
Synthesis of Biologically Active Molecules through Multicomponent Reactions



Abstract
:1. Introduction
2. Some Aspects of the Multicomponent Reactions
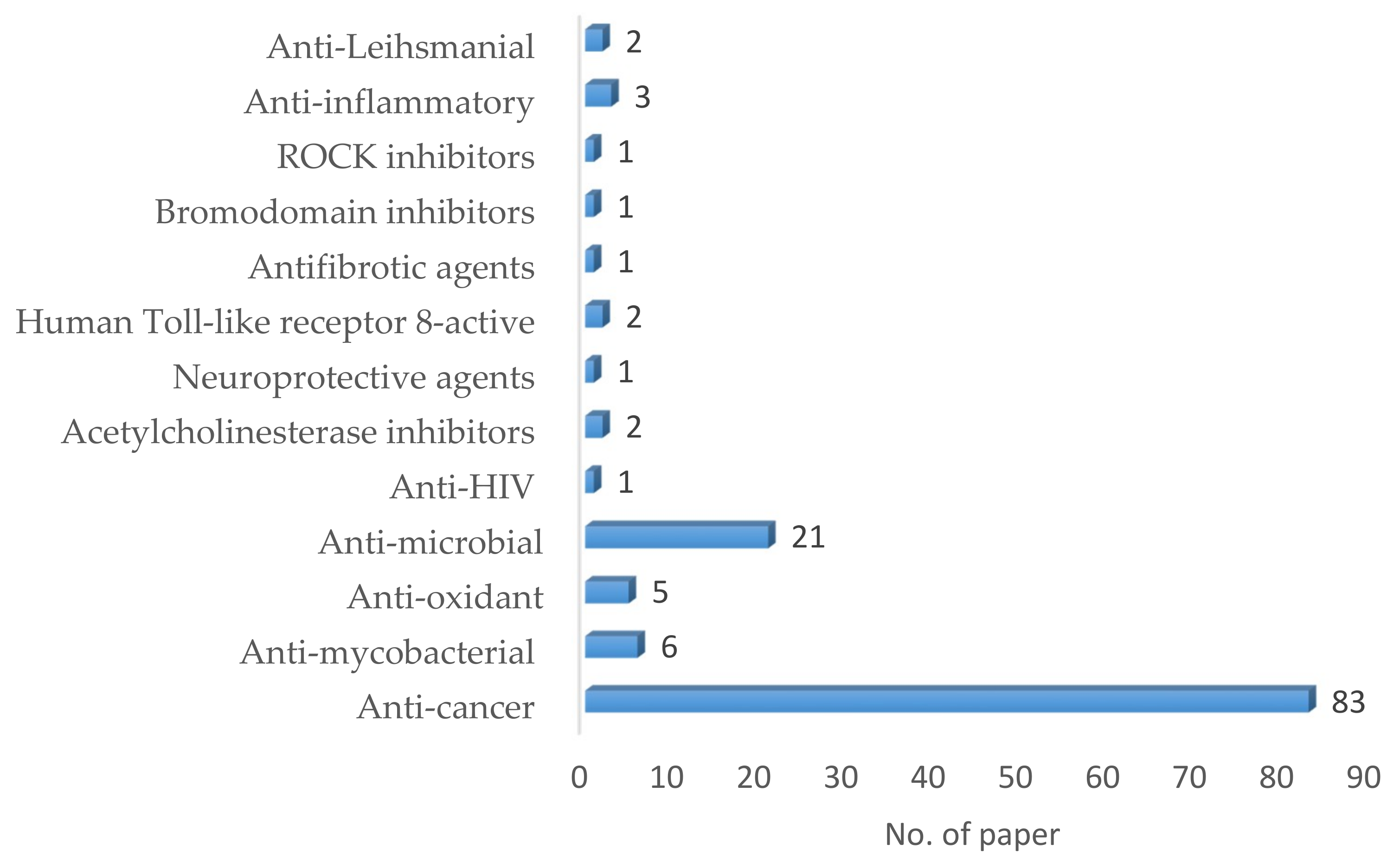
3. Biologically Active Compounds Obtained from Multicomponent Approaches
3.1. Anti-Leihsmanial Activity
3.2. Anti-Inflammatory Activity
3.3. ROCK Inhibitors
3.4. Bromodomain Inhibitors
3.5. Antifibrotic Agents
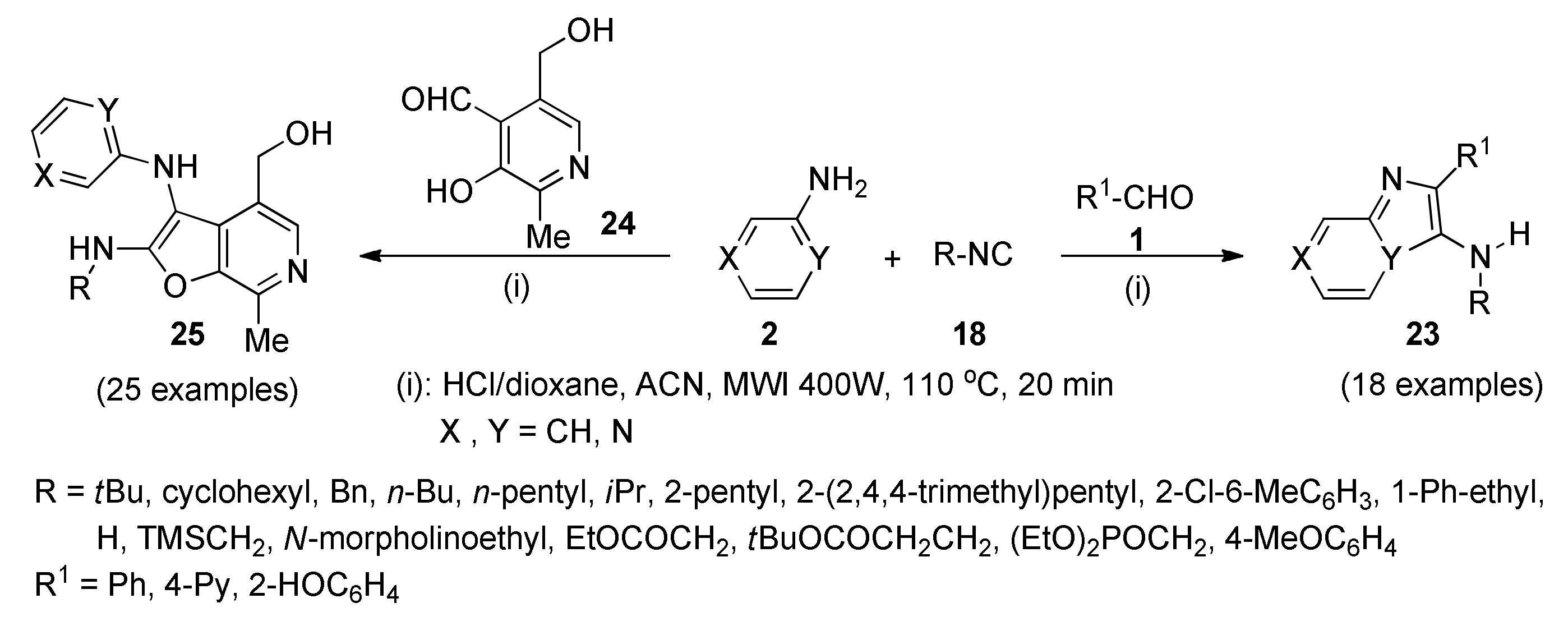
3.6. Human Toll-Like Receptor 8-Active Compounds
3.7. Neuroprotective Agents
3.8. Acetylcholinesterase Inhibitors
3.9. Anti-HIV Activity
3.10. Antimicrobial Activity
3.11. Antioxidant Activity
3.12. Anti-Mycobacterial Activity
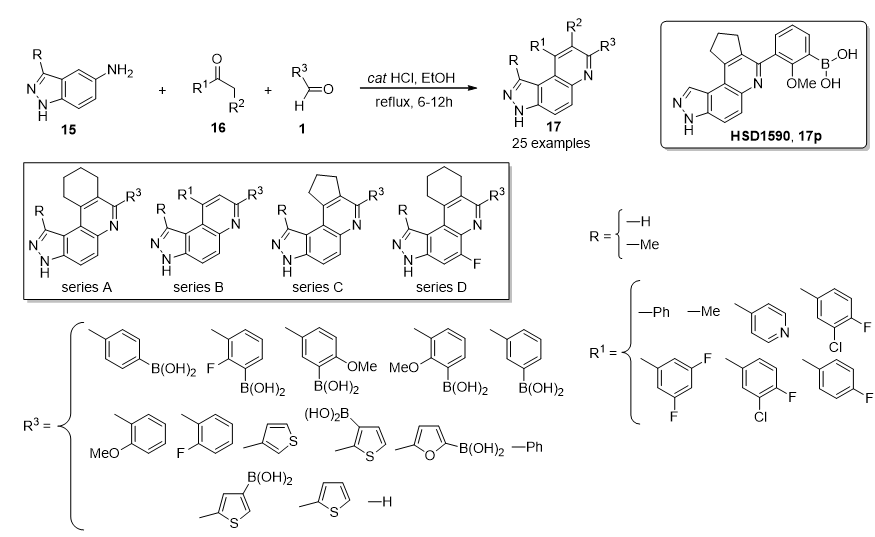
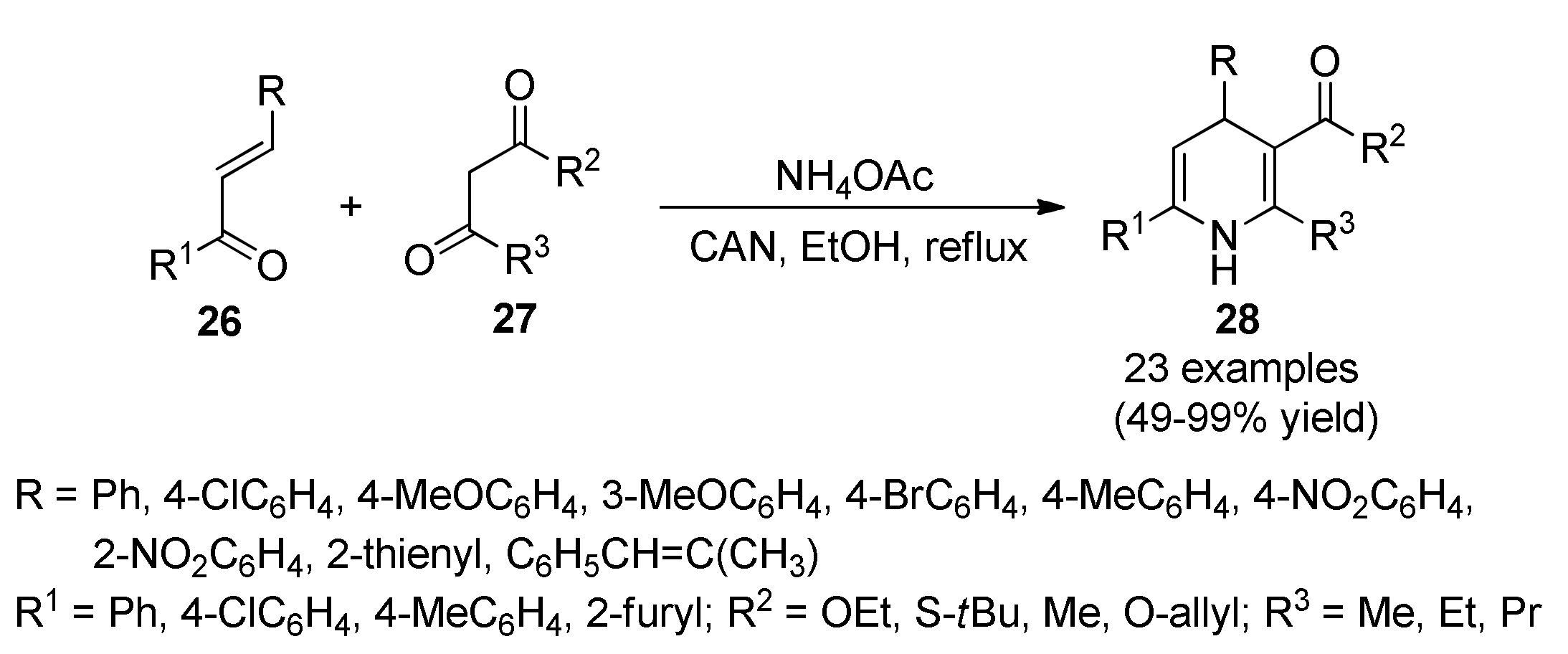
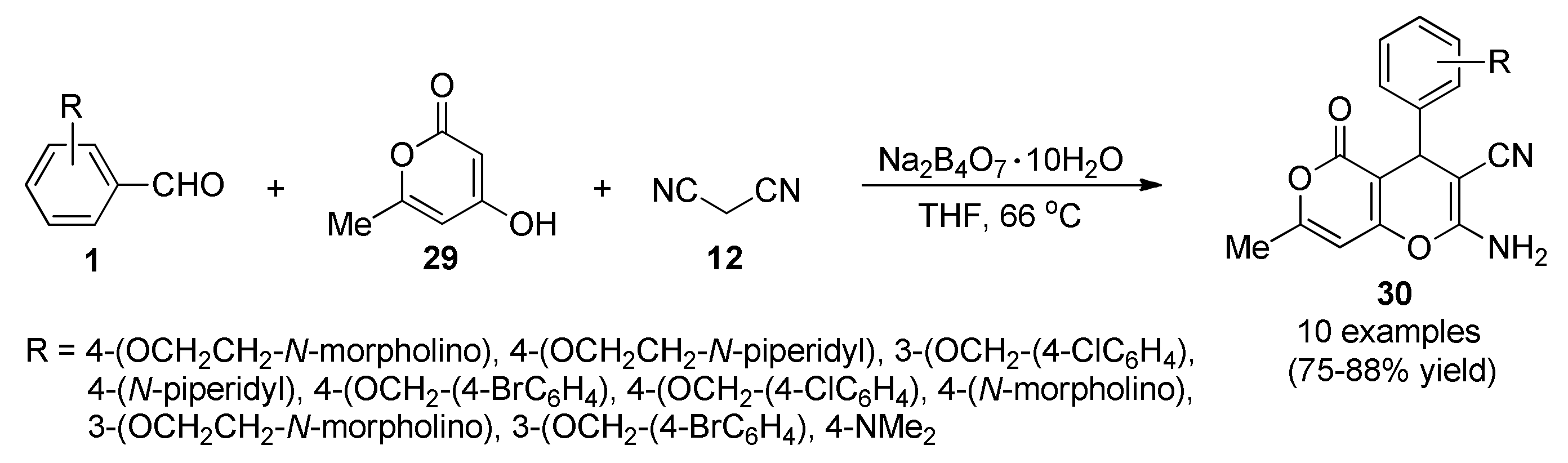
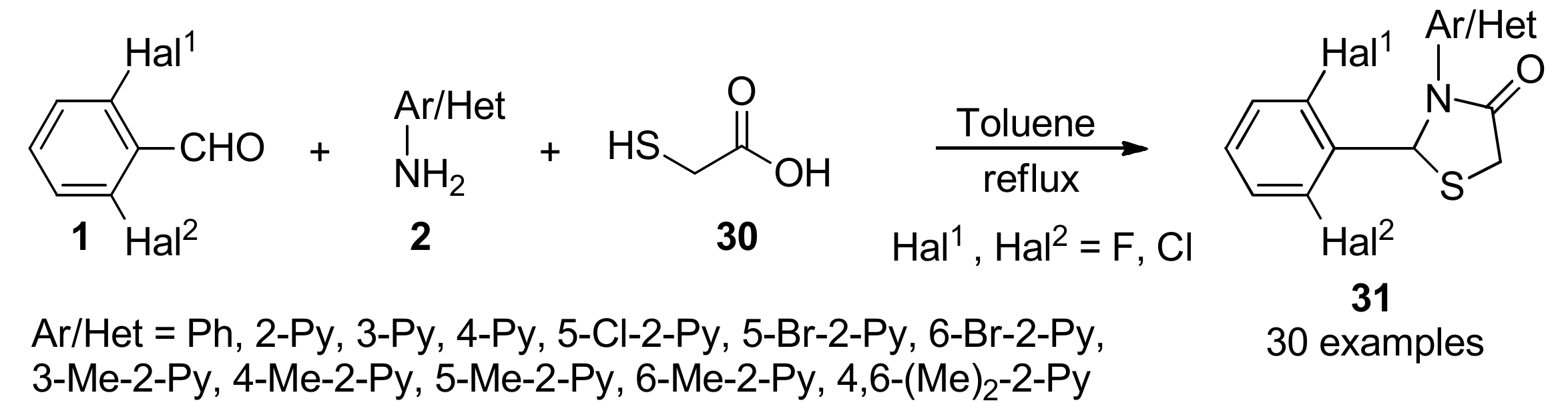
3.13. Anticancer Activity
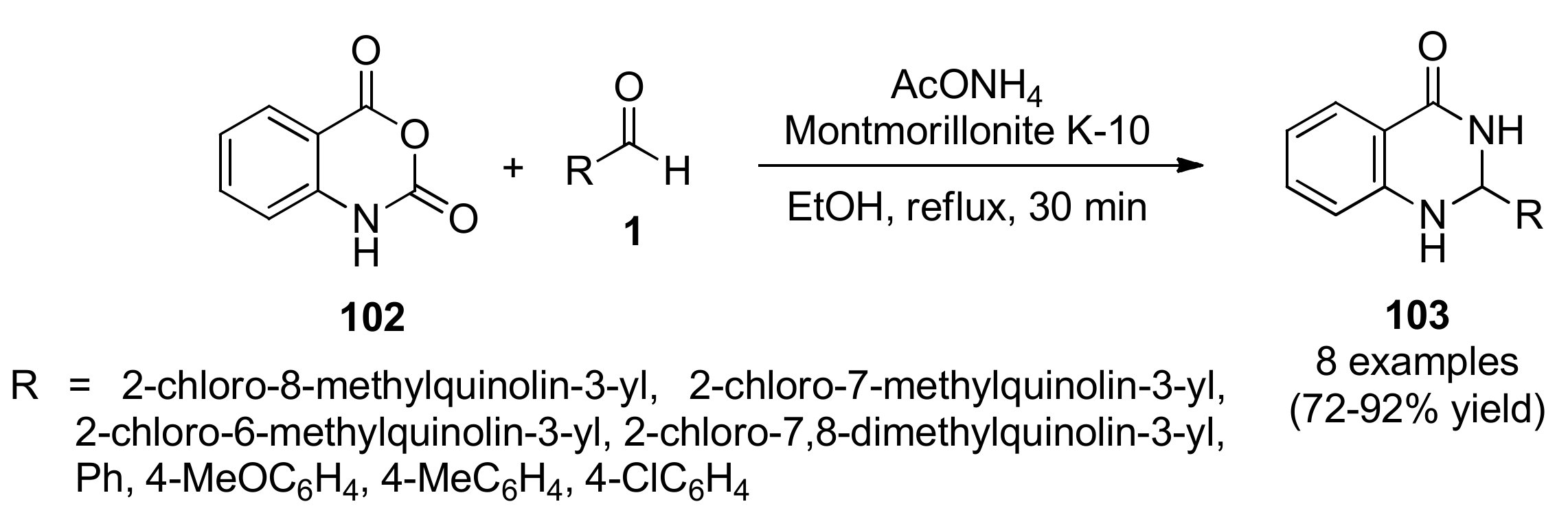
3.13.1. Quinoline Derivatives
3.13.2. Pyrazole Derivatives
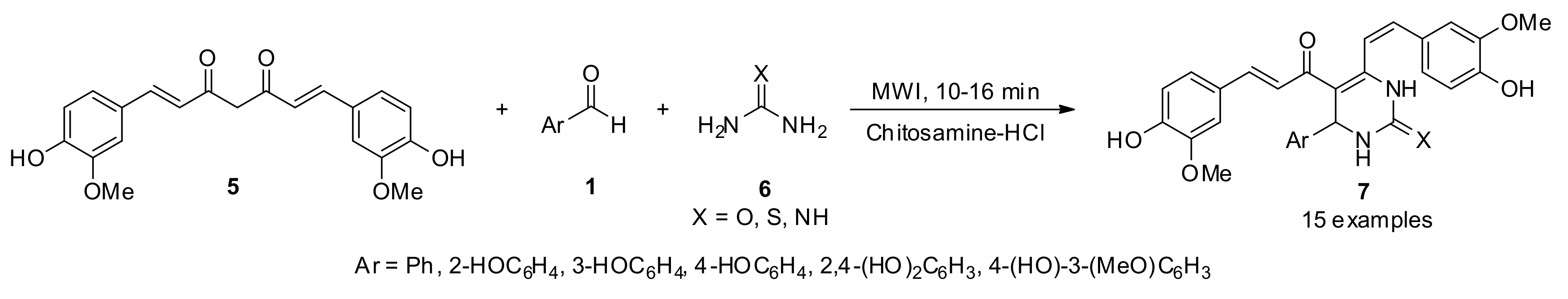
3.13.3. Curcumin Derivatives
3.13.4. Pyrrole Derivatives
3.13.5. Chromone and Chromene Derivatives
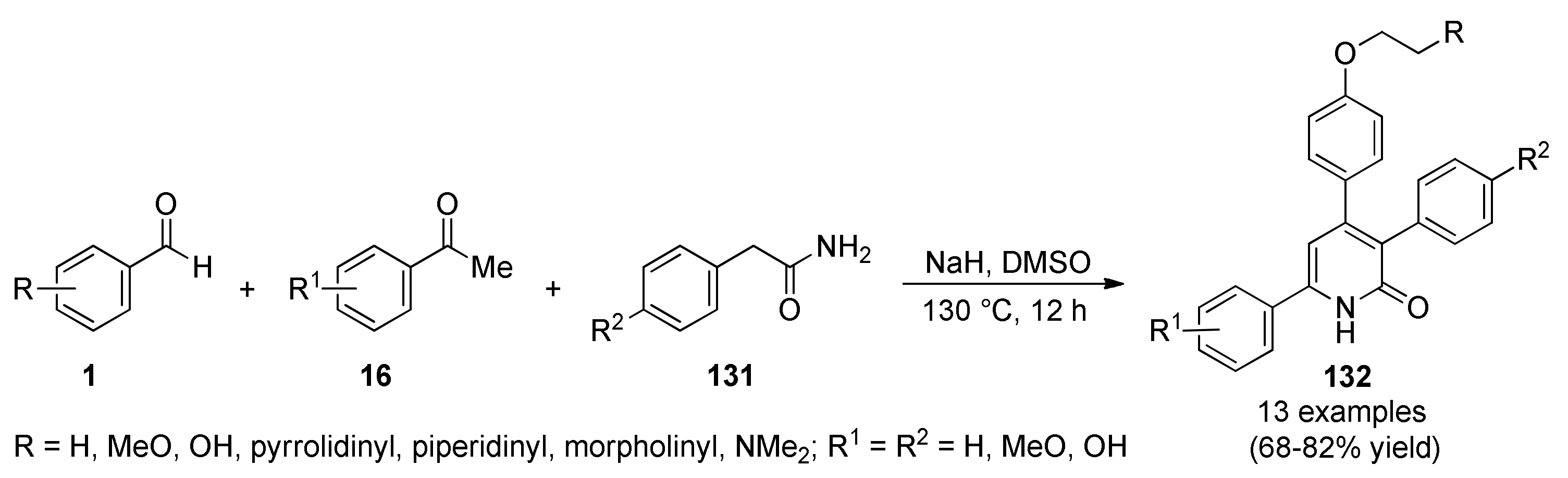
3.13.6. Pyridone Derivatives
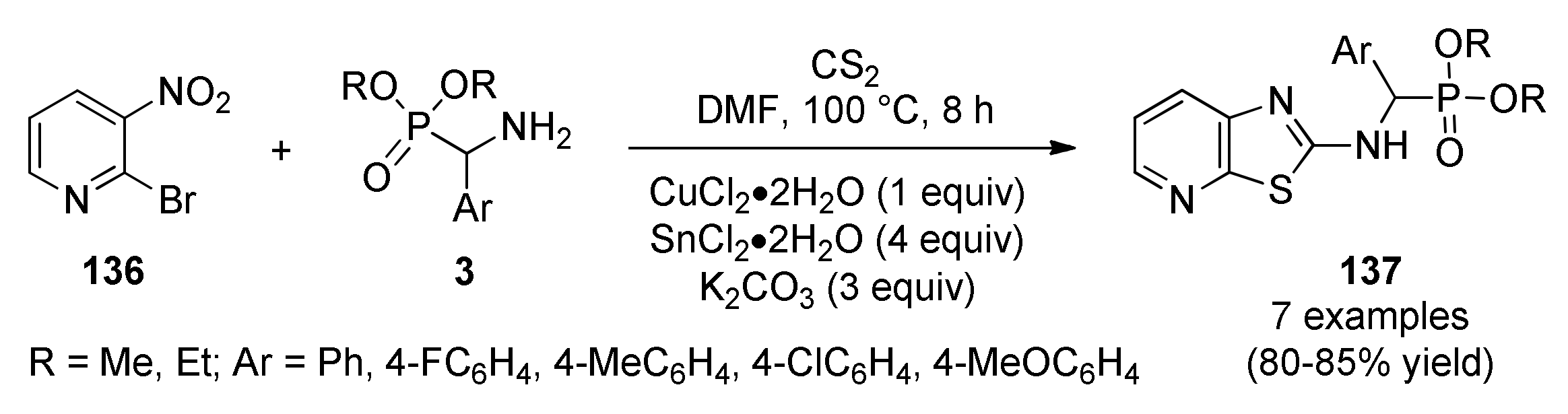
3.13.7. Thiazole Derivatives
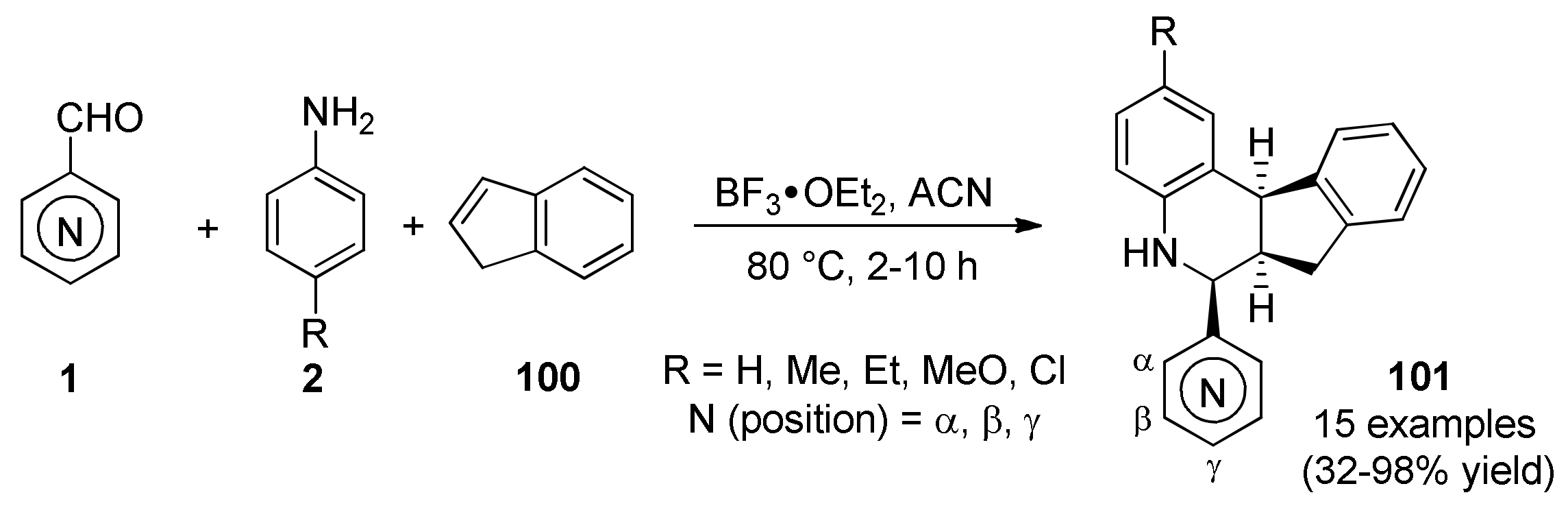
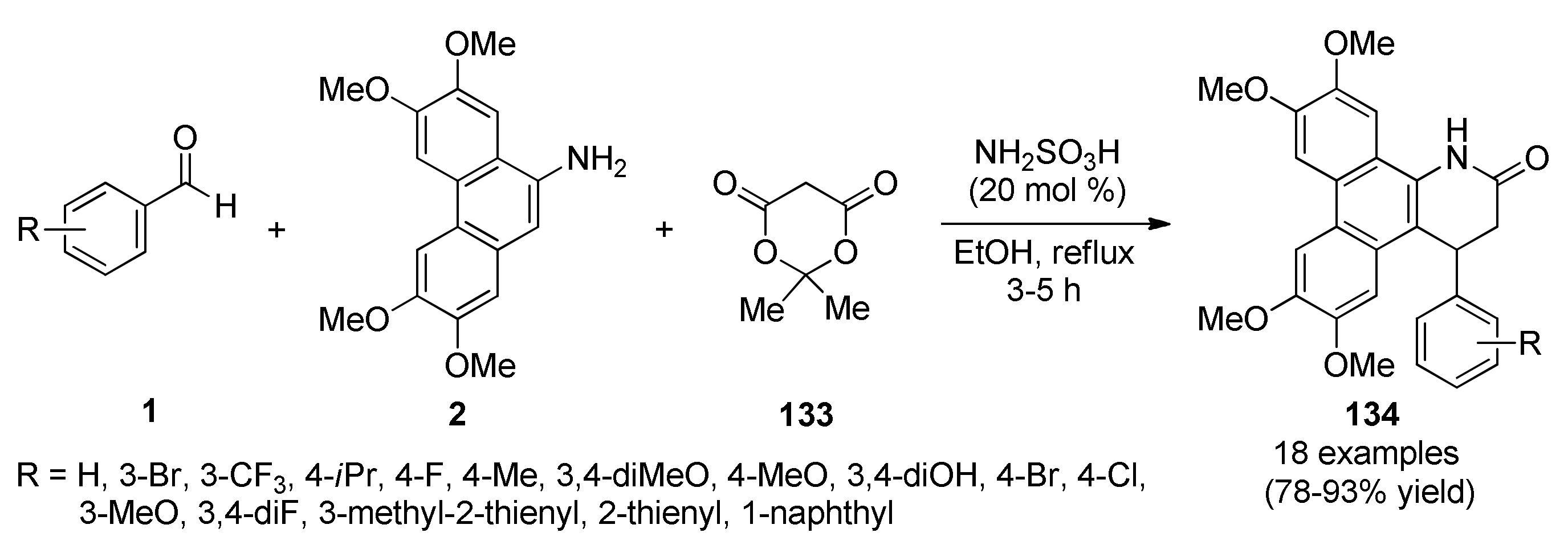
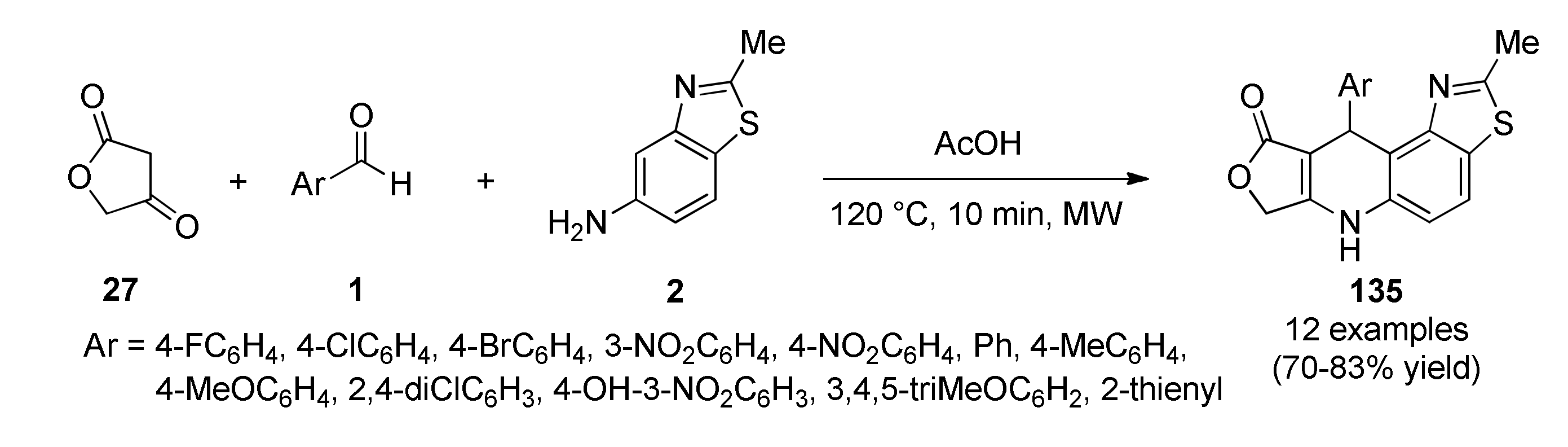
3.13.8. Indole-Based Anticancer Heterocyclic Systems
Indole-Substituted Heterocyclic Derivatives
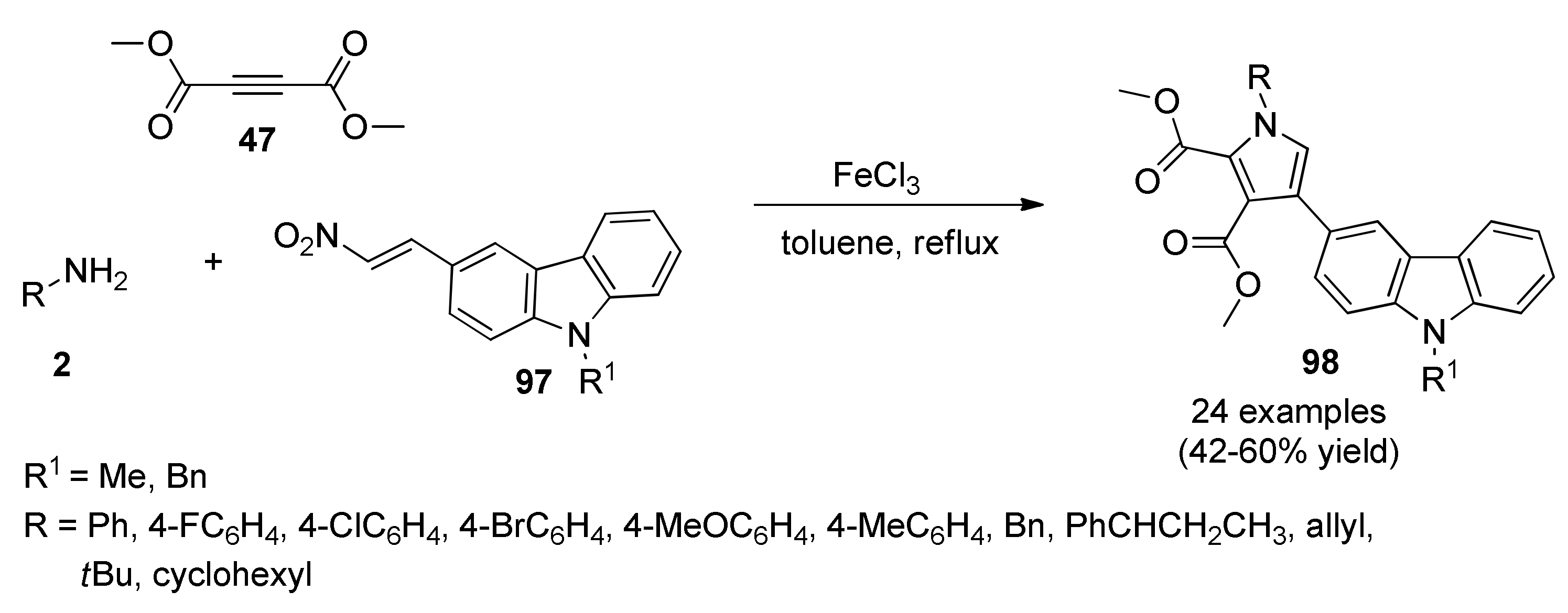
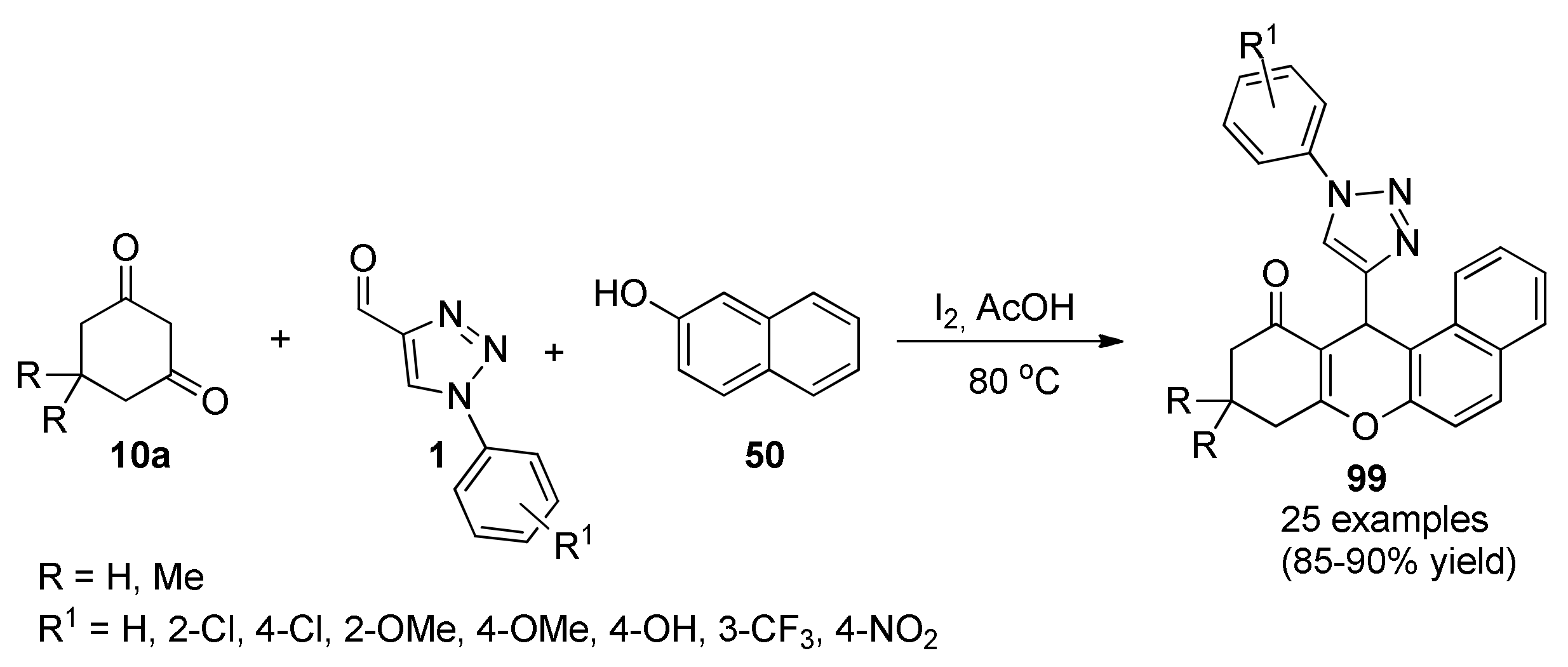
Carbazole (Benzo[b]Indole)-Substituted Heterocyclic Derivatives
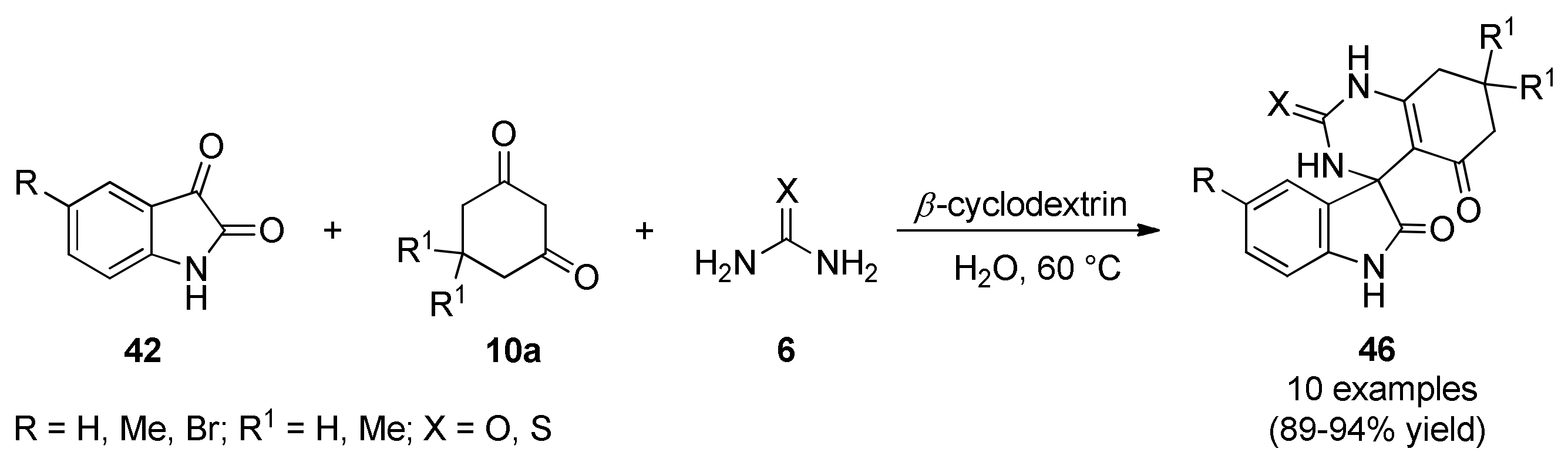
3.13.9. Pyrimidine-Based Anticancer Heterocyclic Systems
Biginelli-Mediated Synthesis of Dihydropyrimidines
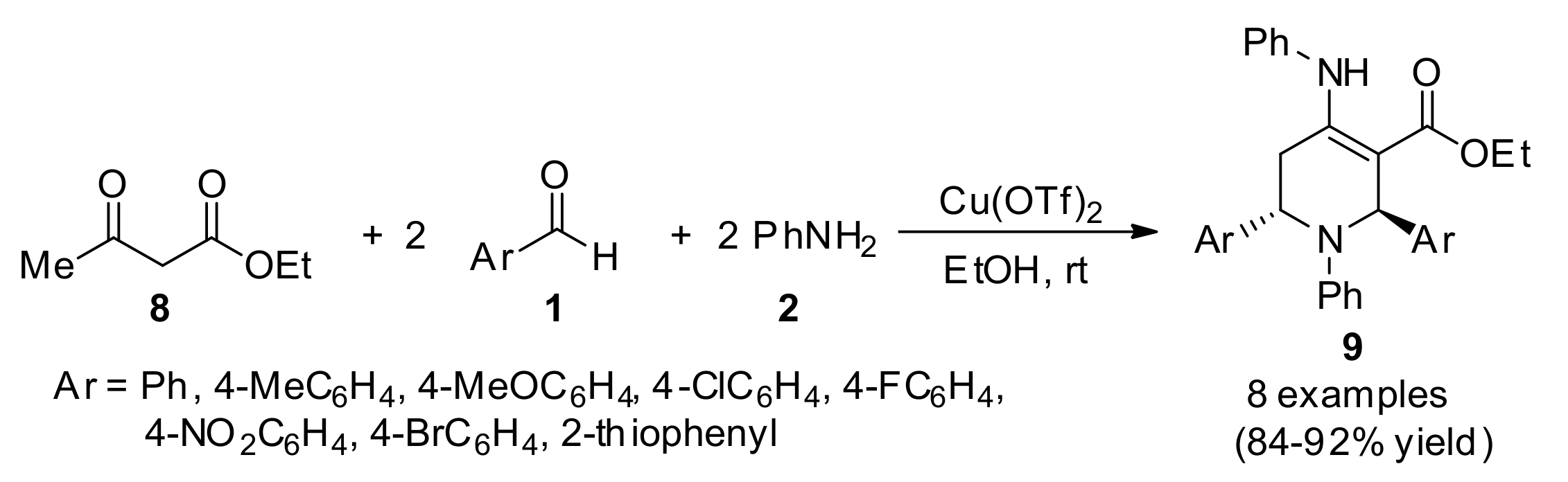
3.13.10. Dihydropyridine-Based Anticancer Heterocyclic Systems
3.13.11. Fused Dihydroquinoline-Based Anticancer Heterocyclic Systems
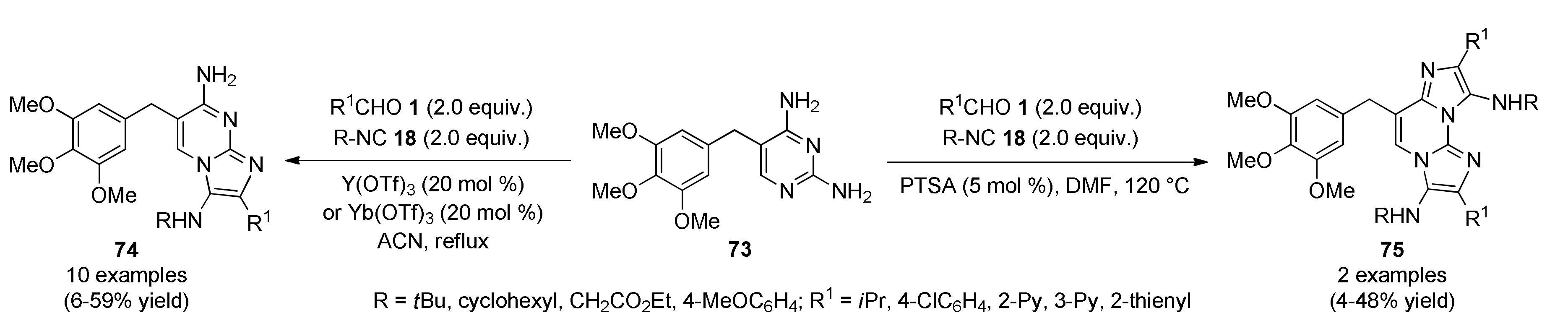
3.13.12. Purine-Like Pyrrolo[2,3-d]Pyrimidine Systems of Anticancer Interest
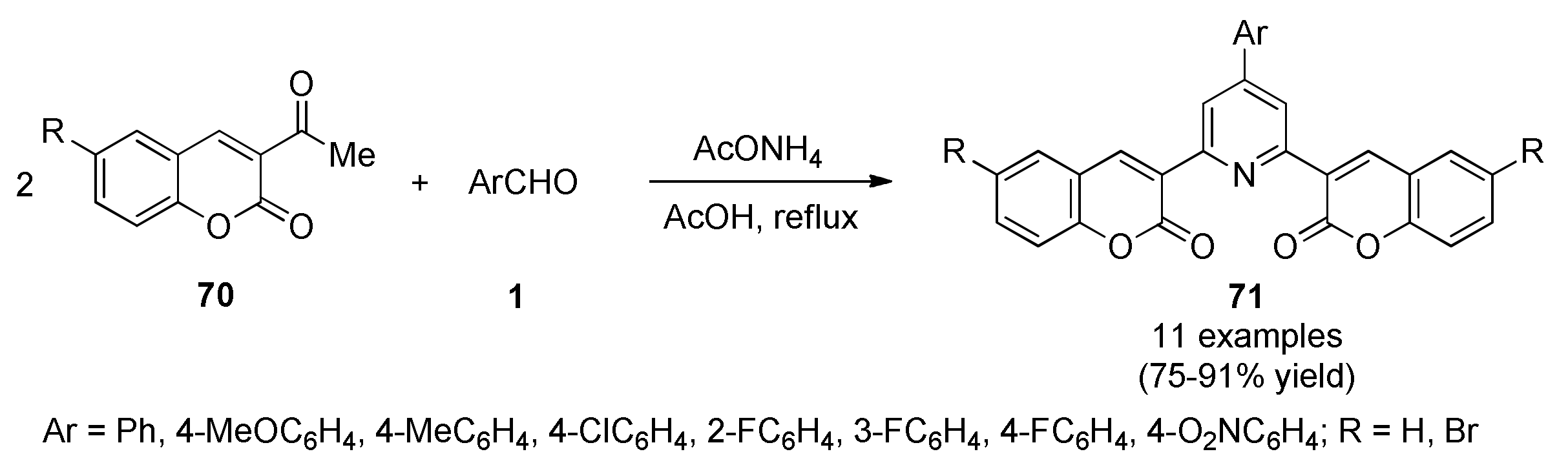
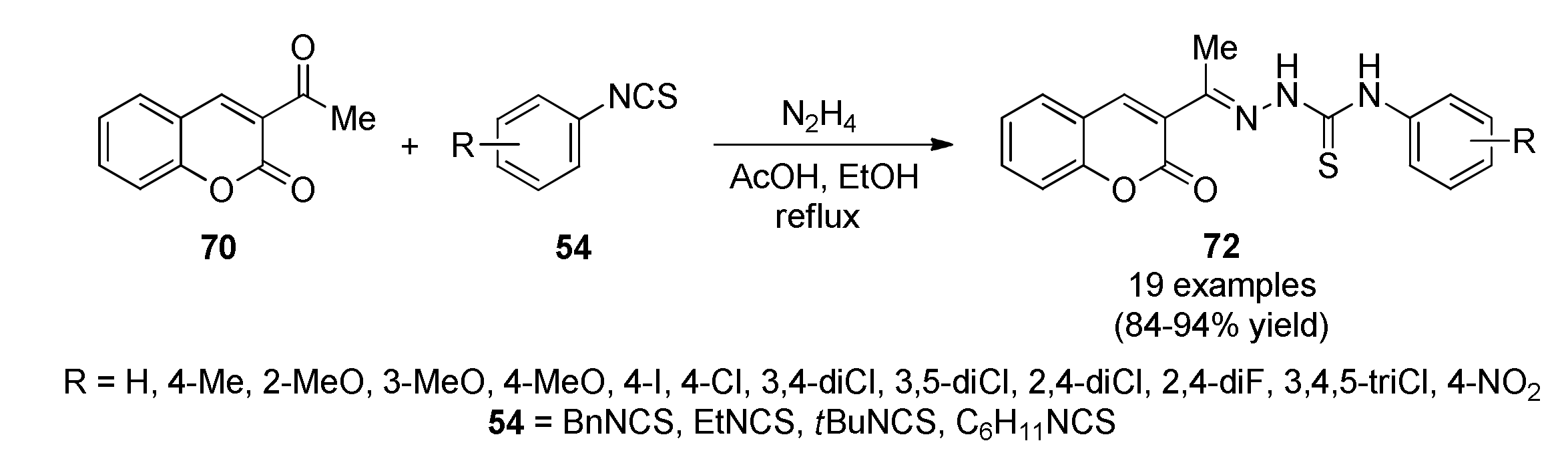
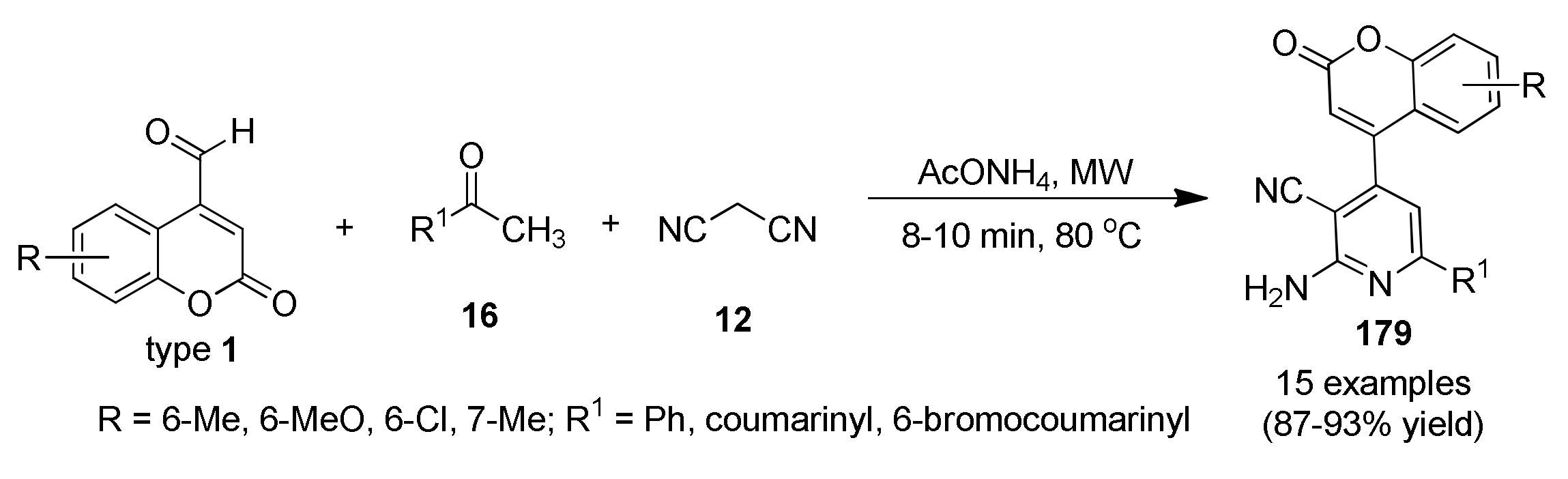
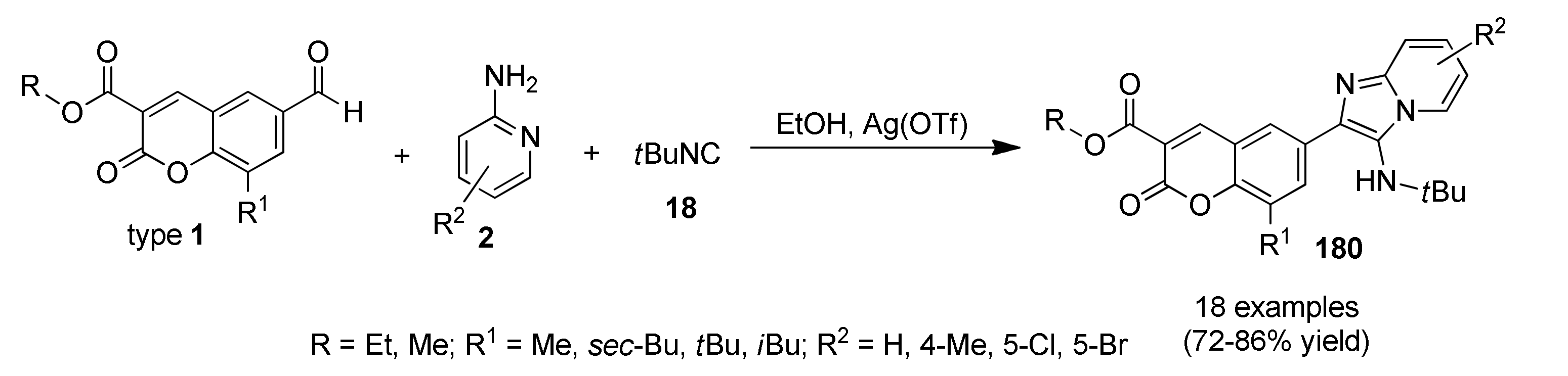
3.13.13. Coumarin Derivatives
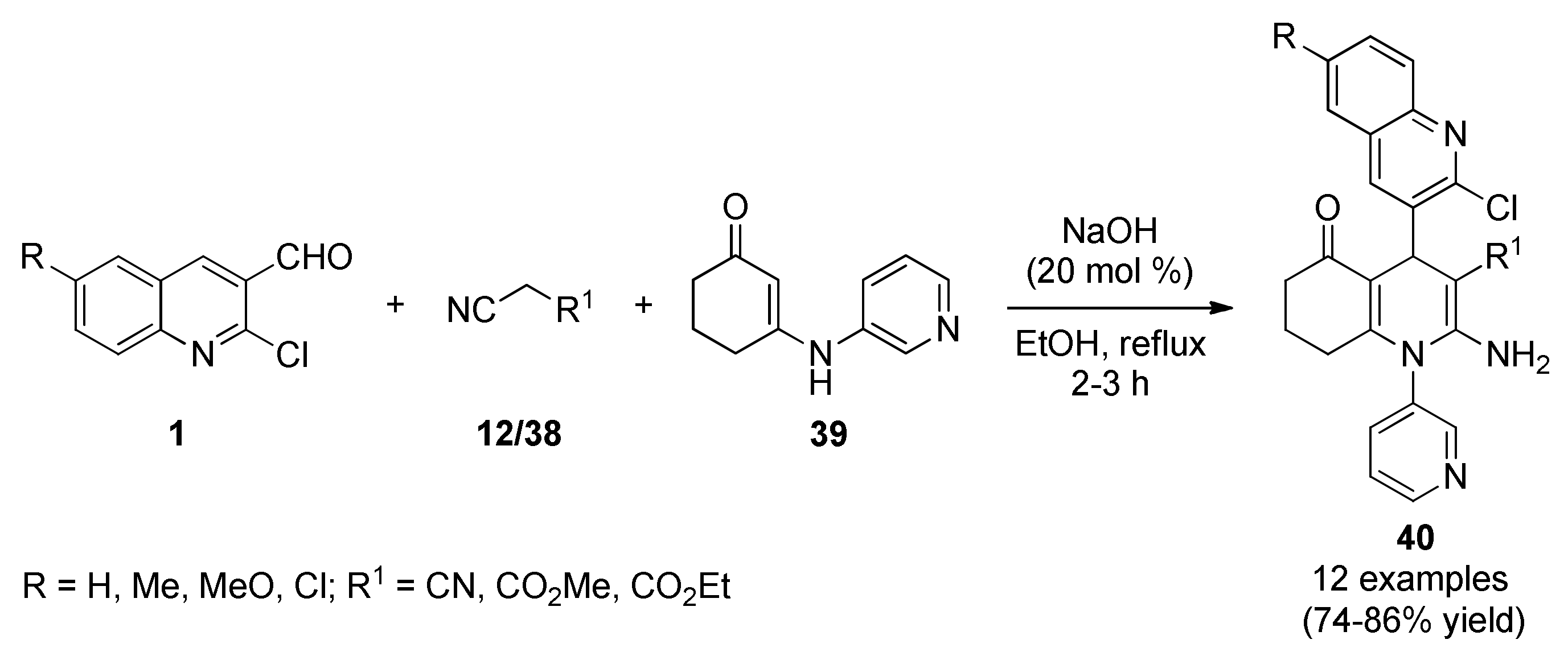
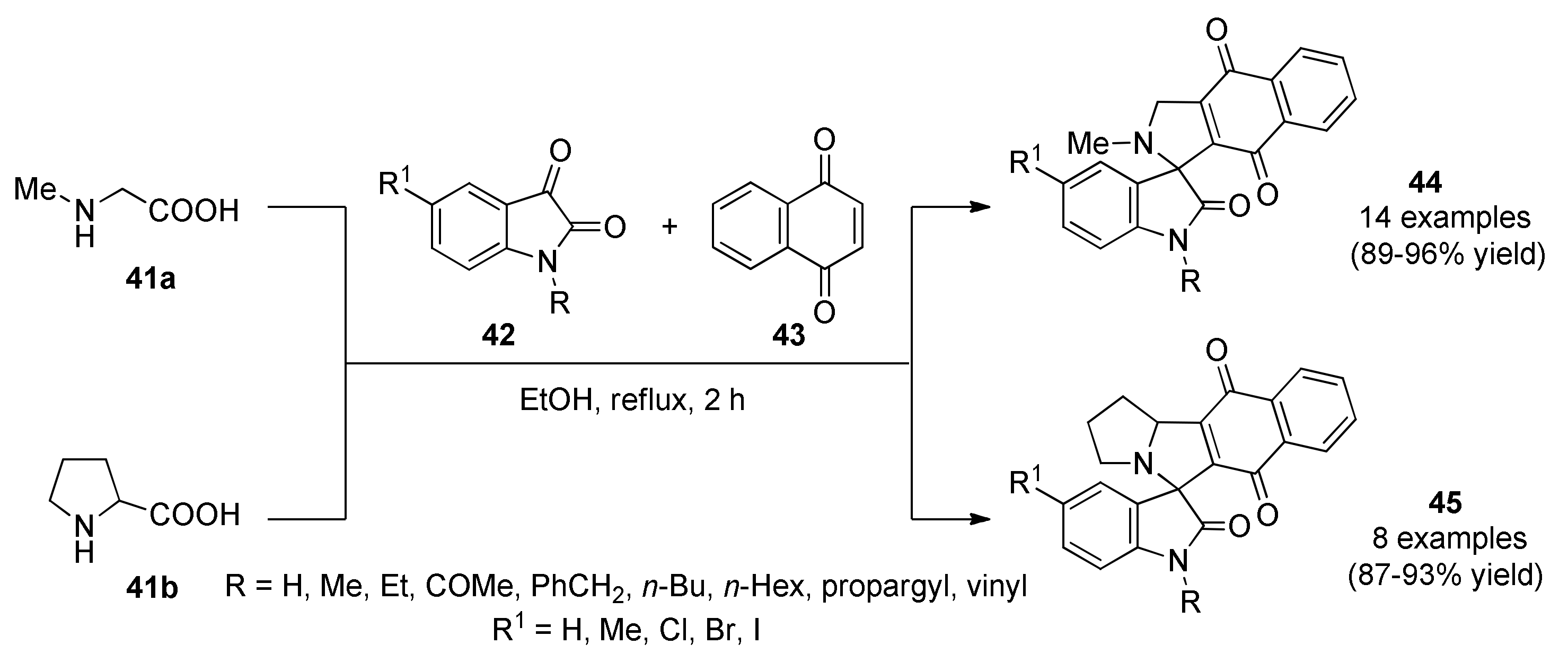
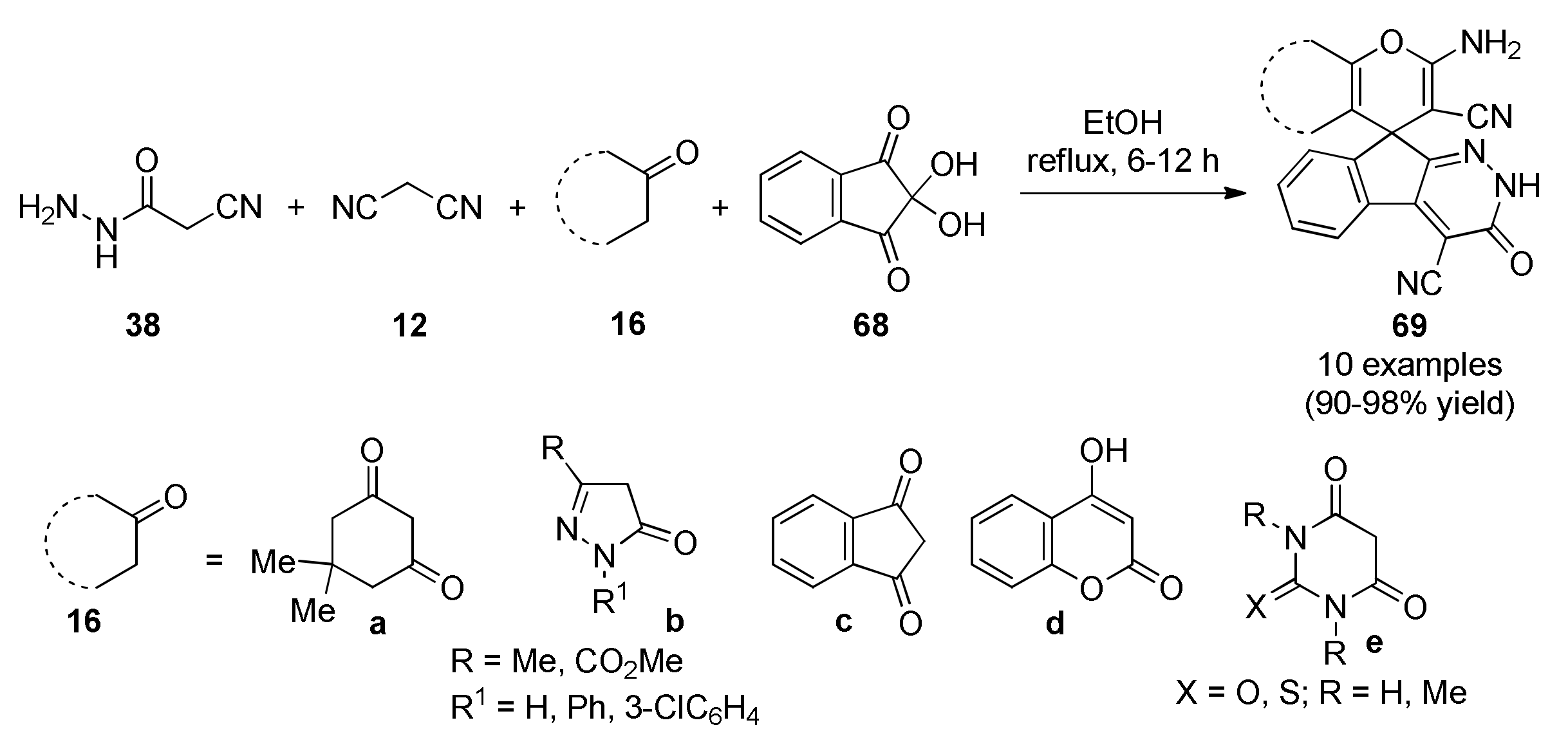
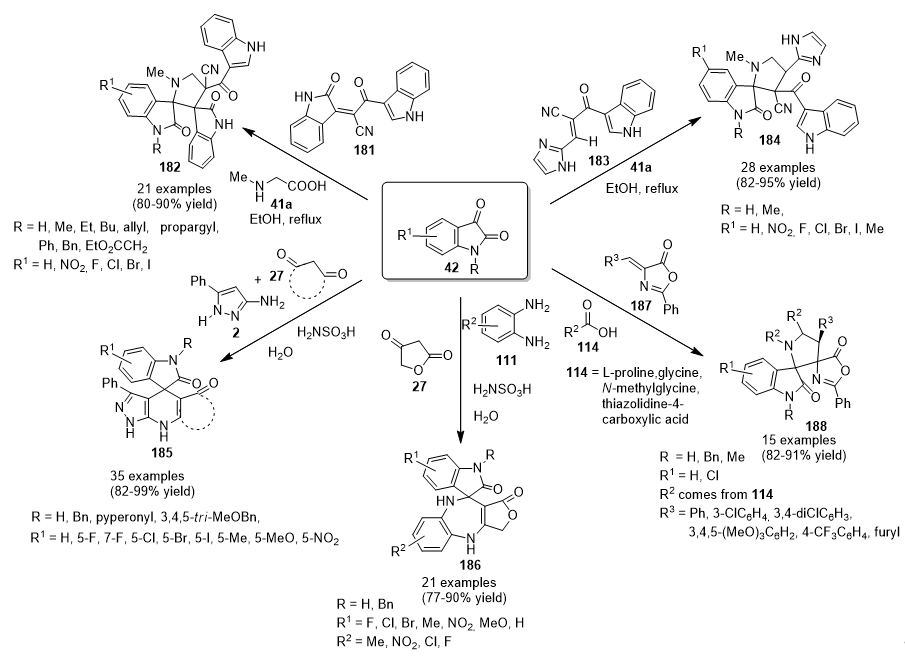
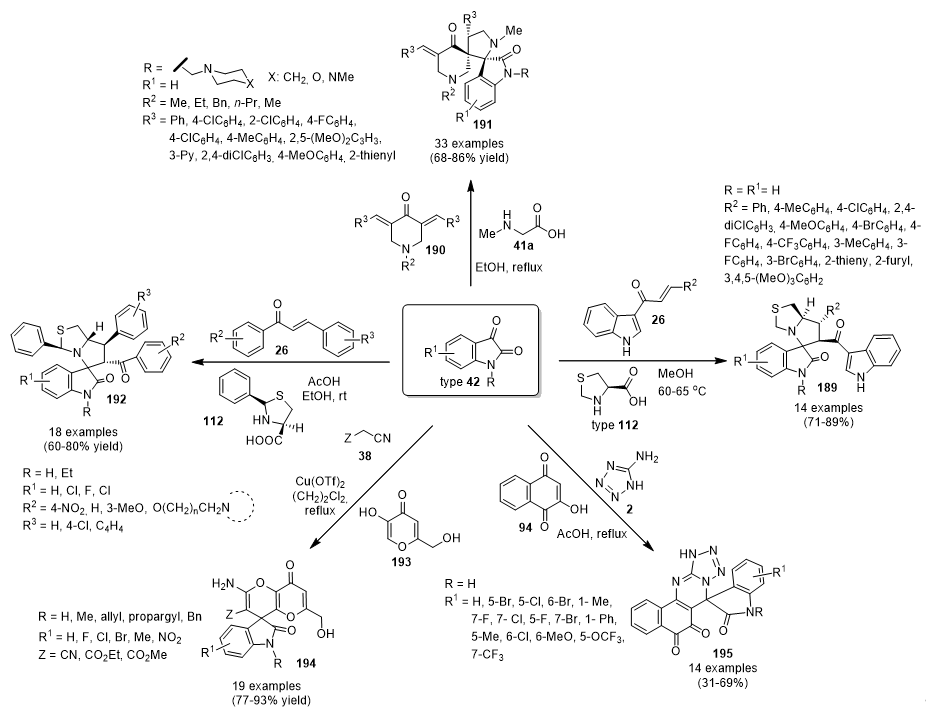
3.13.14. Spiro Derivatives
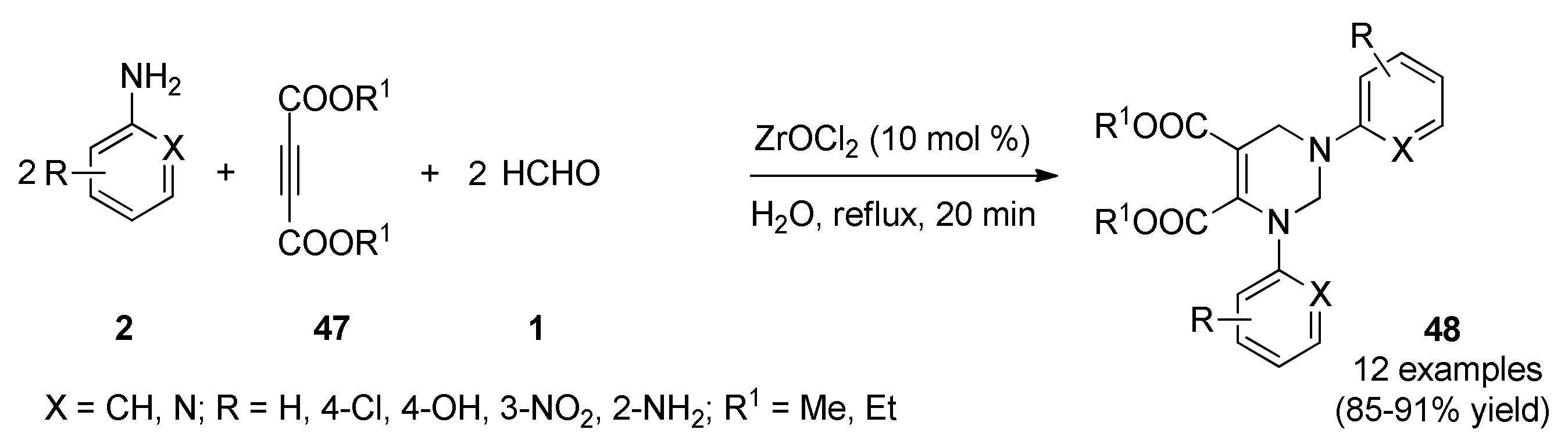
3.13.15. Miscellaneous of Diverse Heterocyclic Systems with Potential Antitumor Activity
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dömling, A.; Ugi, I. Multicomponent reactions with isocyanides. Angew. Chem. Int. Ed. 2000, 39, 3168–3210. [Google Scholar] [CrossRef]
- Rotstein, B.H.; Zaretsky, S.; Rai, V.; Yudin, A.K. Small heterocycles in multicomponent reactions. Chem. Rev. 2014, 114, 8323–8359. [Google Scholar] [CrossRef]
- Dömling, A.; Wang, W.; Wang, K. Chemistry and biology of multicomponent reactions. Chem. Rev 2012, 12, 3083–3135. [Google Scholar] [CrossRef] [Green Version]
- Jarusiewicz, J.; Choe, Y.; Yoo, K.S.; Park, C.P.; Jung, K.W. Efficient three-component Strecker reaction of aldehydes/ketones via NHC-amidate palladium (II) complex catalysis. J. Org. Chem. 2009, 74, 2873–2876. [Google Scholar] [CrossRef] [Green Version]
- Debache, A.; Boulcina, R.; Belfaitah, A.; Rhouati, S.; Carboni, B. One-pot synthesis of 1,4-dihydropyridines via a phenylboronic acid catalyzed Hantzsch three-component reaction. Synlett 2008, 509–512. [Google Scholar] [CrossRef]
- Ryabukhin, S.V.; Plaskon, A.S.; Ostapchuk, E.N.; Volochnyuk, D.M.; Tolmachev, A.A. N-substituted ureas and thioureas in Biginelli reaction promoted by chlorotrimethylsilane: Convenient synthesis of N1-alkyl-, N1-aryl-, and N1,N3-dialkyl-3,4-dihydropyrimidin-2(1H)-(thi)ones. Synthesis 2007, 417–427. [Google Scholar] [CrossRef]
- Cordoba, A.; Watanabe, S.; Tanaka, F.; Notz, W.; Barbas, C.F. A highly enantioselective route to either enantiomer of both α- and β-amino acid derivatives. J. Am. Chem. Soc. 2002, 124, 1866–1867. [Google Scholar] [CrossRef] [PubMed]
- Andrade, C.K.Z.; Takada, S.C.S.; Suarez, P.A.Z.; Alves, M.B. Revisiting the Passerini reaction under eco-friendly reaction conditions. Synlett 2006, 1539–1541. [Google Scholar] [CrossRef]
- Asinger, F. Über die gemeinsame Einwirkung von Schwefel und Ammoniak auf Ketone. Angew. Chem. 1956, 68, 413. [Google Scholar] [CrossRef]
- Akritopoulou-Zanze, I.; Gracias, V.; Djuric, S.W. A versatile synthesis of fused triazolo derivatives by sequential Ugi/alkyne-azide cycloaddition reactions. Tetrahedron Lett. 2004, 45, 8439–8441. [Google Scholar] [CrossRef]
- Tietze, L.F. Domino reactions in organic synthesis. Chem. Rev. 1996, 96, 115–136. [Google Scholar] [CrossRef]
- Coquerel, Y.; Boddaert, T.; Presset, M.; Mailhol, D.; Rodriguez, J. Multiple bond-forming transformations: The key concept toward eco-compatible synthetic organic chemistry. In Ideas in Chemistry and Molecular Sciences: Advances in Synthetic Chemistry; Pignataro, B., Ed.; Wiley-VCH: Weinheim, Germany, 2010; pp. 187–202. [Google Scholar] [CrossRef]
- Ganem, B. Strategies for innovation in multicomponent reaction design. Acc. Chem. Res. 2009, 42, 463–472. [Google Scholar] [CrossRef] [Green Version]
- Răzvan, C.C.; Ruijter, E.; Orru, R.V.A. Multicomponent reactions: Advanced tools for sustainable organic synthesis. Green Chem. 2014, 16, 2958–2975. [Google Scholar] [CrossRef]
- Zhi, S.; Ma, X.; Zhang, W. Consecutive multicomponent reactions for the synthesis of complex molecules. Org. Biomol. Chem. 2019, 17, 7632–7650. [Google Scholar] [CrossRef]
- Saranya, S.; Rohit, K.R.; Radhika, S.; Anilkumar, G. Palladium-catalyzed multicomponent reactions: An overview. Org. Biomol. Chem. 2019; 17, 8048–8061. [Google Scholar] [CrossRef]
- Weber, L. The application of multicomponent reactions in drug discovery. Curr. Med. Chem. 2002; 9, 2085–2093. [Google Scholar] [CrossRef]
- Hulme, C.; Gore, V. Multicomponent reactions: Emerging chemistry in drug discovery” ‘from xylocain to crixivan’. Curr. Med. Chem. 2003, 10, 51–80. [Google Scholar] [CrossRef]
- Chappuis, F.; Sundar, S.; Hailu, A.; Ghalib, H.; Rijal, S.; Peeling, R.W.; Alvar, J.; Boelaert, M. Visceral leishmaniasis: What are the needs for diagnosis, treatment and control? Nat. Rev. Microbiol. 2007, 5, 873–882. [Google Scholar] [CrossRef]
- Desjeux, P. Leishmaniasis: Current situation and new perspectives. Comp. Immunol. Microbiol. Infect. Dis. 2004, 27, 305–318. [Google Scholar] [CrossRef]
- Kobes, T.; Grekov, I.; Lipoldová, M. Leishmaniasis: Prevention, parasite detection and treatment. Curr. Med. Chem. 2012, 19, 1443–1474. [Google Scholar] [CrossRef]
- Fields, E.K. The synthesis of esters of substituted amino phosphonic acids. J. Am. Chem. Soc. 1952, 74, 1528–1531. [Google Scholar] [CrossRef]
- Bhagat, S.; Shah, P.; Garg, S.K.; Mishra, S.; Kaur, P.K.; Singh, S.; Chakraborti, A.K. α-Aminophosphonates as novel anti-leishmanial chemotypes: Synthesis, biological evaluation, and CoMFA studies. Med. Chem. Commun. 2014, 5, 665–670. [Google Scholar] [CrossRef]
- Zayed, M.F.; Hassan, M.H. Synthesis and biological evaluation studies of novel quinazolinone derivatives as antibacterial and anti-inflammatory agents. Saudi Pharm. J. 2014, 22, 157–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, Y.; Sethi, P.; Bansal, G. Coumarin: A potential nucleus for anti-inflammatory molecules. Med. Chem. Res. 2013, 22, 3049–3060. [Google Scholar] [CrossRef]
- Arora, R.K.; Kaur, N.; Bansal, Y.; Bansa, G. Novel coumarin-benzimidazole derivatives as antioxidants and safer anti-inflammatory agents. Acta Pharm. Sin. B. 2014, 4, 368–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madar, J.M.; Shastri, L.A.; Shastri, S.L.; Holiyachi, M.; Naik, N.; Kulkarni, R.; Shaikh, F.; Sungar, V. Design, synthesis, characterization, and biological evaluation of pyrido[1,2-a] pyrimidinone coumarins as promising anti-inflammatory agents. Synth. Commun. 2018, 48, 375–386. [Google Scholar] [CrossRef]
