
Biobased Polymers via Radical Homopolymerization and Copolymerization of a Series of Terpenoid-Derived Conjugated Dienes with exo-Methylene and 6-Membered Ring


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Radical Homopolymerization
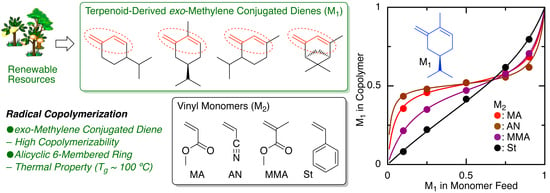
2.2. Radical Copolymerization with Various Common Vinyl Monomers
2.3. Monomer Reactivity Ratio
2.4. Thermal Properties of Copolymers
2.5. Reversible Addition Fragmentation Chain-Transfer (RAFT) Copolymerization of (–)-HCvD and Various Common Vinyl Monomers
3. Materials and Methods
3.1. Materials
3.2. Purification of β-Phellandrene (β-Phe)
3.3. Synthesis of (–)-VnD
3.4. Synthesis of PtD
3.5. RAFT Copolymerization
3.6. Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Moad, G.; Solomon, D.H. The Chemistry of Radical Polymerization: Second Fully Revised Edition; Elsevier: Oxford, UK, 2006. [Google Scholar]
- Hawker, C.J.; Bosman, A.W.; Harth, E. New Polymer Synthesis by Nitroxide Mediated Living Radical Polymerizations. Chem. Rev. 2001, 101, 3661–3688. [Google Scholar] [CrossRef] [PubMed]
- Matyjaszewski, K.; Xia, J.H. Atom Transfer Radical Polymerization. Chem. Rev. 2001, 101, 2921–2990. [Google Scholar] [CrossRef] [PubMed]
- Kamigaito, M.; Ando, T.; Sawamoto, M. Metal-Catalyzed Living Radical Polymerization. Chem. Rev. 2001, 101, 3689–3745. [Google Scholar] [CrossRef] [PubMed]
- Moad, G.; Rizzardo, E.; Thang, S.H. Toward Living Radical Polymerization. Acc. Chem. Res. 2008, 41, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Matyjaszewski, K. Atom Transfer Radical Polymerization (ATRP): Current Status and Future Perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- McKenzie, T.G.; Fu, Q.; Uchiyama, M.; Satoh, K.; Xu, J.; Boyer, C.; Kamigaito, M.; Qiao, G.G. Beyond Traditional RAFT: Alternative Activation of Thiocarbonylthio Compounds for Controlled Polymerization. Adv. Sci. 2016, 3, 1500394. [Google Scholar] [CrossRef]
- Ouchi, M.; Sawamoto, M. 50th Anniversary Perspective: Metal-Catalyzed Living Radical Polymerization: Discovery and Perspective. Macromolecules 2017, 50, 2603–2614. [Google Scholar] [CrossRef]
- Kamigaito, M.; Sawamoto, M. Synergistic Advances in Living Cationic and Radical Polymerizations. Macromolecules 2020, 53, 6749–6753. [Google Scholar] [CrossRef]
- Nothling, M.D.; Fu, Q.; Reyhani, A.; Allison-Logan, S.; Jung, K.; Zhu, J.; Kamigaito, M.; Boyer, C.; Qiao, G.G. Progress and Perspective Beyond Traditional RAFT Polymerization. Adv. Sci. 2020, 7, 2001656. [Google Scholar] [CrossRef]
- Yao, K.; Tang, C. Controlled Polymerization of Next-Generation Renewable Monomers and Beyond. Macromolecules 2013, 46, 1689–1712. [Google Scholar] [CrossRef]
- Iwata, T. Biodegradable and Bio-Based Polymers: Future Prospects of Eco-Friendly Plastics. Angew. Chem. Int. Ed. 2015, 54, 3210–3215. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Romain, C.; Williams, C.K. Sustainable polymers from renewable resources. Nature 2016, 540, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Froidevaux, V.; Negrell, C.; Caillol, S.; Pascault, J.-P.; Boutevin, B. Biobased Amines: From Synthesis to Polymers; Present and Future. Chem. Rev. 2016, 116, 14181–14224. [Google Scholar] [CrossRef] [PubMed]
