
A Pyranoxanthone as a Potent Antimitotic and Sensitizer of Cancer Cells to Low Doses of Paclitaxel






 and
and 
Abstract
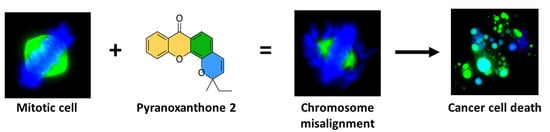
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Synthesis of Pyranoxanthone 2
2.2. The Pyranoxanthone 2 Has a Potent Growth Inhibitory Activity Against Tumor Cells
2.3. The Pyranoxanthone 2 Induces Mitotic Arrest of Cancer Cells
2.4. Treatment with Pyranoxanthone 2 Leads to Chromosome Congression Defects
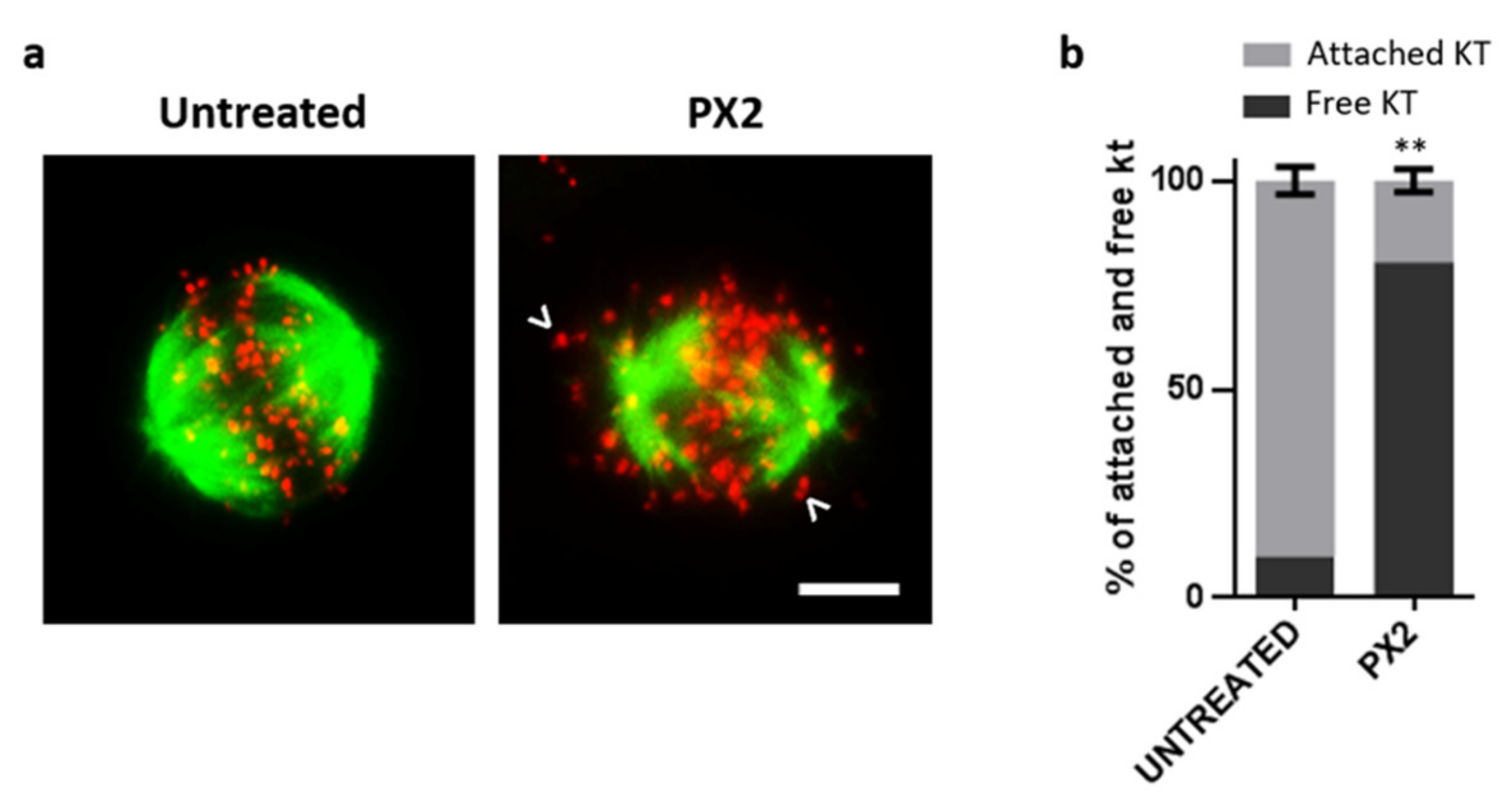
2.5. Treatment with Pyranoxanthone 2 Interferes with Kinetochore-Microtubules Attachments Stability
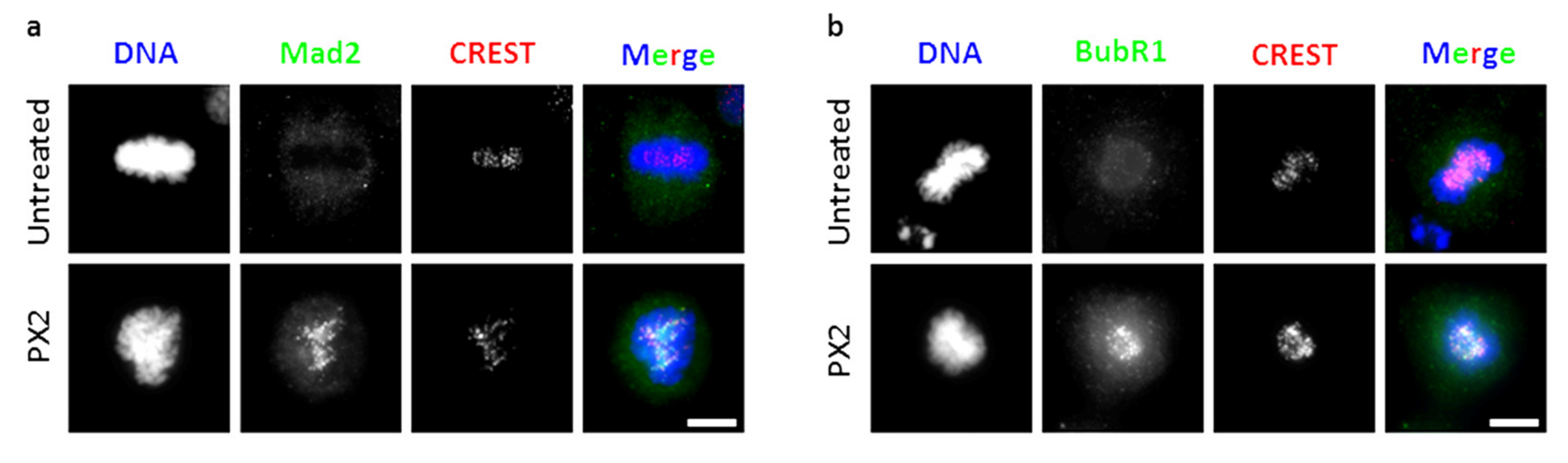
2.6. Treatment with Pyranoxanthone 2 Elicits Spindle Assembly Checkpoint Activation
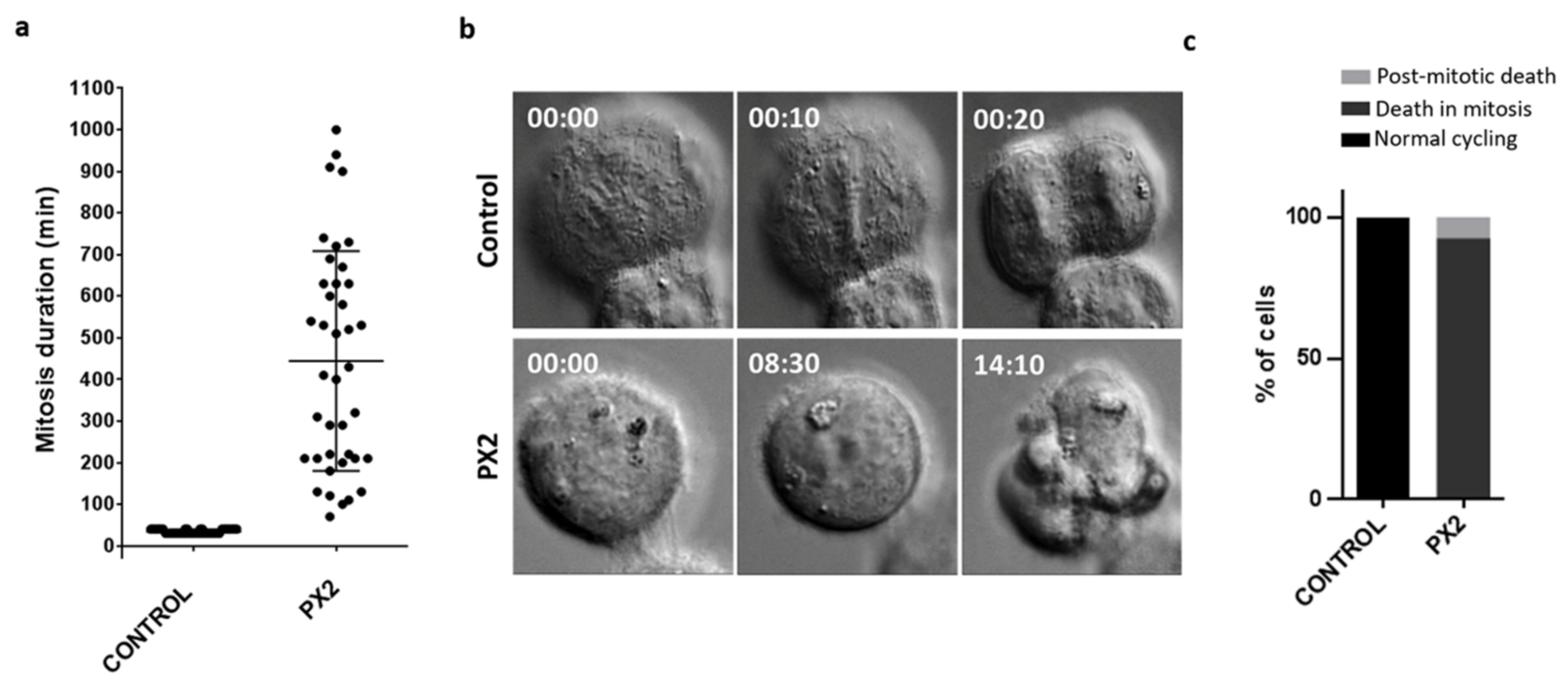
2.7. Cell Fates of Cancer Cells Arrested in Mitosis by the Pyranoxanthone 2
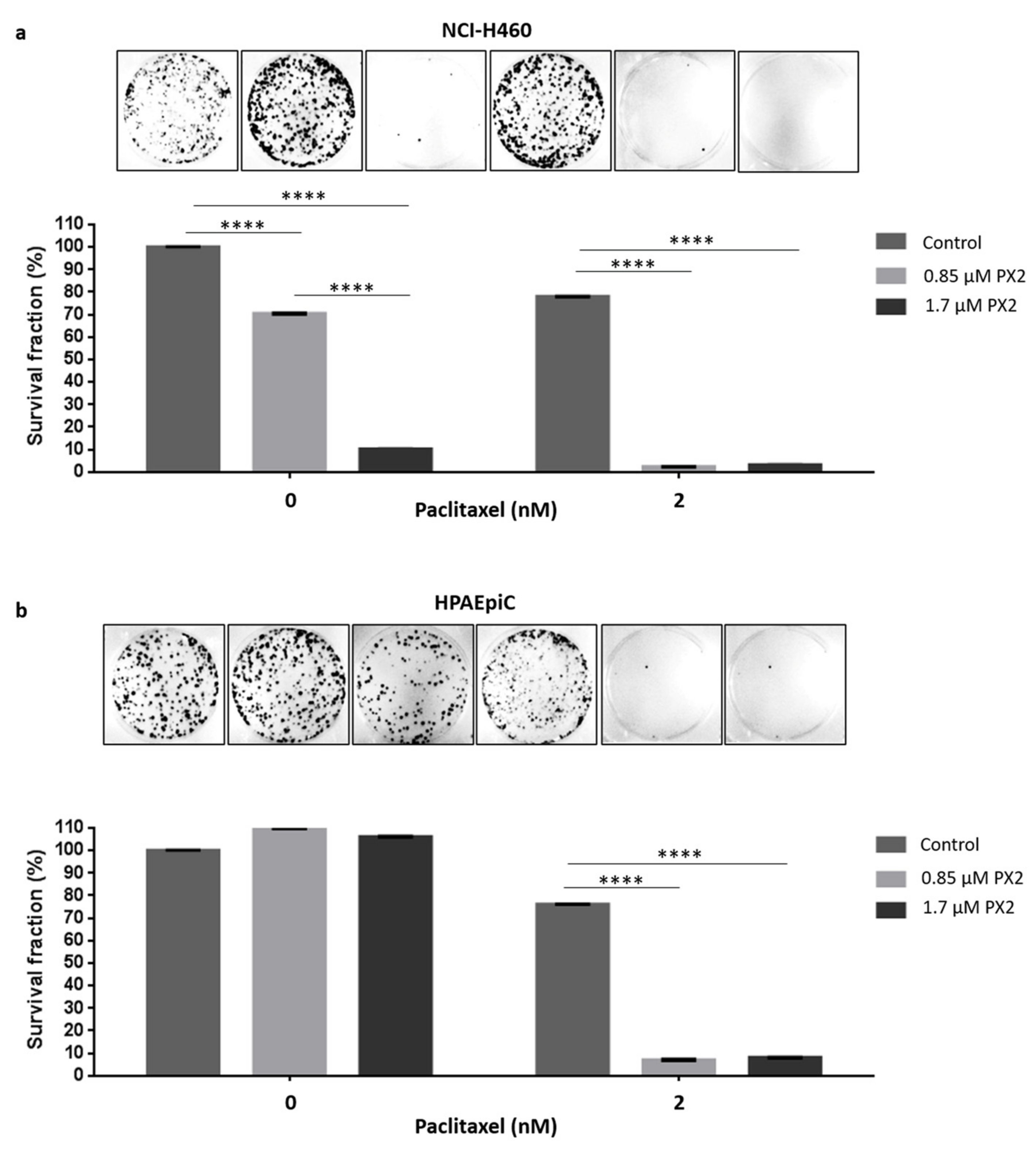
2.8. Treatment with Pyranoxanthone 2 Enhances Paclitaxel Cytotoxicity
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Methods
4.1.2. Synthesis of Methyl 2-(2-Methoxyphenoxy)Benzoate (4)
4.1.3. Synthesis of 4-((3-Methylpent-1-yn-3-yl)oxy)-9H-Xanthen-9-one (8)
4.1.4. Synthesis of 2-Ethyl-2-Methylpyrano[3,2-c]Xanthen-7(2H)-one (2)
4.1.5. Biological Assays
4.2. Cell Culture and Conditions
4.3. Sulforodamine B (SRB) Colorimetric Assay
4.4. Mitotic Index Determination
4.5. Flow Cytometry
4.6. Immunofluorescence
4.7. Functional Assays for Kinetochore-Microtubule (KT-MT) Attachments
4.7.1. Cold Treatment Assay
4.7.2. MG-132 Proteasome Inhibitor Assay
