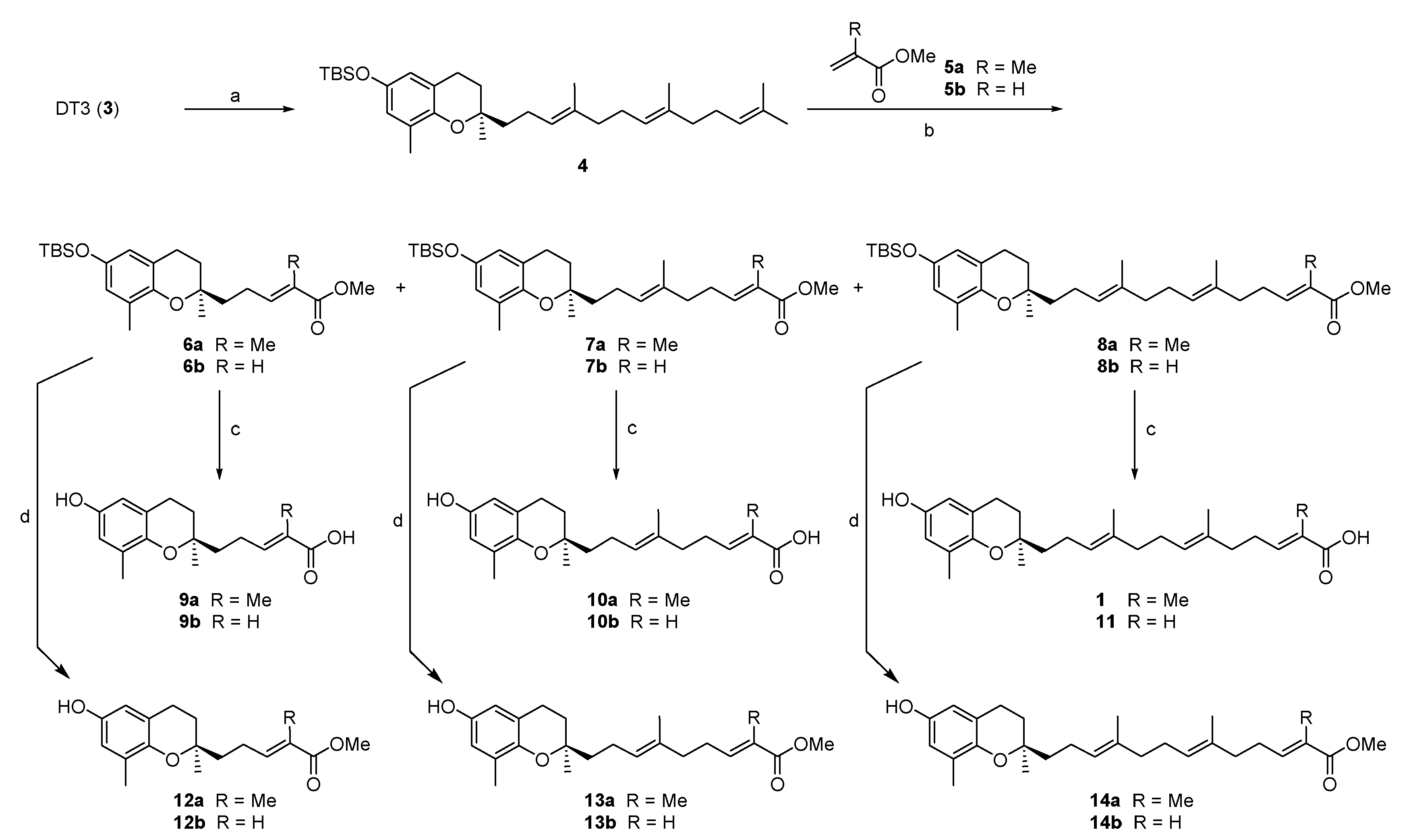
3.2. Chemistry
3.2.1. Synthesis of tert-butyl({(R)-2,8-dimethyl-2-[(3E,7E)-4,8,12-trimethyltrideca-3,7,11-trien-1-yl]-chroman-6-yl}oxy)dimethylsilane (4)
To a solution of δ-tocotrienol (7.9 g, 20.0 mmol) in CH2Cl2 (120 mL) at 0 °C was added imidazole (3.45 g, 50 mmol) and TBSCl (3.66 g, 24 mmol). The resulting mixture was stirred at room temperature for 5 h. The reaction mixture was partitioned between CH2Cl2 (100 mL) and water (75 mL). The aqueous layer was extracted with CH2Cl2 (2 × 50 mL), and the combined organic layers were washed with water (100 mL) and brine (50 mL), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was chromatographed on silica (hexanes/ethyl acetate 9:1) to afford compound 4 (9.5 g, 92%) as a colorless oil; 1H-NMR (CDCl3): δ 6.47 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.37 (d, J = 2.8 Hz, 1H, phenyl C7-H), 5.18–5.03 (m, 3H, C3′-H, C7′-H and C11′-H), 2.70 (t, J = 7.2 Hz, 2H, C4-H2), 2.15–1.96 (m, 13H, C9-H3, C2′-H2, C5′-H2, C6′-H2, C9′-H2 and C10′-H2), 1.81–1.73 (m, 2H, C3-H2), 1.69 (s, 3H, C13′-H3), 1.54–1.46 (m, 11H, C1′-H2, C4′-CH3, C8′-CH3, and C12′-CH3), 1.27 (s, 3H, C10-H3), 0.98 (s, 9H, Si-C(CH3)3), 0.17 (s, 6H, Si(CH3)2); 13C-NMR (CDCl3): δ 147.6 (phenyl C-6), 146.4 (phenyl C-8a), 135.2 (C-8′), 135.0 (C-4′), 131.3 (C-12′), 126.9 (phenyl C-8), 124.5 (C-3′), 124.4 (C-7′), 124.3 (C-11′), 120.9 (phenyl C-7), 120.2 (phenyl C-4a), 117.3 (phenyl C-5), 75.4 (C-2), 39.8 (3C, C-1′, C-5′ and C-9′), 31.6 (C-3), 26.9 (2C, C-6′ and C-10′), 26.7 (C-2′), 25.8 (Si-C(CH3)3), 24.2 (C-10), 22.6 (C-13′), 22.3 (C-4), 18.2 (Si-C(CH3)3), 17.8 (C12′-CH3), 16.2 (C8′-CH3), 16.1 (C4′-CH3), 15.9 (C-9), −4.3 (Si(CH3)2); GC-MS (EI) m/z 510.5 (M+, 100), 291.1, 251.1, 193.1, 69.1.
3.2.2. General Procedure for the Synthesis of 6a, 7a, 8a, 6b, 7b, and 8b
Compound 4 (2.0 g, 4 mmol) and 5a (2.40 g, 24 mmol) or 5b (2.00 g, 24 mmol) were taken in toluene (30 mL) and heated to 80 °C. At this temperature Grubbs II catalyst was added and the reaction was heated at reflux for 24 h. Solvent was removed under reduced pressure and the residue was subjected to column chromatography using hexanes/ethyl acetate (98:2) to afford 6a, 7a, and 8a or 6b, 7b, and 8b as colorless oils.
Methyl 5-{(R)-6-[(tert-butyldimethylsilyl)oxy]-2,8-dimethylchroman-2-yl}-2-methylpent-2E-enoate (6a), colorless oil; Yield 17%; 1H-NMR (CDCl3): δ 6.78 (t, J = 1.2 Hz, 1H, C3′-H), 6.46 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.37 (d, J = 2.8 Hz, 1H, phenyl C7-H), 3.71 (s, 3H, OCH3), 2.78–2.58 (m, 2H, C4-H2), 2.37–2.25 (m, 2H, C2′-H2), 2.10 (s, 3H, C9-H3), 1.82 (s, 3H, C4′-CH3), 1.90–1.57 (m, 4H, C3-H2 and C1′-H2), 1.26 (s, 3H, C10-H3), 0.97 (s, 9H, (Si-C(CH3)3), 0.16 (s, 6H, Si(CH3)2); 13C-NMR (CDCl3): δ 168.7 (C=O), 147.8 (phenyl C-6), 146.1 (phenyl C-8a), 142.6 (C-3′), 127.6 (C-4′), 126.9 (phenyl C-8), 120.7 (phenyl C-7), 120.3 (phenyl C-4a), 117.3 (phenyl C-5), 74.9 (C-2), 51.7 (OCH3), 38.4 (C-1′), 31.6 (C-3), 25.8 (Si-C(CH3)3), 23.9 (C-10), 23.0 (C-2′), 22.4 (C-4), 18.2 (Si-C(CH3)3), 16.1 (C-9), 12.3 (C4′-CH3), -4.33 (Si(CH3)2; GC-MS (EI) m/z 418.3 (M+, 100), 387.3, 361.2, 291.1, 251.2, 225.1, 193.1, 73.1.