- Llevot, A.; Dannecker, P.-K.; von Czapiewski, M.; Over, L.C.; Söyer, Z.; Meier, M.A.R. Renewability is not Enough: Recent Advances in the Sustainable Synthesis of Biomass-Derived Monomers and Polymers. Chem. Eur. J. 2016, 22, 11510–11521. [Google Scholar] [CrossRef]
- Thomsett, M.R.; Storr, T.E.; Monaghan, O.R.; Stockman, R.A.; Howdle, S.M. Progress in the sustainable polymers from terpenes and terpenoids. Green Mater. 2016, 4, 115–134. [Google Scholar] [CrossRef] [Green Version]
- Llevot, A.; Grau, E.; Carlotti, S.; Grelier, S.; Cramail, H. From Lignin-Derived Aromatic Compounds to Novel Biobased Polymers. Macromol. Rapid Commun. 2016, 37, 9–28. [Google Scholar] [CrossRef] [Green Version]
- Schneiderman, D.K.; Hillmyer, M.A. There is a Great Future in Sustainable Polymers. Macromolecules 2017, 50, 3733–3749. [Google Scholar] [CrossRef]
- Nguyen, H.T.H.; Rostagno, P.; Qi, M.; Feteha, A.; Miller, S.A. The quest for high glass transition temperature bioplastics. J. Mater. Chem. A 2018, 6, 9298–9331. [Google Scholar] [CrossRef]
- Tang, X.; Chen, E.Y.-X. Toward Infinity Recyclable Plastics Derived from Renewable Cyclic Esters. Chem 2019, 5, 284–312. [Google Scholar] [CrossRef] [Green Version]
- O’Dea, R.M.; Willie, J.A.; Epps, T.H., III. 100th Anniversary of Macromolecular Science Viewpoint: Polymers from Lignocellulosic Biomass. Current Challenges and Future Opportunities. ACS Macro Lett. 2020, 9, 476–493. [Google Scholar]
- Erman, W.F. Chemistry of the Monoterpenes: An Encyclopedia Handbook; Marcel Dekker, Inc.: New York, NY, USA, 1985. [Google Scholar]
- Connolly, J.D.; Hill, R.A. Dictionary of Terpenoids; Chapman & Hall: London, UK, 1991. [Google Scholar]
- Breitmaier, E. Terpenes: Flavors, Fragrances, Pharmaca, Pheromones; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Handbook of Essential Oils: Science, Technology, and Applications; Başer, K.H.C.; Buchbouer, G. (Eds.) CRC Press: Boca Baton, FL, USA, 2016. [Google Scholar]
- Satoh, K.; Kamigaito, M. New Polymerization Methods for Biobased Polymers. In Bio-Based Polymers; Kimura, Y., Ed.; CMC: Tokyo, Japan, 2013; pp. 95–111. [Google Scholar]
- Satoh, K. Controlled/Living Polymerization of renewable vinyl monomers into bio-based polymers. Polym. J. 2015, 47, 527–536. [Google Scholar] [CrossRef]
- Kamigaito, M.; Satoh, K. Bio-Based Hydrocarbon Polymers, In Encyclopedia of Polymeric Nanomaterials.; Kobayashi, S., Müllen, K., Eds.; Springer: Heidelberg, Germany, 2015; Volume 1, pp. 109–118. [Google Scholar]
- Kamigaito, M.; Satoh, K. Sustainable Vinyl Polymers via Controlled Polymerization of Terpenes. In Sustainable Polymers from Biomass; Tang, C., Ryu, C.Y., Eds.; Wiley-VCH: Weinheim, Germany, 2017; pp. 55–90. [Google Scholar]
