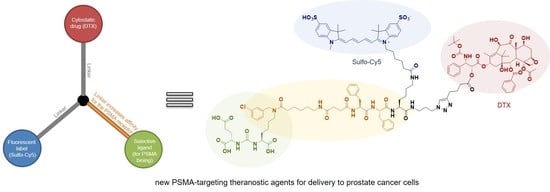
Compound 19. Compound 18 (1 eq.; 14 mg; 6.43 μmol) and DIPEA (8 eq.; 6.7 mg; 51.4 μmol) were dissolved in DMF (2 mL). After, the system was purged with argon. To the mixture, Sulfo-Cy5 NHS-ester (1 eq.; 5 mg; 6.43 μmol) was added. The mixture was stirred for 6 h. After, the solvent was evaporated under reduced pressure. The product was precipitated with MeCN and washed twice with MeCN (2 mL). After, the residue was purified by column chromatography (Puriflash on a column of PF-15C18AQ-F0025 (15μ 25g), eluent: H2O(90%)/MeCN(10%) => H2O(0%)/MeCN(100%) for 20 min after MeCN (100%) for 5 min. Compound 19 was obtained as a blue powder (15.8 mg, yield 92%).
1H-NMR (600 MHz, DMSO-d6, δ): 8.41–8.29 (m, 2H, ArSulfoCy5), 8.24–8.17 (m, 1H, ArSulfoCy5), 7.97 (d, J = 7.2 Hz, 2H, 25 + 29), 7.87 (d, J = 8.9 Hz, 1H, NHBoc), 7.84 (s, 1H, X10Hk), 7.82–7.79 (m, 1H, ArSulfoCy5), 7.79–7.73 (m, 1H, 27), 7.73–7.68 (m, 1H, ArSulfoCy5), 7.68–7.61 (m, 3H, 26 + 28 + ArSulfoCy5), 7.40 (t, J = 7.5 Hz, 2H, 35 + 37), 7.37–7.33 (m, 2H, 34 + 38), 7.34–7.06 (m, 16H, Ph + Ph + X9H(mn) + 36 + SulfoCy5(C=C)), 6.55 (t, J = 12.6 Hz, 1H, SulfoCy5(C=C)), 6.42–6.20 (m, 3H, SulfoCy5(C=C)), 5.82–5.74 (m, 1H, 13), 5.38 (d, J = 6.5 Hz, 1H, 2), 5.12–5.00 (m, 3H, 31 + 10 + 32), 4.95 (br.s., 1H, OH), 4.89 (d, J = 9.4 Hz, 1H, 5), 4.57–4.44 (m, 2H, X9Ha(n) + X9Ha(m)), 4.45–4.37 (m, 2H, OH + F6Ha), 4.35–4.23 (m, 3H, X8Hg + F5Ha), 4.17–3.94 (m, 6H, K7Ha + 7+20a + E1Ha + 20b + K2Ha), 3.62 (d, J = 6.2 Hz, 1H, 3), 3.57 (s, 3H, 28′), 3.36 (br.s, 1H, OH), 3.53–2.95 (m, 12H, 6′ + K2He(mn) + F6Hb(a) + X8Ha+ K7He + F6Hb(b) + X3He(mn)), 2.93–2.84 (m, 1H, F5H(a)), 2.69–2.63 (m, 1H, F5Hb(b)), 2.61 (t, J = 7.0 Hz, 2H, X10Hd), 2.46–2.40 (m, 2H, X10Hb), 2.37–2.11 (m, 9H, X4Hb(mn) + X4Ha(a) + X3Ha(m) + 6Hb + E1Hg + X3Ha(n) +X4Ha(b)), 2.22 (s. 3H, 22), 2.01 (t, J = 7.0 Hz, 2H, 2′), 1.96–1.90 (m, 2H, X8Hb), 1.90–1.84 (m, 2H, X10Hg), 1.84–1.70 (m, 4H, 14Hb + E1Hb(a) + E1Hb(b) + K7Hb(a)), 1.70–1.62 (m. 15H, 18 + 29′ + 30′ + 31′ + 32′), 1.68–1.58 (m, 3H, 14Ha + 6Ha + K2Hb(a)), 1.58–1.31 (m, 10H, K7Hd + K7Hb(b) + K2Hb(b) + X3Hb(m) + X3Hb(n) +X3Hd(mn) + K2Hd(mn)), 1.49 (s. 3H, 19), 1.32 (s. 9H, tBu), 1.30–1.16 (m, 12H, 5′ + 3′ + 4′ + K2Hg + K7Hg +X3Hg), 0.97 (s. 6H, 16 + 17).
ESI-MS C137H172ClN15O35S2: m/z calcd. for [M + 2H+]2+: 1344.57, found: 1345.05.
HRMS (m/z, ESI): calcd. for C137H172ClN15O35S2-[M + 2H+]2+ 1344.5725, found: 1344.5768.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}