4.8. Terminal Deoxynucleotidyl Transferase-Mediated Nick end Labeling (TUNEL) Assay
4.9. Colony Formation Assay
4.10. Live-Cell Imaging
4.11. Image Acquisition and Processing
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Foley, K.; Kim, V.; Jaffee, E.; Zheng, L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett. 2016, 381, 244–251. [Google Scholar] [CrossRef] [Green Version]
- Boegemann, M.; Aydin, A.M.; Bagrodia, A.; Krabbe, L.-M. Prospects and progress of immunotherapy for bladder cancer. Expert Opin. Biol. Ther. 2017, 17, 1417–1431. [Google Scholar] [CrossRef] [PubMed]
- Kaboli, P.J.; Zhang, L.; Xiang, S.; Shen, J.; Li, M.; Zhao, Y.; Wu, X.; Zhao, Q.; Zhang, H.; Lin, L.; et al. Molecular Markers of Regulatory T Cells in Cancer Immunotherapy with Special Focus on Acute Myeloid Leukemia (AML)—A Systematic Review. Curr. Med. Chem. 2020, 27, 4673–4698. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Wu, X.; Yin, J.; Li, M.; Shen, J.; Li, J.; Zhao, Y.; Zhao, Q.; Wu, J.; Wen, Q.; et al. Identification of Genetic Mutations in Cancer: Challenge and Opportunity in the New Era of Targeted Therapy. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, D.P.; Polyak, K. Tumorigenesis: It takes a village. Nat. Rev. Cancer 2015, 15, 473–483. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Silva, P.; Barbosa, J.; Nascimento, A.V.; Faria, J.; Reis, R.; Bousbaa, H. Monitoring the fidelity of mitotic chromosome segregation by the spindle assembly checkpoint. Cell Prolif. 2011, 44, 391–400. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Musacchio, A. The Molecular Biology of Spindle Assembly Checkpoint Signaling Dynamics. Curr. Biol. 2015, 25, R1002–R1018. [Google Scholar] [CrossRef] [Green Version]