- Lal, J.; Gupta, S.K.; Thavaselvam, D.; Agarwal, D.D. Synthesis and pharmacological activity evaluation of curcumin derivatives. Chin. Chem. Lett. 2016, 27, 1067–1072. [Google Scholar] [CrossRef]
- Patil, K.N.; Mane, R.A.; Jadhav, S.B.; Mane, M.M.; Helavi, V.B. One pot multicomponent synthesis of highly functionalized tetrahydropyridine using copper (II) triflate as catalyst and their anti-inflammatory activity. Chem. Data Collect. 2019, 21, 100233. [Google Scholar] [CrossRef]
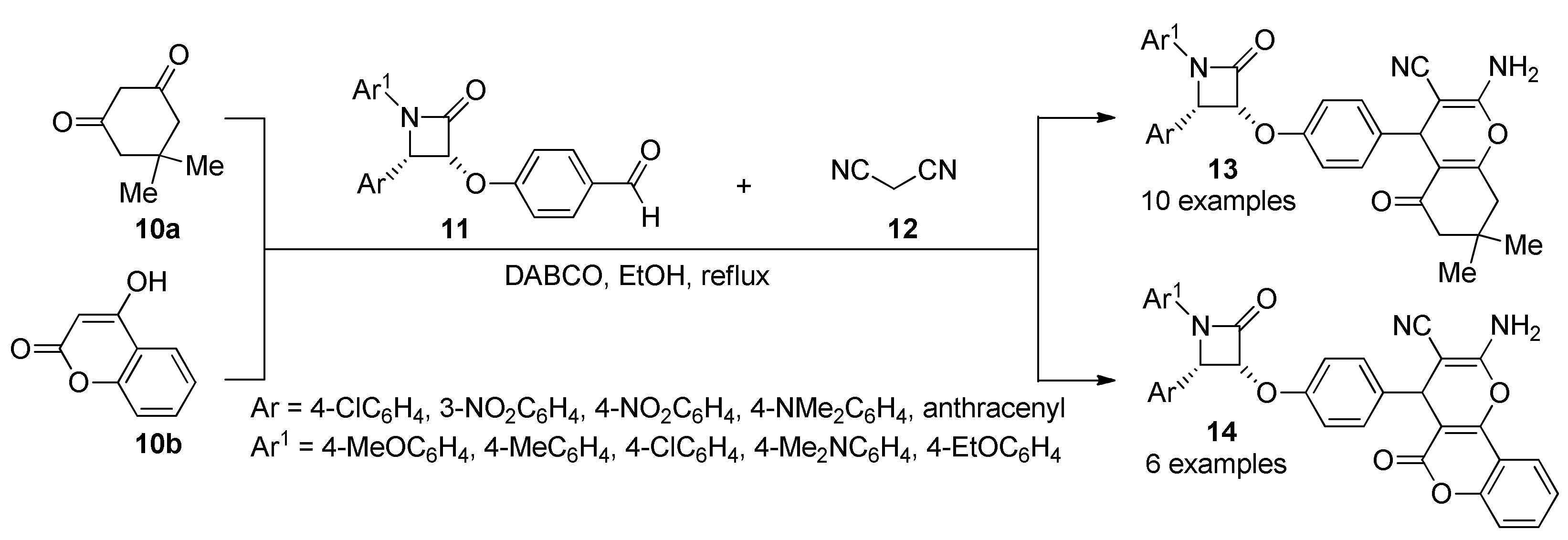
- Borazjani, N.; Sepehri, S.; Behzadi, M.; Jarrahpour, A.M.; Rad, J.A.; Sasanipour, M.; Mohkam, M.; Ghasemi, J.; Akbarizadeh, A.R.; Digiorgio, C.; et al. Three-component synthesis of chromeno β-lactam hybrids for inflammation and cancer screening. Eur. J. Med. Chem. 2019, 179, 389–403. [Google Scholar] [CrossRef]
- Olson, M.F. Applications for ROCK kinase inhibition. Curr. Opin. Cell Biol. 2008, 20, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Hoy, S.M. Netarsudil ophthalmic solution 0.02%: First global approval. Drugs 2018, 78, 389–396. [Google Scholar] [CrossRef]
- Dayal, N.; Mikek, C.G.; Hernandez, D.; Naclerio, G.A.; Yin Chu, E.F.; Carter-Cooper, B.A.; Lapidus, R.G.; Sintim, H.O. Potently inhibiting cancer cell migration with novel 3H-pyrazolo[4,3-f]quinoline boronic acid ROCK inhibitors. Eur. J. Med. Chem. 2019, 180, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKeown, M.R.; Shaw, D.L.; Fu, H.; Liu, S.; Xu, X.; Marineau, J.J.; Huang, Y.; Zhang, X.; Buckley, D.L.; Kadam, A.; et al. Biased multicomponent reactions to develop novel bromodomain inhibitors. J. Med. Chem. 2014, 57, 9019–9027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.D.; Roberts, L.R. Hepatocellular carcinoma: A global view. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 448–458. [Google Scholar] [CrossRef] [Green Version]
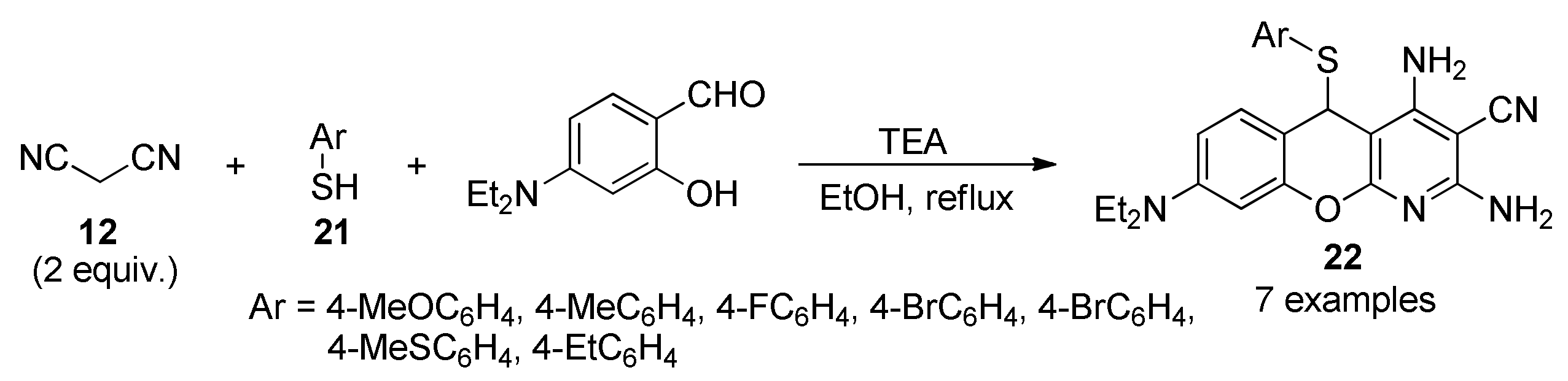
- Patil, R.; Ghosh, A.; Cao, P.S.; Sommer, R.D.; Grice, K.A.; Waris, G.; Patil, S. Novel 5-arylthio-5H-chromenopyridines as a new class of anti-fibrotic agents. Bioorg. Med. Chem. Lett. 2017, 27, 1129–1135. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Regulation of adaptive immunity by the innate immune system. Science 2010, 327, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Kumagai, Y.; Takeuchi, O.; Akira, S. Pathogen recognition by innate receptors. J. Infect. Chemother. 2008, 14, 86–92. [Google Scholar] [CrossRef]
- Takeda, K.; Akira, S. Toll-like receptors. Curr. Protoc. Immunol. 2015, 109, 1–10. [Google Scholar] [CrossRef]
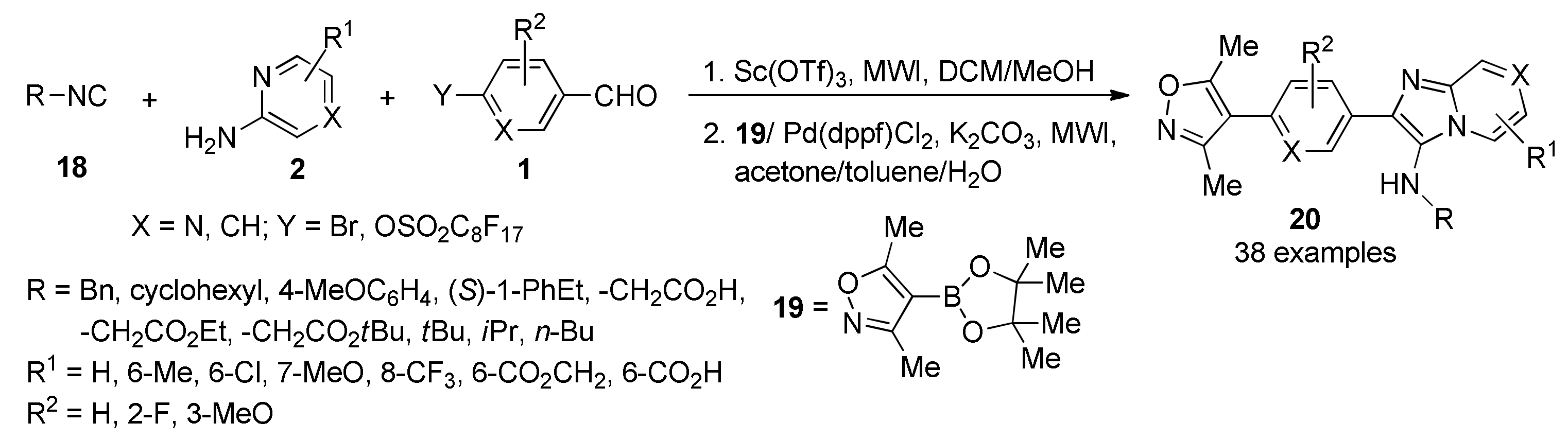
- Shaaban, S.; Abdel-Wahab, B.F. Groebke-Blackburn-Bienaymé multicomponent reaction: Emerging chemistry for drug discovery. Mol. Divers. 2016, 20, 233–254. [Google Scholar] [CrossRef]
- Salunke, D.B.; Yoo, E.; Shukla, N.M.; Balakrishna, R.; Malladi, S.S.; Serafin, K.J.; Day, V.W.; Wang, X.; David, S.A. Structure-activity relationships in human Toll-like receptor 8-active 2,3-diamino-furo[2,3-c]pyridines. J. Med. Chem. 2012, 55, 8137–8151. [Google Scholar] [CrossRef] [Green Version]
- Jaffer, H.; Morris, V.B.; Stewart, D.; Labhasetwar, V. Advances in stroke therapy. Drug Deliv. Transl. Res. 2011, 1, 409–419. [Google Scholar] [CrossRef] [Green Version]
- Tenti, G.; Parada, P.; León, R.; Egea, J.; Martínez-Revelles, S.; Briones, A.M.; Sridharan, V.; López, M.G.; Ramos, M.T.; Menéndez, J.C. New 5-unsubstituted dihydropyridines with improved CaV1.3 selectivity as potential neuroprotective agents against ischemic injury. J. Med. Chem. 2014, 57, 4313–4323. [Google Scholar] [CrossRef]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitt, J.; Irons, R.; Al-Obaidi, M.; Missouris, C. A case of donepezil-related torsades de pointes. BMJ Case Rep. 2015, 1–3. [Google Scholar] [CrossRef]
- Pourshojaei, Y.; Abiri, A.; Eskandari, R.; Dourandish, F.; Eskandari, K.; Asadipour, A. Synthesis, biological evaluation, and computational studies of novel fused six-membered O-containing heterocycles as potential acetylcholinesterase inhibitors. Comp. Biol. Chem. 2019, 80, 249–258. [Google Scholar] [CrossRef]
- Jonckheere, H.; Anné, J.; De Clercq, E. The HIV-1 reverse transcription (RT) process as target for RT inhibitors. Med. Res. Rev. 2000, 20, 129–154. [Google Scholar] [CrossRef]
- De Clercq, E. Novel compounds in preclinical/early clinical development for the treatment of HIV infections. Rev. Med. Virol. 2000, 10, 255–277. [Google Scholar] [CrossRef]
- Barreca, M.L.; Balzarini, J.; Chimirri, A.; De Clercq, E.; De Luca, L.; Höltje, H.D.; Höltje, M.; Monforte, A.M.; Monforte, P.; Pannecouque, C.; et al. Design, synthesis, structure-activity relationships, and molecular modeling studies of 2,3-diaryl-1,3-thiazolidin-4-ones as potent anti-HIV agents. J. Med. Chem. 2002, 45, 5410–5413. [Google Scholar] [CrossRef]
- Woodford, N. Novel agents for the treatment of resistant Gram-positive infections. Expert Opin. Investig. Drugs 2003, 12, 117–137. [Google Scholar] [CrossRef]
- Ma, Z.; Lynch, A.S. Development of a dual-acting antibacterial agent (TNP-2092) for the treatment of persistent bacterial infections. J. Med. Chem. 2016, 59, 6645–6657. [Google Scholar] [CrossRef] [Green Version]
- Lakshmi, N.V.; Thirumurugan, P.; Noorulla, K.M.; Perumal, P.T. InCl3 mediated one-pot multicomponent synthesis, anti-microbial, antioxidant and anticancer evaluation of 3-pyranyl indole derivatives. Bioorg. Med. Chem. Lett. 2010, 20, 5054–5061. [Google Scholar] [CrossRef] [PubMed]
- Mungra, D.C.; Patel, M.P.; Rajani, D.P.; Patel, R.G. Synthesis and identification of β-aryloxyquinolines and their pyrano[3,2-c]chromene derivatives as a new class of antimicrobial and antituberculosis agents. Eur. J. Med. Chem. 2011, 46, 4192–4200. [Google Scholar] [CrossRef] [PubMed]
- Vijesh, A.M.; Isloor, A.M.; Peethambar, S.K.; Shivananda, K.N.; Arulmoli, T.; Isloor, N.A. Hantzsch reaction: Synthesis and characterization of some new 1,4-dihydropyridine derivatives as potent antimicrobial and antioxidant agents. Eur. J. Med. Chem. 2011, 46, 5591–5597. [Google Scholar] [CrossRef] [PubMed]
- El-borai, M.A.; Rizk, H.; Abd-Aal, M.F.; El-Deeb, I.Y. Synthesis of pyrazolo[3,4-b] pyridines under microwave irradiation in multicomponent reactions and their antitumor and antimicrobial activities. Eur. J. Med. Chem. 2012, 48, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.M.; Patel, M.P.; Patel, R.G. Synthesis of a novel class of some biquinoline pyridine hybrids via one-pot, three-component reaction and their antimicrobial activity. J. Chem. Sci. 2012, 124, 669–677. [Google Scholar] [CrossRef] [Green Version]
- Bhaskar, G.; Arun, Y.; Balachandran, C.; Saikumar, C.; Perumal, P.T. Synthesis of novel spirooxindole derivatives by one pot multicomponent reaction and their antimicrobial activity. Eur. J. Med. Chem. 2012, 51, 79–91. [Google Scholar] [CrossRef]
- Singh, S.B.; Tiwari, K.; Verma, P.K.; Srivastava, M.; Tiwari, K.P.; Singh, J. A new eco-friendly strategy for the synthesis of novel antimicrobial spiro-oxindole derivatives via supramolecular catalysis. Supramol. Chem. 2013, 25, 255–262. [Google Scholar] [CrossRef]
- Darandale, S.N.; Pansare, D.N.; Mulla, N.A.; Shinde, D.B. Green synthesis of tetrahydropyrimidine analogues and evaluation of their antimicrobial activity. Bioorg. Med. Chem. Lett. 2013, 23, 2632–2635. [Google Scholar] [CrossRef]
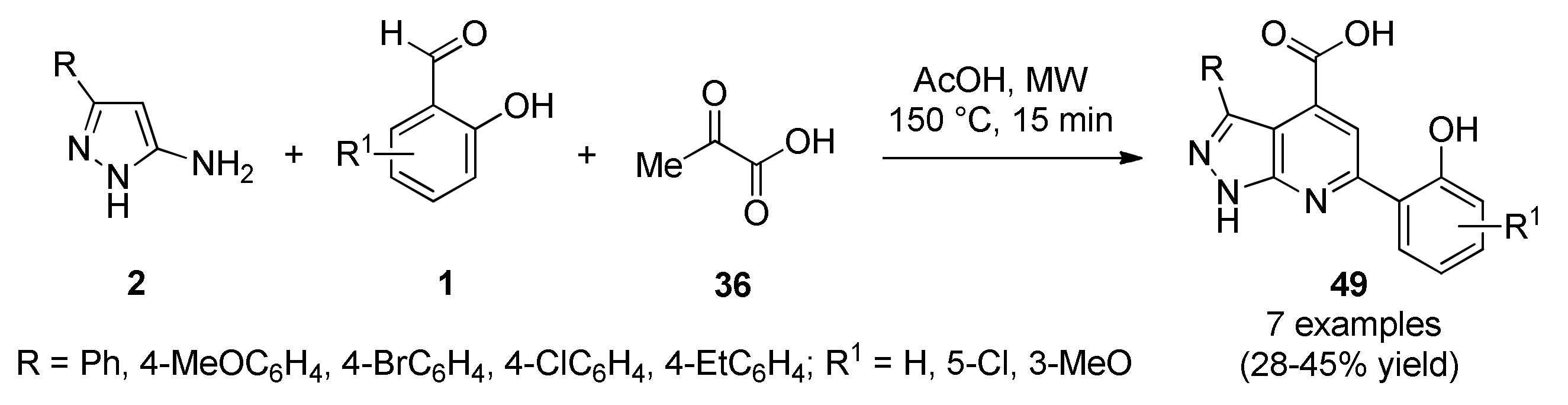
- Murlykina, M.V.; Sakhno, Y.I.; Desenko, S.M.; Konovalova, I.S.; Shishkin, O.V.; Sysoiev, D.A.; Kornet, M.N.; Chebanov, V.A. Features of switchable multicomponent heterocyclizations of salicylic aldehydes and 5-aminopyrazoles with pyruvic acids and antimicrobial activity of the reaction products. Tetrahedron 2013, 69, 9261–9269. [Google Scholar] [CrossRef]
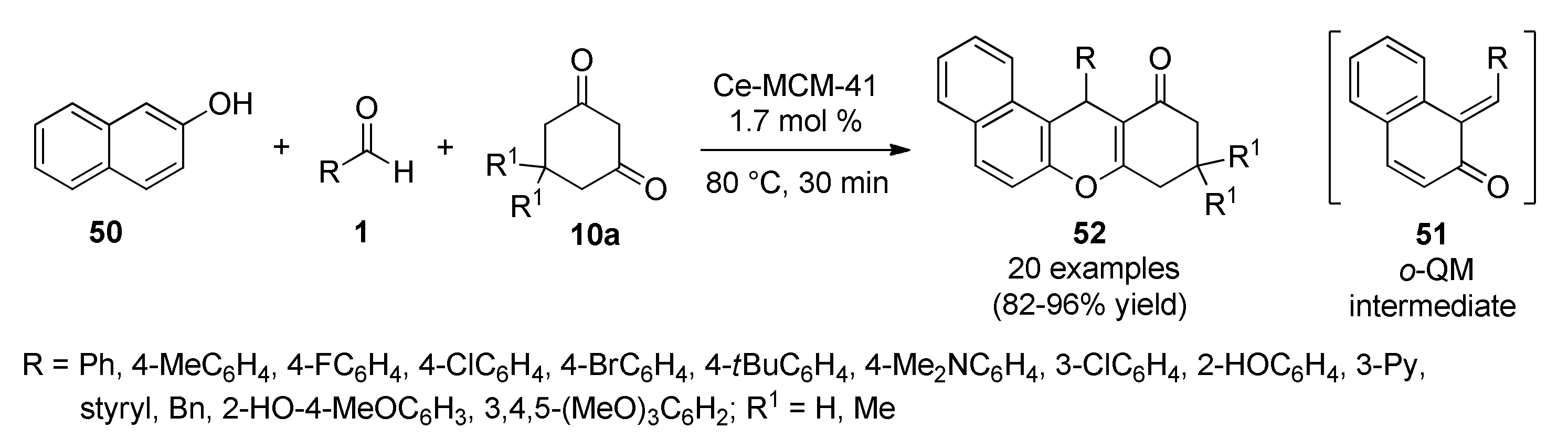
- Akondi, A.M.; Kantam, M.L.; Trivedi, R.; Sreedhar, B.; Buddana, S.K.; Prakasham, R.S.; Bhargava, S. Formation of benzoxanthenones and benzochromenones viacerium-impregnated-MCM-41 catalyzed, solvent-free, three-component reaction and their biological evaluation as antimicrobial agents. J. Mol. Catal. A Chem. 2014, 386, 49–60. [Google Scholar] [CrossRef]