Methyl 9-{(R)-6-[(tert-butyldimethylsilyl)oxy]-2,8-dimethylchroman-2-yl}-2E,6E-dimethylnona-2E,6E-dienoate (7a), colorless oil; Yield 15%; 1H-NMR (CDCl3): δ 6.73 (t, J = 1.6 Hz, 1H, C7′-H), 6.43 (d, J = 2.0 Hz, 1H, phenyl C5-H), 6.36 (d, J = 2.0 Hz, 1H, phenyl C7-H), 5.16 (t, J = 6.4 Hz, 1H, C3′-H), 3.72 (s, 3H, OCH3), 2.66 (t, J = 5.2 Hz, 2H, C4-H2), 2.65–2.04 (m, 9H, C9-H3, C2′-H2, C5′-H2 and C6′-H2), 1.82 (s, 3H, C8′-CH3), 1.81–1.45 (m, 7H, C1′-H2, C3-H2 and C4′-CH3), 1.25 (s, 3H, C10-H3), 0.96 (s, 9H, Si-C(CH3)3), 0.15 (s, 6H, Si(CH3)2); 13C-NMR (CDCl3): δ 168.8 (C=O), 147.7 (phenyl C-6), 146.4 (phenyl C-8a), 142.3 (C-7′), 134.1 (C-4′), 127.6 (C-8′), 126.9 (phenyl C-8), 125.4 (C-3′), 120.9 (phenyl C-7), 120.2 (phenyl C-4a), 117.3 (phenyl C-5), 75.3 (C-2), 51.8 (OCH3), 39.7 (C-1′), 38.3 (C-5′), 31.6 (C-3), 27.4 (C-6′), 25.8 (Si-C(CH3)3), 24.1 (C-10), 22.6 (C-2′), 22.3 (C-4), 18.2 (Si-C(CH3)3), 16.2 (C-9), 15.9 (C4′-CH3), 12.5 (C8′-CH3), -4.30 (Si(CH3)2); GC-MS (EI) m/z 486.4 (M+, 100), 455.3, 429.3, 291.1, 251.1 225.1, 193.1, 73.1.
Methyl 13-{(R)-6-[(tert-butyldimethylsilyl)oxy]-2,8-dimethylchroman-2-yl}-2,6,10-trimethyltrideca-2E,6E,10E-trienoate (8a), colorless oil; Yield 11%; 1H-NMR (CDCl3): δ 6.74 (t, J = 1.6 Hz, 1H, C11′-H), 6.45 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.36 (d, J = 2.8 Hz, 1H, phenyl C7-H), 5.11 (t, J = 7.2 Hz, 2H, C3′-H and C7′-H), 3.72 (s, 3H, OCH3), 2.66 (t, J = 5.6 Hz, 2H, C4-H2), 2.35–2.22 (m, 2H, C10′-H2), 2.15–1.95 (m, 11H, C9-H3, C2′-H2, C5′-H2, C6′-H2 and C9′-H2), 1.83 (s, 3H, C12′-CH3), 1.80–1.45 (m, 10H, C1′-H2, C3-H2, C4′-CH3 and C8′-CH3), 1.25 (s, 3H, C10-H3), 0.96 (s, 9H, Si-C(CH3)3), 0.15 (s, 6H, Si(CH3)2); 13C-NMR (CDCl3): δ 168.8 (C=O), 147.6 (phenyl C-6), 146.4 (phenyl C-8a), 142.4 (C-11′), 135.1 (C-8′), 134.0 (C-4′), 127.5 (C-12′), 126.9 (phenyl C-8), 125.2 (C-3′), 124.5 (C-7′), 120.9 (phenyl C-7), 120.2 (phenyl C-4a), 117.3 (phenyl C-5), 75.3 (C-2), 51.8 (OCH3), 39.8 (C-5′), 39.7 (C-1′), 38.3 (C-9′), 31.6 (C-3), 27.5 (C-10′), 26.7 (C-6′), 25.8 (Si-C(CH3)3), 24.2 (C-10), 22.6 (C-2′), 22.3 (C-4), 18.2 (Si-C(CH3)3), 16.2 (C-9), 16.1 (C4′-CH3), 15.9 (C8′-CH3), 12.5 (C12′-CH3), −4.30 (Si(CH3)2); GC-MS (EI) m/z 554.5 (M+), 523.4, 497.4, 291.1, 251.1 225.1, 193.1, 73.1.
Methyl 5-{(R)-6-[(tert-butyldimethylsilyl)oxy]-2,8-dimethylchroman-2-yl}pent-2E-enoate (6b), colorless oil; Yield 54%; 1H-NMR (CDCl3): δ 7.04–6.96 (m, 1H, C3′-H), 6.45 (d, J = 2.4 Hz, 1H, phenyl C5-H), 6.36 (d, J = 2.4 Hz, 1H, phenyl C7-H), 5.83 (d, J = 16.0 Hz, 1H, C4′-H), 3.71 (s, 3H, OCH3), 2.66 (t, J = 5.6 Hz, 2H, C4-H2), 2.25–2.18 (m, 2H, C2′-H2), 2.09 (s, 3H, C9-H3), 1.82–1.57 (m, 4H, C3-H2 and C1′-H2), 1.25 (s, 3H, C10-H3), 0.95 (s, 9H, Si-C(CH3)3), 0.15 (s, 6H, Si(CH3)2); 13C-NMR (CDCl3): δ 167.2 (C=O), 149.8 (C-3′), 147.9 (phenyl C-6), 146.1 (phenyl C-8a), 127.0 (phenyl C-8), 120.9 (C-4′), 120.7 (phenyl C-7), 120.3 (phenyl C-4a), 117.3 (phenyl C-5), 74.8 (C-2), 51.5 (OCH3), 38.2 (C-1′), 31.7 (C-3), 25.8 (Si-C(CH3)3), 23.9 (C-10), 22.6 (C-2′), 22.5 (C-4), 18.2 (Si-C(CH3)3), 16.2 (C-9), −4.3 (Si-(CH3)2); GC-MS (EI) m/z 404.3 (M+, 100), 373.3, 347.2, 291.1, 251.2, 225.1, 193.1, 73.1.
Methyl 9-{(R)-6-[(tert-butyldimethylsilyl)oxy]-2,8-dimethylchroman-2-yl}-6-methylnona-2E,6E-dienoate (7b), colorless oil; Yield 14%; 1H-NMR (CDCl3): δ 6.99–6.85 (m, 1H, C7′-H), 6.45 (d, J = 2.8, 1H, phenyl C5-H), 6.36 (d, J = 2.8, 1H, phenyl C7-H), 5.88 (d, J = 15.4 Hz, 1H, C8′-H), 5.16 (t, J = 1.2 Hz, 1H, C3′-H), 3.71 (s, 3H, OCH3), 2.75–2.62 (m, 2H, C4-H2), 2.35–2.18 (m, 2H, C6′- H2), 2.16–2.03 (m, 7H, C9-H3, C2′-H2, and C5′-H2), 1.82–1.45 (m, 7H, C1′-H2, C3-H2 and C4′-CH3), 1.25 (s, 3H, C10-H3), 0.96 (s, 9H, Si-C(CH3)3), 0.16 (s, 6H, Si(CH3)2); 13C-NMR (CDCl3): δ 167.2 (C=O), 149.4 (C-7′), 147.6 (phenyl C-6), 146.4 (phenyl C-8a), 133.6 (C-4′), 126.9 (phenyl C-8), 125.6 (C-3′), 121.0 (C-8′), 120.9 (phenyl C-7), 120.2 (phenyl C-4a), 117.3 (phenyl C-5), 75.2 (C-2), 51.5 (OCH3), 39.7 (C-1′), 38.0 (C-5′), 31.6 (C-3), 30.8 (C-6′), 25.8 (Si-C(CH3)3), 24.1 (C-10), 22.5 (C-2′), 22.2 (C-4), 18.2 (Si-C(CH3)3), 16.2 (C-9), 15.8 (C4′-CH3), −4.3 (Si-(CH3)2); GC-MS (EI) m/z 472.4 (M+, 100), 441.3, 415.3, 291.1, 251.2, 225.1, 193.1, 73.1.