- Akkapeddi, M.K. Poly(α-methylene-γ-butyrolactone) Synthesis, Configurational Structure, and Properties. Macromolecules 1979, 12, 546–551. [Google Scholar] [CrossRef]
- Akkapeddi, M.K. The free radical copolymerization characteristics of α-methylene γ-butyrolactone. Polymer 1979, 20, 1215–1216. [Google Scholar] [CrossRef]
- Mosnacek, J.; Matyjaszewski, K. Atom Transfer Radical Polymerization of Tulipalin A: A Naturally Renewable Monomer. Macromolecules 2008, 41, 5509–5511. [Google Scholar] [CrossRef]
- Mosnacek, J.; Yoon, J.A.; Juhari, A.; Koynov, K.; Matyjaszewski, K. Synthesis, morphology and mechanical properties of linear triblock copolymers based on poly(α-methylene-γ-butyrolactone). Polymer 2009, 50, 2087–2094. [Google Scholar] [CrossRef]
- Zhang, Y.; Miyake, G.M.; Chen, E.Y.-X. Alane-Based Classical and Frustrated Lewis Pairs in Polymer Synthesis: Rapid Polymerization of MMA and Naturally Renewable Methylene Butyrolactones to High Molecular Weight Polymers. Angew. Chem. Int. Ed. 2010, 49, 10158–10162. [Google Scholar] [CrossRef]
- Schmitt, M.; Falivene, L.; Caporaso, L.; Cavallo, L.; Chen, E.Y.-X. High-speed organocatalytic polymerization of a renewable methylene butyrolactone by a phospazene superbase. Polym. Chem. 2014, 5, 3261–3270. [Google Scholar] [CrossRef]
- Pfelifer, V.F.; Vojnovich, C.; Heger, E.N. Itaconic Acid by Fermentation with Aspergillus Terreus. Ind. Chem. Eng. 1952, 44, 2975–2980. [Google Scholar] [CrossRef]
- Kertes, A.S.; King, C.J. Extraction chemistry of fermentation product carboxylic acids. Biotechnol. Bioeng. 1986, 28, 269–282. [Google Scholar] [CrossRef]
- Tate, B.E. Polymerization of itaconic acid and derivatives. Adv. Polym. Sci. 1967, 5, 214–232. [Google Scholar]
- Ishida, S.; Saito, S. Polymerization of Itaconic Acid Derivatives. J. Polym. Sci. Part. A-1 Polym. Chem. 1967, 5, 689–705. [Google Scholar] [CrossRef]
- Satoh, K.; Lee, D.-H.; Nagai, K.; Kamigaito, M. Precision Synthesis of Bio-Based Acrylic Thermoplastic Elastomer by RAFT Polymerization of Itaconic Acid Derivatives. Macromol. Rapid Commun. 2014, 35, 161–167. [Google Scholar] [PubMed]
- Gowda, R.R.; Chen, E.Y.-X. Synthesis of β-methyl-α-methylene-γ-butyrolactone from biorenewable itaconic acid. Org. Chem. Fornt. 2014, 1, 230–234. [Google Scholar] [CrossRef]
- Nonoyama, Y.; Satoh, K.; Kamigaito, M. Renewable β-methylstyrenes for bio-based heat-resistant styrenic copolymers: Radical copolymerization enhanced by fluoroalcohol and controlled/living copolymerization by RAFT. Polym. Chem. 2014, 5, 3182–3189. [Google Scholar]
- Terao, Y.; Satoh, K.; Kamigaito, M. Controlled Radical Copolymerization of Cinnamic Derivatives as Renewable Vinyl Monomers with Both Acrylic and Styrenic Substituents: Reactivity, Regioselectivity, Properties, and Functions. Biomacromolecules 2019, 20, 192–203. [Google Scholar] [CrossRef]
- Terao, Y.; Satoh, K.; Kamigaito, M. 1:3 ABAA sequence-regulated substituted polymethylenes via alternating radical copolymerization of methyl cinnamate and maleic anhydride followed by post-polymerization reactions. Eur. Polym. J. 2019, 120, 109225. [Google Scholar] [CrossRef]