- Corbett, K.D. Molecular Mechanisms of Spindle Assembly Checkpoint Activation and Silencing. In Centromeres and Kinetochores; Black, E.B., Ed.; Springer International Publishing AG: Cham, Switzerland, 2017; Volume 56, pp. 429–455. [Google Scholar]
- Silva, P.M.A.; Reis, R.M.; Bolanos-Garcia, V.M.; Florindo, C.; Tavares, Á.A.; Bousbaa, H. Dynein-dependent transport of spindle assembly checkpoint proteins off kinetochores toward spindle poles. FEBS Lett. 2014, 588, 3265–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriques, A.C.; Ribeiro, D.; Pedrosa, J.; Sarmento, B.; Silva, P.M.A.; Bousbaa, H. Mitosis inhibitors in anticancer therapy: When blocking the exit becomes a solution. Cancer Lett. 2019, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Dias, T.A.; Brito, A.; Proença, F. Biological importance of structurally diversified chromenes. Eur. J. Med. Chem. 2016, 123, 487–507. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, D.R.P.; Soares, J.X.; Costa, J.C.; Magalhães, Á.F.; Azevedo, C.M.G.; Pinto, M.M.M.; Afonso, C.M.M. Structures, Activities and Drug-Likeness of Anti-Infective Xanthone Derivatives Isolated from the Marine Environment: A Review. Molecules 2019, 24, 243. [Google Scholar] [CrossRef] [Green Version]
- Masters, K.-S.; Bräse, S. Xanthones from Fungi, Lichens, and Bacteria: The Natural Products and Their Synthesis. Chem. Rev. 2012, 112, 3717–3776. [Google Scholar] [CrossRef]