Methyl 13-{(R)-6-[(tert-butyldimethylsilyl)oxy]-2,8-dimethylchroman-2-yl}-6,10-dimethyltrideca-2E,6E,10E-trienoate (8b), colorless oil; Yield 5%; 1H-NMR (CDCl3): δ 6.99–6.85 (m, 1H, C11′-H), 6.45 (d, J = 2.4 Hz, 1H, phenyl C5-H), 6.36 (d, J = 2.4 Hz, 1H, phenyl C7-H), 5.81 (d, J = 15.6 Hz, 1H, C12′-H), 5.12 (t, J = 6.4 Hz, 2H, C3′-H and C7′-H), 3.71 (s, 3H, OCH3), 2.67 (t, J = 6.8 Hz, 2H, C4-H2), 2.18–1.45 (m, 23H, C9-H3, C3-H2, C1′-H2, C2′-H2, C5′-H2, C6′-H2, C9′-H2, C10′-H2, C4′-CH3 and C8′-CH3), 1.25 (s, 3H, C10-H3), 0.96 (s, 9H, Si-C(CH3)3), 0.16 (s, 6H, Si(CH3)2); 13C-NMR (CDCl3): δ 167.2 (C=O), 149.4 (C-11′), 147.6 (phenyl C-6), 146.4 (phenyl C-8a), 135.0 (C-8′), 133.5 (C-4′), 126.9 (phenyl C-8), 125.4 (C-3′), 124.6 (C-7′), 121.0 (C-12′), 120.9 (phenyl C-7), 120.2 (phenyl C-4a), 117.3 (phenyl C-5), 75.3 (C-2), 51.5 (OCH3), 39.8 (C-5′), 39.6 (C-1′), 38.0 (C-9′), 31.5 (C-3), 26.6 (C-6′), 26.0 (C-10′), 25.8 (Si-C(CH3)3), 24.2 (C-10), 22.6 (C-2′), 22.2 (C-4), 18.2 (Si-C(CH3)3), 16.2 (C-9), 16.0 (C4′-CH3), 15.9 (C8′-CH3), −4.3 (Si(CH3)2); GC-MS (EI) m/z 540.5 (M+, 100), 509.4, 483.4, 291.1, 251.2, 225.1, 193.1, 135.1, 107.1, 73.1.
3.2.3. General Procedure for the Synthesis of 9a, 10a, 1, 9b, 10b, and 11
To a stirred solution of compound 6a, 7a, 8a, 6b, 7b, or 8b (0.1 mmol) in THF-MeOH-H2O (3:1:1, 4 mL) was added (0.3 mmol) of LiOH. The reaction mixture was heated at 40 °C under N2 for 14 h. After cool to room temperature, the reaction mixture was diluted with ethyl acetate (5 mL) and treated with of 1 N HCl (5 mL). The aqueous layer was extracted with ethyl acetate (2 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column using 1:1 hexanes-ethyl acetate to afford compound 9a, 10a, 1, 9b, 10b, and 11, respectively, as colorless oils.
5-[(R)-6-Hydroxy-2,8-dimethylchroman-2-yl]-2-methylpent-2E-enoic acid (9a), colorless oil; Yield 88%; 1H-NMR (CDCl3): δ 6.92 (t, J = 1.2 Hz, 1H, C3′-H), 6.48 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.39 (d, J = 2.8 Hz, 1H, Phenyl C7-H), 2.78–2.58 (m, 2H, C4-H2), 2.40–2.26 (m, 2H, C2′-H2), 2.12 (s, 3H, C9-H3), 1.86–1.61 (m, 7H, C1′-H2, C3-H2 and C4′-CH3), 1.26 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 173.2 (C=O), 148.0 (phenyl C-6), 145.8 (C-3′), 145.2 (phenyl C-8a), 127.5 (C-4′), 127.1 (phenyl C-8), 121.1 (phenyl C-4a), 115.9 (phenyl C-7), 112.7 (phenyl C-5), 75.0 (C-2), 38.3 (C-1′), 31.6 (C-3), 23.9 (C-10), 23.3 (C-4), 22.5 (C-2′), 16.1 (C-9), 12.0 (C4′-CH3); HRMS (ESI) m/z = 313.1410 calcd. for C17H22O4Na [M + Na]+; found: 313.1405.
9-[(R)-6-Hydroxy-2,8-dimethylchroman-2-yl]-2,6-dimethylnona-2E,6E-dienoic acid (10a), colorless oil; Yield 84%; 1H-NMR (CDCl3): δ 6.86 (t, J = 6.8 Hz, 1H, C7′-H), 6.47 (d, J = 2.4 Hz, 1H, phenyl C5-H), 6.38 (d, J = 2.4 Hz, 1H, phenyl C7-H), 5.16 (t, J = 6.4 Hz, 1H, C3′-H), 2.68 (t, J = 6.4 Hz, 2H, C4-H2), 2.34–2.21 (m, 2H, C6′-H2), 2.17–2.01 (m, 7H, C9-H3, C2′-H2 and C5′-CH3), 1.82–1.48 (m, 10H, C1′-H2, C3-H2, C4′-CH3 and C8′-CH3), 1.27 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ173.1 (C=O), 147.8 (phenyl C-6), 146.0 (C-7′), 145.0 (phenyl C-8a), 133.9 (C-4′), 127.4 (C-8′), 127.0 (phenyl C-8), 125.5 (C-3′), 121.3 (phenyl C-4a), 115.7 (phenyl C-7), 112.7 (phenyl C-5), 75.3 (C-2), 39.5 (C-5′), 38.1 (C-1′), 31.5 (C-3), 27.5 (C-6′), 24.1 (C-10), 22.5 (C-4), 22.2 (C-2′), 16.2 (C-9), 15.9 (C4′-CH3), 12.1 (C8′-CH3); HRMS (ESI) m/z = 381.2036 calcd. for C22H30O4Na [M + Na]+; found: 381.2036.
13-[(R)-6-Hydroxy-2,8-dimethylchroman-2-yl]-2,6,10-trimethyltrideca-2E,6E,10E-trienoic acid (
1), colorless oil; Yield 87%;
1H-NMR (CDCl
3): δ 6.86 (t,
J = 6.0 Hz, 1H, C11′-H), 6.47 (d,
J = 2.4 Hz, 1H, phenyl C5-H), 6.38 (d,
J = 2.4 Hz, 1H, phenyl C7-H), 5.12 (t,
J = 7.2 Hz, 2H, C3′-H and C7′-H), 2.69 (t,
J = 6.8 Hz, 2H, C4-H
2), 2.35–2.24 (m, 2H, C10′-H
2), 2.16–1.92 (m, 11H, C2′-H
2, C5′-H
2, C6′-H
2, C9′-H
2 and C9-H
3), 1.85–1.45 (m, 13H, C1′-H
2, C3-H
2, C4′-CH
3, C8′-CH
3 and C12′- CH
3), 1.26 (s, 3H, C10-H
3);
13C-NMR (CDCl
3): δ 172.0 (C=O), 147.8 (phenyl C-6), 146.1 (C-11′), 145.0 (phenyl C-8a), 135.0 (C-8′), 133.8 (C-4′), 127.5 (C-12′), 126.8 (phenyl C-8), 125.3 (C-3′), 124.6 (C-7′), 121.3 (phenyl C-4a), 115.7 (phenyl C-7), 112.7 (phenyl C-5), 75.4 (C-2), 39.8 (C-5′), 39.6 (C-1′), 38.1 (C-9′), 31.5 (C-3), 27.6 (C-6′), 26.6 (C-10′), 24.2 (C-10), 22.6 (C-4), 22.3 (C-2′), 16.2 (C-9), 16.1 (C8′-
CH
3), 15.9 (C4′-
CH
3), 12.2 (C12′-
CH
3); MS (ESI)
m/
z 425.2 [M − H]
−; HRMS (ESI)
m/
z = 449.2662 calcd. for C
27H
38O
4Na [M + Na]
+; found: 449.2662. The NMR data are in line with those reported [
10].