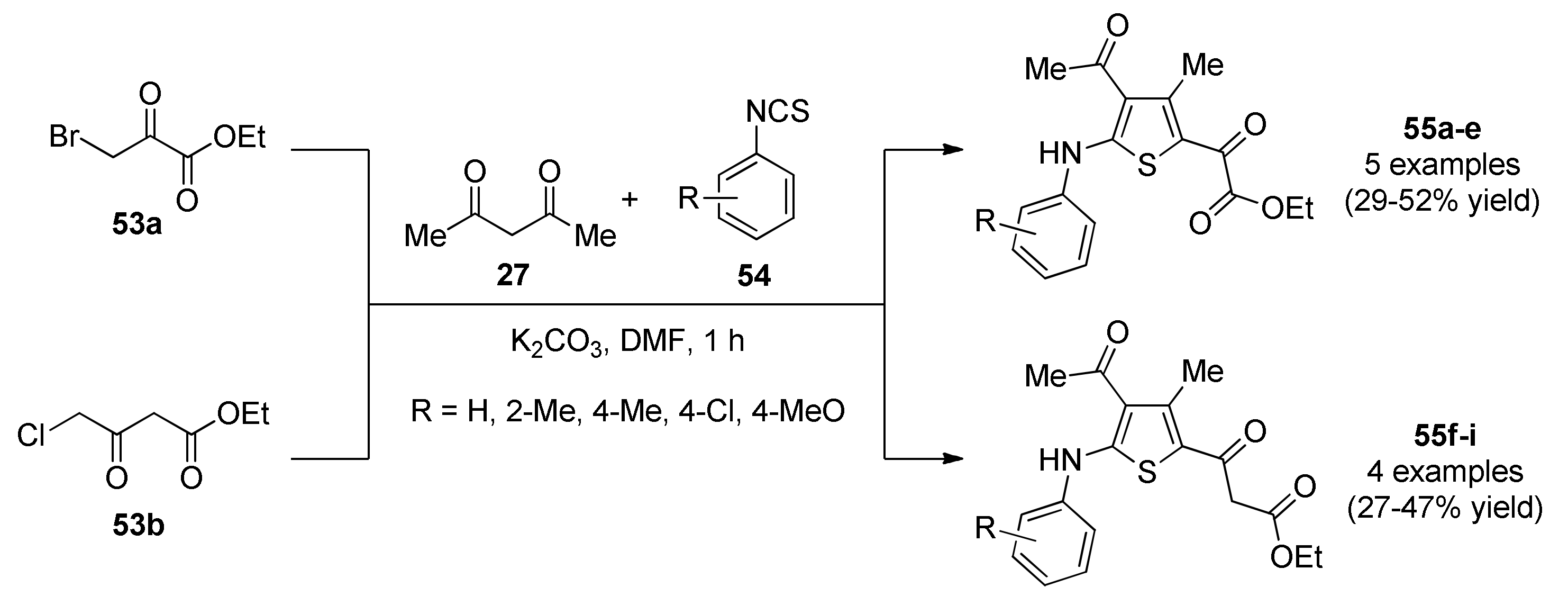
- Sable, P.N.; Ganguly, S.; Chaudhari, P.D. An efficient one-pot three-component synthesis and antimicrobial evaluation of tetra substituted thiophene derivatives. Chin. Chem. Lett. 2014, 25, 1099–1103. [Google Scholar] [CrossRef]
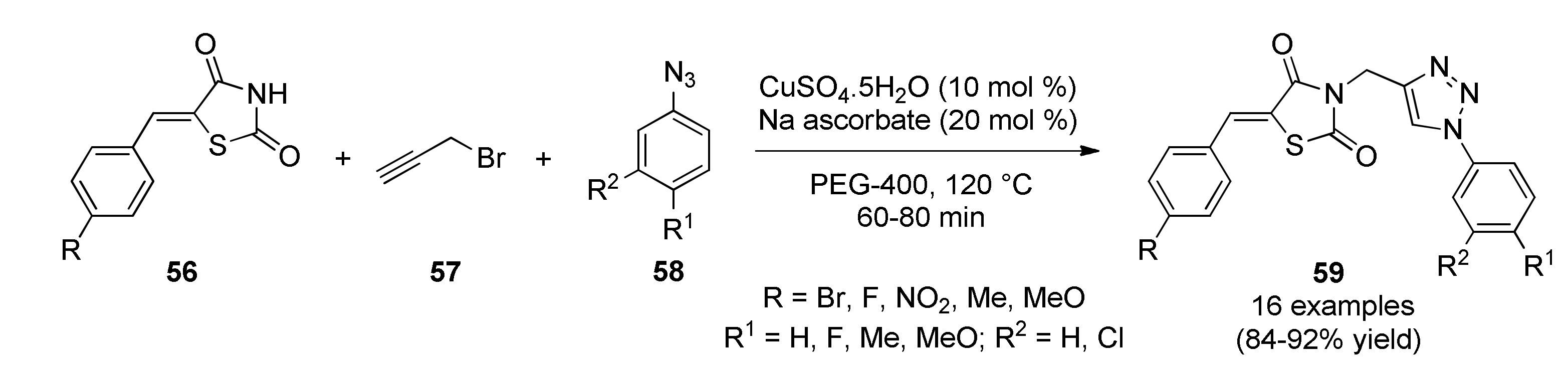
- Sindhu, J.; Singh, H.; Khurana, J.M.; Sharma, C.; Aneja, K.R. Multicomponent domino process for the synthesis of some novel 5-(arylidene)-3-((1-aryl-1H-1,2,3-triazol-4-yl)methyl)-thiazolidine-2,4-diones using PEG-400 as an efficient reaction medium and their antimicrobial evaluation. Chin. Chem. Lett. 2015, 26, 50–54. [Google Scholar] [CrossRef]
- Boltjes, A.; Dömling, A. The Groebke-Blackburn-Bienaymé reaction. Eur. J. Org. Chem. 2019, 2019, 7007–7049. [Google Scholar] [CrossRef]
- Aouali, M.; Mhalla, D.; Allouche, F.; El Kaim, L.; Tounsi, S.; Trigui, M.; Chabchoub, F. Synthesis, antimicrobial and antioxidant activities of imidazotriazoles and new multicomponent reaction toward 5-amino-1-phenyl [1,2,4] triazole derivatives. Med. Chem. Res. 2015, 24, 2732–2741. [Google Scholar] [CrossRef]
- Dhinakaran, I.; Padmini, V.; Ganesan, K.; Selvarasu, K. A one-pot four-component and microwave-assisted synthesis of pyrrolo [1,10] phenanthrolines. ChemistrySelect 2017, 2, 6154–6158. [Google Scholar] [CrossRef]
- Meena, K.; Kumari, S.; Khurana, J.M.; Malik, A.; Sharma, C.; Panwar, H. One-pot three-component synthesis of spiro[indolo-3,10’-indeno[1,2-b]quinolin]-2,4,11’-triones as a new class of antifungal and antimicrobial agents. Chin. Chem. Lett. 2017, 28, 136–142. [Google Scholar] [CrossRef]
- Ashok, D.; Devulapally, M.G.; Aamate, V.K.; Gundu, S.; Adam, S.; Murthy, S.D.S.; Balasubramanian, S.; Naveen, B.; Parthasarathy, T. Novel pyrano [3,2-b] xanthen-7(2H)-ones: Synthesis, antimicrobial, antioxidant and molecular docking studies. J. Mol. Struct. 2019, 1177, 215–228. [Google Scholar] [CrossRef]
- Safari, F.; Hosseini, H.; Bayat, M.; Ranjbar, A. Synthesis and evaluation of antimicrobial activity, cytotoxic and pro-apoptotic effects of novel spiro-4H-pyran derivatives. RSC Adv. 2019, 9, 24843–24851. [Google Scholar] [CrossRef] [Green Version]
- Kenchappa, R.; Bodke, Y.D.; Chandrashekar, A.; Telkar, S.; Manjunatha, K.S.; Sindhe, M.A. Synthesis of some 2,6-bis(1-coumarin-2-yl)-4-(4-substituted phenyl) pyridine derivatives as potent biological agents. Arab. J. Chem. 2017, 10, S1336–S1344. [Google Scholar] [CrossRef] [Green Version]
- Vekariya, R.V.; Patel, K.D.; Rajani, D.P.; Rajani, S.D.; Patel, H.D. A one pot, three component synthesis of coumarin hybrid thiosemicarbazone derivatives and their antimicrobial evolution. J. Assoc. Arab. Univ. Basic Appl. Sci. 2017, 23, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Pedrola, M.; Jorba, M.; Jardas, E.; Jardi, F.; Ghashghaei, O.; Viñas, M.; Lavilla, R. Multicomponent reactions upon the known drug trimethoprim as a source of novel antimicrobial agents. Front. Chem. 2019, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tsolaki, E.; Nobelos, P.; Geronikaki, A.; Rekka, E.A. Selected heterocyclic compounds as antioxidants. Synthesis and biological evaluation. Curr. Top. Med. Chem. 2014, 14, 2462–2477. [Google Scholar] [CrossRef] [PubMed]
- Dangolani, S.K.; Panahi, F.; Tavaf, Z.; Nourisefat, M.; Yousefi, R.; Khalafi-Nezhad, A. Synthesis and antioxidant activity evaluation of some novel aminocarbonitrile derivatives incorporating carbohydrate moieties. ACS Omega 2018, 3, 10341–10350. [Google Scholar] [CrossRef]
- Khansole, G.S.; Prasad, D.; Angulwar, J.A.; Atar, A.B.; Nagaraja, B.M.; Jadhav, A.H.; Bhosalee, V.N. Tetrabutylammonium hydrogen sulfate mediated three-component reaction for the synthesis of thiadiazolo[2,3-b]quinazolin-6-(7H)-ones and antioxidant activity. Mater. Today Proc. 2019, 9, 653–660. [Google Scholar] [CrossRef]
- Ezzatzadeh, E.; Hossaini, Z.; Moradi, A.V.; Salimifard, M.; Abad, S.A.S. Copper iodide and ZnO nanoparticles catalyzed multicomponent synthesis of 1,3-cyclopentadiene: Study of antioxidant activity. Can. J. Chem. 2019, 97, 270–276. [Google Scholar] [CrossRef]
- Cohen, J. New TB drug promises shorter, simpler treatment. Science 2004, 306, 1872. [Google Scholar] [CrossRef]
- Dye, C.; Watt, C.J.; Bleed, D.M.; Hosseini, S.M.; Raviglione, M.C. Evolution of tuberculosis control and prospects for reducing tuberculosis incidence, prevalence, and deaths globally. J. Am. Med. Assoc. 2005, 293, 2767–2775. [Google Scholar] [CrossRef]
- Ramprasad, J.; Nayak, N.; Dalimba, U.; Yogeeswari, P.; Sriram, D.; Peethambar, S.K.; Achur, R.; Kumar, H.S.S. Synthesis and biological evaluation of new imidazo [2,1-b] [1,3,4] thiadiazole-benzimidazole derivatives. Eur. J. Med. Chem. 2015, 95, 49–63. [Google Scholar] [CrossRef]
- Chandrasekera, N.S.; Alling, T.; Bailey, M.A.; Files, M.; Early, J.V.; Ollinger, J.; Ovechkina, Y.; Masquelin, T.; Desai, P.V.; Cramer, J.W.; et al. Identification of phenoxyalkylbenzimidazoles with antitubercular activity. J. Med. Chem. 2015, 58, 7273–7285. [Google Scholar] [CrossRef]
- Kalalbandi, V.K.A.; Seetharamappa, J.; Katrahalli, U.; Bhat, K.G. Synthesis, crystal studies, anti-tuberculosis and cytotoxic studies of 1-[(2E)-3-phenylprop-2-enoyl]-1H-benzimidazole derivative. Eur. J. Med. Chem. 2014, 79, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.; Kulkarni, M.V.; Joshi, S.D.; Dixit, S.R. One pot Click chemistry: A three component reaction for the synthesis of 2-mercaptobenzimidazole linked coumarinyl triazoles as anti-tubercular agents. Bioorg. Med. Chem. Lett. 2016, 26, 4709–4713. [Google Scholar] [CrossRef] [PubMed]
- Ramani, A.V.; Monika, A.; Indira, V.L.; Karyavardhi, G.; Venkatesh, J.; Jeankumar, V.U.; Manjashetty, T.H.; Yogeeswari, P.; Sriram, D. Synthesis of highly potent novel anti-tubercular isoniazid analogues with preliminary pharmacokinetic evaluation. Bioorg. Med. Chem. Lett. 2012, 22, 2764–2767. [Google Scholar] [CrossRef] [PubMed]
- Dandia, A.; Singh, R.; Saini, D. Ionic liquid-mediated three-component synthesis of fluorinated spiro-thiazine derivatives and their antimycobacterial and DNA cleavage activities. J. Chem. Sci. 2013, 125, 1045–1053. [Google Scholar] [CrossRef] [Green Version]
- Quiroga, J.; Diaz, Y.; Bueno, J.; Insuasty, B.; Abonia, R.; Ortiz, A.; Nogueras, M.; Cobo, J. Microwave induced three-component synthesis and antimycobacterial activity of benzopyrazolo [3,4-b] quinolindiones. Eur. J. Med. Chem. 2014, 74, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Surineni, G.; Yogeeswari, P.; Sriram, D.; Kantevari, S. Design and synthesis of novel carbazole tethered pyrrole derivatives as potent inhibitors of Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 2015, 25, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Goud, G.L.; Ramesh, S.; Ashok, D.; Reddy, V.P.; Yogeeswari, P.; Sriram, D.; Saikrishna, B.; Manga, V. Design, synthesis, molecular-docking and antimycobacterial evaluation of some novel 1,2,3-triazolyl xanthenones. Med. Chem. Commun. 2017, 8, 559–570. [Google Scholar] [CrossRef]
- Abonia, R.; Insuasty, D.; Castillo, J.; Insuasty, B.; Quiroga, J.; Nogueras, M.; Cobo, J. Synthesis of novel quinoline-2-one based chalcones of potential anti-tumor activity. Eur. J. Med. Chem. 2012, 57, 29–40. [Google Scholar] [CrossRef]
- Insuasty, B.; Montoya, A.; Becerra, D.; Quiroga, J.; Abonia, R.; Robledo, S.; Vélez, I.V.; Upegui, Y.; Nogueras, M.; Cobo, J. Synthesis of novel analogs of 2-pyrazoline obtained from [(7-chloroquinolin-4-yl)amino]chalcones and hydrazine as potential antitumor and antimalarial agents. Eur. J. Med. Chem. 2013, 67, 252–262. [Google Scholar] [CrossRef]
- Bianchini, G.; Ribelles, P.; Becerra, D.; Ramos, M.T.; Menéndez, J.C. Efficient synthesis of 2-acylquinolines based on an aza-vinylogous Povarov reaction. Org. Chem. Front. 2016, 3, 412–422. [Google Scholar] [CrossRef]
- Nainwal, L.M.; Tasneem, S.; Akhtar, W.; Verma, G.; Khan, M.F.; Parvez, S.; Shaquiquzzaman, M.; Akhter, M.; Alam, M.M. Green recipes to quinoline: A review. Eur. J. Med. Chem. 2019, 164, 121–170. [Google Scholar] [CrossRef] [PubMed]