- Imada, M.; Takenaka, Y.; Hatanaka, H.; Tsuge, T.; Abe, H. Unique acrylic resins with aromatic side chains by homopolymerization of cinnamic monomers. Commun. Chem. 2019, 2, 109. [Google Scholar] [CrossRef] [Green Version]
- Takeshima, H.; Satoh, K.; Kamigaito, M. Bio-Based Functional Styrene Monomers Derived from Naturally Occurring Ferulic Acid for Poly(vinylcatechol) and Poly(vinylguaicol) via Controlled Radical Polymerization. Macromolecules 2017, 50, 4206–4216. [Google Scholar] [CrossRef]
- Takeshima, H.; Satoh, K.; Kamigaito, M. Scalable Synthesis of Bio-Based Functional Styrene: Protected Vinyl Catechol from Caffeic Acid and Controlled Radical and Anionic Polymerizations Thereof. ACS Sustainable Chem. Eng. 2018, 6, 13681–13686. [Google Scholar] [CrossRef]
- Takeshima, H.; Satoh, K.; Kamigaito, M. Bio-Based Vinylphenol Family: Synthesis via Decarboxylation of Naturally Occurring Cinnamic Acids and Living Radical Polymerization for Functionalized Polystyrenes. J. Polym. Sci. 2020, 58, 91–100. [Google Scholar] [CrossRef]
- Satoh, K.; Matsuda, M.; Nagai, K.; Kamigaito, M. AAB-Sequence Living Radical Chain Copolymerization of Naturally-Occurring Limonene with Maleimide: An End-to-End Sequence-Regulated Copolymer. J. Am. Chem. Soc. 2010, 132, 10003–10005. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Satoh, K.; Kamigaito, M. Periodically Functionalized and Grafted Copolymers via 1:2-Sequence-Regulated Radical Copolymerization of Naturally Occurring Functional Limonene and Maleimide Derivatives. Macromolecules 2013, 46, 5473–5482. [Google Scholar] [CrossRef]
- Matsuda, M.; Satoh, K.; Kamigaito, M. Controlled Radical Copolymerization of Naturally-Occurring Terpenes with Acrylic Monomers in Fluorinated Alcohol. KGK Kaut. Gummi Kunstst. 2013, 66, 51–56. [Google Scholar]
- Matsuda, M.; Satoh, K.; Kamigaito, M. 1:2-Sequence-Regulated Radical Copolymerization of Naturally Occurring Terpenes with Maleimide Derivatives in Fluorinated Alcohol. J. Polym. Sci. Part. A Polym. Chem. 2013, 51, 1774–1785. [Google Scholar] [CrossRef]
- Miyaji, H.; Satoh, K.; Kamigaito, M. Bio-Based Polyketones by Selective Ring-Opening Radical Polymerization of α-Pinene Derived Pinocarvone. Angew. Chem. Int. Ed. 2016, 55, 1372–1376. [Google Scholar] [CrossRef]
- Ojika, M.; Satoh, K.; Kamigaito, M. BAB-random-C Monomer Sequence via Radical Terpolymerization of Limonene (A), Maleimide (B), and Methacrylate (C): Terpene Polymers with Randomly Distributed Periodic Sequences. Angew. Chem. Int. Ed. 2017, 56, 1789–1793. [Google Scholar] [CrossRef]
- Hashimoto, H.; Takeshima, H.; Nagai, T.; Uchiyama, M.; Satoh, K.; Kamigaito, M. Valencene as a naturally occurring sesquiterpene monomer for radical copolymerization with maleimide to induce concurrent 1:1 and 1:2 propagation. Polym. Degrad. Stab. 2019, 161, 183–190. [Google Scholar] [CrossRef]