5-[(R)-6-Hydroxy-2,8-dimethylchroman-2-yl]pent-2E-enoic acid (9b), colorless oil; Yield 83%; 1H-NMR (CDCl3): δ 7.11–7.02 (m, 1H, C3′-H), 6.47 (s, 1H, phenyl C5-H), 6.38 (s, 1H, phenyl C7-H), 5.83 (d, J = 16 Hz, 1H, C4′-H), 2.78–2.58 (m, 2H, C4-H2), 2.44–2.39 (m, 2H, C2′-H2), 2.10 (s, 3H, C9-H3), 1.85–1.59 (m, 4H, C1′-H2 and C3-H2), 1.25 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 171.6 (C=O), 152.5 (C-3′), 148.0 (phenyl C-6), 145.7 (phenyl C-8a), 127.5 (phenyl C-8), 121.1 (phenyl C-4a), 120.5 (C-4′), 115.9 (phenyl C-7), 112.7 (phenyl C-5), 74.8 (C-2), 38.0 (C-1′), 31.6 (C-3), 26.7 (C-10), 23.9 (C-4), 22.4 (C-2′), 16.2 (C-9); HRMS (ESI) m/z = 299.1254 calcd. for C16H20O4Na [M + Na]+; found: 299.1244.
9-[(R)-6-Hydroxy-2,8-dimethylchroman-2-yl]-6-methylnona-2E,6E-dienoic acid (10b), colorless oil; Yield 85% yield; 1H-NMR (CDCl3): δ 7.07–6.95 (m, 1H, C7′-H), 6.47 (d, J = 2.4 Hz, 1H, phenyl C5-H), 6.38 (d, J = 2.4 Hz, 1H, phenyl C7-H), 5.80 (d, J = 15.6 Hz, 1H, C8′-H), 5.16 (t, J = 6.8 Hz, 1H, C3′-H), 2.68 (t, J = 7.6 Hz, 2H, C4-H2), 2.37–2.25 (m, 2H, C6′-H2), 2.11–2.04 (m, 7H, C9-H3, C2′-H2 and C5′-H2), 1.82–1.45 (m, 7H, C1′-H2, C3-H2 and C4′-CH3), 1.25 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 171.6 (C=O), 152.0 (C-7′), 147.8 (phenyl C-6), 146.0 (phenyl C-8a), 133.5 (C-4′), 127.4 (phenyl C-8), 125.7 (C-8′), 121.3 (phenyl C-4a), 120.7 (C-3′), 115.7 (phenyl C-7), 112.7 (phenyl C-5), 75.3 (C-2), 39.5 (C-5′), 37.8 (C-1′), 31.5 (C-3), 30.9 (C-6′), 24.1 (C-10), 22.5 (C-4), 22.2 (C-2′), 16.2 (C-9), 15.9 (C4′-CH3); HRMS (ESI) m/z = 367.1880 calcd. for C21H28O4Na [M + Na]+; found: 367.1865.
13-[(R)-6-Hydroxy-2,8-dimethylchroman-2-yl]-6,10-dimethyltrideca-2E,6E,10E-trienoic acid (11), colorless oil; Yield 83%; 1H-NMR (CDCl3): δ 7.07–6.96 (m, 1H, C11′-H), 6.46 (d, J = 2.4, 1H, phenyl C5-H), 6.37 (d, J = 2.4 Hz, 1H, phenyl C7-H), 5.80 (d, J = 15.6 Hz, 1H, C12′-H), 5.11 (t, J = 6.8 Hz, 2H, C3′-H and C7′-H), 2.68 (t, J = 6.8 Hz, 2H, C4-H2), 2.35–2.18 (m, 2H, C10′-H2), 2.25–1.92 (m, 11H, C9-H3, C2′-H2, C5′-H2, C6′-H2 and C9′-H2), 1.85–1.51 (m, 10H, C1′-H2, C3-H2, C4′-CH3 and C8′-CH3), 1.25 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 171.1 (C=O), 152.2 (C-11′), 147.8 (phenyl C-6), 146.0 (phenyl C-8a), 134.9 (C-8′), 133.4 (C-4′), 127.5 (phenyl C-8), 125.5 (C-3′), 124.6 (C-7′), 121.3 (phenyl C-4a), 120.5 (C-12′), 115.8 (phenyl C-7), 112.7 (phenyl C-5), 75.4 (C-2), 39.6 (C-5′), 39.5 (C-9′), 37.8 (C-1′), 31.5 (C-3), 31.0 (C-6′), 26.5 (C-10′), 24.2 (C-10), 22.6 (C-4), 22.3 (C-2′), 16.2 (C8′-CH3), 16.0 (C4′-CH3), 15.9 (C4′-CH3); HRMS (ESI) m/z = 435.2506 calcd. for C26H36O4Na [M + Na]+; found: 435.2511.
3.2.4. General Procedure for the Synthesis of Compounds 12a, 13a, 14a, 12b, 13b, and 14b
To a stirred solution of compound 6a, 7a, 8a, 6b, 7b, or 8b (0.1 mmol) in anhydrous THF (3 mL) at 0 °C under argon atmosphere was added TBAF (0.2 mL, 1M in THF, 0.2 mmol) dropwise, and allowed to stir at room temperature until TLC revealed full consumption of the starting material (2 h). Reaction mixture was quenched by adding water (2 mL) and extracted with ethyl acetate (2 × 5 mL). Combined organic layers were dried over anhydrous Na2SO4, and solvents were removed under reduced pressure. The residue was purified by silica gel column using hexanes-ethyl acetate (6:4) to afford compound 12a, 13a, 14a, 12b, 13b, and 14b, respectively, as colorless oils.
Methyl 5-[(R)-6-hydroxy-2,8-dimethylchroman-2-yl]-2-methylpent-2E-enoate (12a), colorless oil; Yield 91%; 1H-NMR (CDCl3): δ 6.79 (dt, J = 1.6, 7.6 Hz, 1H, C3′-H), 6.48 (d, J = 3.2 Hz, 1H, phenyl C5-H), 6.39 (d, J = 3.2 Hz, 1H, phenyl C7-H), 4.70 (br s, 1H, OH), 3.72 (s, 3H, OCH3), 2.78–2.58 (m, 2H, C4-H2), 2.38–2.26 (m, 2H, C2′-H2), 2.11 (s, 3H, C9-H3), 1.83 (s, 3H, C4′-CH3), 1.82–1.52 (m, 4H, C3-H2 and C1′-H2), 1.26 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 168.9 (C=O), 148.1 (phenyl C-6), 145.7 (phenyl C-8a), 142.8 (C-3′), 127.6 (C-4′), 127.4 (phenyl C-8), 121.1 (phenyl C-7), 115.9 (phenyl C-4a), 112.7 (phenyl C-5), 75.0 (C-2), 51.8 (OCH3), 38.4 (C-1′), 31.6 (C-3), 23.9 (C-10), 23.1 (C-4), 22.5 (C-2′), 16.1 (C-9), 12.3 (C4′-CH3); HRMS (ESI) m/z = 327.1567 calcd. for C18H24O4Na [M + Na]+; found: 327.1577.