- Kouznetsov, V.V.; Ochoa, C.; Romero, A.R.; Zacchino, S.A.; Sortino, M.; Gupta, M.; Vázquez, Y.; Bahsas, A.; Amaro-Luis, J. A straightforward synthetic approach to antitumoral pyridinyl substituted 7H-indeno [2,1-c] quinoline derivatives via three-component imino Diels-Alder reaction. Lett. Org. Chem. 2006, 3, 300–304. [Google Scholar] [CrossRef]
- Roopan, S.M.; Khan, F.N.; Jin, J.S.; Kumar, R.S. An efficient one pot–three component cyclocondensation in the synthesis of 2-(2-chloroquinolin-3-yl)-2,3-dihydroquinazolin-4(1H)-ones: Potential antitumor agents. Res. Chem. Intermed. 2011, 37, 919–927. [Google Scholar] [CrossRef]
- Sangani, C.B.; Makawana, J.A.; Duan, Y.-T.; Yin, Y.; Teraiya, S.B.; Thumar, N.J.; Zhu, H.-L. Design, synthesis and molecular modeling of biquinoline–pyridine hybrids as a new class of potential EGFR and HER-2 kinase inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 4472–4476. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.; Fuertes, M.; Martín-Encinas, E.; Selas, A.; Rubiales, G.; Tesauro, C.; Knudssen, B.K.; Palacios, F. Novel topoisomerase I inhibitors. Syntheses and biological evaluation of phosphorus substituted quinoline derivates with antiproliferative activity. Eur. J. Med. Chem. 2018, 149, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.-C.; Jimenez, E.; Portilla, J.; Insuasty, B.; Quiroga, J.; Moreno-Fuquen, R.; Kennedy, A.R.; Abonia, R. Application of a catalyst-free Domino Mannich/Friedel-Crafts alkylation reaction for the synthesis of novel tetrahydroquinolines of potential antitumor activity. Tetrahedron 2018, 74, 932–947. [Google Scholar] [CrossRef]
- Abonia, R.; Castillo, J.; Insuasty, B.; Quiroga, J.; Nogueras, M.; Cobo, J. Efficient catalyst-free four-component synthesis of novel γ-aminoethers mediated by a Mannich type reaction. ACS Comb. Sci. 2013, 15, 2–9. [Google Scholar] [CrossRef]
- Mani, K.S.; Murugesapandian, B.; Kaminsky, W.; Rajendrana, S.P. Enantioselective approach towards the synthesis of spiro-indeno [1,2-b] quinoxaline pyrrolothiazoles as antioxidant and antiproliferative. Tetrahedron Lett. 2018, 59, 2921–2929. [Google Scholar] [CrossRef]
- Taheri, S.; Nazifi, M.; Mansourian, M.; Hosseinzadeh, L.; Shokoohiniad, Y. Ugi efficient synthesis, biological evaluation and molecular docking of coumarin-quinoline hybrids as apoptotic agents through mitochondria-related pathways. Bioorg. Chem. 2019, 91, 103147. [Google Scholar] [CrossRef]
- Castillo, J.-C.; Portilla, J. Recent advances in the synthesis of new pyrazole derivatives. Targets Heterocycl. Syst. 2018, 22, 194–223. [Google Scholar] [CrossRef]
- Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.N.; Al-Aizari, F.A.; Ansar, M. Synthesis and pharmacological activities of pyrazole derivatives: A review. Molecules 2018, 23, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, J.C.; Quiroga, J.; Abonia, R.; Rodriguez, J.; Coquerel, Y. The aryne aza-Diels–Alder reaction: Flexible syntheses of isoquinolines. Org. Lett. 2015, 17, 3374–3377. [Google Scholar] [CrossRef] [PubMed]
- Galvez, J.; Castillo, J.C.; Quiroga, J.; Rajzmann, M.; Rodriguez, J.; Coquerel, Y. Divergent chemo-, regio-, and diastereoselective normal electron-demand Povarov-type reactions with α-oxo-ketene dienophiles. Org. Lett. 2014, 16, 4126–4129. [Google Scholar] [CrossRef] [PubMed]
- Abonia, R.; Castillo, J.; Insuasty, B.; Quiroga, J.; Nogueras, M.; Cobo, J. An efficient synthesis of 7-(arylmethyl)-3-tert-butyl-1-phenyl-6,7-dihydro-1H,4H-pyrazolo[3,4-d][1,3]oxazines. Eur. J. Org. Chem. 2010, 33, 6454–6463. [Google Scholar] [CrossRef]
- Kamble, A.A.; Kamble, R.R.; Chougala, L.S.; Kadadevarmath, J.S.; Maidur, S.R.; Patil, P.S.; Kumbar, M.N.; Marganakop, S.B. Photophysical, electrochemical studies of novel pyrazol-4-yl-2,3-dihydroquinazolin-4(1H)-ones and their anticancer activity. ChemistrySelect 2017, 2, 6882–6890. [Google Scholar] [CrossRef]
- Nimbalkar, U.D.; Seijas, J.A.; Vazquez-Tato, M.P.; Damale, M.G.; Sangshetti, J.N.; Nikalje, A.P.G. Ionic liquid-catalyzed green protocol for multi-component synthesis of dihydropyrano[2,3-c]pyrazoles as potential anticancer scaffolds. Molecules 2017, 22, 1628. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Pi, C.; Ye, Y.; Zhao, L.; Wei, Y. Recent advances of analogues of curcumin for treatment of cancer. Eur. J. Med. Chem. 2019, 180, 524–535. [Google Scholar] [CrossRef]
- Noureddin, S.A.; El-Shishtawy, R.M.; Al-Footy, K.O. Curcumin analogues and their hybrid molecules as multifunctional drugs. Eur. J. Med. Chem. 2019, 182, 111631. [Google Scholar] [CrossRef]
- Rodrigues, F.C.; Kumar, N.V.A.; Thakur, G. Developments in the anticancer activity of structurally modified curcumin: An up-to-date review. Eur. J. Med. Chem. 2019, 177, 76–104. [Google Scholar] [CrossRef]
- Khor, P.Y.; Moh-Aluwi, M.F.F.; Rullah, K.; Lam, K.W. Insights on the synthesis of asymmetric curcumin derivatives and their biological activities. Eur. J. Med. Chem. 2019, 183, 111704. [Google Scholar] [CrossRef]
- Sharma, R.; Jadav, S.S.; Yasmin, S.; Bhatia, S.; Khalilullah, H.; Ahsan, M.J. Simple, efficient, and improved synthesis of Biginelli-type compounds of curcumin as anticancer agents. Med. Chem. Res. 2015, 24, 636–644. [Google Scholar] [CrossRef]
- Bhuvaneswari, K.; Sivaguru, P.; Lalitha, A. Synthesis, biological evaluation and molecular docking of novel curcumin derivatives as Bcl-2 inhibitors targeting human breast cancer MCF-7 cells. ChemistrySelect 2017, 2, 11552–11560. [Google Scholar] [CrossRef]
- Estévez, V.; Villacampa, M.; Menéndez, J.C. Recent advances in the synthesis of pyrroles by multicomponent reactions. Chem. Soc. Rev. 2014, 43, 4633–4657. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Alam, O.; Naim, M.J.; Shaquiquzzaman, M.; Alam, M.M.; Iqbal, M. Pyrrole: An insight into recent pharmacological advances with structure activity relationship. Eur. J. Med. Chem. 2018, 157, 527–561. [Google Scholar] [CrossRef] [PubMed]
- Abonia, R.; Insuasty, B.; Quiroga, J.; Kolshorn, H.; Meier, H. A versatile synthesis of 4,5-dihydropyrrolo [1,2-a] quinoxalines. J. Heterocycl. Chem. 2001, 38, 671–674. [Google Scholar] [CrossRef]
- Abonia, R.; Cuervo, P.; Insuasty, B.; Quiroga, J.; Nogueras, M.; Cobo, J. A simple two-step sequence for the synthesis of novel 4-aryl-4,5-dihydro-6H-[1,3] dioxolo [4,5-h] pyrrolo[1,2-a][1]benzazepin-6-ones from 6-amino-3,4-methylenedioxyacetophenone. Eur. J. Org. Chem. 2008, 27, 4684–4689. [Google Scholar] [CrossRef]
- Castillo, J.C.; Abonia, R.; Cobo, J.; Glidewell, C. A chain of [π]-stacked molecules in 4-(2-chloro-phenyl)pyrrolo[1,2-a]quinoxaline and a hydrogen-bonded sheet in (4RS)-4-(1,3-benzodioxol-6-yl)-4,5-dihydropyrrolo[1,2-a]quinoxaline. Acta Cryst. 2013, 69, 544–548. [Google Scholar] [CrossRef]
- Castillo, J.C.; Tigreros, A.; Coquerel, Y.; Rodríguez, J.; Macías, M.A.; Portilla, J. Synthesis of pyrrolo[2,3-c]isoquinolines via the cycloaddition of benzyne with arylideneaminopyrroles: Photophysical and crystallographic Study. ACS Omega 2019, 4, 17326–17339. [Google Scholar] [CrossRef]
- Magedov, I.V.; Luchetti, G.; Evdokimov, N.M.; Manpadi, M.; Steelant, W.F.A.; Van Slambrouck, S.; Tongwa, P.; Antipin, M.Y.; Kornienko, A. Novel three-component synthesis and antiproliferative properties of diversely functionalized pyrrolines. Bioorg. Med. Chem. Lett. 2008, 18, 1392–1396. [Google Scholar] [CrossRef] [Green Version]
- Pagadala, R.; Kommidi, D.R.; Kankala, S.; Maddila, S.; Singh, P.; Moodley, B.; Koorbanally, N.A.; Jonnalagadda, S.B. Multicomponent one-pot synthesis of highly functionalized pyrrole-3-carbonitriles in aqueous medium and computational study. Org. Biomol. Chem. 2015, 13, 1800–1806. [Google Scholar] [CrossRef]
- Sharma, S.K.; Kumar, S.; Chand, K.; Kathuria, A.; Gupta, A.; Jain, R. An update on natural occurrence and biological activity of chromones. Curr. Med. Chem. 2011, 18, 3825–3852. [Google Scholar] [CrossRef] [PubMed]
- Keri, R.S.; Budagumpi, S.; Pai, R.K.; Balakrishna, R.G. Chromones as a privileged scaffold in drug discovery: A review. Eur. J. Med. Chem. 2014, 78, 340–374. [Google Scholar] [CrossRef] [PubMed]