- Adler, M.J.; Baldwin, S.W. Direct, regioselective synthesis of 2,2-dimethyl-2H-chromenes. Total syntheses of octandrenolone and precocenes I and II. Tetrahedron Lett. 2009, 50, 5075–5079. [Google Scholar] [CrossRef]
- Pinto, M.M.M.; Sousa, M.E.; Nascimento, M.S.J. Xanthone Derivatives: New Insights in Biological Activities. Curr. Med. Chem. 2005, 12, 2517–2538. [Google Scholar] [CrossRef]
- Kampkötter, A.; Nkwonkam, C.G.; Zurawski, R.F.; Timpel, C.; Chovolou, Y.; Wätjen, W.; Kahl, R. Investigations of protective effects of the flavonoids quercetin and rutin on stress resistance in the model organism Caenorhabditis elegans. Toxicology 2007, 234, 113–123. [Google Scholar] [CrossRef]
- Klein-Júnior, L.C.; Campos, A.; Niero, R.; Corrêa, R.; Vander Heyden, Y.; Filho, V.C. Xanthones and Cancer: From Natural Sources to Mechanisms of Action. Chem. Biodivers. 2020, 17. [Google Scholar] [CrossRef]
- Azevedo, C.M.G.; Afonso, C.M.M.; Soares, J.X.; Reis, S.; Sousa, D.; Lima, R.T.; Vasconcelos, M.H.; Pedro, M.; Barbosa, J.; Gales, L.; et al. Pyranoxanthones: Synthesis, growth inhibitory activity on human tumor cell lines and determination of their lipophilicity in two membrane models. Eur. J. Med. Chem. 2013, 69, 798–816. [Google Scholar] [CrossRef]
- Azevedo, C.M.G.; Afonso, C.M.M.; Sousa, D.; Lima, R.T.; Vasconcelos, M.H.; Pedro, M.; Barbosa, J.; Corrêa, A.G.; Reis, S.; Pinto, M.M.M. Multidimensional optimization of promising antitumor xanthone derivatives. Bioorg. Med. Chem. 2013, 21, 2941–2959. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Cai, Q. Copper/Amino Acid Catalyzed Cross-Couplings of Aryl and Vinyl Halides with Nucleophiles †. Acc. Chem. Res. 2008, 41, 1450–1460. [Google Scholar] [CrossRef] [PubMed]