Methyl 9-[(R)-6-hydroxy-2,8-dimethylchroman-2-yl]-2,6-dimethylnona-2E,6E-dienoate (13a). colorless oil; Yield 90%; 1H-NMR (CDCl3): δ 6.72 (t, J = 1.2 Hz, 1H, C7′-H), 6.48 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.38 (d, J = 2.8 Hz, 1H, phenyl C7-H), 5.16 (t, J = 1.2 Hz, 1H, C3′-H), 4.27 (br s, 1H, OH), 3.73 (s, 3H, OCH3), 2.74–2.65 (m, 2H, C4-H2), 2.31–2.20 (m, 2H, C6′-H2), 2.17–2.02 (m, 7H, C9-H3, C2′-H2 and C5′-H2), 1.84–1.51 (m, 10H, C3-H2, C1′-H2, C4′-CH3 and C8′-CH3), 1.26 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 168.8 (C=O), 147.9 (phenyl C-6), 146.1 (phenyl C-8a), 142.4 (C-7′), 134.1 (C-4′), 127.6 (C-8′), 127.5 (phenyl C-8), 125.3 (C-3′), 121.3 (phenyl C-7), 115.7 (phenyl C-4a), 112.7 (phenyl C-5), 75.4 (C-2), 51.8 (OCH3), 39.7 (C-1′), 38.3 (C-5′), 31.5 (C-3), 27.4 (C-6′), 24.1 (C-10), 22.6 (C-2′), 22.3 (C-4), 16.2 (C-9), 15.9 (C4′-CH3), 12.5 (C8′-CH3); HRMS (ESI) m/z = 395.2193 calcd. for C23H32O4Na [M + Na]+; found: 395.2191.
Methyl 13-[(R)-6-hydroxy-2,8-dimethylchroman-2-yl]-2,6,10-trimethyltrideca-2E,6E,10E-trienoate (14a), colorless oil; Yield 91%; 1H-NMR (CDCl3): δ 6.74 (t, J = 6.8 Hz, 1H, C11′-H), 6.48 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.38 (d, J = 2.8 Hz, 1H, phenyl C7-H), 5.16–5.08 (m, 2H, C3′-H and C7′-H), 4.49 (br s, 1H, OH), 3.73 (s, 3H, OCH3), 2.68 (t, J = 6.8 Hz, 2H, C4-H2), 2.28–2.21 (m, 2H, C10′-H2), 2.15–1.92 (m, 11H, C9-H3, C2′-H2, C5′-H2, C6′-H2 and C9′-H2), 1.82 (s, 3H, C12′-CH3), 1.81–1.50 (m, 10H, C3-H2, C1′-H2, C4′-CH3 and C8′-CH3), 1.26 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 168.9 (C=O), 147.9 (phenyl C-6), 146.0 (phenyl C-8a), 142.6 (C-11′), 135.0 (C-8′), 134.0 (C-4′), 127.5 (C-12′), 127.4 (phenyl C-8), 125.1 (C-3′), 124.6 (C-7′), 121.3 (phenyl C-7), 115.8 (phenyl C-4a), 112.7 (phenyl C-5), 75.4 (C-2), 51.8 (OCH3), 39.6 (2C, C-1′ and C-5′), 38.3 (C-9′), 31.5 (C-3), 27.5 (C-10′), 26.6 (C-6′), 24.2 (C-10), 22.6 (C-2′), 22.3 (C-4), 16.2 (C-9), 16.1 (C4′-CH3), 15.9 (C8′-CH3), 12.5 (C12′-CH3); HRMS (ESI) m/z = 463.2819 calcd. for C28H40O4Na [M + Na]+; found: 463.2825.
Methyl 5-[(R)-6-hydroxy-2,8-dimethylchroman-2-yl]pent-2E-enoate (12b), colorless oil; Yield 91%; 1H-NMR (CDCl3): δ 7.14–6.91 (m, 1H, C3′-H), 6.48 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.38 (d, J = 2.8 Hz, 1H, phenyl C7-H), 5.82 (d, J = 16.0 Hz, 1H, C4′-H), 4.51 (br s, 1H, OH), 3.72 (s, 3H, OCH3), 2.75–2.56 (m, 2H, C4-H2), 2.46–2.38 (m, 2H, C2′-H2), 2.11 (s, 3H, C9-H3), 1.86–1.56 (m, 4H, C1′-H2 and C3′-H2), 1.25 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 167.3 (C=O), 149.8 (C-3′), 148.1 (phenyl C-6), 145.7 (phenyl C-8a), 127.5 (phenyl C-8), 121.1 (C-4′), 120.9 (phenyl C-7), 115.9 (phenyl C-4a), 112.7 (phenyl C-5), 74.9 (C-2), 51.6 (OCH3), 38.1 (C-1′), 31.6 (C-3), 26.6 (C-2′), 23.9 (C-10), 22.4 (C-4), 16.1 (C-9); HRMS (ESI) m/z = 313.1410 calcd. for C17H22O4Na [M + Na]+; found: 313.1416.
Methyl 9-[(R)-6-hydroxy-2,8-dimethylchroman-2-yl]-6-methylnona-2E,6E-dienoate (13b), colorless oil; Yield 90%; 1H-NMR (CDCl3): δ 6.99–6.85 (m, 1H, C7′-H), 6.48 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.38 (d, J = 2.8 Hz, 1H, phenyl C7-H), 5.82 (d, J = 15.6, 1H, H-8′), 5.15 (t, J = 7.2 Hz, 1H, H-3′), 4.94 (brs, 1H, OH), 3.72 (s, 3H, OCH3), 2.71–2.62 (m, 2H, C4-H2), 2.36–2.23 (m, 2H, H-6′), 2.17–2.03 (m, 7H, H-2′, 5′ and 9), 1.82–1.49 (m, 7H, H-1′, 3 and 4′-CH3), 1.24 (s, 3H, H-10); 13C-NMR (CDCl3): δ 167.5 (C=O), 149.6 (C-7′), 148.1 (phenyl C-6), 145.8 (phenyl C-8a), 133.6 (C-4′), 127.3 (phenyl C-8), 125.5 (C-3′), 121.2 (C-8′), 120.9 (phenyl C-7), 115.8 (phenyl C-4a), 112.7 (phenyl C-5), 75.3 (C-2), 51.6 (OCH3), 39.6 (C-1′), 37.9 (C-5′), 31.5 (C-3), 30.8 (C-6′), 24.1 (C-10), 22.5 (C-2′), 22.2 (C-4), 16.1 (C-9), 15.9 (C4′-CH3); HRMS (ESI) m/z = 381.2036 calcd. for C22H30O4Na [M + Na]+; found: 381.2033.
Methyl 13-[(R)-6-hydroxy-2,8-dimethylchroman-2-yl]-6,10-dimethyltrideca-2E,6E,10E-trienoate (14b), colorless oil; Yield 90%; 1H-NMR (CDCl3): δ 6.99–6.95 (m, 1H, C11′-H), 6.48 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.38 (d, J = 2.8 Hz, 1H, phenyl C7-H), 5.81(d, J = 15.6 Hz, 1H, C12′-H), 5.11 (d, J = 1.2 Hz, 2H, C3′-H and C7′-H), 4.73 (br s, 1H, OH), 3.72 (s, 3H, OCH3), 2.66 (t, J = 7.2 Hz, 2H, C4-H2), 2.31–2.23 (m, 2H, C10′-H2), 2.08–1.92 (m, 11H, C9-H3, C2′-H2, C5′-H2, C6′-H2 and C9′-H2), 1.92–1.51 (m, 10H, C1′-H2, C3-H2, C4′-CH3 and C8′-CH3), 1.26 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 167.4 (C=O), 149.7 (C-11′), 148.0 (phenyl C-6), 146.0 (phenyl C-8a), 134.9 (C-8′), 133.5 (C-4′), 127.4 (phenyl C-8), 125.3 (C-3′), 124.6 (C-7′), 121.3 (C-12′), 120.8 (phenyl C-7), 115.8 (phenyl C-4a), 112.7 (phenyl C-5), 75.4 (C-2), 51.6 (OCH3), 39.6 (C-5′), 39.5 (C-1′), 37.9 (C-9′), 31.5 (C-3), 30.9 (C-10′), 26.5 (C-6′), 24.3 (C-10), 22.6 (C-2′), 22.2 (C-4), 16.2 (C-9), 16.0 (C4′-CH3), 15.9 (C8′-CH3); HRMS (ESI) m/z = 449.2662 calcd. for C27H38O4Na [M + Na]+; found: 449.2675.