- Pratap, R.; Ram, V.J. Natural and synthetic chromenes, fused chromenes, and versatility of dihydrobenzo[h]chromenes in organic synthesis. Chem. Rev. 2014, 114, 10476–10526. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, N.; Paul, N.D.; Mandal, S.; de Bruin, B.; Wulff, W.D. Catalytic synthesis of 2H-chromenes. ACS Catal. 2015, 5, 2329–2366. [Google Scholar] [CrossRef]
- Costa, M.; Dias, T.A.; Brito, A.; Proença, F. Biological importance of structurally diversified chromenes. Eur. J. Med. Chem. 2016, 123, 487–507. [Google Scholar] [CrossRef]
- Huang, W.; Ding, Y.; Miao, Y.; Liu, M.-Z.; Li, Y.; Yang, G.-F. Synthesis and antitumor activity of novel dithiocarbamate substituted chromones. Eur. J. Med. Chem. 2009, 44, 3687–3696. [Google Scholar] [CrossRef]
- Kumar, A.; Sharma, S.; Maurya, R.A.; Sarkar, J. Diversity oriented synthesis of benzoxanthene and benzochromene libraries via one-pot, tricomponent reactions and their anti-proliferative activity. J. Comb. Chem. 2010, 12, 20–24. [Google Scholar] [CrossRef]
- Paliwal, P.K.; Jetti, S.R.; Jain, S. Green approach towards the facile synthesis of dihydropyrano(c)chromene and pyrano[2,3-d]pyrimidine derivatives and their biological evaluation. Med. Chem. Res. 2013, 22, 2984–2990. [Google Scholar] [CrossRef]
- Kalla, R.M.N.; Choi, J.-S.; Yoo, J.-W.; Byeon, S.J.; Heo, M.S.; Kim, I. Synthesis of 2-amino-3-cyano-4H-chromen-4-ylphosphonates and their anticancer properties. Eur. J. Med. Chem. 2014, 76, 61–66. [Google Scholar] [CrossRef]
- Perumal, M.; Sengodu, P.; Venkatesan, S.; Srinivasan, R.; Paramsivam, M. Environmentally benign copper triflate-mediated multicomponent one-pot synthesis of novel benzo[g]chromenes possess potent anticancer activity. ChemistrySelect 2017, 2, 5068–5072. [Google Scholar] [CrossRef]
- Torres, M.; Gil, S.; Parra, M. New synthetic methods to 2-pyridone rings. Curr. Org. Chem. 2005, 9, 1757–1779. [Google Scholar] [CrossRef]
- Hamama, W.S.; Waly, M.; El-Hawary, I.; Zoorob, H.H. Developments in the chemistry of 2-pyridone. Synth. Commun. 2014, 44, 1730–1759. [Google Scholar] [CrossRef]
- Hirano, K.; Miura, M. A lesson for site-selective C–H functionalization on 2-pyridones: Radical, organometallic, directing group and steric controls. Chem. Sci. 2018, 9, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Prendergast, A.M.; McGlacken, G.P. Transition metal mediated C–H activation of 2-pyrones, 2-pyridones, 2-coumarins and 2-quinolones. Eur. J. Org. Chem. 2018, 44, 6068–6082. [Google Scholar] [CrossRef]
- Abonia, R.; Castillo, J.; Cuervo, P.; Insuasty, B.; Quiroga, J.; Ortíz, A.; Nogueras, M.; Cobo, J. A simple one-pot synthesis of new imidazol-2-yl-1H-quinolin-2-ones from the direct reaction of 2-chloroquinolin-3-carbaldehyde with aromatic o-diamines. Eur. J. Org. Chem. 2010, 2, 317–325. [Google Scholar] [CrossRef]
- Magedov, I.V.; Manpadi, M.; Ogasawara, M.A.; Dhawan, A.S.; Rogelj, S.; Van Slambrouck, S.; Steelant, W.F.A.; Evdokimov, N.M.; Uglinskii, P.Y.; Elias, E.M.; et al. Structural simplification of bioactive natural products with multicomponent synthesis. 2. Antiproliferative and antitubulin activities of pyrano[3,2-c]pyridones and pyrano[3,2-c]quinolones. J. Med. Chem. 2008, 51, 2561–2570. [Google Scholar] [CrossRef] [Green Version]
- Ansari, M.I.; Arun, A.; Hussain, M.K.; Konwar, R.; Hajela, K. Discovery of 3,4,6-triaryl-2-pyridones as potential anticancer agents that promote ROS-independent mitochondrial-mediated apoptosis in human breast carcinoma cells. ChemistrySelect 2016, 1, 4255–4264. [Google Scholar] [CrossRef]
- Kumar, N.P.; Thatikonda, S.; Tokala, R.; Kumari, S.S.; Lakshmi, U.J.; Godugu, C.; Shankaraiah, N.; Kamal, A. Sulfamic acid promoted one-pot synthesis of phenanthrene fused-dihydrodibenzo-quinolinones: Anticancer activity, tubulin polymerization inhibition and apoptosis inducing studies. Bioorg. Med. Chem. 2018, 26, 1996–2008. [Google Scholar] [CrossRef]
- Ayati, A.; Emami, S.; Asadipour, A.; Shafiee, A.; Foroumadi, A. Recent applications of 1,3-thiazole core structure in the identification of new lead compounds and drug discovery. Eur. J. Med. Chem. 2015, 97, 699–718. [Google Scholar] [CrossRef]
- Jain, S.; Pattnaik, S.; Pathak, K.; Kumar, S.; Pathak, D.; Jain, S.; Vaidya, A. Anticancer potential of thiazole derivatives: A retrospective review. Mini Rev. Med. Chem. 2018, 18, 640–655. [Google Scholar] [CrossRef]
- Pathania, S.; Narang, R.K.; Rawal, R.K. Role of sulphur-heterocycles in medicinal chemistry: An update. Eur. J. Med. Chem. 2019, 180, 486–508. [Google Scholar] [CrossRef]
- Abonia, R.; Castillo, J.; Insuasty, B.; Quiroga, J.; Sortino, M.; Nogueras, M.; Cobo, J. Catalyst-, solvent- and desiccant-free three-component synthesis of novel C-2,N-3 disubstituted thiazolidin-4-ones. Arab. J. Chem. 2019, 12, 122–133. [Google Scholar] [CrossRef]
- Shi, F.; Zeng, X.-N.; Zhang, G.; Ma, N.; Jiang, B.; Tu, S. Facile synthesis of new 4-aza-podophyllotoxin analogs via microwave-assisted multi-component reactions and evaluation of their cytotoxic activity. Bioorg. Med. Chem. Lett. 2011, 21, 7119–7123. [Google Scholar] [CrossRef]
- Gu, L.; Jin, C. Synthesis and antitumor activity of α-aminophosphonates containing thiazole[5,4-b]pyridine moiety. Org. Biomol. Chem. 2012, 10, 7098–7102. [Google Scholar] [CrossRef] [PubMed]
- Bathula, C.; Tripathi, S.; Srinivasan, R.; Jha, K.K.; Ganguly, A.; Chakraborty, G.; Singh, S.; Munshi, P.; Sen, S. Synthesis of novel 5-arylidenethiazolidinones with apoptotic properties via a three-component reaction using piperidine as a bifunctional reagent. Org. Biomol. Chem. 2016, 14, 8053–8063. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.V.; Seijas, J.A.; Vazquez-Tato, M.P.; Sarkate, A.P.; Lokwani, D.K.; Nikalje, A.P.G. Ultrasound mediated one-pot, three-component synthesis, docking and ADME prediction of novel 5-amino-2-(4-chlorophenyl)-7-substituted phenyl-8,8a-dihydro-7H-(1,3,4)thiadiazolo(3,2-α)pyrimidine-6-carbonitrile derivatives as anticancer agents. Molecules 2016, 21, 894. [Google Scholar] [CrossRef] [Green Version]
- Semenov, V.V.; Lichitsky, B.V.; Komogortsev, A.N.; Dudinov, A.A.; Krayushkin, M.M.; Konyushkin, L.D.; Atamanenko, O.P.; Karmanova, I.B.; Strelenko, Y.A.; Shor, B.; et al. Synthesis and anti-mitotic activity of 6,7-dihydro-4H-isothiazolo[4,5-b]pyridin-5-ones: In vivo and cell-based studies. Eur. J. Med. Chem. 2017, 125, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Yakaiah, S.; Kumar, P.S.V.; Rani, P.B.; Prasad, K.D.; Aparna, P. Design, synthesis and biological evaluation of novel pyrazolooxothiazolidine derivatives as antiproliferative agents against human lung cancer cell line A549. Bioorg. Med. Chem. Lett. 2018, 28, 630–636. [Google Scholar] [CrossRef]
- Griglio, A.; Torre, E.; Serafini, M.; Bianchi, A.; Schmid, R.; Zabetta, G.C.; Massarotti, A.; Sorba, G.; Pirali, T.; Fallarini, S. A multicomponent approach in the discovery of indoleamine 2,3-dioxygenase 1 inhibitors: Synthesis, biological investigation and docking studies. Bioorg. Med. Chem. Lett. 2018, 28, 651–657. [Google Scholar] [CrossRef]
- Wu, Y.-J. New indole-containing medicinal compounds. In Heterocyclic Scaffolds II: Reactions and Applications of Indoles; Gribble, G., Ed.; Topics in Heterocyclic Chemistry; Springer: Berlin/Heidelberg, Germany, 2010; Volume 26, pp. 1–29. [Google Scholar] [CrossRef]
- Sakemi, S.; Sun, H.H. Nortopsentins, A, B, and C. Cytotoxic and antifungal imidazolediylbis [indoles] from the sponge Spongosorites ruetzleri. J. Org. Chem. 1991, 56, 4304–4307. [Google Scholar] [CrossRef]
- Wijeratne, E.M.K.; Turbyville, T.J.; Zhang, Z.; Bigelow, D.; Pierson, L.S.; VanEtten, H.D.; Whitesell, L.; Canfield, L.M.; Gunatilaka, A.A.L. Cytotoxic constituents of Aspergillus terreus from the rhizosphere of Opuntia versicolor of the Sonoran desert. J. Nat. Prod. 2003, 66, 1567–1573. [Google Scholar] [CrossRef] [PubMed]