- Maiti, D.; Buchwald, S.L. Cu-Catalyzed Arylation of Phenols: Synthesis of Sterically Hindered and Heteroaryl Diaryl Ethers. J. Org. Chem. 2010, 75, 1791–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azevedo, C.; Afonso, C.; Pinto, M. Routes to Xanthones: An Update on the Synthetic Approaches. Curr. Org. Chem. 2012, 16, 2818–2867. [Google Scholar] [CrossRef]
- Logarinho, E.; Resende, T.; Torres, C.; Bousbaa, H. The Human Spindle Assembly Checkpoint Protein Bub3 Is Required for the Establishment of Efficient Kinetochore–Microtubule Attachments. Mol. Biol. Cell 2008, 19, 1798–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebucci, M.; Michiels, C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem. Pharmacol. 2013, 85, 1219–1226. [Google Scholar] [CrossRef]
- Silk, A.D.; Zasadil, L.M.; Holland, A.J.; Vitre, B.; Cleveland, D.W.; Weaver, B.A. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc. Natl. Acad. Sci. USA 2013, 110, E4134–E4141. [Google Scholar] [CrossRef] [Green Version]
- Topham, C.H.; Taylor, S.S. Mitosis and apoptosis: How is the balance set? Curr. Opin. Cell Biol. 2013, 25, 780–785. [Google Scholar] [CrossRef]
- Ikui, A.E.; Yang, C.-P.H.; Matsumoto, T.; Horwitz, S.B. Low Concentrations of Taxol Cause Mitotic Delay Followed by Premature Dissociation of p55CDC from Mad2 and BubR1 and Abrogation of the Spindle checkpoint, Leading to Aneuploidy. Cell Cycle 2005, 4, 1385–1388. [Google Scholar] [CrossRef] [Green Version]