- Runckel, W.J.; Goldblatt, L.A. Inhibition of Myrcene Polymerization during Storage. Ind. Eng. Chem. 1946, 38, 749–751. [Google Scholar] [CrossRef]
- Johanson, A.J.; McKennon, F.L.; Goldblatt, L.A. Emulsion Polymerization of Myrcene. Ind. Eng. Chem. 1948, 401, 500–502. [Google Scholar] [CrossRef]
- Marvel, C.S.; Hwa, C.C.L. Polymyrcene. J. Polym. Sci. 1960, 45, 25–34. [Google Scholar] [CrossRef]
- Cawse, J.L.; Stanford, J.L.; Still, R.H. Polymers from Renewable Resources. III. Hydroxy-Terminated Myrcene Polymers. J. Appl. Polym. Sci. 1986, 31, 1963–1975. [Google Scholar] [CrossRef]
- Trumbo, D.L. Free radical copolymerization behavior of myrcene I. Copolymers with styrene, methyl methacrylate or p-fluorostyrene. Polym. Bull. 1993, 31, 629–636. [Google Scholar] [CrossRef]
- Sarkar, P.; Bhowmick, A.K. Synthesis, characterization and properties of a bio-based elastomer: Polymyrcene. RSC Adv. 2014, 41, 61343–61354. [Google Scholar] [CrossRef]
- Hilschmann, J.; Kali, G. Bio-based polymyrcene with highly ordered structure via solvent free controlled radical polymerization. Eur. Polym. J. 2015, 73, 363–373. [Google Scholar] [CrossRef]
- Kamigaito, M.; Satoh, K.; Suzuki, S.; Kori, Y.; Eguchi, Y.; Iwasa, K.; Shiroto, H. β-Phellandrene polymer, production method for same, and molded article. WO 2015/060310 A1; filed 21 October 2014 and issued 30 April 2015,
- Nishida, T.; Satoh, K.; Nagano, S.; Seki, T.; Tamura, M.; Li, Y.; Tomishige, K.; Kamigaito, M. Biobased Cycloolefin Polymers: Carvone-Derived Cyclic Conjugated Diene with Reactive exo-Methylene Group for Regioselective and Stereospecific Living Cationic Polymerization. ACS Macro Lett. 2020, 9, 1178–1183. [Google Scholar] [CrossRef]
- Matsumoto, A.; Yamamoto, D. Radical Copolymerization of N-Phenylmaleimide and Diene Monomers in Competition with Diels–Alder Reaction. J. Polym. Sci., Part. A Polym. Chem. 2016, 54, 3616–3625. [Google Scholar] [CrossRef]
- Li, Y.; Padias, A.B.; Hall, H.K. Evidence for 2-Hexene-1,6-diyl Diradicals Accompanying the Concerted Diels-Alder Cycloaddition of Acrylonitrile with Nonpolar 1,3-Dienes. J. Org. Chem. 1993, 58, 7049–7058. [Google Scholar] [CrossRef]
- Trumbo, D.L. Synthesis and Polymerization of 1-Methyl-4-isopropenyl-6-methylene-1-cyclohexene. J. Polym. Sci.: Part. A Polym. Chem. 1995, 33, 599–601. [Google Scholar] [CrossRef]
- Kobayashi, S.; Lu, C.; Hoye, T.R.; Hillmyer, M.A. Controlled Polymerization of a Cyclic Diene Prepared from the Ring-Closing Metathesis of a Naturally Occurring Monoterpene. J. Am. Chem. Soc. 2009, 131, 7960–7961. [Google Scholar] [CrossRef]
- Yamamoto, D.; Matsumoto, A. Controlled Radical Polymerization of 3-Methylenecyclopentene with N-Substituted Maleimides To Yield Highly Alternating and Regiospecific Copolymers. Macromolecules 2013, 46, 9526–9536. [Google Scholar] [CrossRef]
- Polymer Handbook, 4th ed.; Brandrup, J.; Immergut, E.H.; Grulke, E.A. (Eds.) John Wiley & Sons: New York, NY, USA, 1999. [Google Scholar]