- Radwan, M.A.A.; Ragab, E.A.; Sabry, N.M.; El-Shenawy, S.M. Synthesis and biological evaluation of new 3-substituted indole derivatives as potential anti-inflammatory and analgesic agents. Bioorg. Med. Chem. 2007, 15, 3832–3841. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Z.; Drewe, J.; Tseng, B.; Kasibhatla, S.; Cai, S.X. Discovery and SAR of indole-2-carboxylic acid benzylidene-hydrazides as a new series of potent apoptosis inducers using a cell-based HTS assay. Bioorg. Med. Chem. 2004, 12, 3649–3655. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, N.S.; Shirazi, A.N.; El-Meligy, M.G.; El-Ziaty, A.K.; Rowley, D.; Sun, J.; Nagib, Z.A.; Parang, K. Synthesis of 4-aryl-6-indolylpyridine-3-carbonitriles and evaluation of their antiproliferative activity. Tetrahedron Lett. 2014, 55, 1154–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sashidhara, K.V.; Modukuri, R.K.; Singh, S.; Bhaskara Rao, K.; Aruna Teja, G.; Gupta, S.; Shukla, S. Design and synthesis of new series of coumarin-aminopyran derivatives possessing potential anti-depressant-like activity. Bioorg. Med. Chem. Lett. 2015, 25, 337–341. [Google Scholar] [CrossRef]
- Gupta, S.; Maurya, P.; Upadhyay, A.; Kushwaha, P.; Krishna, S.; Siddiqi, M.I.; Sashidhara, K.V.; Banerjee, D. Synthesis and bio-evaluation of indole-chalcone based benzopyrans as promising antiligase and antiproliferative agents. Eur. J. Med. Chem. 2018, 143, 1981–1996. [Google Scholar] [CrossRef]
- Knölker, H.J.; Reddy, K.R. Isolation and synthesis of biologically active carbazole alkaloids. Chem. Rev. 2002, 102, 4303–4427. [Google Scholar] [CrossRef]
- Indumathi, T.; Ahamed, V.S.J.; Moon, S.S.; Fronczek, F.R.; Prasad, K.J.R. L-Proline anchored multicomponent synthesis of novel pyrido[2,3-a]carbazoles; investigation of in vitro antimicrobial, antioxidant, cytotoxicity and structure activity relationship studies. Eur. J. Med. Chem. 2011, 46, 5580–5590. [Google Scholar] [CrossRef]
- Reddy, P.N.; Padmaja, P.; Reddy, B.R.; Jadav, S.S. Synthesis, in vitro antiproliferative activity, antioxidant activity and molecular modeling studies of new carbazole Mannich bases. Med. Chem. Res. 2017, 26, 2243–2259. [Google Scholar] [CrossRef]
- Sathiyachandran, P.; Manogaran, P.; Nesterov, V.N.; Padma, V.V.; Rajendra Prasad, K.J. Design and synthesis of novel pyrrolo[2,3-a]carbazoles: 7-Chloro-2-oxo-3a-(2′oxo-2′,3′-dihydro-1′H-indol-3′-yl)-2,3,3a,4,5,10-hexahydro-pyrrolo[3,2-a]carbazole-1-carbonitrile as an efficient anticancer agent. Eur. J. Med. Chem. 2018, 150, 851–863. [Google Scholar] [CrossRef]
- Canto, R.F.S.; Bernardi, A.; Battastini, A.M.O.; Russowsky, D.; Eifler-Lima, V.L. Synthesis of dihydropyrimidin-2-one/thione library and cytotoxic activity against the human U138-MG and Rat C6 glioma cell lines. J. Braz. Chem. Soc. 2011, 22, 1379–1388. [Google Scholar] [CrossRef]
- Kappe, C.O. Recent advances in the Biginelli dihydropyrimidine synthesis. New tricks from an old dog. Acc. Chem. Res. 2000, 33, 879–888. [Google Scholar] [CrossRef]
- Climent, M.J.; Corma, A.; Iborra, S. Homogeneous and heterogeneous catalysts for multicomponent reactions. RSC Adv. 2012, 2, 16–58. [Google Scholar] [CrossRef] [Green Version]
- Biginelli, P. Synthesis of 3,4-dihydropyrimidin-2(1H)-ones. Gazz. Chim. Ital. 1893, 23, 360–416. [Google Scholar]
- Da Silva, D.L.; Reis, F.S.; Muniz, D.R.; Ruiz, A.L.T.G.; de Carvalho, J.E.; Sabino, A.A.; Modolo, L.V.; de Fátima, A. Free radical scavenging and antiproliferative properties of Biginelli adducts. Bioorg. Med. Chem. 2012, 20, 2645–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadlapalli, R.K.; Chourasia, O.P.; Vemuri, K.; Sritharan, M.; Perali, R.S. Synthesis and in vitro anticancer and antitubercular activity of diarylpyrazole ligated dihydropyrimidines possessing lipophilic carbamoyl group. Bioorg. Med. Chem. Lett. 2012, 22, 2708–2711. [Google Scholar] [CrossRef] [PubMed]
- Ramos, L.M.; Guido, B.C.; Nobrega, C.C.; Corrêa, J.R.; Silva, R.G.; de Oliveira, H.C.B.; Gomes, A.F.; Gozzo, F.C.; Neto, B.A.D. The Biginelli reaction with an imidazolium–tagged recyclable iron catalyst: Kinetics, mechanism, and antitumoral activity. Chem. Eur. J. 2013, 19, 4156–4168. [Google Scholar] [CrossRef]
- Silva, G.C.O.; Correa, J.R.; Rodrigues, M.O.; Alvim, H.G.O.; Guido, B.C.; Gatto, C.C.; Wanderley, K.A.; Fioramonte, M.; Gozzo, F.C.; de Souza, R.O.M.A.; et al. The Biginelli reaction under batch and continuous flow conditions: Catalysis, mechanism and antitumoral activity. RSC Adv. 2015, 5, 48506–48515. [Google Scholar] [CrossRef]
- Matiasa, M.; Camposa, G.; Santosa, A.O.; Falcao, A.; Silvestre, S.; Alves, G. Potential antitumoral 3,4-dihydropyrimidin-2-(1H)-ones: Synthesis, in vitro biological evaluation and QSAR studies. RSC Adv. 2016, 6, 84943–84958. [Google Scholar] [CrossRef]
- Carosati, E.; Ioan, P.; Micucci, M.; Broccatelli, F.; Cruciani, G.; Zhorov, B.S.; Chiarini, A.; Budriesi, R. 1,4-Dihydropyridine scaffold in medicinal chemistry, the story so far and perspectives (part 2): Action in other targets and antitargets. Curr. Med. Chem. 2012, 19, 4306–4323. [Google Scholar] [CrossRef]
- Gadotti, V.M.; Bladen, C.; Zhang, F.X.; Chen, L.; Gündüz, M.G.; Şimşek, R.; Şafak, C.; Zamponi, G.W. Analgesic effect of a broad-spectrum dihydropyridine inhibitor of voltage-gated calcium channels. Pflug. Arch. 2015, 467, 2485–2493. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.E.; Manganiello, V.; Degerman, E. Re-discovering PDE3 inhibitors--new opportunities for a long neglected target. Curr. Top. Med. Chem. 2007, 7, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Abadi, A.H.; Abouel-Ella, D.A.; Lehmann, J.; Tinsley, H.N.; Gary, B.D.; Piazza, G.A.; Abdel-Fattah, M.A.O. Discovery of colon tumor cell growth inhibitory agents through a combinatorial approach. Eur. J. Med. Chem. 2010, 45, 90–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radadiya, A.; Khedkar, V.; Bavishi, A.; Vala, H.; Thakrar, S.; Bhavsar, D.; Shah, A.; Coutinho, E. Synthesis and 3D-QSAR study of 1,4-dihydropyridine derivatives as MDR cancer reverters. Eur. J. Med. Chem. 2014, 74, 375–387. [Google Scholar] [CrossRef]
- Kumari, A.K.; Reddy, V.H.; Reddy, G.M.; Reddy, Y.V.R.; Leelavathi, S. Synthesis of dihydropyridine derivatives under eco-friendly approach and investigation of cytotoxic activity. J. Heterocycl. Chem. 2019, 56, 1661–1666. [Google Scholar] [CrossRef]
- Safak, C.; Simsek, R. Fused 1,4-dihydropyridines as potential calcium modulatory compounds. Mini Rev. Med. Chem. 2006, 6, 747–755. [Google Scholar] [CrossRef]
- Sharma, V.K.; Singh, S.K. Synthesis, utility and medicinal importance of 1,2- and 1,4-dihydropyridines. RSC Adv. 2017, 7, 2682–2732. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhang, C.; Wu, L. L-Proline catalyzed three-component synthesis of para-naphthoquinone-4-aza-podophyllotoxin hybrids as potent antitumor agents. RSC Adv. 2015, 5, 18945–18951. [Google Scholar] [CrossRef]
- Kumar, N.P.; Sharma, P.; Reddy, T.S.; Nekkanti, S.; Shankaraiah, N.; Lalita, G.; Sujanakumari, S.; Bhargava, S.K.; Naidu, V.G.M.; Kamal, A. Synthesis of 2,3,6,7-tetramethoxyphenanthren-9-amine: An efficient precursor to access new 4-aza-2,3-dihydropyridophenanthrenes as apoptosis inducing agents. Eur. J. Med. Chem. 2017, 127, 305–317. [Google Scholar] [CrossRef]
- Kumar, N.P.; Sharma, P.; Reddy, T.S.; Shankaraiah, N.; Bhargava, S.K.; Kamal, A. Microwave-assisted one-pot synthesis of new phenanthrene fused-tetrahydrodibenzoacridinones as potential cytotoxic and apoptosis inducing agents. Eur. J. Med. Chem. 2018, 151, 173–185. [Google Scholar] [CrossRef]
- Relling, M.V.; Gardner, E.E.; Sandborn, W.J.; Schmiegelow, K.; Pui, C.H.; Yee, S.W.; Stein, C.M.; Carrillo, M.; Evans, W.E.; Hicks, J.K.; et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin. Pharm. 2013, 93, 324–325. [Google Scholar] [CrossRef] [PubMed]
- D´Souza, P.F.; Ashoka, S.; Rajan, M.S.; Shabaraya, A.R. Antitumor activity of mercaptopurine in combination with trikatu and gomutra on 20-methylcholantrene induced carcinogenesis. J. Appl. Pharm. Sci. 2013, 3, 20–24. [Google Scholar] [CrossRef]