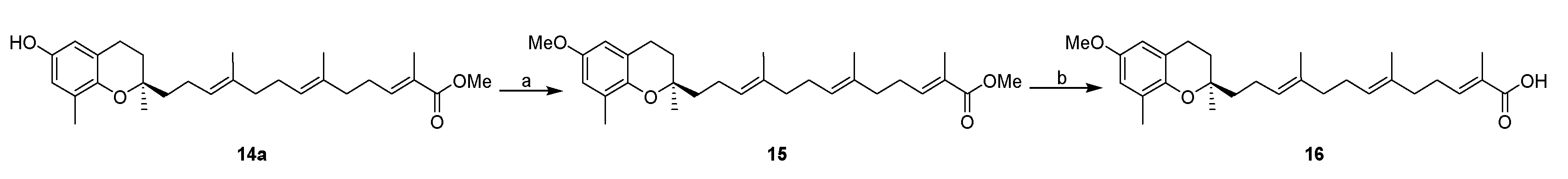
3.2.5. Synthesis of Methyl 13-[(R)-6-methoxy-2,8-dimethylchroman-2-yl]-2,6,10-trimethyltrideca-2E,6E,10E-trienoate (15)
To a stirred solution of NaH (4.6 mg, 0.2 mmol, 60%) in anhydrous THF (2 mL) at 0 °C under argon atmosphere was added 14a (44 mg, 0.1 mmol) and stirred at room temperature for 15 min. The reaction mixture was cooled to 0 °C, methyl iodide (0.13 mL, 0.2 mml) was added dropwise, and stirred at room temperature for 8 h. The reaction mixture was quenched by adding cold water (2 mL) and extracted with ethyl acetate (2 × 5 mL). The combined organic layers were dried over anhydrous Na2SO4, and solvents were removed under reduced pressure. The residue was purified by silica gel column using hexanes-ethyl acetate as eluents to afford compound 15 (41 mg, yield 90%) as colorless oil; 1H-NMR (CDCl3): δ 6.73 (t, J = 1.6 Hz, 1H, C11′-H), 6.56 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.43 (d, J = 2.8 Hz, 1H, phenyl C7-H), 5.13 (t, J = 6.4 Hz, 2H, C3′-H and C7′-H), 3.72 (s, 6H, OCH3 and COOCH3), 2.76–2.70 (m, 2H, C4-H2), 2.26–2.21 (m, 2H, C10′-H2), 2.17–2.18 (m, 11H, C9-H3, C2′-H2, C5′-H2, C6′-H2 and C9′-H2), 1.85–1.47 (m, 13H, C3-H2, C1′-H2, C4′-CH3, C8′-CH3 and C12′-CH3), 1.25 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 168.8 (C=O), 152.2 (phenyl C-6), 146.2 (phenyl C-8a), 142.4 (C-11′), 135.1 (C-8′), 134.0 (C-4′), 127.5 (C-12′), 127.3 (phenyl C-8), 125.2 (C-3′), 124.5 (C-7′), 121.0 (phenyl C-7), 114.9 (phenyl C-4a), 111.1 (phenyl C-5), 75.4 (C-2), 55.7 (OCH3), 51.8 (COOCH3), 39.8 (C-5′), 39.7 (C-1′), 38.3 (C-9′), 31.5 (C-3), 27.5 (C-10′), 26.7 (C-6′), 22.8 (C-4), 22.3 (C-2′), 16.3 (C-9), 16.1 (C4′-CH3), 16.0 (C8′-CH3), 12.5 (C12′-CH3); HRMS (ESI) m/z = 477.2975 calcd. for C29H42O4Na [M + Na]+; found: 477.2995.
3.2.6. Synthesis of 13-[(R)-6-methoxy-2,8-dimethylchroman-2-yl]-2,6,10-trimethyltrideca-2E,6E,10E-trienoic Acid (16)
To a stirred solution of compound 15 (27 mg, 0.06 mmol) in THF-MeOH-H2O (3:1:1, 4 mL) was added LiOH (7 mg, 0.3 mmol). The reaction mixture was heated at 40 °C under N2 for 12 h, then cooled to room temperature, diluted with ethyl acetate (10 mL), and acidified with 1N HCl (5 mL). The aqueous layer was extracted with ethyl acetate (2 × 15 mL). The combined organic layer was washed with brine (15 mL) and dried over anhydrous Na2SO4. Solvents were removed under reduced pressure and the residue was purified by silica gel column using hexanes-ethyl acetate (6:4) as eluents to afford compound 16 (23 mg, yield 88%) as colorless oil; 1H-NMR (CDCl3): δ 6.87 (t, J = 1.2 Hz, 1H, C11′-H), 6.56 (d, J = 2.8 Hz, 1H, phenyl C5-H), 6.43 (d, J = 2.8 Hz, 1H, phenyl C7-H), 5.13 (t, J = 6.4 Hz, 2H, C3′-H and C7′-H), 3.72 (s, 3H, OCH3), 2.72 (t, J = 6.0 Hz, 2H, C4-H2), 2.35–2.21 (m, 2H, C10′-H2), 2.19–2.10 (m, 9H, C5′-H2, C6′-H2, C9′-H2 and C9-H3), 2.01–1.93 (m, 2H, C2′-H2), 1.83–1.45 (m, 13H, C1′-H2, C3-H2, C4′-CH3, C8′-CH3 and C12′-CH3), 1.26 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 172.8 (COOH), 152.2 (C-11′), 146.2 (phenyl C-6), 145.0 (phenyl C-8a), 135.0 (C-8′), 133.8 (C-4′), 127.3 (C-12′), 126.9 (phenyl C-8), 125.3 (C-3′), 124.5 (C-7′), 121.0 (phenyl C-4a), 114.9 (phenyl C-7), 111.1 (phenyl C-5), 75.4 (C-2), 55.7 (OCH3), 39.8 (C-5′), 39.6 (C-1′), 38.1 (C-9′), 31.5 (C-3), 27.6 (C-6′), 26.6 (C-10′), 24.1 (C-10), 22.8 (C-4), 22.3 (C-2′), 16.3 (C-9), 16.1 (C8′-CH3), 16.0 (C4′-CH3), 12.1 (C12′-CH3); MS (ESI) m/z 439.2 [M − H]−.