- Thang, S.H.; Chong, Y.K.; Mayadunne, R.T.A.; Moad, G.; Rizzardo, E. A novel synthesis of functional dithioesters, dithiocarbamates, xanthates, and trithiocarbonates. Tetrahedron Lett. 1999, 40, 2435–2438. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Monomer | Temp. (°C) | Conv. (%) b | Mn(SEC) c | Mw/Mnc | 1,4-/1,2- d | Tg (°C) e |
|---|---|---|---|---|---|---|---|
| 1 | β-Phe | 60 | 2 | 3700 | 2.82 | 93/7 | n.d. f |
| 2 | β-Phe | 100 | 27 | 17,700 | 1.79 | 83/17 | 66 |
| 3 | HCvD | 60 | 0 | 10,800 | 3.03 | >99/0 | n.d. f |
| 4 | HCvD | 100 | 32 | 34,400 | 1.89 | >99/0 | 105 |
| 5 | PtD | 60 | 0 | 3200 | 2.47 | >99/0 | n.d. f |
| 6 | PtD | 100 | 29 | 10,700 | 1.69 | >99/0 | 113 |
| 7 | VnD | 60 | 0 | 240 | 1.10 | n.d. | n.d. f |
| 8 | VnD | 100 | 2 | 300 | 1.15 | n.d. | n.d. f |
| Entry | M1 | M2 | Time (h) | Conv.(%) b M1/M2 | Mn (SEC) c | 1,4-/1,2- d | M1/M2 (NMR) d | M1/M2 (Calcd) e | r1f | r2f |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | β-Phe | MA | 90 | 38/27 | 7900 | 83/17 | 61/39 | 58/42 | 0.66 | 0.04 |
| 2 | HCvD | MA | 20 | 44/36 | 26,000 | 95/5 | 52/48 | 55/45 | 0.14 | 0.08 |
| 3 | PtD | MA | 20 | 43/35 | 23,800 | 96/4 | 51/49 | 55/45 | 0.11 | 0.08 |
| 4 | β-Phe | AN | 40 | 51/45 | 14,600 | 84/16 | 52/48 | 53/47 | 0.17 | 0.03 |
| 5 | HCvD | AN | 10 | 44/50 | 42,100 | 95/5 | 53/47 | 47/53 | 0.09 | 0.03 |
| 6 | PtD | AN | 10 | 45/50 | 42,300 | 99/1 | 50/50 | 47/53 | 0.02 | 0.05 |
| 7 | β-Phe | MMA | 150 | 36/31 | 7800 | 89/11 | 60/40 | 54/46 | 0.68 | 0.09 |
| 8 | HCvD | MMA | 50 | 44/51 | 12,900 | 95/5 | 47/53 | 46/54 | 0.17 | 0.31 |
| 9 | PtD | MMA | 120 | 55/62 | 13,500 | >99/0 | 46/54 | 47/53 | 0.19 | 0.25 |
| 10 | β-Phe | St | 120 | 38/29 | 6700 | 85/15 | 60/40 | 57/43 | 1.53 | 0.43 |
| 11 | HCvD | St | 100 | 40/54 | 19,000 | 95/5 | 43/57 | 43/57 | 0.40 | 1.09 |
| 12 | PtD | St | 50 | 29/35 | 10,900 | >99/0 | 41/59 | 45/55 | 0.48 | 0.96 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishida, T.; Satoh, K.; Kamigaito, M. Biobased Polymers via Radical Homopolymerization and Copolymerization of a Series of Terpenoid-Derived Conjugated Dienes with exo-Methylene and 6-Membered Ring. Molecules 2020, 25, 5890. https://doi.org/10.3390/molecules25245890
Nishida T, Satoh K, Kamigaito M. Biobased Polymers via Radical Homopolymerization and Copolymerization of a Series of Terpenoid-Derived Conjugated Dienes with exo-Methylene and 6-Membered Ring. Molecules. 2020; 25(24):5890. https://doi.org/10.3390/molecules25245890
Chicago/Turabian StyleNishida, Takenori, Kotaro Satoh, and Masami Kamigaito. 2020. "Biobased Polymers via Radical Homopolymerization and Copolymerization of a Series of Terpenoid-Derived Conjugated Dienes with exo-Methylene and 6-Membered Ring" Molecules 25, no. 24: 5890. https://doi.org/10.3390/molecules25245890
APA StyleNishida, T., Satoh, K., & Kamigaito, M. (2020). Biobased Polymers via Radical Homopolymerization and Copolymerization of a Series of Terpenoid-Derived Conjugated Dienes with exo-Methylene and 6-Membered Ring. Molecules, 25(24), 5890. https://doi.org/10.3390/molecules25245890