- Kowalska, A.; Latocha, M.; Pluta, K. Synthesis and anticancer activity of thiosubstituted purines. Med. Chem. Res. 2015, 24, 3107–3116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frolova, L.V.; Magedov, I.V.; Romero, A.E.; Karki, M.; Otero, I.; Hayden, K.; Evdokimov, N.M.; Banuls, L.M.; Rastogi, S.K.; Smith, W.R.; et al. Exploring natural product chemistry and biology with multicomponent reactions. 5. Discovery of a novel tubulin-targeting scaffold derived from the rigidin family of marine alkaloids. J. Med. Chem. 2013, 56, 6886–6900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, A.; Singla, R.; Jaitak, V. Coumarins as anticancer agents: A review on synthetic strategies, mechanism of action and SAR studies. Eur. J. Med. Chem. 2015, 101, 476–495. [Google Scholar] [CrossRef]
- Bubols, G.B.; Da Rocha Vianna, D.; Medina-Remon, A.; von Poser, G.; Lamuela-Raventos, R.M.; Eifler-Lima, V.L.; Garcia, S.C. The antioxidant activity of coumarins and flavonoids. Mini Rev. Med. Chem. 2013, 13, 318–334. [Google Scholar] [CrossRef]
- Hu, Y.Q.; Xu, Z.; Zhang, S.; Wu, X.; Ding, J.W.; Lv, Z.S.; Feng, L.S. Recent developments of coumarin-containing derivatives and their anti-tubercular activity. Eur. J. Med. Chem. 2017, 136, 122–130. [Google Scholar] [CrossRef]
- Hadjipavlou-Litina, D.; Litinas, K.; Kontogiorgis, C. The anti-inflammatory effect of coumarin and its derivatives. Anti Inflamm. Anti Allergy Agents Med. Chem. 2007, 6, 293–306. [Google Scholar] [CrossRef]
- Tan, N.; Yazıcı-Tütüniş, S.; Bilgin, M.; Tan, E.; Miski, M. Antibacterial activities of pyrenylated coumarins from the roots of Prangos hulusii. Molecules 2017, 22, 1098. [Google Scholar] [CrossRef]
- Al-Amiery, A.A.; Kadhum, A.A.H.; Mohamad, A.B. Antifungal activities of new coumarins. Molecules 2012, 17, 5713–5723. [Google Scholar] [CrossRef]
- Chen, Z.; Bi, J.; Su, W. Synthesis and antitumor activity of novel coumarin derivatives via a three-component reaction in water. Chin. J. Chem. 2013, 31, 507–514. [Google Scholar] [CrossRef]
- Vodnala, S.; Bhavani, A.K.D.; Kamutam, R.; Naidu, V.G.M.; Prabhakar, C. DABCO-catalyzed one-pot three component synthesis of dihydropyrano[3,2-c]chromene substituted quinazolines and their evaluation towards anticancer activity. Bioorg. Med. Chem. Lett. 2016, 26, 3973–3977. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.R.; Manikandan, A.; Sivakumar, A.; Dhayabaran, V.V. An eco-friendly catalytic system for multicomponent, one-pot synthesis of novel spiro-chromeno indoline-triones and their anti-prostate cancer potentials evaluated via alkaline phosphatase inhibition mechanism. Bioorg. Chem. 2018, 81, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Kavitha, K.; Srikrishna, D.; Aparna, P. An efficient one-pot three-component synthesis of 2-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)-3-arylacrylonitriles and their cytotoxic activity evaluation with molecular docking. J. Saudi Chem. Soc. 2019, 23, 325–337. [Google Scholar] [CrossRef]
- Vaarla, K.; Kesharwani, R.K.; Santosh, K.; Vedula, R.R.; Kotamraju, S.; Toopurani, M.K. Synthesis, biological activity evaluation and molecular docking studies of novel coumarin substituted thiazolyl-3-aryl-pyrazole-4-carbaldehydes. Bioorg. Med. Chem. Lett. 2015, 25, 5797–5803. [Google Scholar] [CrossRef]
- Shaikh, S.K.J.; Sannaikar, M.S.; Kumbar, M.N.; Bayannavar, P.K.; Kamble, R.R.; Inamdar, S.R.; Joshi, S.D. Microwave-expedited green synthesis, photophysical, computational studies of coumarin-3-yl-thiazol-3-yl-1,2,4-triazolin-3-ones and their anticancer activity. ChemistrySelect 2018, 3, 4448–4462. [Google Scholar] [CrossRef]
- Chougala, B.M.; Shastri, S.L.; Holiyachi, M.; Naik, N.S.; Shastri, L.A.; Dodamani, S.S.; Jalalpure, S.S.; Dixit, S.R.; Joshi, S.D.; Sunagar, V.A. Microwave synthesis of coumarinyl substituted pyridine derivatives as potent anticancer agents and molecular docking studies. ChemistrySelect 2017, 2, 5234–5242. [Google Scholar] [CrossRef]
- Sashidhara, K.V.; Singh, L.R.; Choudhary, D.; Arun, A.; Gupta, S.; Adhikary, S.; Palnati, G.R.; Konwar, R.; Trivedi, R. Design, synthesis and in vitro evaluation of coumarin–imidazo[1,2-a]pyridine derivatives against cancer induced osteoporosis. RSC Adv. 2016, 6, 80037–80048. [Google Scholar] [CrossRef]
- Saxena, R.; Gupta, G.; Manohar, M.; Debnath, U.; Popli, P.; Prabhakar, Y.S.; Konwar, R.; Kumar, S.; Kumar, A.; Dwivedi, A. Spiro-oxindole derivative 5-chloro-4′,5′-diphenyl-3′-(4-(2-(piperidin-1-yl) ethoxy) benzoyl) spiro[indoline-3,2′-pyrrolidin]-2-one triggers apoptosis in breast cancer cells via restoration of p53 function. Int. J. Biochem. Cell Biol. 2016, 70, 105–117. [Google Scholar] [CrossRef]
- Barmak, A.; Niknam, K.; Mohebbi, G.; Pournabi, H. Antibacterial studies of hydroxyspiro[indoline-3,9-xanthene]trione against spiro[indoline3,9-xanthene]trione and their use as acetyl and butyrylcholinesterase inhibitors. Microb. Pathog. 2019, 130, 95–99. [Google Scholar] [CrossRef]
- Guardia, A.; Baiget, J.; Cacho, M.; Pérez, A.; Ortega-Guerra, M.; Nxumalo, W.; Khanye, S.D.; Rullas, J.; Ortega, F.; Jiménez, E. Easy-to-synthesize spirocyclic compounds possess remarkable in vivo activity against Mycobacterium tuberculosis. J. Med. Chem. 2018, 61, 11327–11340. [Google Scholar] [CrossRef] [PubMed]
- Maurya, R.; Soni, A.; Anand, D.; Ravi, M.; Raju, K.S.R.; Taneja, I.; Naikade, N.K.; Puri, S.K.; Wahajuddin Kanojiya, S.; Yadav, P.P. Synthesis and antimalarial activity of 3,3-spiroanellated 5,6-disubstituted 1,2,4-trioxanes. ACS Med. Chem. Lett 2013, 4, 165–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajanarendar, E.; Ramakrishna, S.; Govardhan Reddy, K.; Nagaraju, D.; Reddy, Y.N. A facile synthesis, anti-inflammatory and analgesic activity of isoxazolyl-2,3-dihydrospiro[benzo[f]isoindole-1,3′-indoline]-2′,4,9-triones. Bioorg. Med. Chem. Lett. 2013, 23, 3954–3958. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Ismail, R.; Choon, T.S.; Yoon, Y.K.; Wei, A.C.; Pandian, S.; Kumar, R.S.; Osman, H.; Manogaran, E. Substituted spiro[2.3′]oxindolespiro[3.2′′]-5,6-dimethoxy-indane-1′′-one-pyrrolidine analogue as inhibitors of acetylcholinesterase. Bioorg. Med. Chem. Lett. 2010, 20, 7064–7066. [Google Scholar] [CrossRef] [PubMed]
- Arun, Y.; Bhaskar, G.; Balachandran, C.; Ignacimuthu, S.; Perumal, P.T. Facile one-pot synthesis of novel dispirooxindole-pyrrolidine derivatives and their antimicrobial and anticancer activity against A549 human lung adenocarcinoma cancer cell line. Bioorg. Med. Chem. Lett. 2013, 23, 1839–1845. [Google Scholar] [CrossRef] [PubMed]
- Arun, Y.; Saranraj, K.; Balachandran, C.; Perumal, P.T. Novel spirooxindole–pyrrolidine compounds: Synthesis, anticancer and molecular docking studies. Eur. J. Med. Chem. 2014, 74, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Babu, K.S.; Vishnu Vardhan, M.V.P.S.; Hussaini, S.M.A.; Mahesh, R.; Shaik, S.P.; Alarifi, A. Sulfamic acid promoted one-pot three-component synthesis and cytotoxic evaluation of spirooxindoles. Bioorg. Med. Chem. Lett. 2015, 25, 2199–2202. [Google Scholar] [CrossRef]
- Nagaraju, B.; Kovvuri, J.; Babu, K.S.; Adiyala, P.R.; Nayak, V.L.; Alarifi, A.; Kamal, A. A facile one-pot C-C and C-N bond formation for the synthesis of spiro-benzodiazepines and their cytotoxicity. Tetrahedron 2017, 73, 6969–6976. [Google Scholar] [CrossRef]
- Dong, H.; Song, S.; Li, J.; Xu, C.; Zhang, H.; Ouyang, L. The discovery of oxazolones-grafted spirooxindoles via three-component diversity oriented synthesis and their preliminary biological evaluation. Bioorg. Med. Chem. Lett. 2015, 25, 3585–3591. [Google Scholar] [CrossRef]
- Kumar, A.; Gupta, G.; Srivastava, S.; Bishnoi, A.K.; Saxena, R.; Kant, R.; Khanna, R.S.; Maulik, P.R.; Dwivedi, A. Novel diastereoselective synthesis of spiropyrrolidine-oxindole derivatives as anti-breast cancer agents. RSC Adv. 2013, 3, 4731–4735. [Google Scholar] [CrossRef]
- George, R.F.; Panda, S.S.; Shalaby, E.-S.M.; Srour, A.M.; Farag, I.S.A.; Girgis, A.S. Synthesis and molecular modeling studies of indole-based antitumor agents. RSC Adv. 2016, 6, 45434–45451. [Google Scholar] [CrossRef]