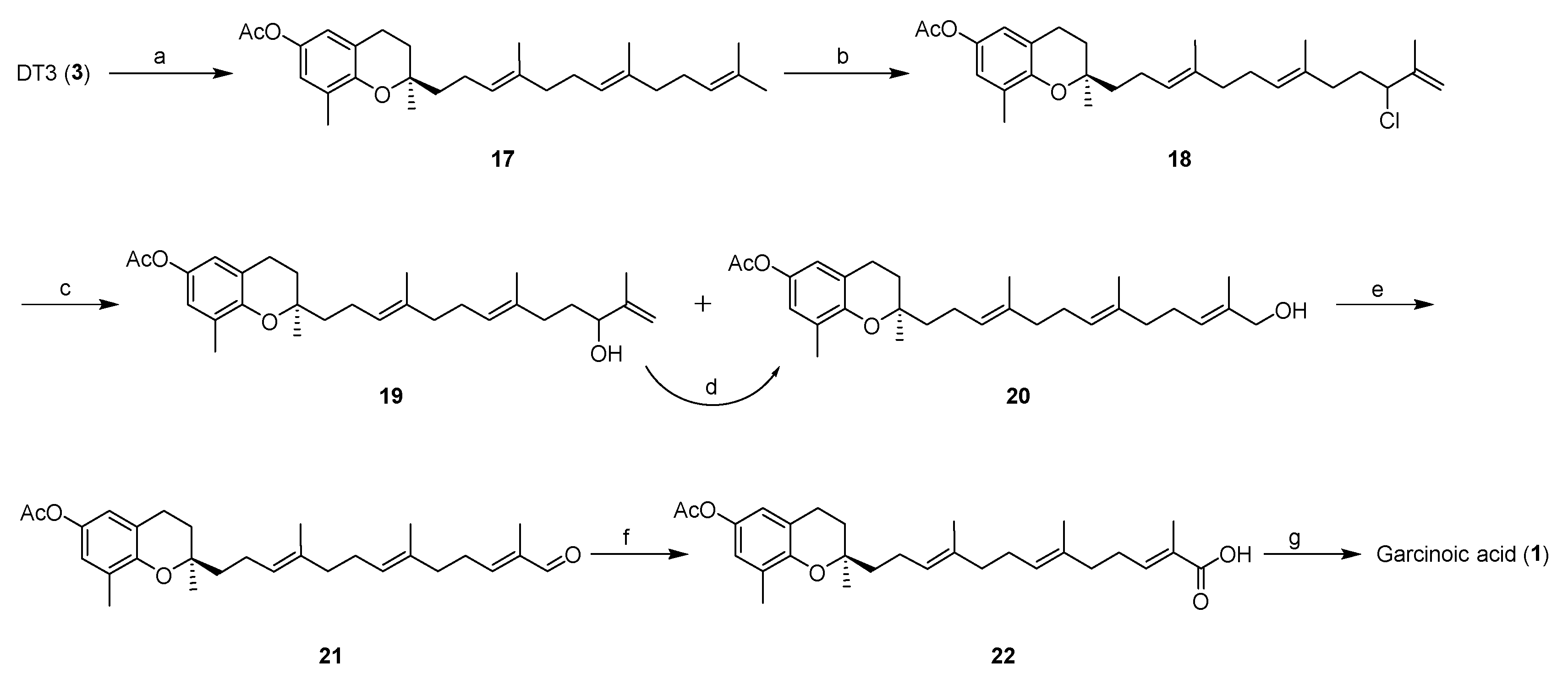
3.2.7. Synthesis of (R)-2,8-dimethyl-2-[(3E,7E)-4,8,12-trimethyltrideca-3,7,11-trien-1-yl]chroman-6-yl Acetate (17)
To a solution of δ-tocotrienol (3) (2 g, 5 mmol) in CH2Cl2 (25 mL) at 0 °C was added triethylamine (1.35 mL, 10 mmol) and DMAP (100 mg). The resulting mixture was stirred at room temperature for 15 min, and cooled to 0 °C. At the same temperature Ac2O (0.71 mL, 7.5 mmol) was added and continue stirring at room temperature for 5 h. The reaction mixture was partitioned between CH2Cl2 (25 mL) and water (50 mL). The aqueous layer was extracted with CH2Cl2 (2 × 25 mL), and the combined organic layers were washed with water (50 mL) and brine (50 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was chromatographed on silica (hexanes-ethyl acetate 9:1) to afford compound 17 (1.97 g, 90%) as colorless oil: 1H-NMR (CDCl3): δ 6.67 (d, J = 2.4 Hz, 1H, phenyl C5-H), 6.61 (d, J = 2.4 Hz, 1H, phenyl C7-H), 5.17–5.05 (m, 3H, C3′-H, C7′-H and C11′-H), 2.74–2.68 (m, 2H, C4-H2), 2.24 (s, 3H, COCH3), 2.16–1.92 (m, 13H, C9-H3, C2′-H2, C5′-H2, C6′-H2, C9′-H2 and C10′-H2), 1.83–1.54 (m, 16H, C3-H2, C1′-H2, C4′-CH3, C8′-CH3, C12′-CH3 and C13′-H3), 1.27 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 170.3 (COCH3), 149.8 (phenyl C-6), 142.6 (phenyl C-8a), 135.3 (C-8′), 135.0 (C-4′), 131.3 (C-12′), 127.4 (phenyl C-8), 124.5 (C-3′), 124.3 (C-7′), 124.2 (C-11′), 121.2 (phenyl C-7), 120.9 (phenyl C-4a), 119.1 (phenyl C-5), 75.9 (C-2), 40.0 (C-1′), 39.8 (2C, C-5′ and C-9′), 31.0 (C-3), 26.8 (C-10′), 26.7 (C-8′), 25.8 (C-13′), 24.2 (C-10), 22.5 (C-2′), 22.2 (C-4), 21.2 (COCH3), 17.7 (C12′-CH3), 16.2 (C-9), 16.1 (C8′-CH3), 16.0 (C4′-CH3); GC-MS (EI) m/z 438.3 (M+, 100), 423.2, 219.1, 177.1, 69.1.
3.2.8. Synthesis of Compounds 19 and 20
To a stirred solution of compound 17 (613 mg, 1.4 mmol) in anhydrous CH2Cl2 (12 mL) was added PhSeCl (27 mg, 0.14 mmol) under an Argon atmosphere. To this solution NCS (205 mg, 1.54 mmol) was added and resulting mixture was stirred for 4.5 h (monitored by TLC). Solvent was removed under reduced pressure, and to the residue was added Et2O (15 mL). The ether layer was decanted from the solid, and the organic layer was washed with H2O (2 × 10 mL), brine (10 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude was dissolved in acetone-H2O (1:1, 20 mL), followed by adding 2,4,6-collidine (0.65 mL, 4.9 mmol) and AgBF4 (681 mg, 3.5 mmol). The resulting mixture was heated at 70 °C for 6 h (monitored by TLC), then cooled to room temperature and filtered through a pad of celite. Acetone was removed from the filtrate under reduced pressure and the residue was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with 2 M HCl (3 × 10 mL) and brine (15 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography (hexane-ethyl acetate, 7:3) on silica gel to afford 19 and 20.
(2R)-2-[(3E,7E)-11-Hydroxy-4,8,12-trimethyltrideca-3,7,12-trien-1-yl]-2,8-dimethylchroman-6-yl acetate (19), 165 mg, colorless oil; Yield 26%; 1H-NMR (CDCl3): δ 6.66 (d, J = 2.4 Hz, 1H, phenyl C5-H), 6.61 (d, J = 2.4 Hz, 1H, phenyl C7-H), 5.15–5.05 (m, 2H, C3′-H and C7′-H), 4.92 (s, 1H, C13′-H), 4.82 (s, 1H, C13′-H), 4.04 (t, J = 6.4 Hz, 1H, C11′-H), 2.73 (t, J = 3.2 Hz, 2H, C4-H2), 2.24 (s, 3H, COCH3), 2.17–1.91 (m, 11H, C9-H3, C2′-H2, C5′-H2, C6′-H2 and C9′-H2), 1.82–1.45 (m, 15H, C3-H2, C1′-H2, C10′-H2, C4′-CH3, C8′-CH3 and 12′-CH3), 1.27 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 170.3 (COCH3), 149.8 (phenyl C-6), 147.5 (C-12′), 142.5 (phenyl C-8a), 135.1 (C-4′), 134.8 (C-8′), 127.4 (phenyl C-8), 124.7 (C-7′), 124.3 (C-3′), 121.2 (phenyl C-7), 120.9 (phenyl C-4a), 119.1 (phenyl C-5), 111.0 (C-13′), 75.9 (C-2), 75.6 (C-11′), 39.9 (C-5′), 39.6 (C-1′), 35.7 (C-9′), 33.2 (C-10′), 31.0 (C-3), 26.5 (C-6′), 24.2 (C-10), 22.5 (C-4), 22.2 (C-2′), 21.1 (COCH3), 17.7 (C12′-CH3), 16.2 (C-9), 16.0 (C8′-CH3), 15.9 (C4′-CH3); GC-MS (EI) m/z 454.3 (M+), 439.2, 412.3, 219.1, 177.1, 137.1 (100), 93.1, 55.1.