- Zasadil, L.M.; Andersen, K.A.; Yeum, D.; Rocque, G.B.; Wilke, L.G.; Tevaarwerk, A.J.; Raines, R.T.; Burkard, M.E.; Weaver, B.A. Cytotoxicity of Paclitaxel in Breast Cancer Is due to Chromosome Missegregation on Multipolar Spindles. Sci. Transl. Med. 2014, 6, 229ra43. [Google Scholar] [CrossRef] [Green Version]









Sample Availability: Samples of the compounds are not available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
França, F.; Silva, P.M.A.; Soares, J.X.; Henriques, A.C.; Loureiro, D.R.P.; Azevedo, C.M.G.; Afonso, C.M.M.; Bousbaa, H. A Pyranoxanthone as a Potent Antimitotic and Sensitizer of Cancer Cells to Low Doses of Paclitaxel. Molecules 2020, 25, 5845. https://doi.org/10.3390/molecules25245845
França F, Silva PMA, Soares JX, Henriques AC, Loureiro DRP, Azevedo CMG, Afonso CMM, Bousbaa H. A Pyranoxanthone as a Potent Antimitotic and Sensitizer of Cancer Cells to Low Doses of Paclitaxel. Molecules. 2020; 25(24):5845. https://doi.org/10.3390/molecules25245845
Chicago/Turabian StyleFrança, Fábio, Patrícia M. A. Silva, José X. Soares, Ana C. Henriques, Daniela R. P. Loureiro, Carlos M. G. Azevedo, Carlos M. M. Afonso, and Hassan Bousbaa. 2020. "A Pyranoxanthone as a Potent Antimitotic and Sensitizer of Cancer Cells to Low Doses of Paclitaxel" Molecules 25, no. 24: 5845. https://doi.org/10.3390/molecules25245845
APA StyleFrança, F., Silva, P. M. A., Soares, J. X., Henriques, A. C., Loureiro, D. R. P., Azevedo, C. M. G., Afonso, C. M. M., & Bousbaa, H. (2020). A Pyranoxanthone as a Potent Antimitotic and Sensitizer of Cancer Cells to Low Doses of Paclitaxel. Molecules, 25(24), 5845. https://doi.org/10.3390/molecules25245845