(R)-2-[(3E,7E,11E)-13-Hydroxy-4,8,12-trimethyltrideca-3,7,11-trien-1-yl]-2,8-dimethylchroman-6-yl acetate (20), 204 mg, colorless oil; Yield 32%; 1H-NMR (CDCl3): δ 6.66 (d, J = 2.4 Hz, 1H, phenyl C5-H), 6.61 (d, J = 2.4 Hz, 1H, phenyl C7-H), 5.42–5.37 (m, 1H, C11′-H), 5.17–5.05 (m, 2H, C3′-H and C7′-H), 3.97 (s, 2H, C13′-H2), 2.72–2.70 (m, 2H, C4-H2), 2.24 (s, 3H, COCH3), 2.16–1.95 (m, 13H, C9-H3, C2-H2, C5′-H2, C6′-H2, C9′-H2 and C10′-H2), 1.74–1.54 (m, 13H, C1′-H2, C3-H2, C4′-CH3, C8′-CH3 and C12′-CH3), 1.27 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 170.3 (COCH3), 149.8 (phenyl C-6), 142.6 (phenyl C-8a), 135.2 (C-12′), 134.7 (C-8′), 133.7 (C-4′), 127.4 (phenyl C-8), 126.2 (C-11′), 124.5 (C-7′), 124.3 (C-3′), 121.2 (phenyl C-7), 120.9 (phenyl C-4a), 119.1 (phenyl C-5), 75.9 (C-2), 69.1 (C-13′), 40.0 (C-5′), 39.7 (C-1′), 39.4 (C-9′), 31.0 (C-3), 26.6 (C-6′), 26.3 (C-10′), 24.2 (C-10), 22.5 (C-4), 22.2 (C-2′), 21.1 (COCH3), 16.2 (C-9), 16.1 (C8′-CH3), 16.0 (C4′-CH3), 13.7 (C12′-CH3); MS (ESI) m/z 453.2 [M − H]−.
3.2.9. Conversion of 19 to 20
To a solution of 19 (228 mg, 0.5 mmol) in pyridine (5 mL) at 0 °C was added DMAP (10 mg). After 10 min, MsCl (0.08 mL, 3.0 mmol) was added. The reaction mixture was stirred for 1 h and then quenched with sat. NaHCO3 solution (15 mL) and extracted with ethyl acetate (2 × 20 mL). The combined organic layers were washed with 2 M HCl (15 mL), sat. NaHCO3 solution (15 mL), and brine (15 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was dissolved in acetone (5 mL) and H2O (2 mL), NaOAc (410 mg in 1 mL water) was added, and the mixture was heated at reflux for 2 h. Acetone was removed under reduced pressure and the resulting crude was extracted with ethyl acetate (2 × 10 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography (hexane-ethylacetate, 7:3) on silica gel to afford mixture of 20 (78 mg, yield 34%) and 19 (73 mg, yield 32%).
3.2.10. Synthesis of (R)-2,8-dimethyl-2-[(3E,7E,11E)-4,8,12-trimethyl-13-oxotrideca-3,7,11-trien-1-yl]chroman-6-yl Acetate (21)
To a stirred solution of 20 (45 mg, 0.1 mmol) in CH2Cl2 (3 mL) at 0 °C was added Dess-Martin reagent (51 mg, 0.12 mmol). The reaction was continued at room temperature for another 2 h, and then quenched with saturated aqueous Na2S2O3 solution (3 mL) and saturated aqueous NaHCO3 solution (3 mL). Stirred for another 20 min and extracted with CH2Cl2 (2 × 50 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude residue was purified by column chromatography (hexane-ethyl acetate 8:2) on silica gel to afford 21 (41 mg, yield 92%) as colorless oil; 1H-NMR (CDCl3): δ 9.37 (s, 1H, aldehyde), 6.66 (d, J = 2.4 Hz, 1H, phenyl C5-H), 6.60 (d, J = 2.4 Hz, 1H, phenyl C7-H), 6.52–6.41 (m, 1H, C11′-H), 5.16–5.12 (m, 2H, C3′-H and C7′-H), 2.79–2.69 (m, 2H, C4-H2), 2.49–2.36 (m, 2H, C10′-H2), 2.24 (s, 3H, COCH3), 2.12–1.91 (m, 11H, C9-H3, C2′-H2, C5′-H2, C6′-H2 and C9′-H2), 1.86–1.52 (m, 13H, C1′-H2, C3-H2, C4′-CH3, C8′-CH3 and C12′-CH3), 1.27 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 195.3 (aldehyde), 170.3 (COCH3), 154.5 (C-11′), 149.7 (phenyl C-6), 142.5 (phenyl C-8a), 139.3 (C-12′), 135.0 (C-8′), 133.4 (C-4′), 127.3 (phenyl C-8), 125.6 (C-3′), 124.4 (C-7′), 121.2 (phenyl C-7), 120.9 (phenyl C-4a), 119.1 (phenyl C-5), 75.9 (C-2), 39.9 (C-5′), 39.6 (C-1′), 38.0 (C-9′), 31.0 (C-3), 27.5 (C-6′), 26.6 (C-10′), 24.2 (C-10), 22.4 (C-4), 22.2 (C-2′), 21.1 (COCH3), 16.2 (C-9), 16.0 (C8′-CH3), 15.9 (C4′-CH3), 9.29 (C12′-CH3); MS (ESI) m/z 453.2 [M + H]+.
3.2.11. Synthesis of 13-[(R)-6-acetoxy-2,8-dimethylchroman-2-yl]-2,6,10-trimethyltrideca-2E,6E,10E-trienoic Acid (22)
To a solution of aldehyde 21 (23 mg, 0.05 mmol) in t-BuOH (3.0 mL) was added NaH2PO4 (29 mg, 0.48 mmol) and 2-methyl-2-butene (0.5 mL) successively. The reaction mixture was cooled to 0 °C. A freshly prepared solution of sodium chlorite (47 mg, 0.52 mmol in 0.5 mL H2O) was added to the mixture. The resulting mixture was stirred for 2 h at room temperature (monitored by TLC). Water (1.0 mL) was added, and the aqueous layer was extracted with ethyl acetate (3 × 5 mL). The combined organic layers were washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, the residue was purified by silica gel column with hexanes-ethyl acetate (1:1) to give 22 (19 mg, yield 80%) as colorless oil. 1H-NMR (CDCl3): δ 6.87 (t, J = 1.2 Hz, 1H, C11′-H), 6.66 (d, J = 2.4 Hz, 1H, phenyl C5-H), 6.61 d, J = 2.4 Hz, 1H, phenyl C5-H), 5.13 (t, J = 6.0 Hz, 2H, C3′-H and C7′-H), 2.72 (t, J = 3.6 Hz, 2H, C4-H2), 2.36–2.24 (m, 5H, C10′-H2 and COCH3), 2.09–1.91 (m, 11H, C9-H3, C2′-H2, C5′-H2, C6′-H2, and C9′-H2), 1.86–1.56 (m, 13H, C1′-H2, C3-H2, C4′-CH3, C8′-CH3 and C12′-CH3), 1.27 (s, 3H, C10-H3); 13C-NMR (CDCl3): δ 172.9 (C13-C=O), 170.4 (COCH3), 149.8 (phenyl C-6), 145.0 (C-11′), 142.6 (phenyl C-8a), 135.1 (C-8′), 133.8 (C-4′), 127.4 (C-12′), 127.0 (phenyl C-8), 125.3 (C-3′), 124.4 (C-7′), 121.2 (2C, phenyl C-4a and C-7), 119.2 (phenyl C-5), 76.0 (C-2), 40.0 (C-5′), 39.6 (C-1′), 38.1 (C-9′), 31.1 (C-3), 27.6 (C-6′), 26.6 (C-10′), 24.2 (C-10), 22.5 (C-4), 22.2 (C-2′), 21.2 (COCH3), 16.2 (C-9), 16.1 (C8′-CH3), 16.0 (C4′-CH3), 12.1 (C12′-CH3). MS (ESI) m/z 467.2 [M − H]−.
3.2.12. Synthesis of 13-[(R)-6-Hydroxy-2,8-dimethylchroman-2-yl]-2,6,10-trimethyltrideca-2E,6E,10E-trienoic acid (1, garcinoic acid)
To a stirred solution of 22 (23 mg, 0.05 mmol) in MeOH (2 mL) was added K2CO3 (21 mg, 0.15 mmol). Reaction was continued at room temperature under N2 until TLC showed complete conversion. The mixture was filtered and the solids was rinsed with ethyl acetate (5 mL) and acidified by adding 1 N HCl (3 mL). The aqueous layer was extracted with ethyl acetate (2 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column using hexanes-ethyl acetate (1:1) to afford compound 1 (14 mg, yield 85%) as a colorless oil.









{kind=link}
{kind=link}
{kind=link}
{kind=link}