- Islam, M.S.; Ghawas, H.M.; El-Senduny, F.F.; Al-Majid, A.M.; Elshaier, Y.A.M.M.; Badria, F.A.; Barakat, A. Synthesis of new thiazolo-pyrrolidine–(spirooxindole) tethered to 3-acylindole as anticancer agents. Bioorg. Chem. 2019, 82, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, K.; Praveen, C.; Balachandran, C.; Senthil Kumar, P.; Ignacimuthu, S.; Perumal, P.T. Cu(OTf)2 catalyzed three component reaction: Efficient synthesis of spiro[indoline-3,4′-pyrano[3,2-b]pyran derivatives and their anticancer potency towards A549 human Lung cancer cell lines. Bioorg. Med. Chem. Lett. 2013, 23, 2708–2713. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Liu, Y.; Li, Y. Synthesis of spirooxindole-O-naphthoquinone-tetrazolo[1,5-a]pyrimidine hybrids as potential anticancer agents. Molecules 2018, 23, 2330. [Google Scholar] [CrossRef] [Green Version]
- Chavan, P.V.; Desai, U.V.; Wadgaonkar, P.P.; Tapase, S.R.; Kodam, K.M.; Choudhari, A.; Sarkar, D. Click chemistry based multicomponent approach in the synthesis of spirochromenocarbazole tethered 1,2,3-triazoles as potential anticancer agents. Bioorg. Chem. 2019, 85, 475–486. [Google Scholar] [CrossRef]
- Liu, X.-L.; Feng, T.-T.; Jiang, W.-D.; Yang, C.; Tian, M.-Y.; Jiang, Y.; Lin, B.; Zhao, Z.; Zhou, Y. Molecular hybridization-guided 1,3-dipolar cycloaddition reaction enabled pyrimidine-fused spiropyrrolidine oxindoles synthesis as potential anticancer agents. Tetrahedron Lett. 2016, 57, 4411–4416. [Google Scholar] [CrossRef]
- Sudhapriya, N.; Perumal, P.T.; Balachandran, C.; Ignacimuthu, S.; Sangeetha, M.; Doble, M. Synthesis of new class of spirocarbocycle derivatives by multicomponent domino reaction and their evaluation for antimicrobial, anticancer activity and molecular docking studies. Eur. J. Med. Chem. 2014, 83, 190–207. [Google Scholar] [CrossRef]
- Pirali, T.; Faccio, V.; Mossetti, R.; Grolla, A.A.; Di Micco, S.; Bifulco, G.; Genazzani, A.A.; Tron, G.C. Synthesis, molecular docking and biological evaluation as HDAC inhibitors of cyclopeptide mimetics by a tandem three-component reaction and intramolecular [3+2] cycloaddition. Mol. Divers. 2010, 14, 109–121. [Google Scholar] [CrossRef]
- Baviskar, A.T.; Madaan, C.; Preet, R.; Mohapatra, P.; Jain, V.; Agarwal, A.; Guchhait, S.K.; Kundu, C.N.; Banerjee, U.C.; Bharatam, P.V. N-Fused imidazoles as novel anticancer agents that inhibit catalytic activity of topoisomerase IIr and induce apoptosis in G1/S phase. J. Med. Chem. 2011, 54, 5013–5030. [Google Scholar] [CrossRef]
- Le Floch, C.; Le Gall, E.; Léonel, E.; Martens, T.; Cresteil, T. Synthesis and cytotoxic evaluation of novel paraconic acid analogs. Bioorg. Med. Chem. Lett. 2011, 21, 7054–7058. [Google Scholar] [CrossRef]
- Shi, F.; Zeng, X.-N.; Cao, X.-D.; Zhang, S.; Jiang, B.; Zheng, W.-F.; Tu, S.-J. Design and diversity-oriented synthesis of novel 1,4-thiazepan-3-ones fused with bioactive heterocyclic skeletons and evaluation of their antioxidant and cytotoxic activities. Bioorg. Med. Chem. Lett. 2012, 22, 743–746. [Google Scholar] [CrossRef] [PubMed]
- Kou, L.; Wang, M.-J.; Wang, L.-T.; Zhao, X.-B.; Nan, X.; Yang, L.; Liu, Y.-Q.; Morris-Natschke, S.L.; Lee, K.-H. Toward synthesis of third-generation spin-labeled podophyllotoxin derivatives using isocyanide multicomponent reactions. Eur. J. Med. Chem. 2014, 75, 282–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EL Massry, A.M.; Asal, A.M.; Khattab, S.N.; Haiba, N.S.; Awney, H.A.; Helmy, M.; Langer, V.; Amer, A. Synthesis and structure elucidation of novel fused 1,2,4-triazine derivatives as potent inhibitors targeting CYP1A1 activity. Bioorg. Med. Chem. 2012, 20, 2624–2637. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.-R.; Du, L.-J.; Qin, X.-C.; Xu, L.; Wang, J.-F.; Zhou, X.-F.; Tu, Z.-F.; Li, L.; Liu, Y.-H. Site selective synthesis of cytotoxic 1,3,6-trisubstituted 3,6-diunsaturated (3Z,6Z)-2,5-diketopiperazines via a one-pot multicomponent method. Tetrahedron 2016, 72, 1051–1057. [Google Scholar] [CrossRef]
- Raghav, N.; Jangra, S.; Kumar, A.; Bhattacharyya, S.; Wadhwa, D.; Sindhu, J. Cathepsin B, H and L inhibitors as cell proliferating agents: Design, synthesis, computational and pharmacological studies of some novel 2-(2-naphthoyl)-6,6-dimethyl-3-aryl-2,3,6,7-tetrahydrobenzofuran-4(5H)-ones. RSC Adv. 2016, 6, 34588–34599. [Google Scholar] [CrossRef]
- Gohel, J.N.; Lunagariya, K.S.; Kapadiya, K.M.; Khunt, R.C. An efficient protocol for the synthesis of 1, 5-disubstituted tetrazole derivatives via a TMS-N3 based Ugi reaction and their anti-cancer activity. ChemistrySelect 2018, 3, 11657–11662. [Google Scholar] [CrossRef]
- He, L.-J.; Yang, D.-L.; Li, S.-K.; Zhang, Y.-J.; Tang, Y.; Lei, J.; Frett, B.; Lin, H.-K.; Li, H.-Y.; Chen, Z.Z.; et al. Facile construction of fused benzimidazole-isoquinolinones that induce cell-cycle arrest and apoptosis in colorectal cancer cells. Bioorg. Med. Chem. 2018, 26, 3899–3908. [Google Scholar] [CrossRef]
- Mohareb, R.M.; Hassaneen, H.M.; Ahmed, N.A.M. New approaches for the synthesis of thiophenes, thiazole, pyran derivatives, and their antitumor evaluation. Jacobs J. Org. Chem. 2016, 1, 1–13. [Google Scholar]
- Pelliccia, S.; Amato, J.; Capasso, D.; Di Gaetano, S.; Massarotti, A.; Piccolo, M.; Irace, C.; Tron, G.C.; Pagano, B.; Randazzo, A.; et al. Bio-inspired dual-selective BCL-2/c-MYC G-quadruplex binders: Design, synthesis, and anticancer activity of drug-like imidazo[2,1-i]purine derivatives. J. Med. Chem. 2019. [Google Scholar] [CrossRef]































































































{kind=link}
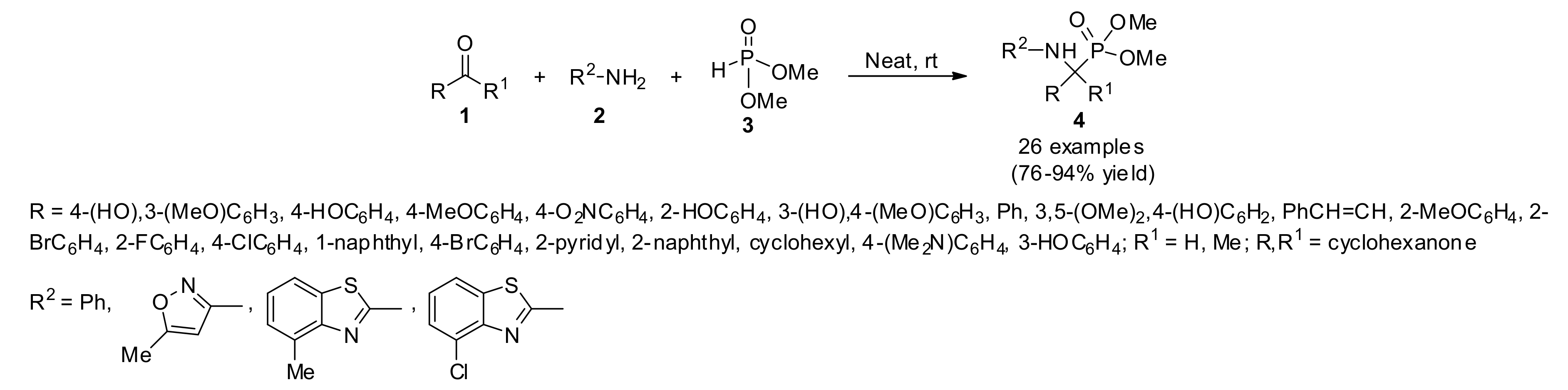{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
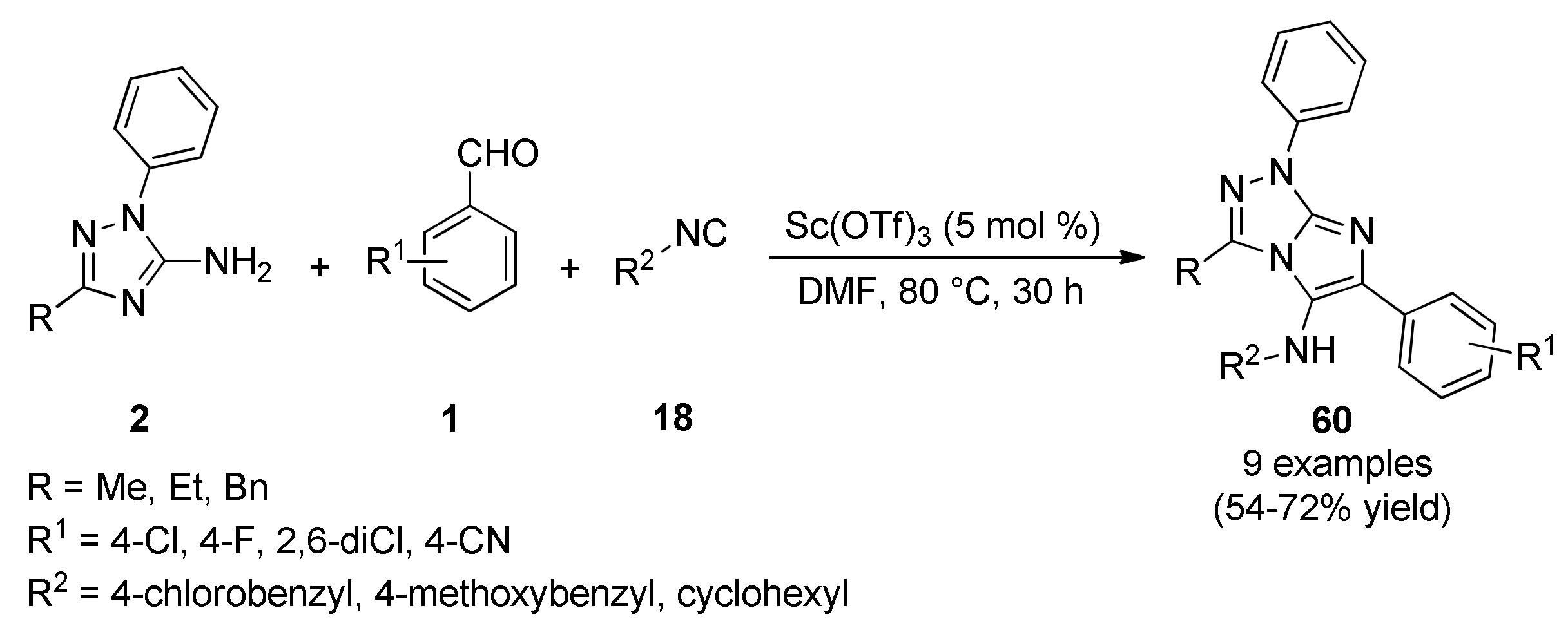{kind=link}
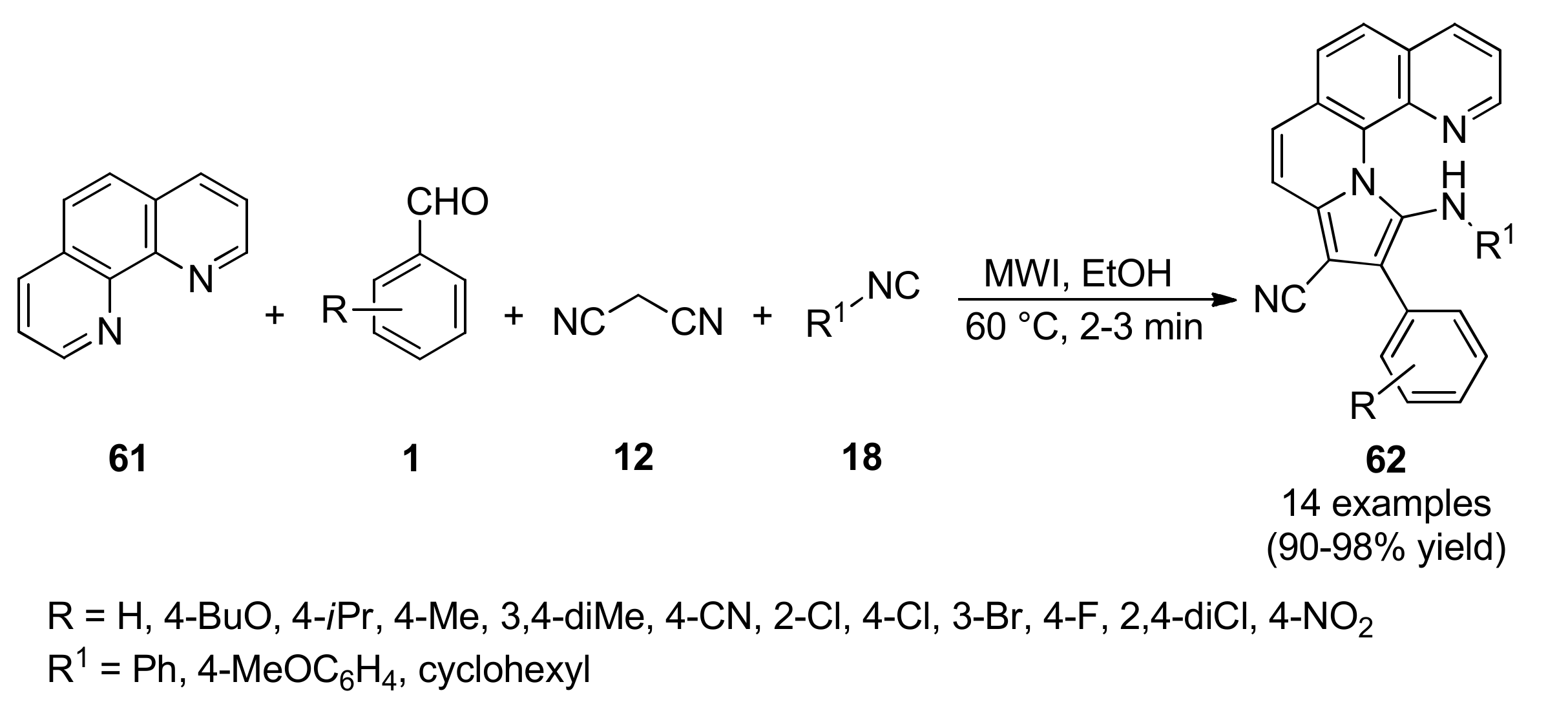{kind=link}
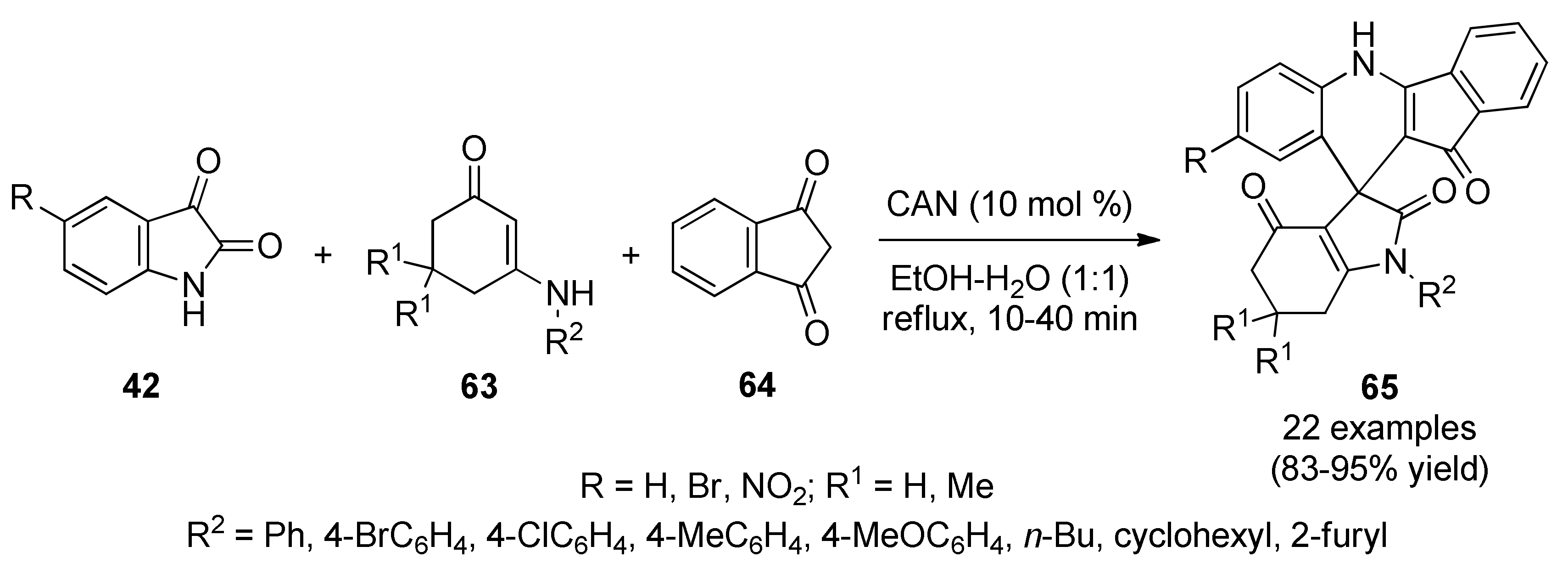{kind=link}
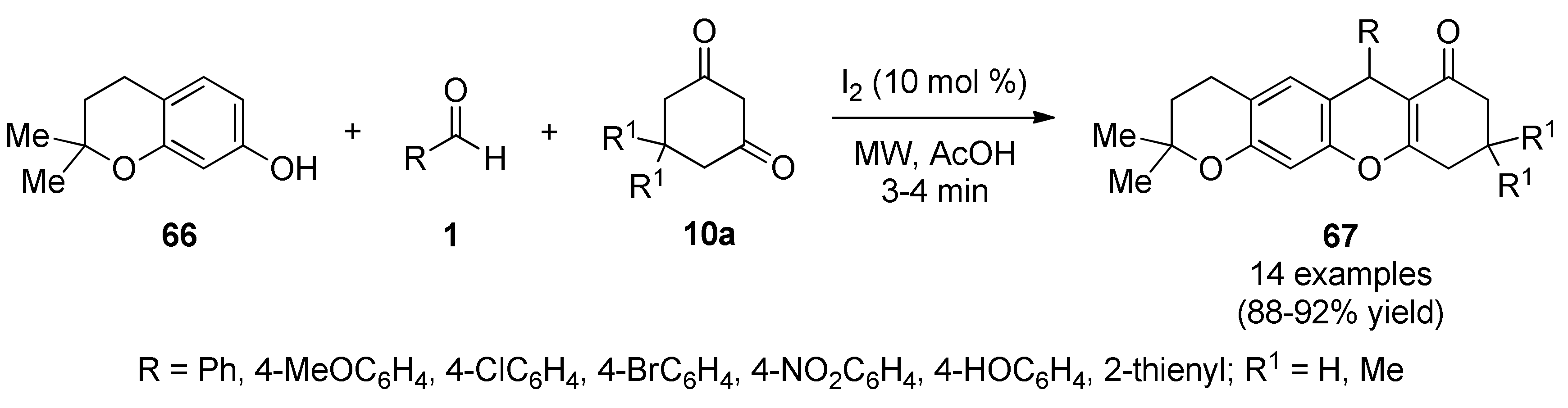{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
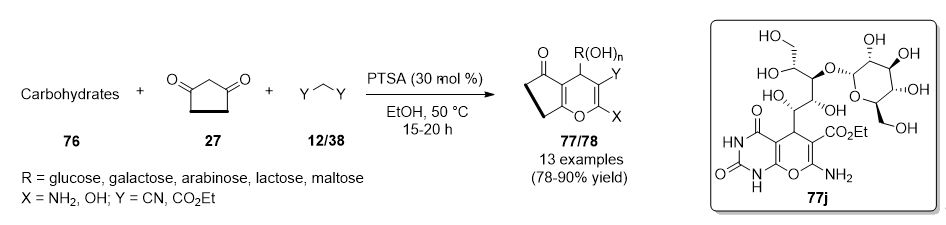{kind=link}
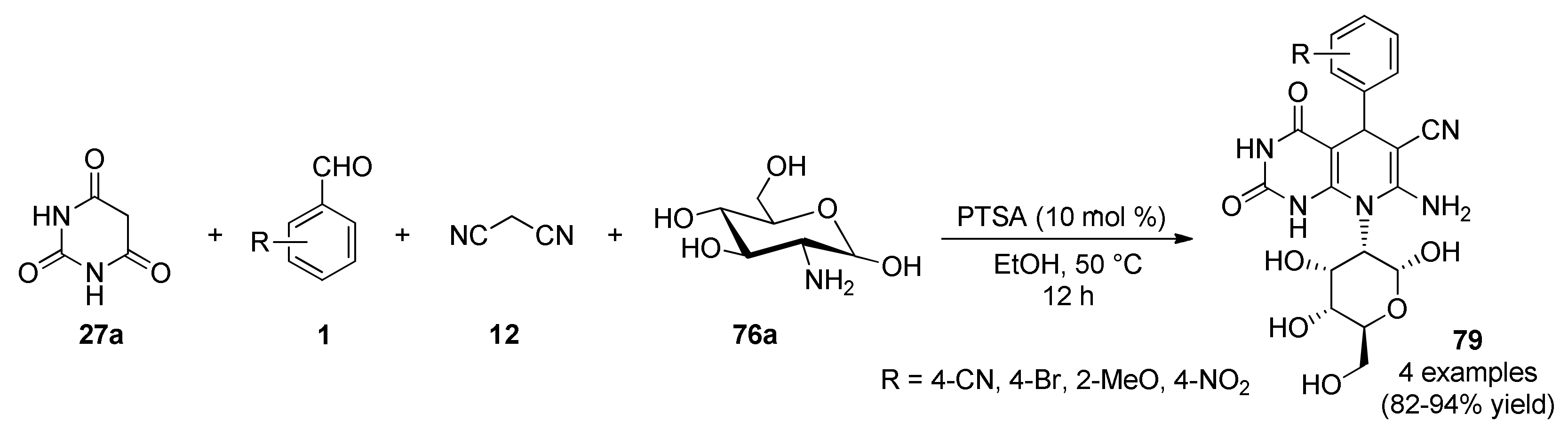{kind=link}
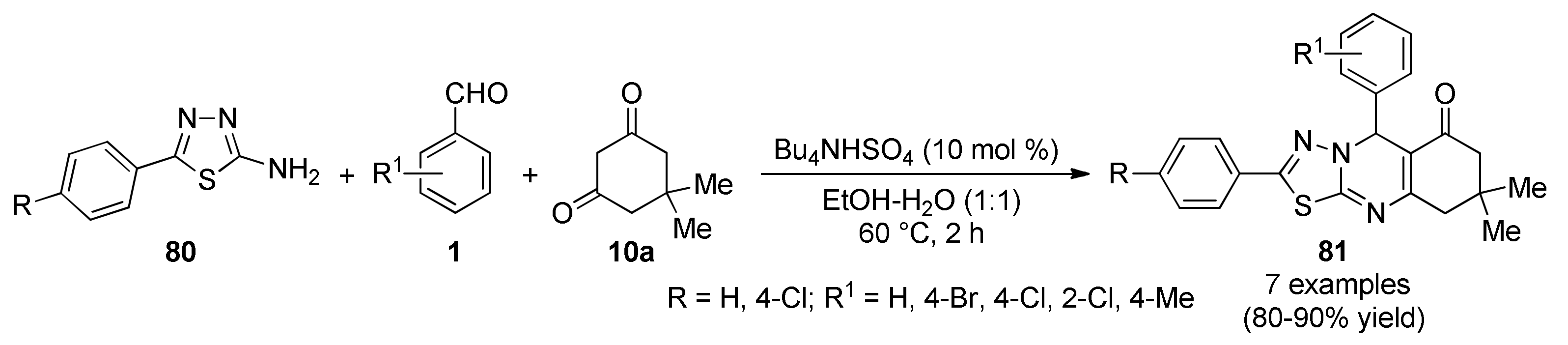{kind=link}
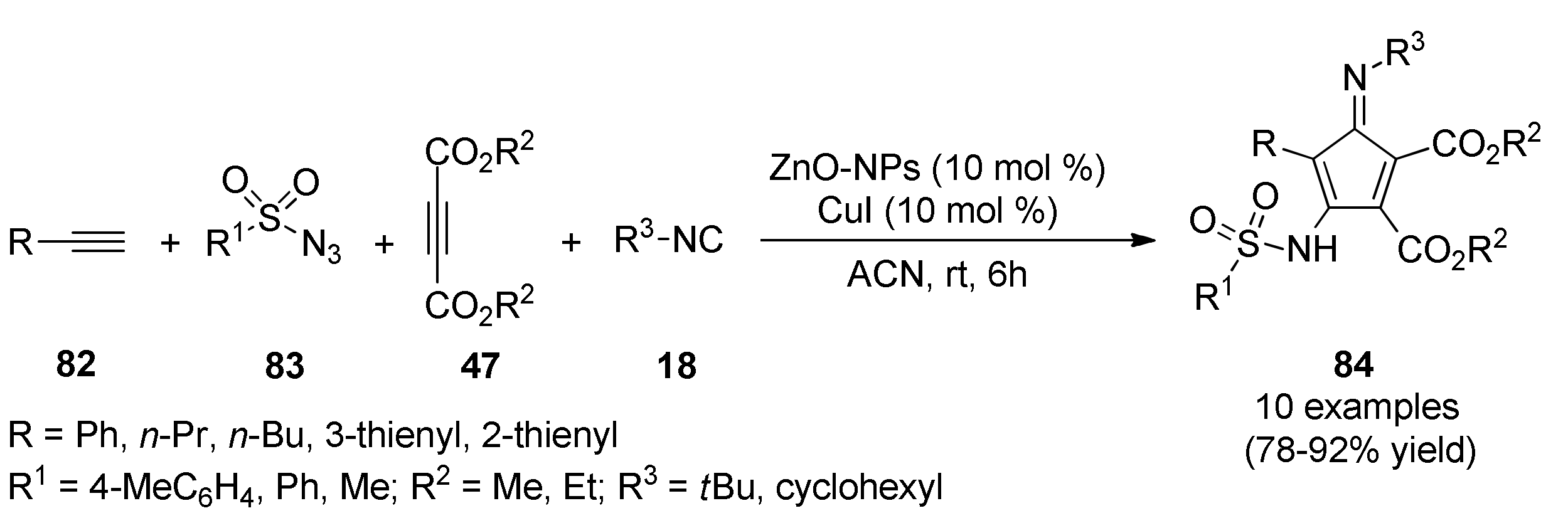{kind=link}
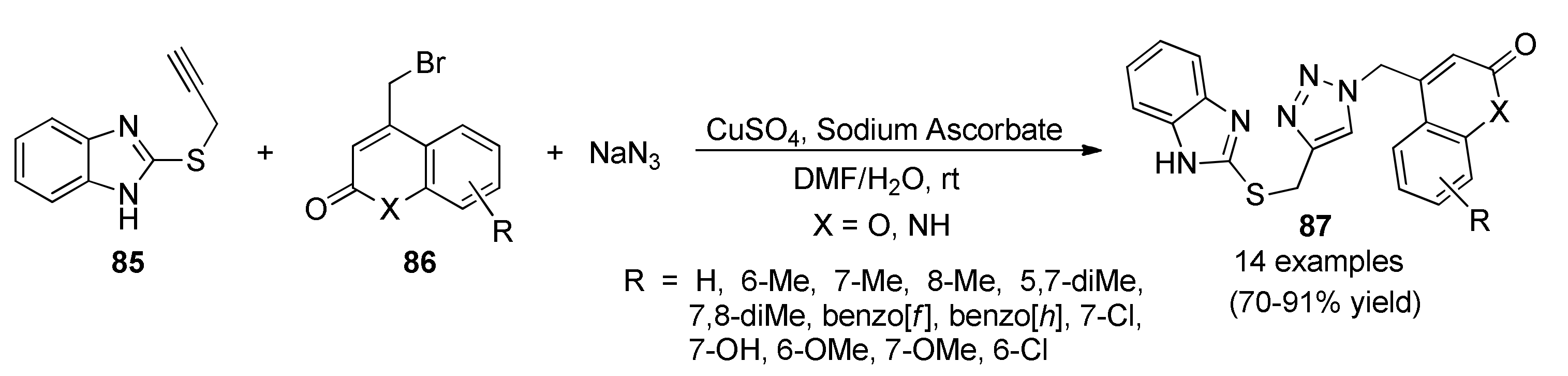{kind=link}
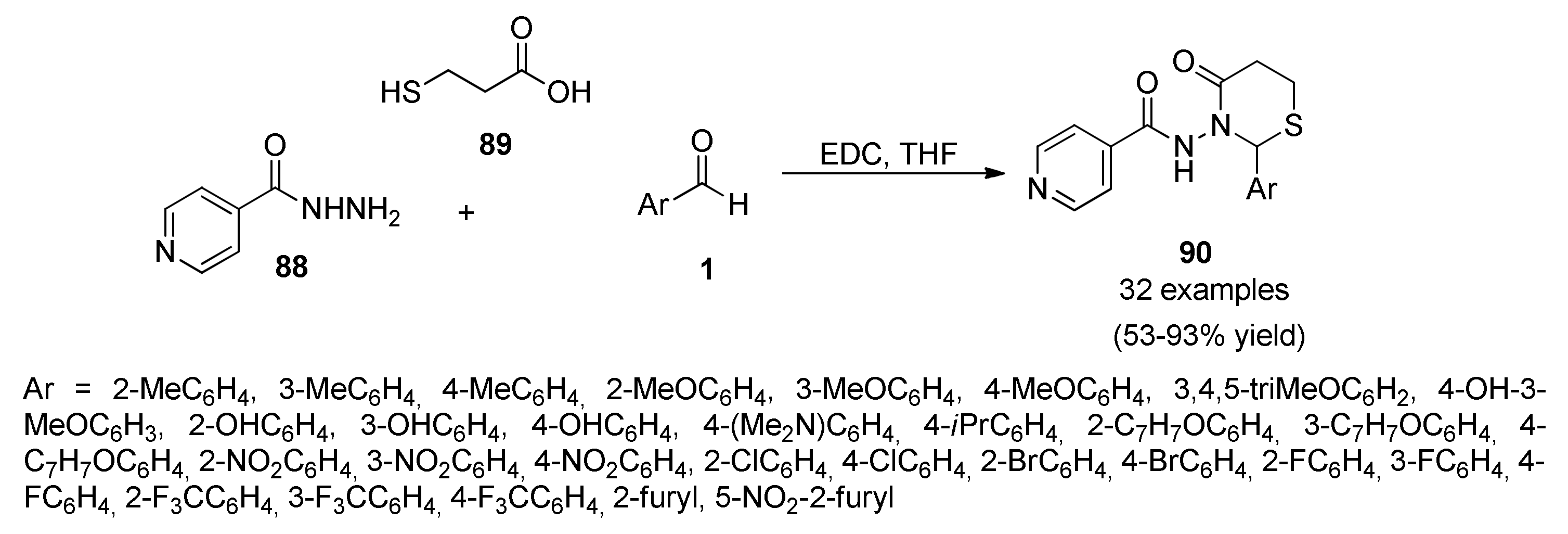{kind=link}
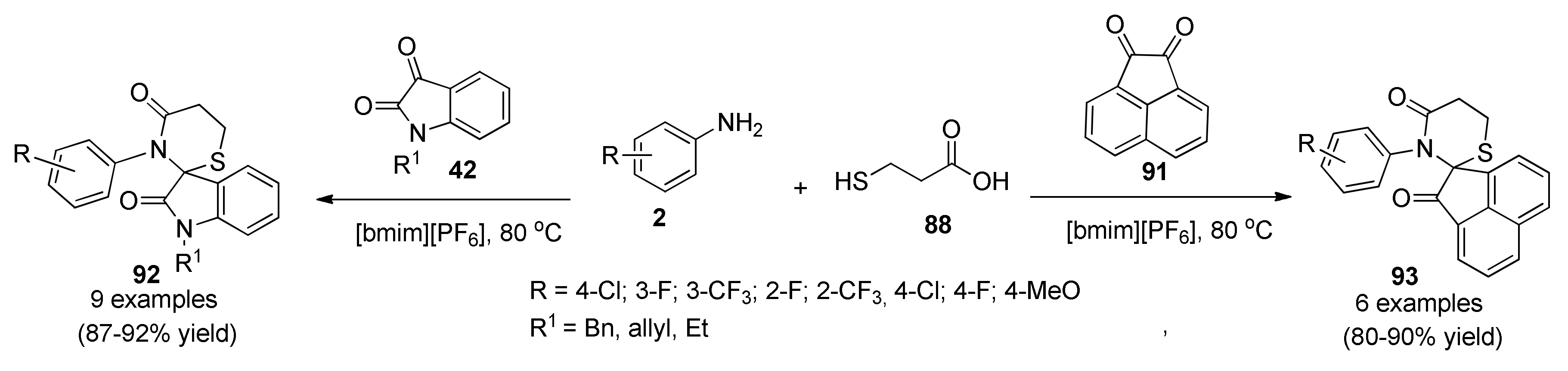{kind=link}
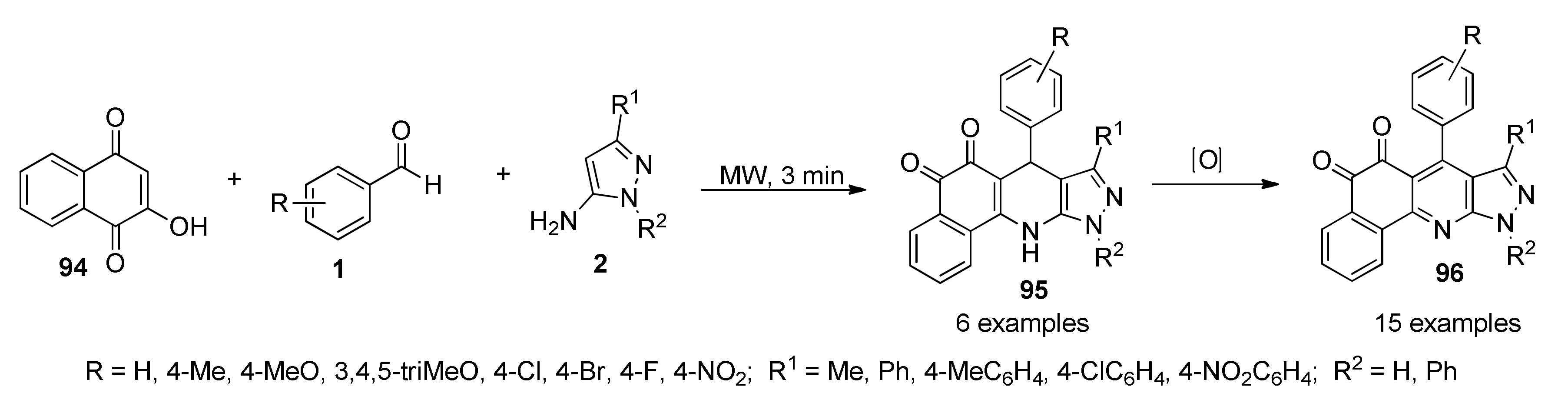{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
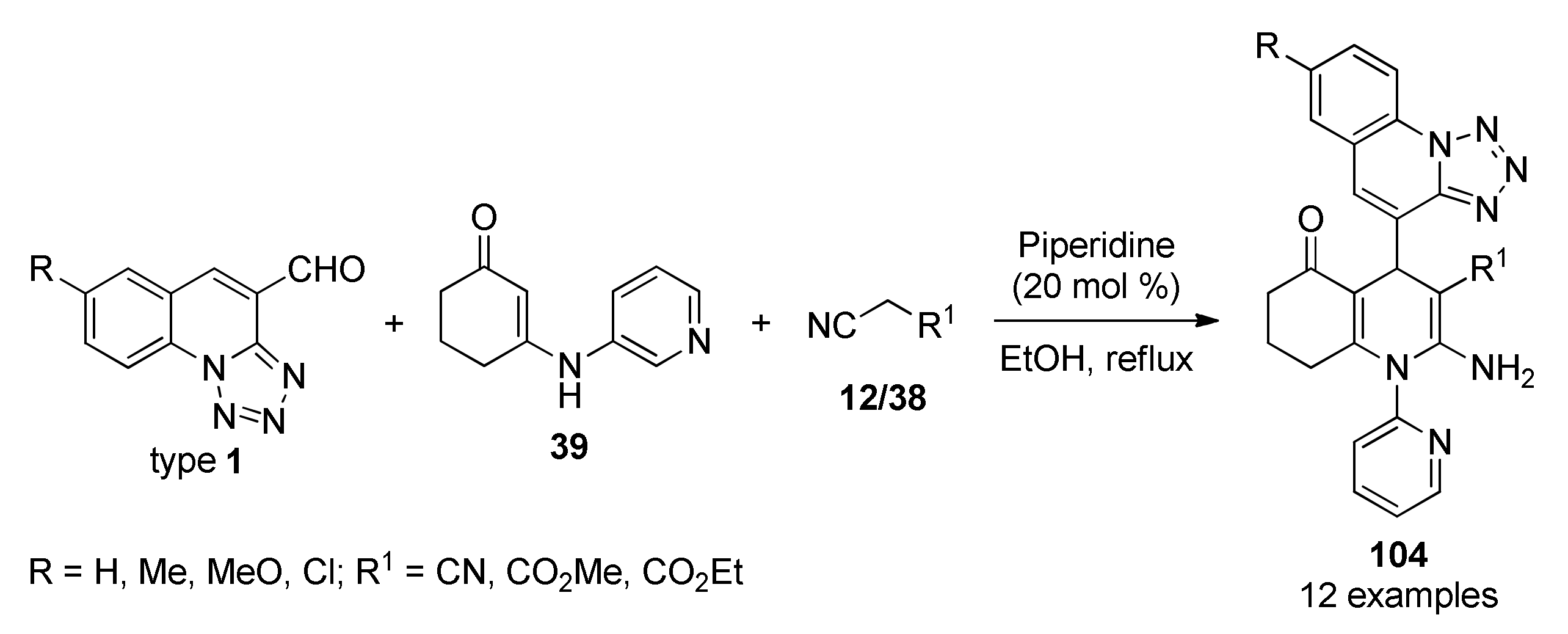{kind=link}
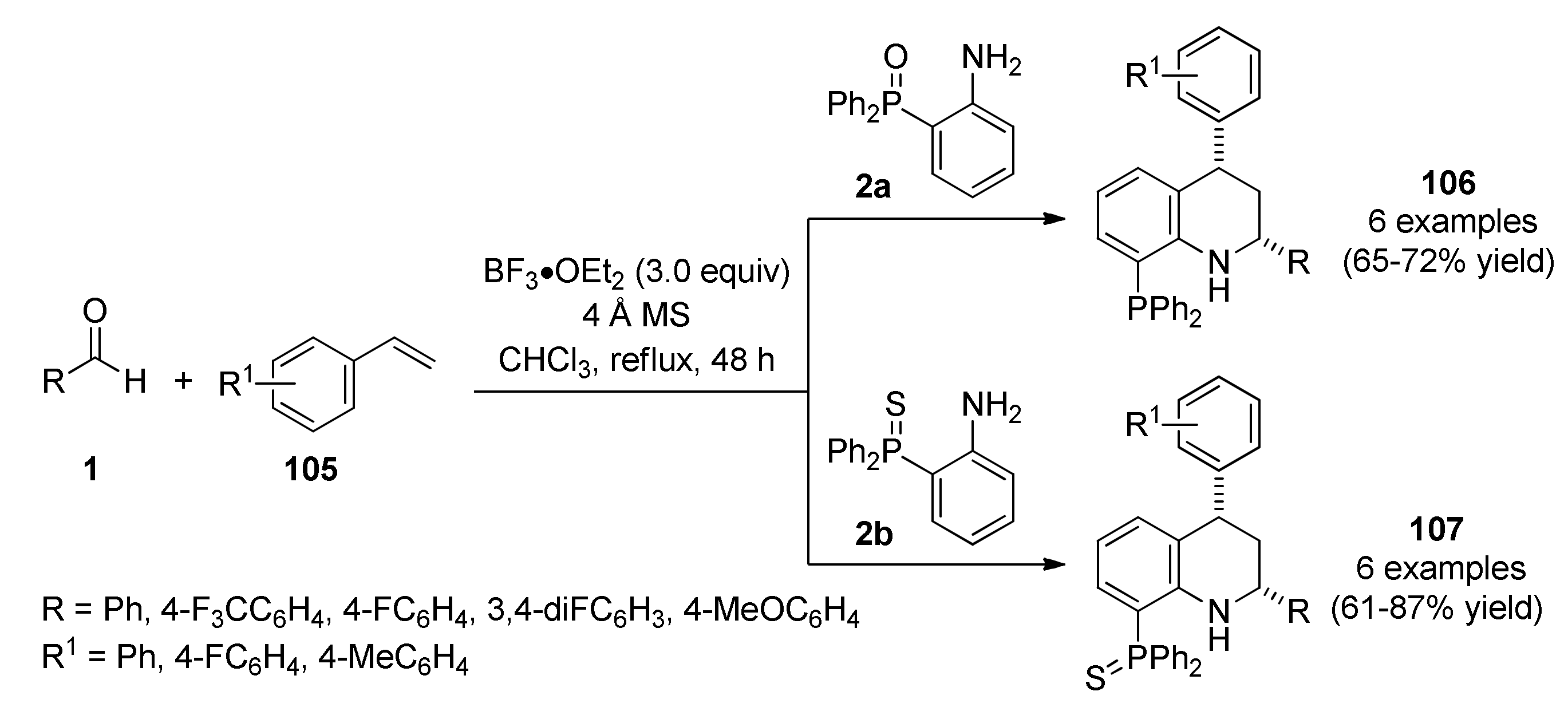{kind=link}
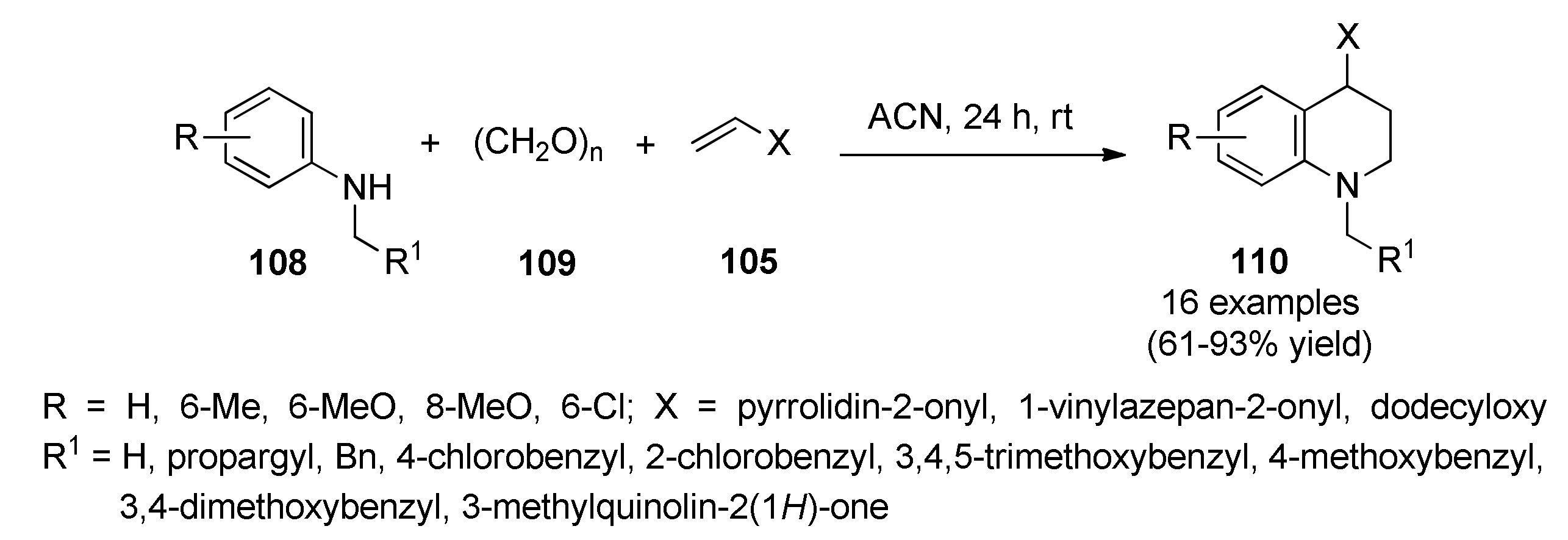{kind=link}
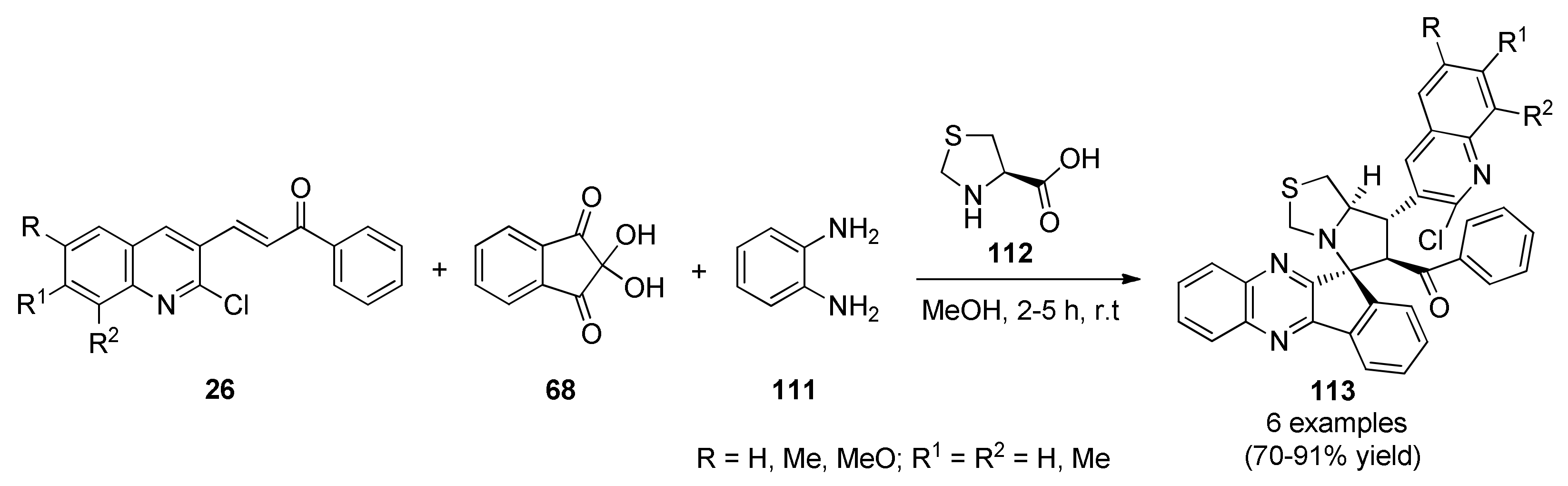{kind=link}
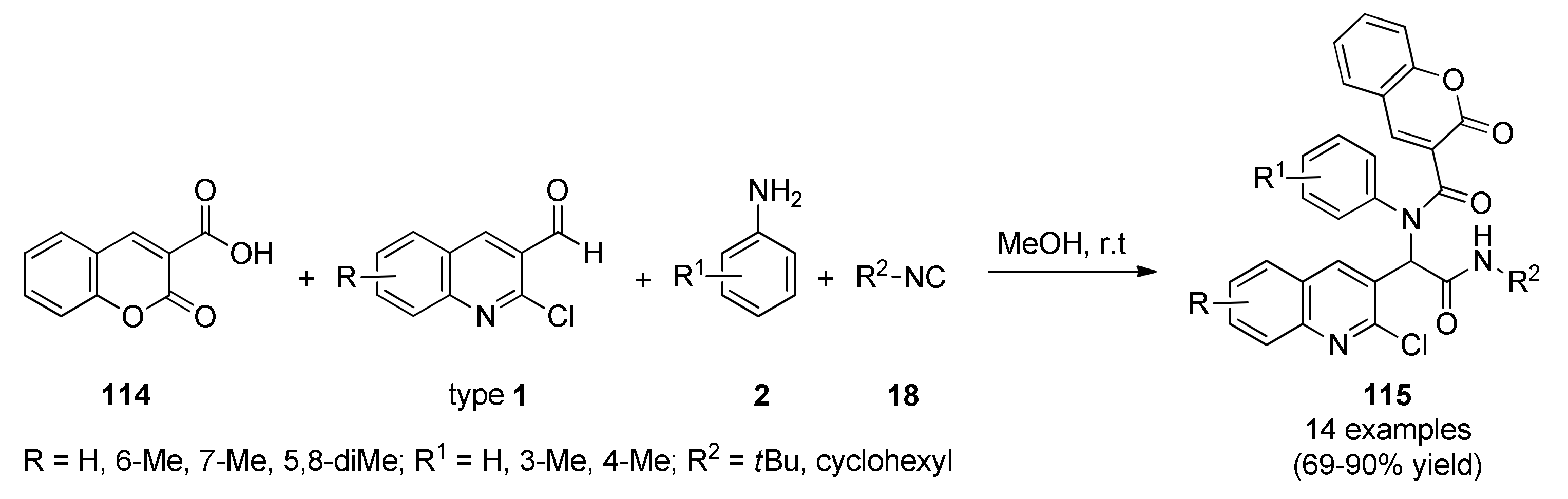{kind=link}
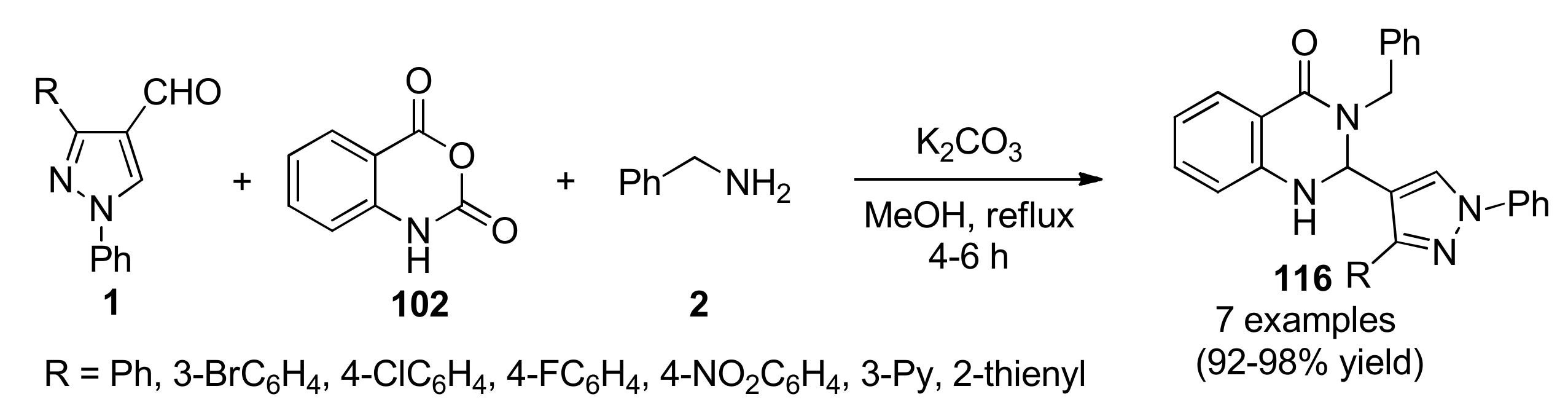{kind=link}
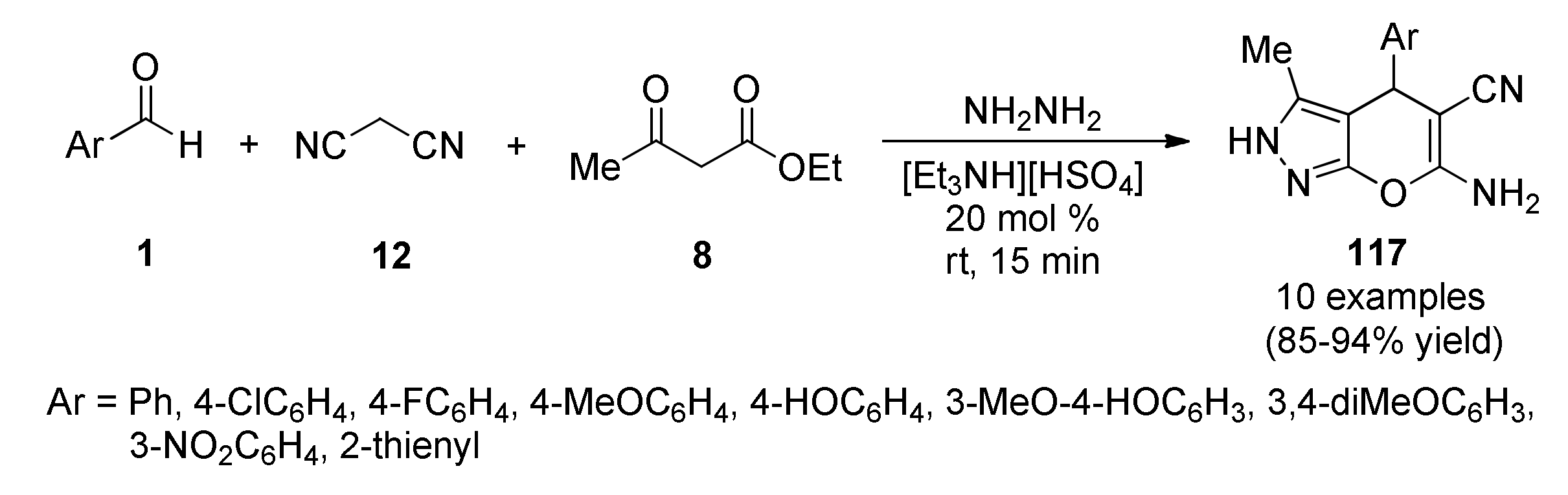{kind=link}
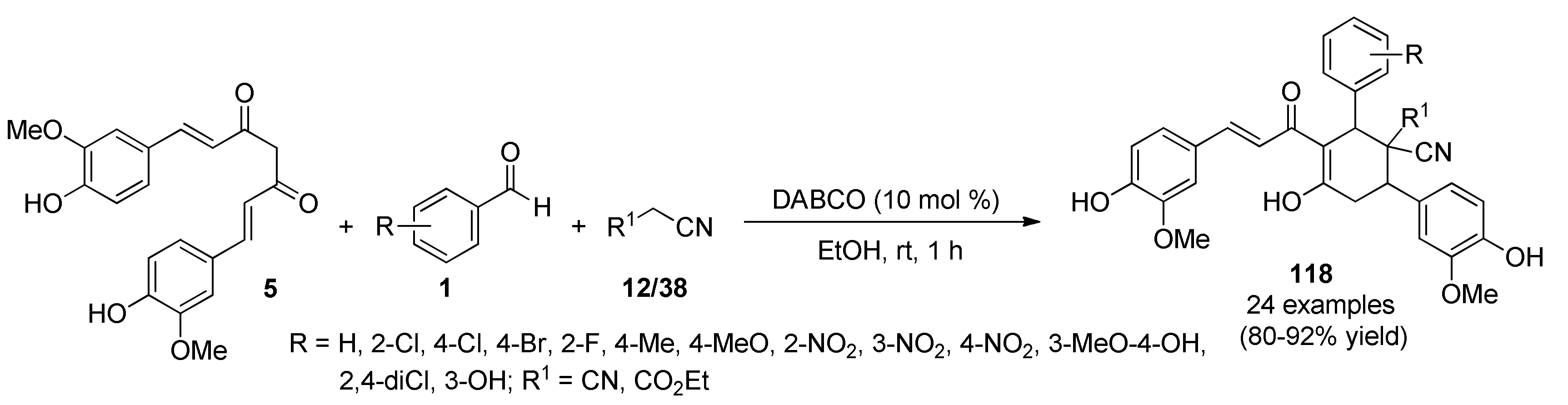{kind=link}
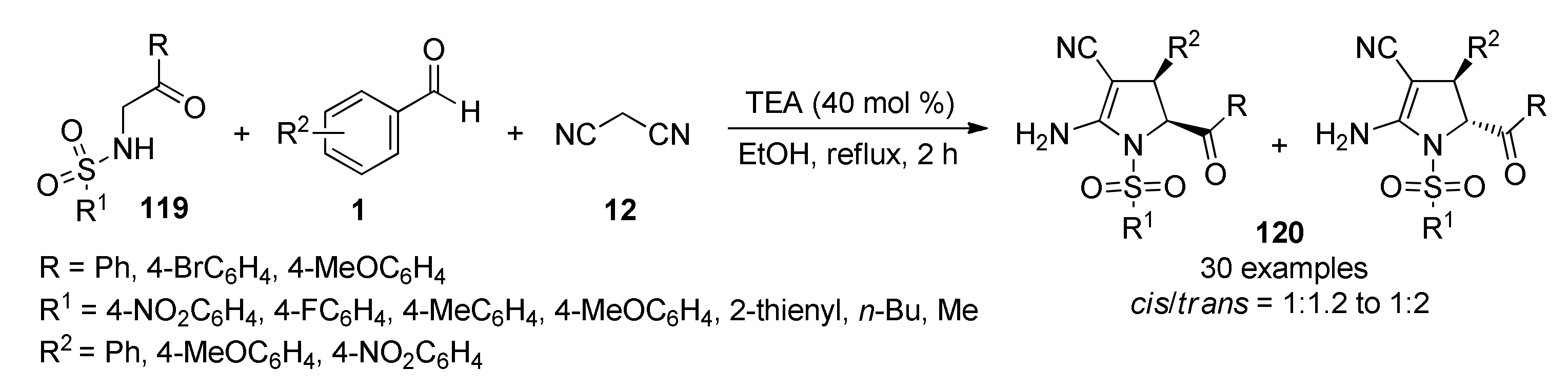{kind=link}
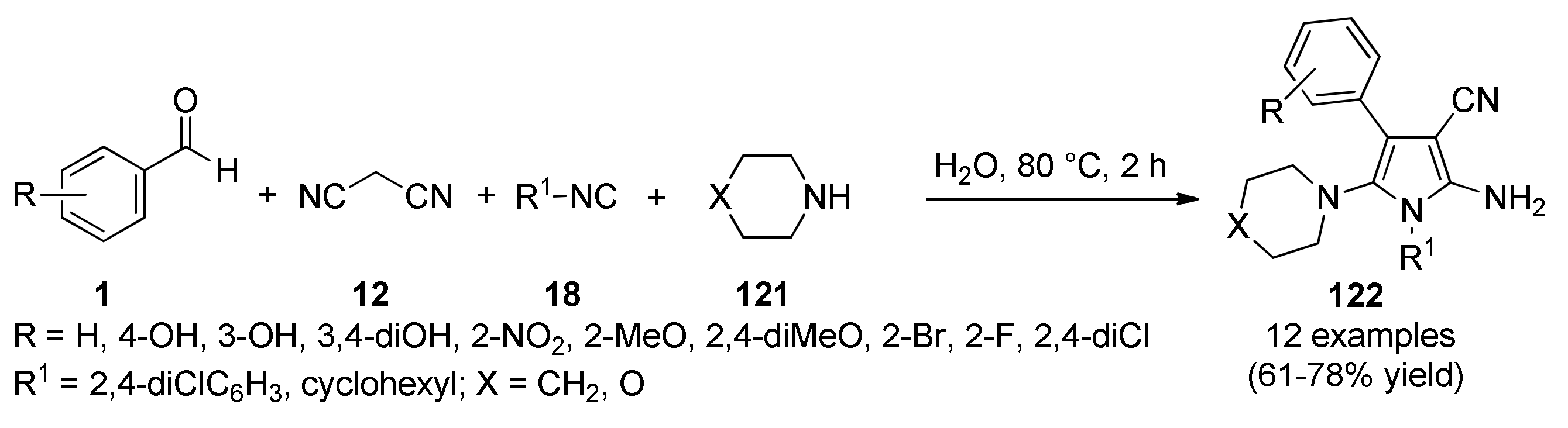{kind=link}
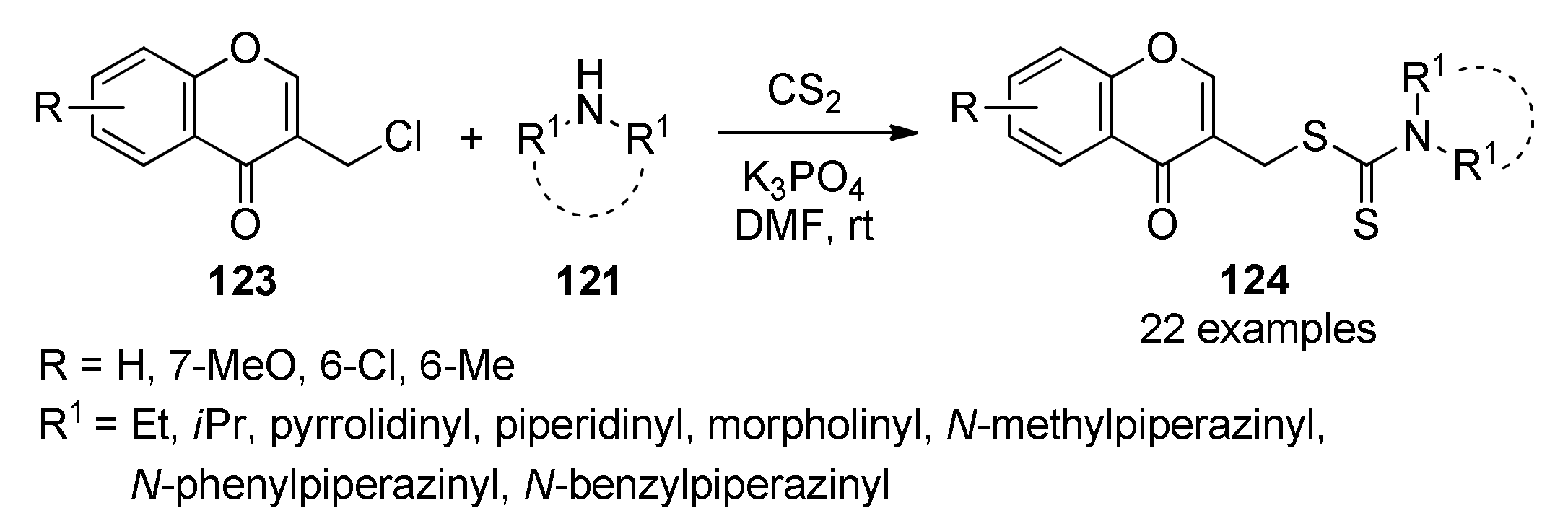{kind=link}
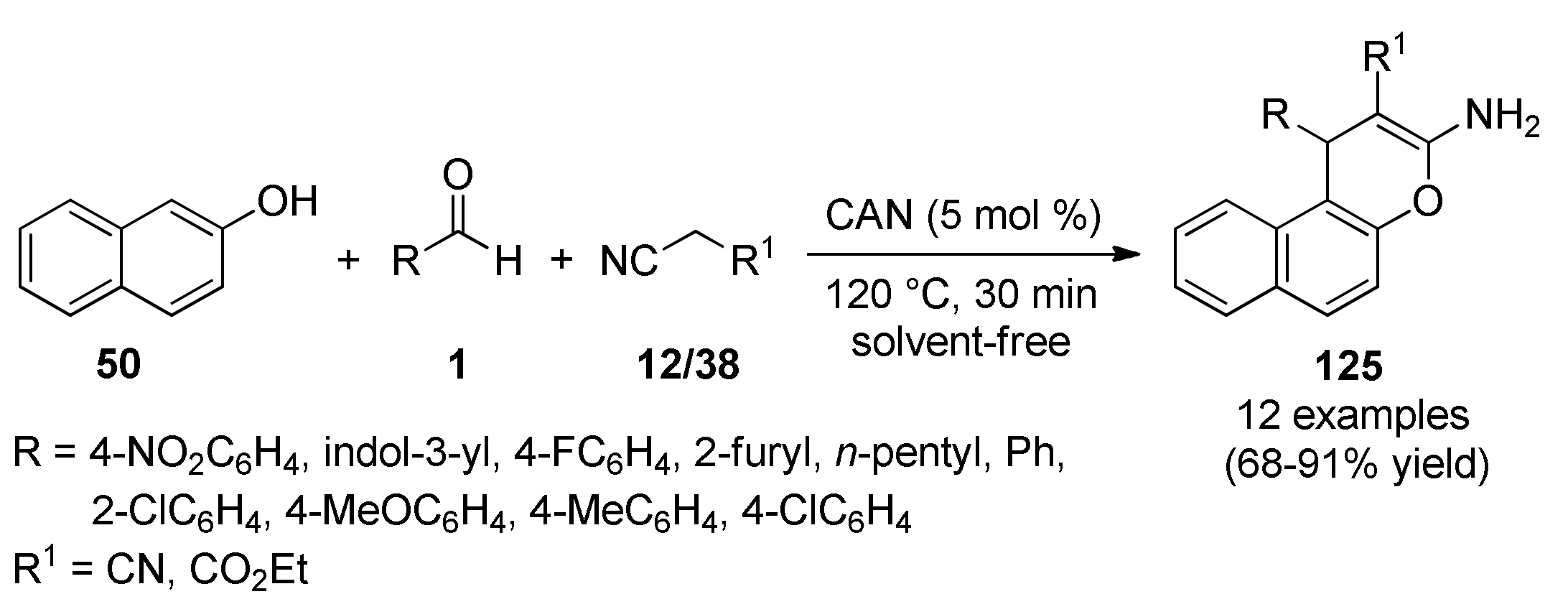{kind=link}
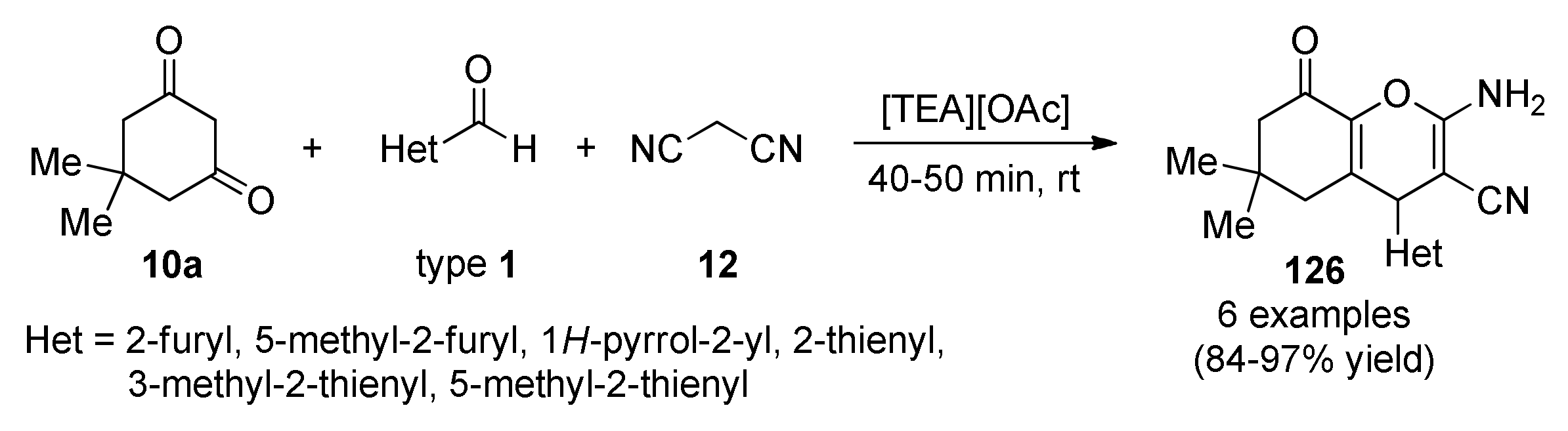{kind=link}
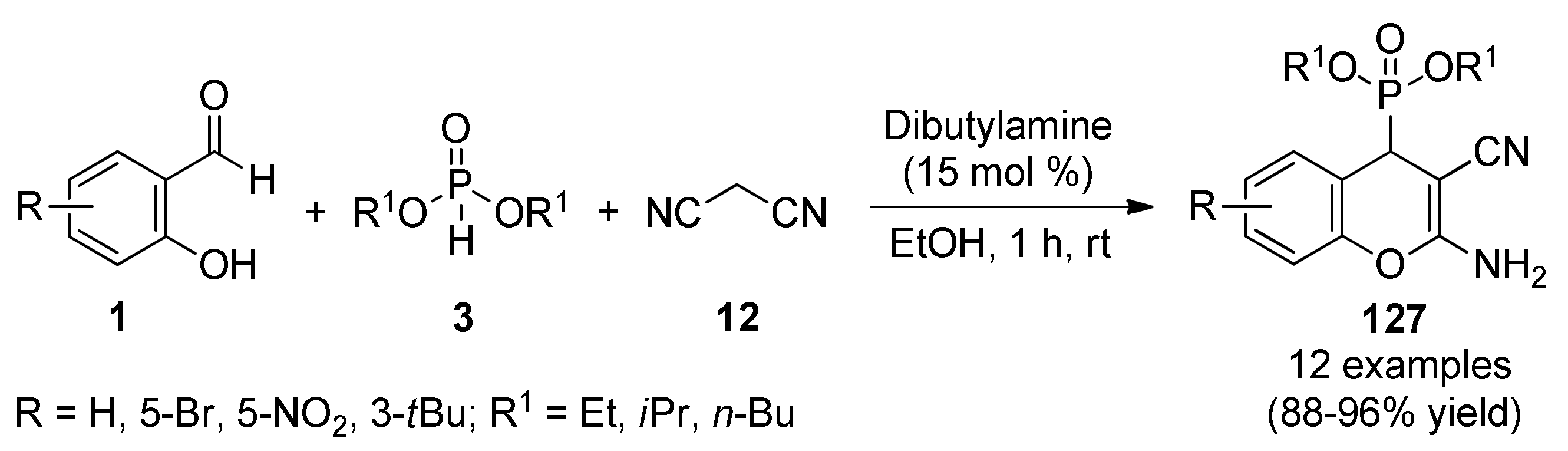{kind=link}
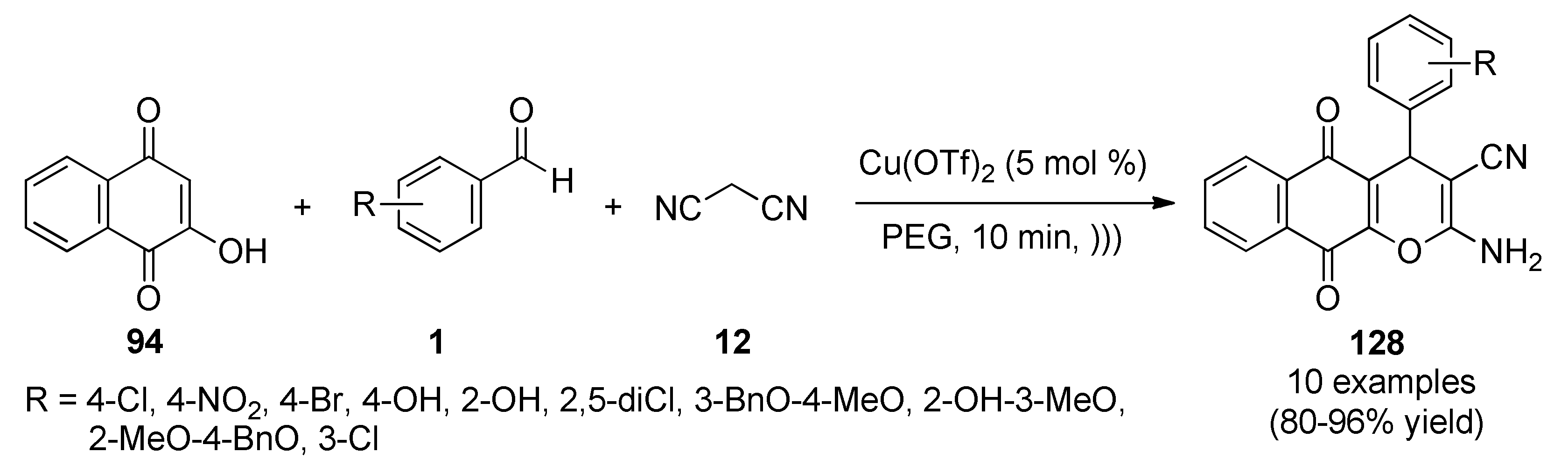{kind=link}
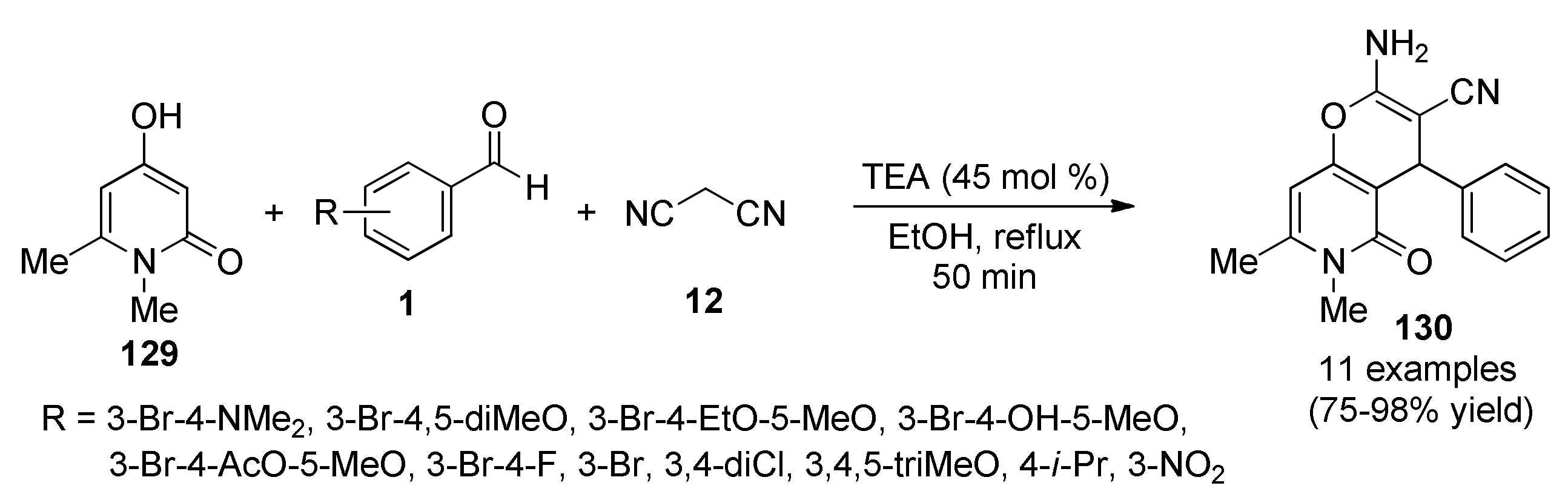{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
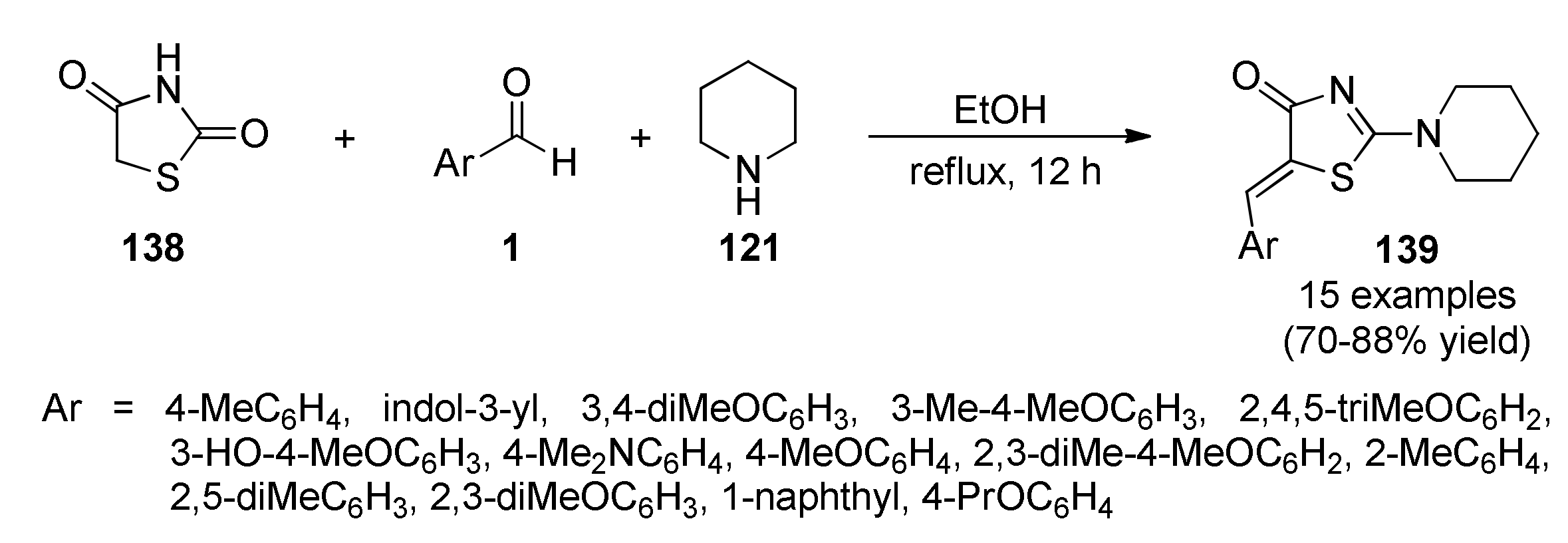{kind=link}
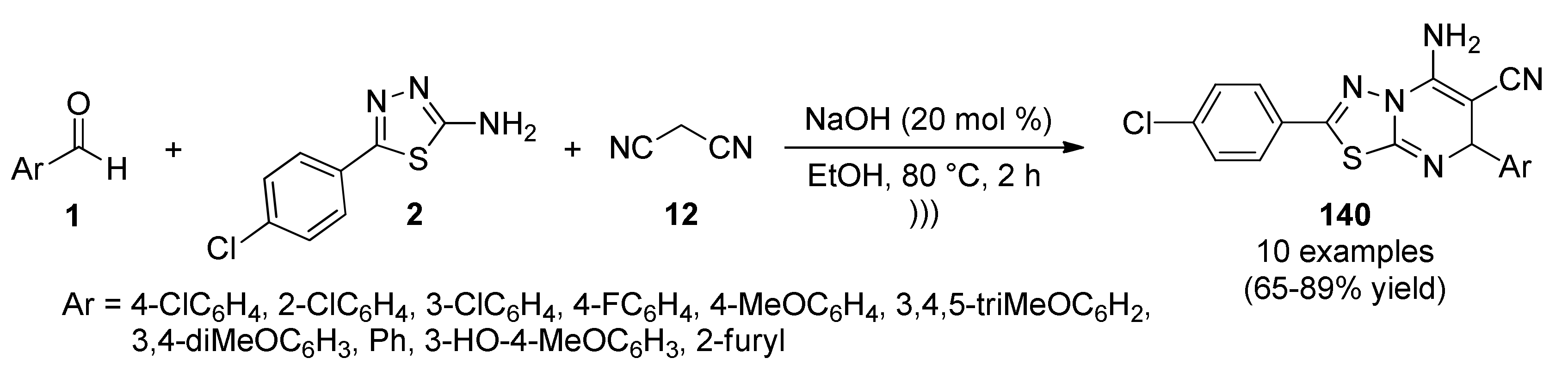{kind=link}
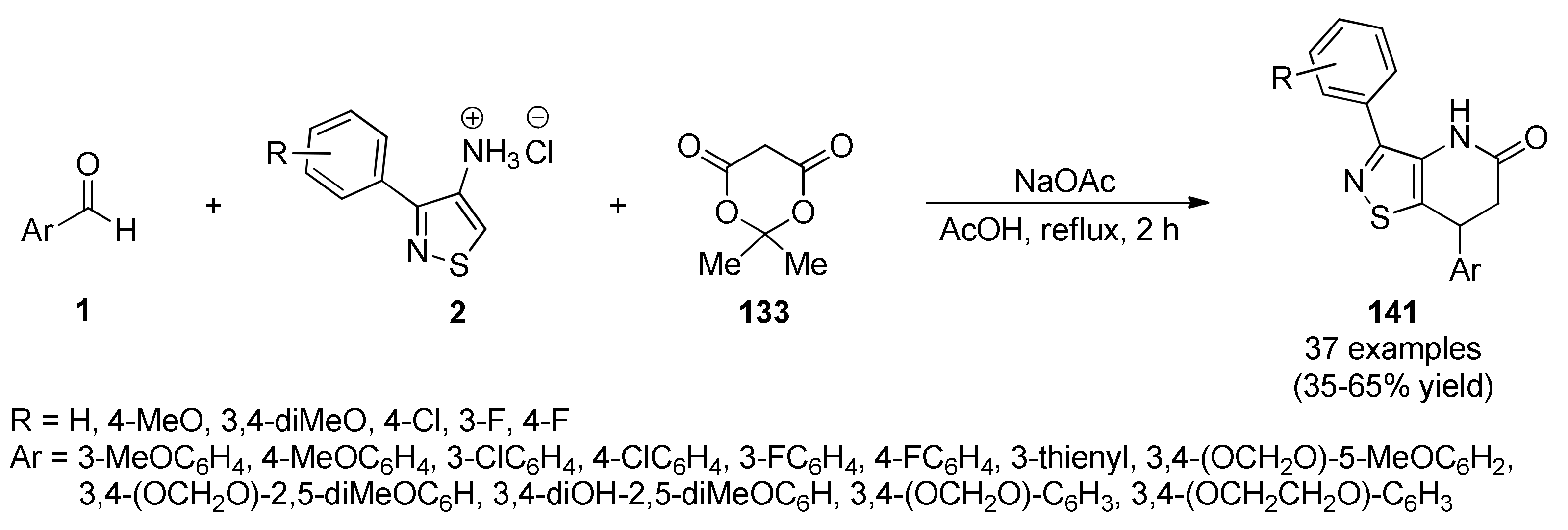{kind=link}
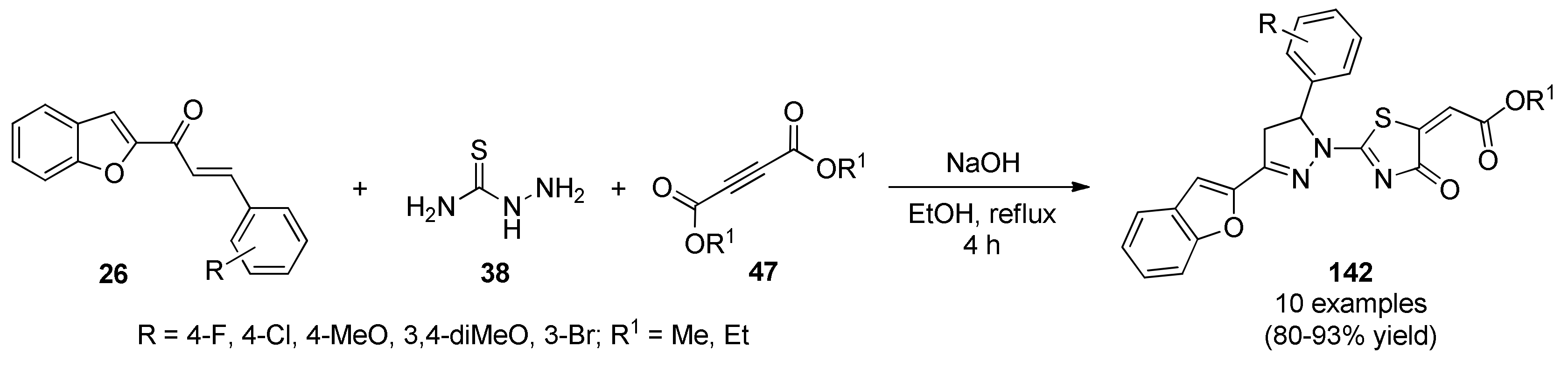{kind=link}
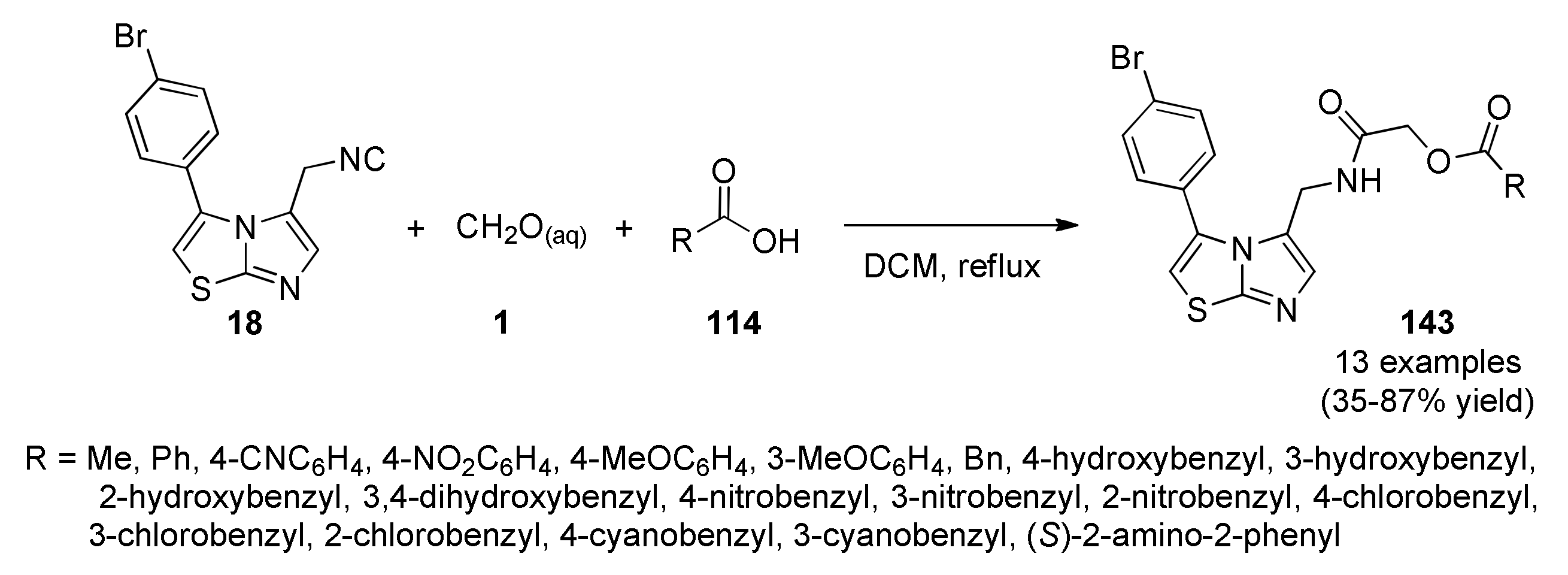{kind=link}
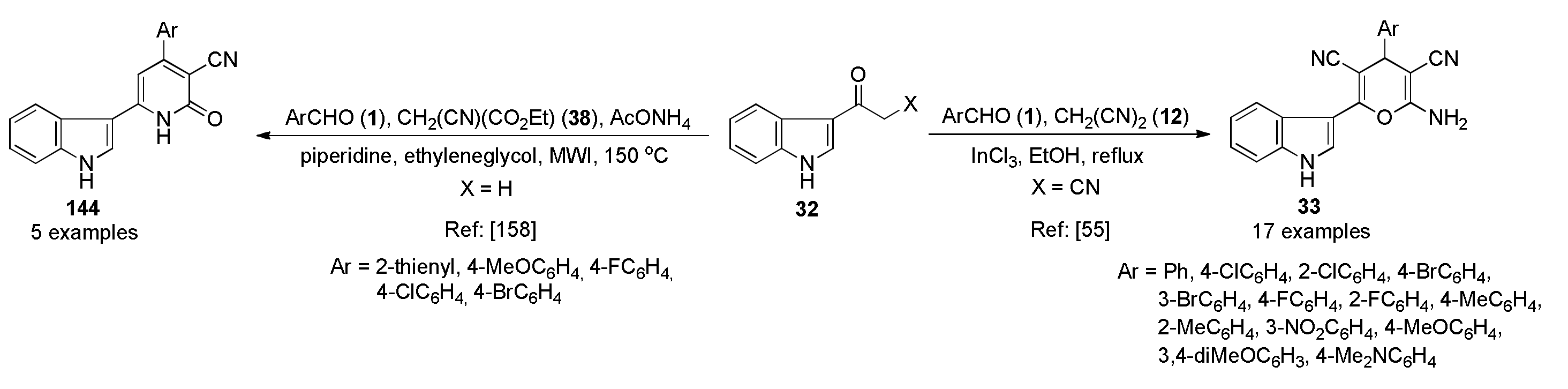{kind=link}
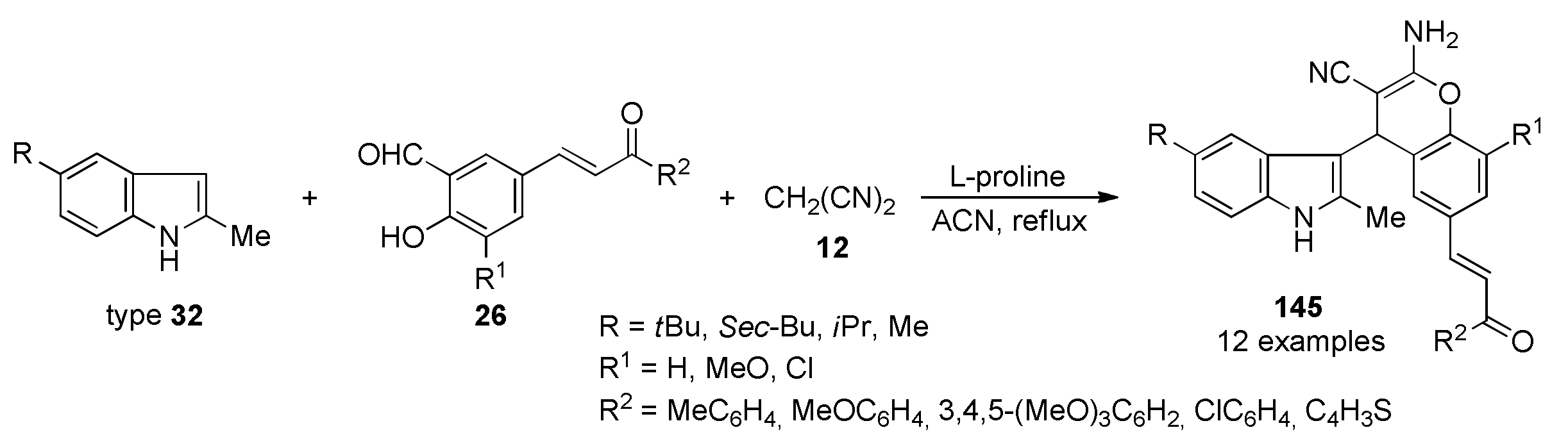{kind=link}
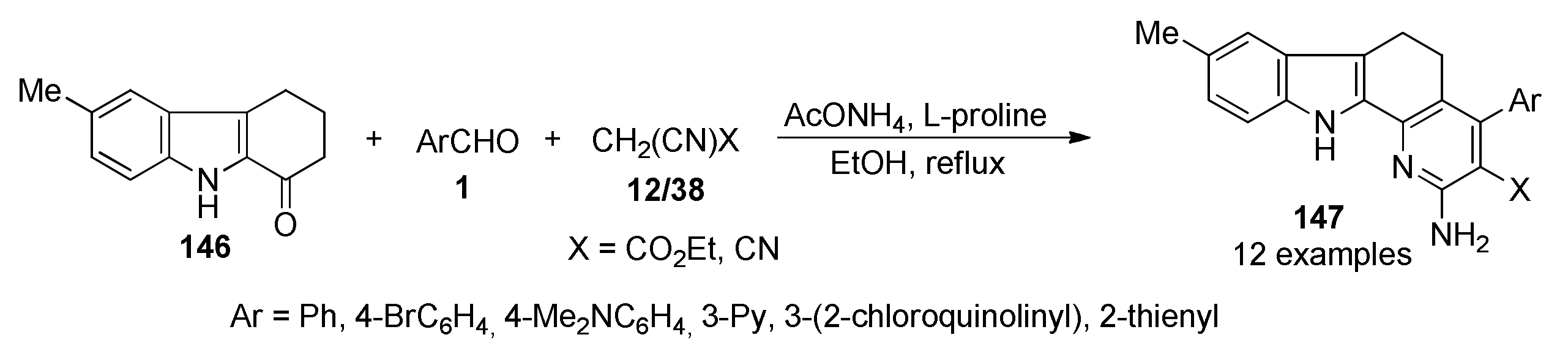{kind=link}
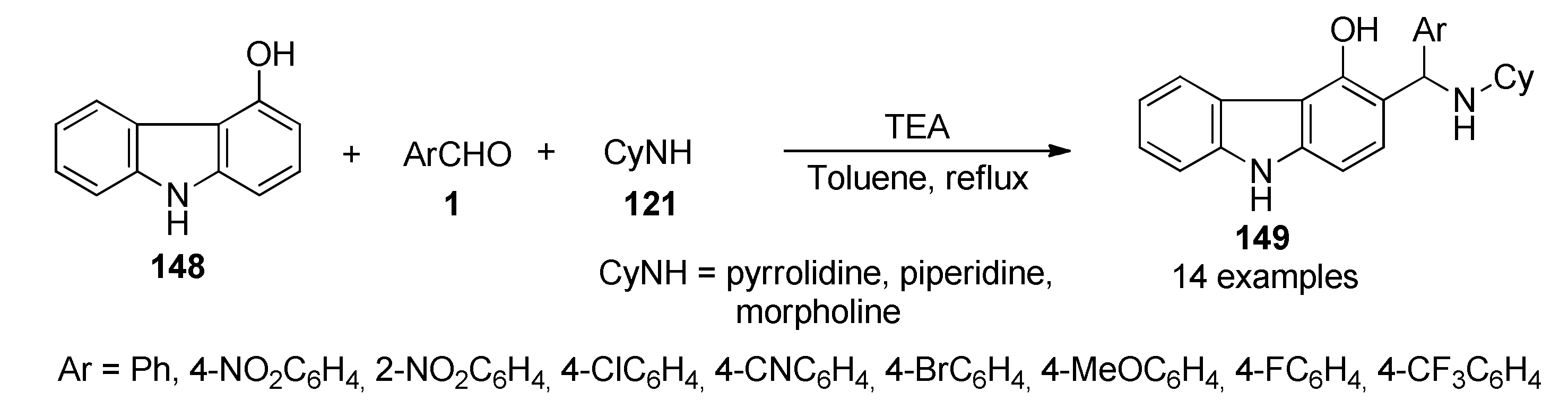{kind=link}
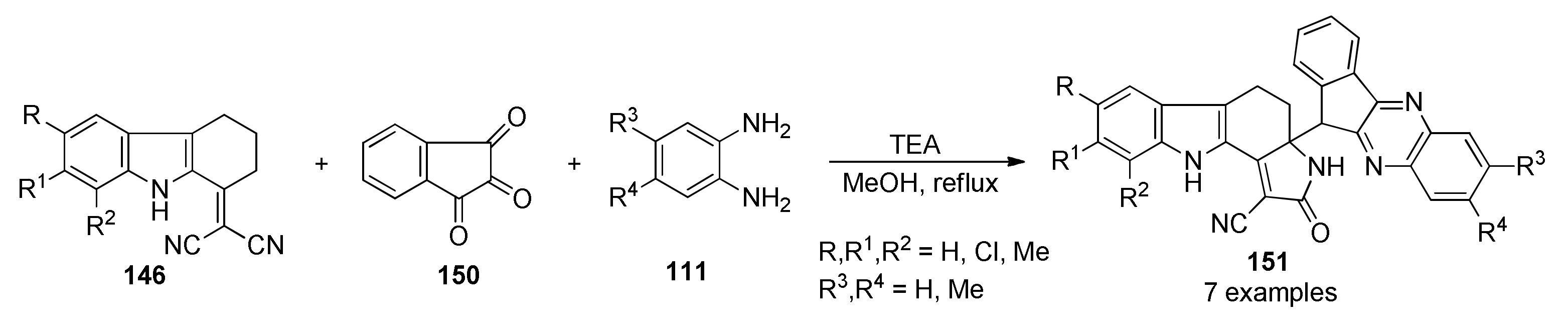{kind=link}
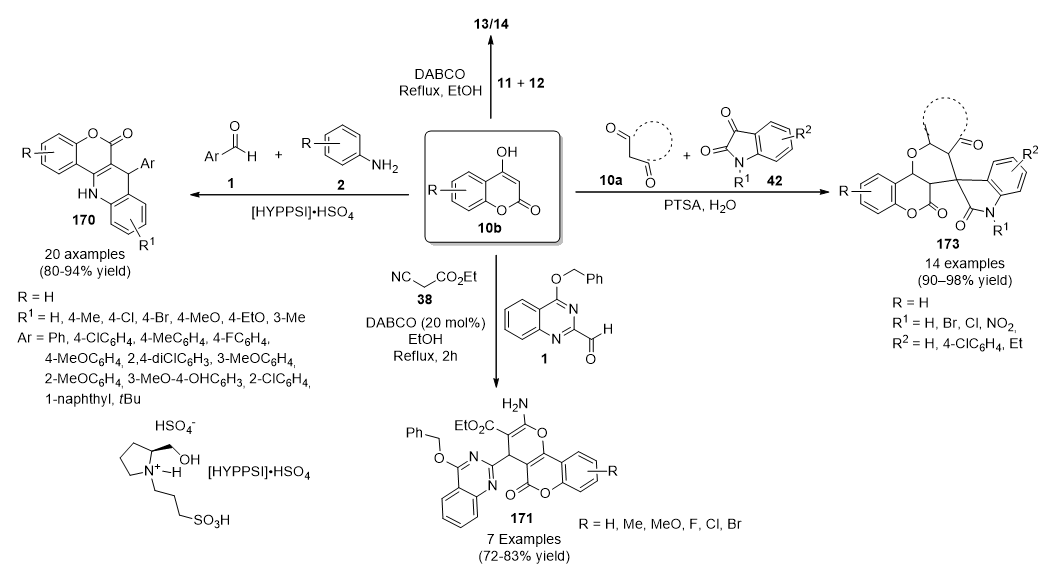{kind=link}
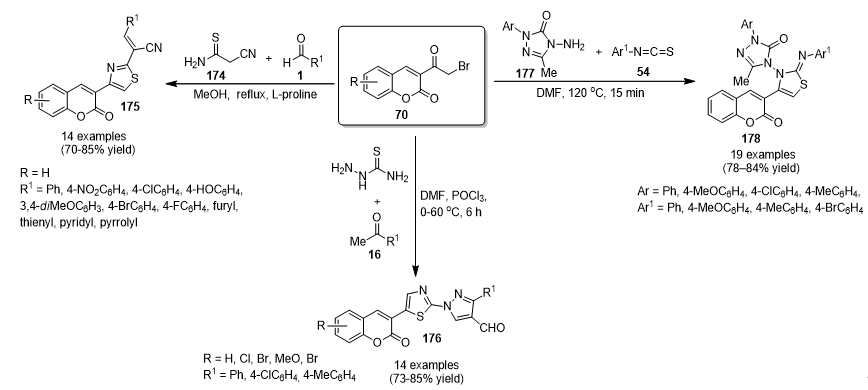{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
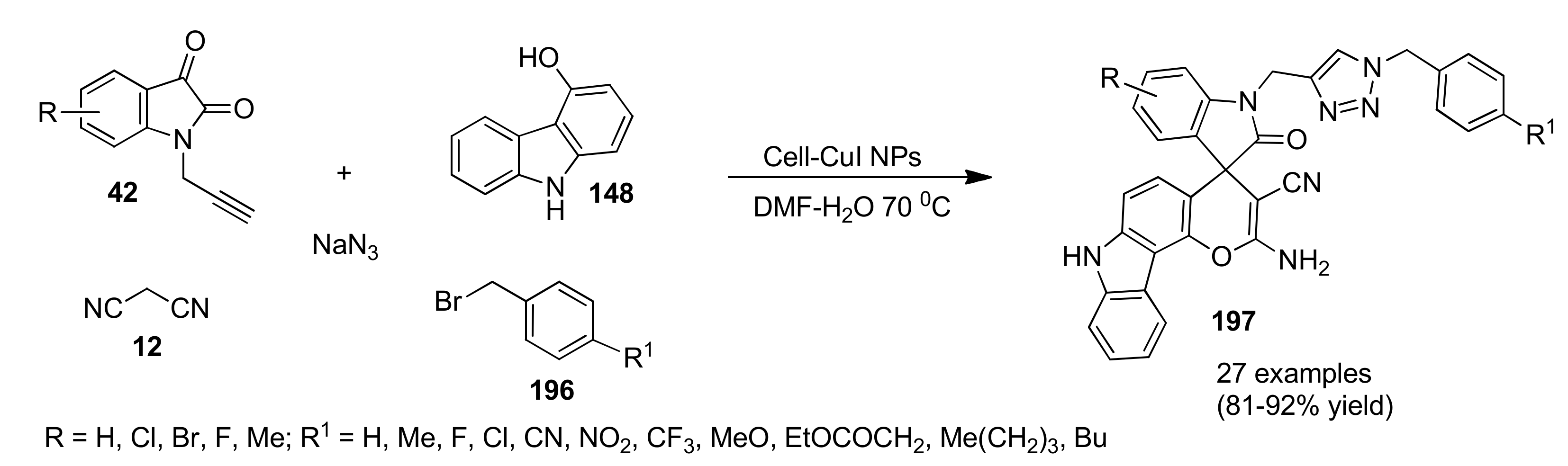{kind=link}
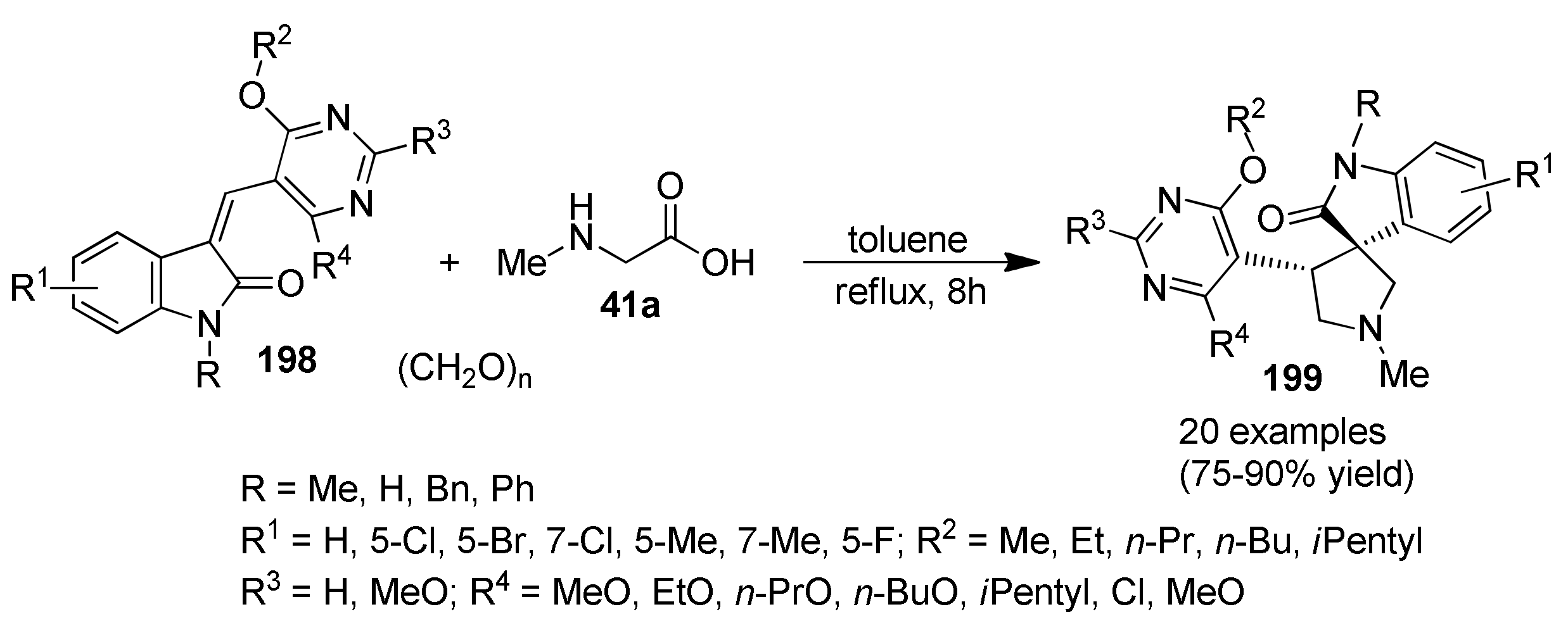{kind=link}
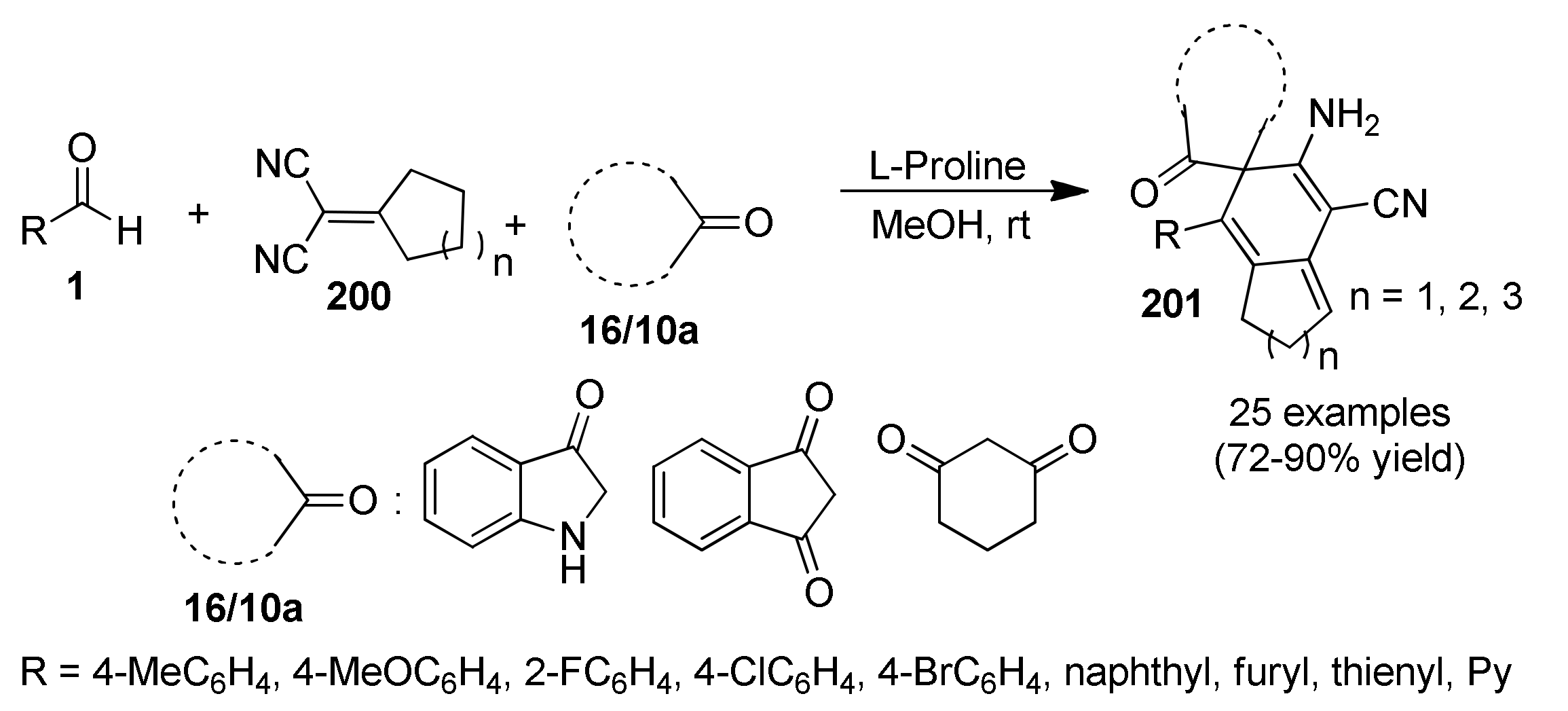{kind=link}
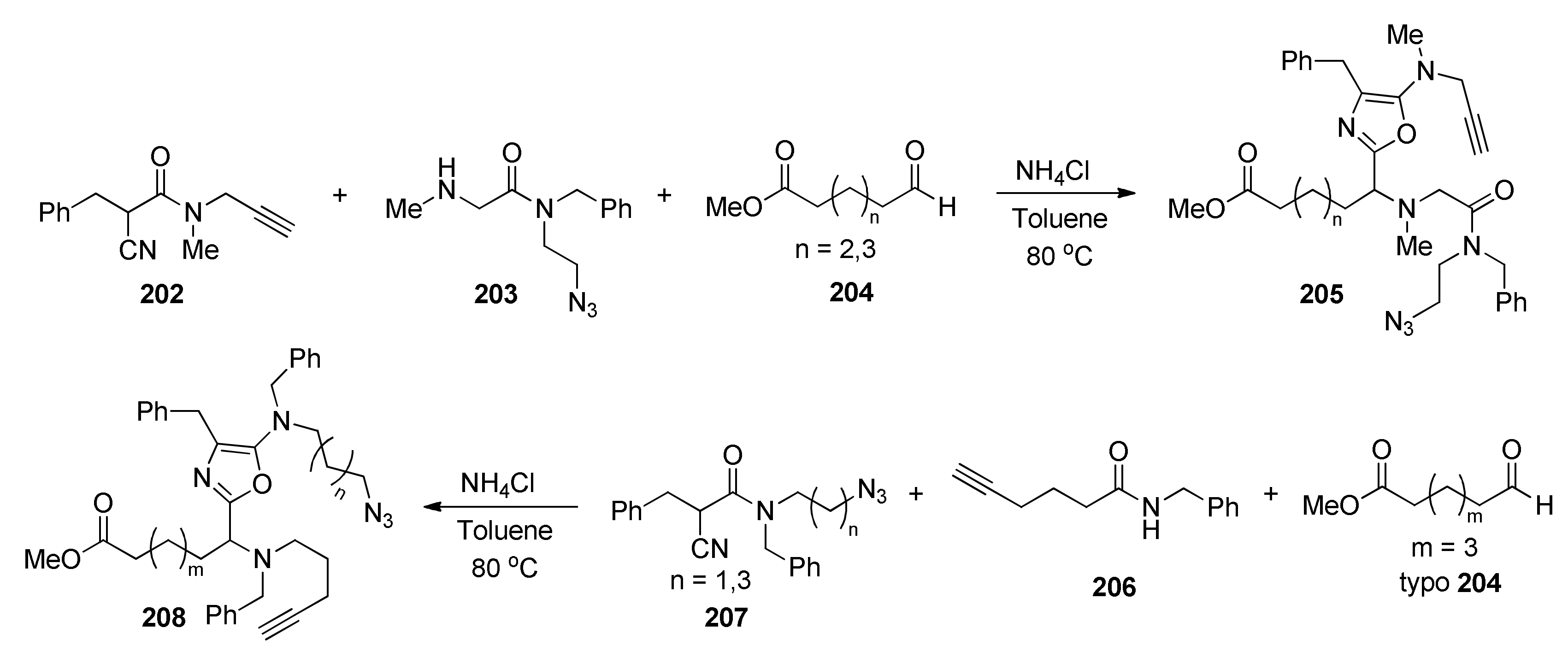{kind=link}
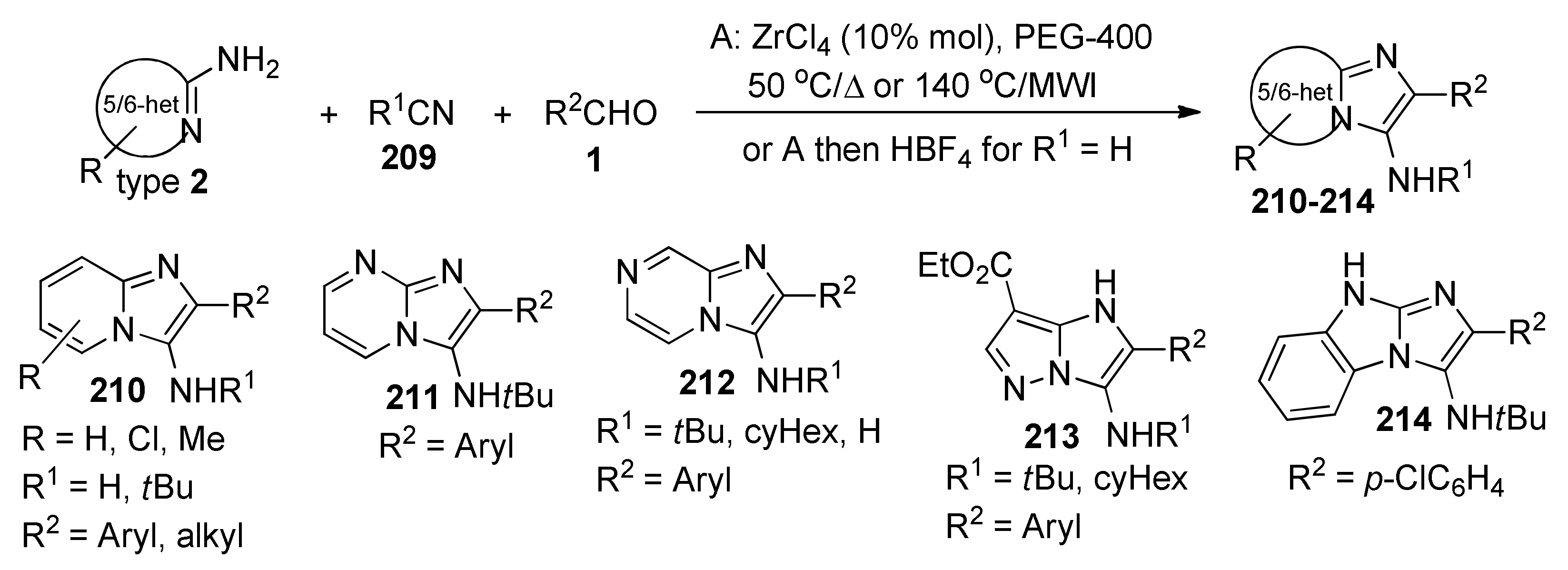{kind=link}
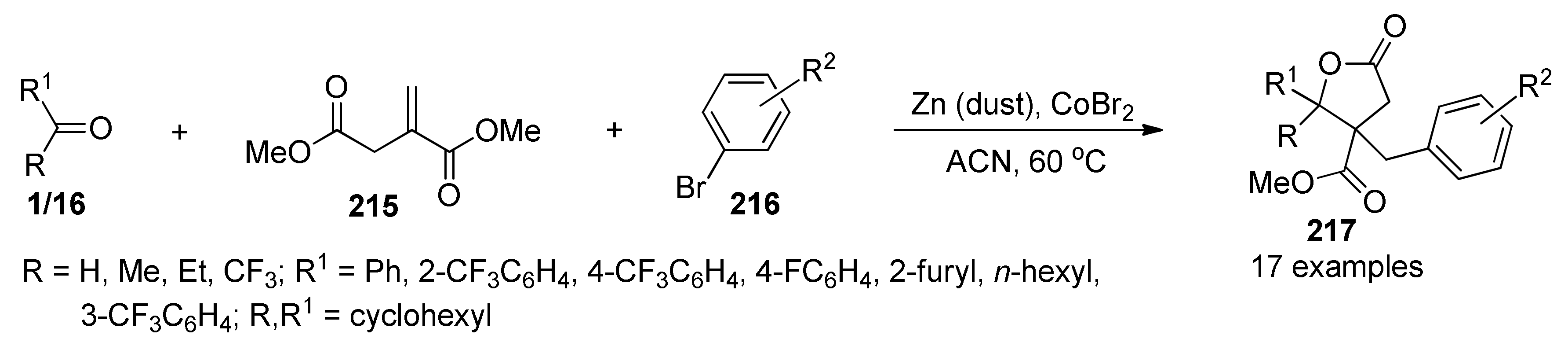{kind=link}
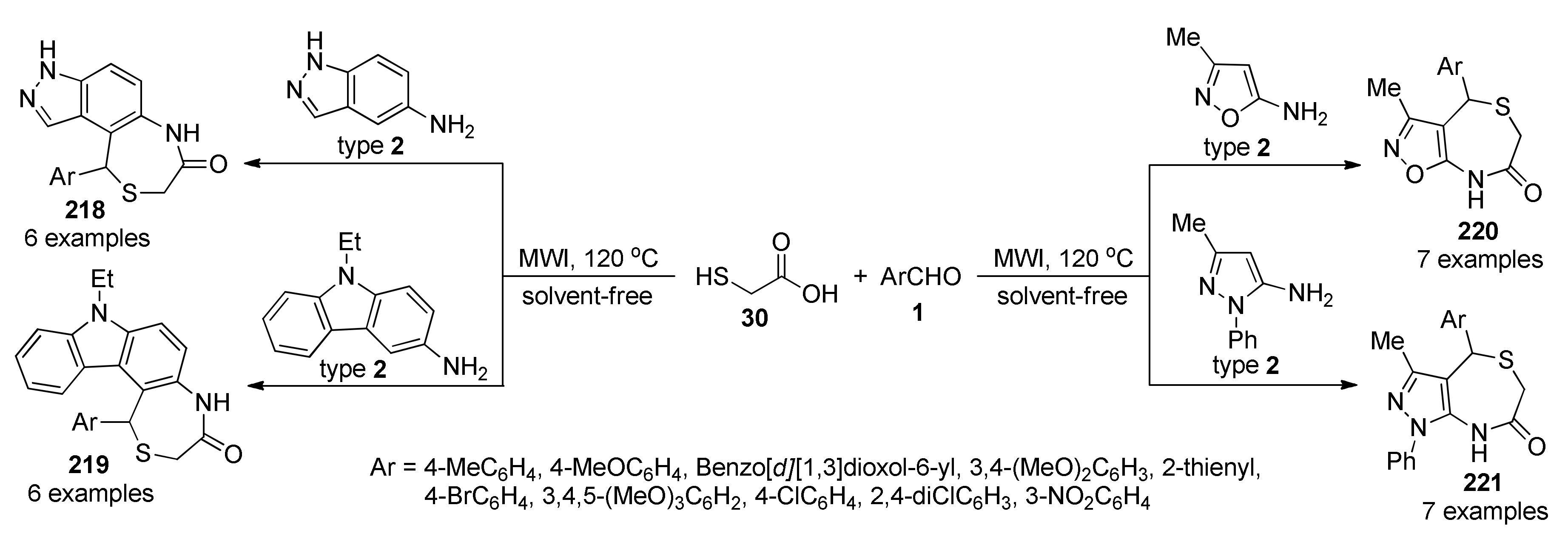{kind=link}
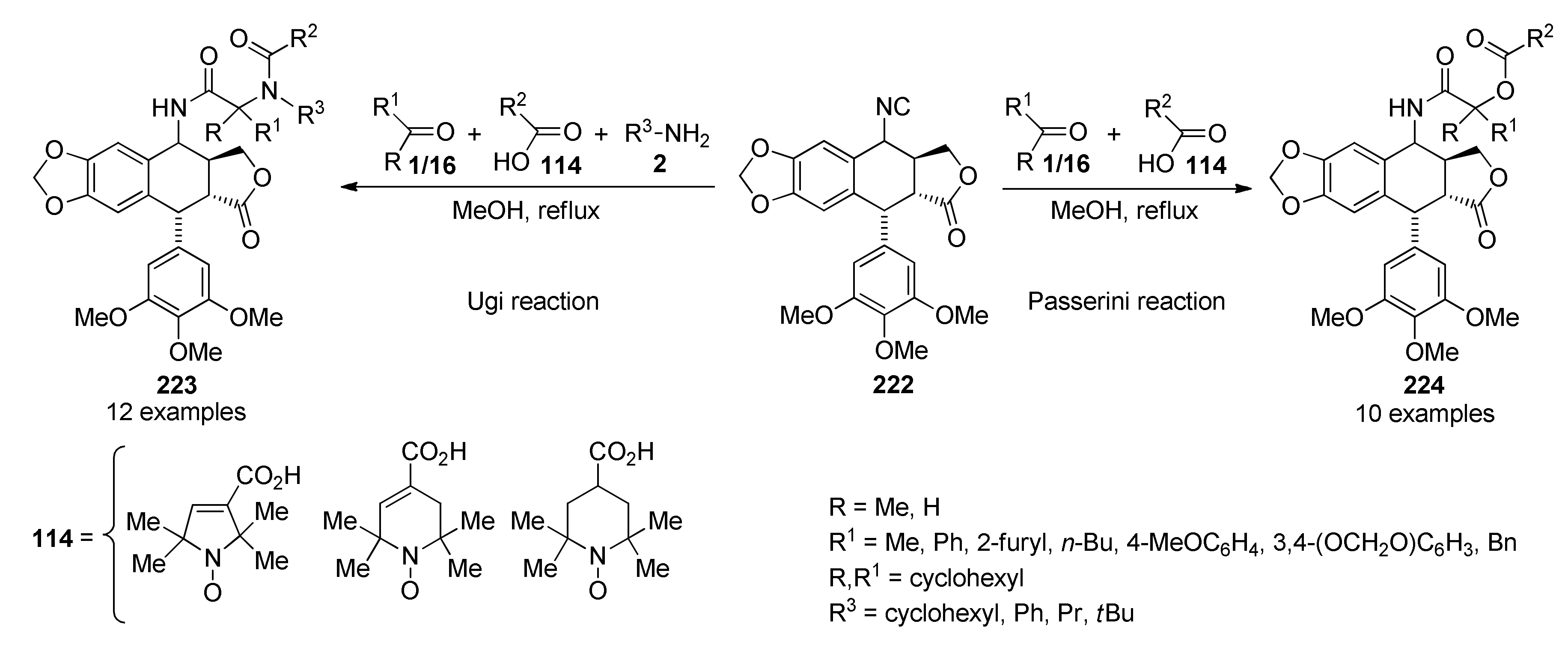{kind=link}
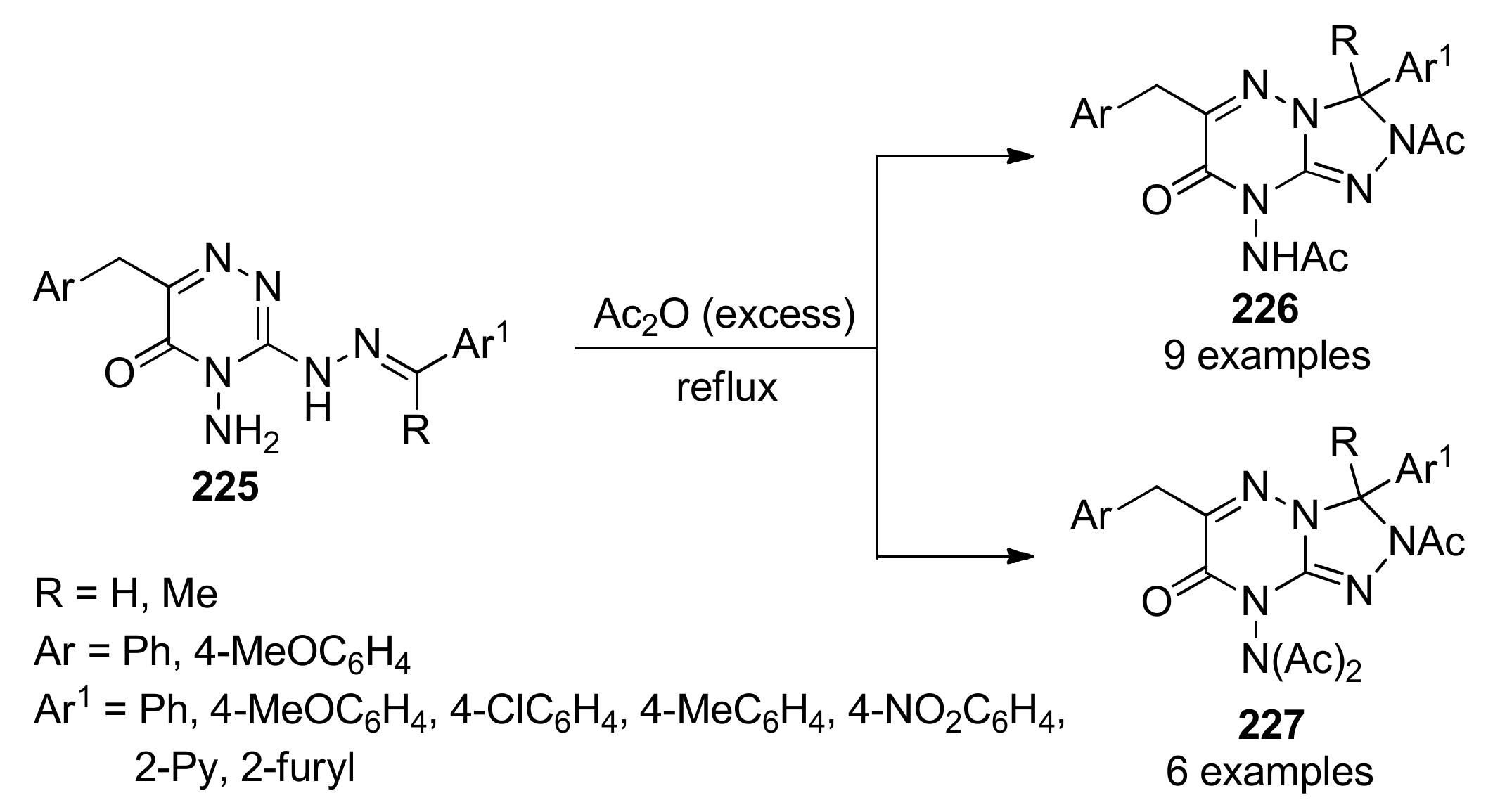{kind=link}
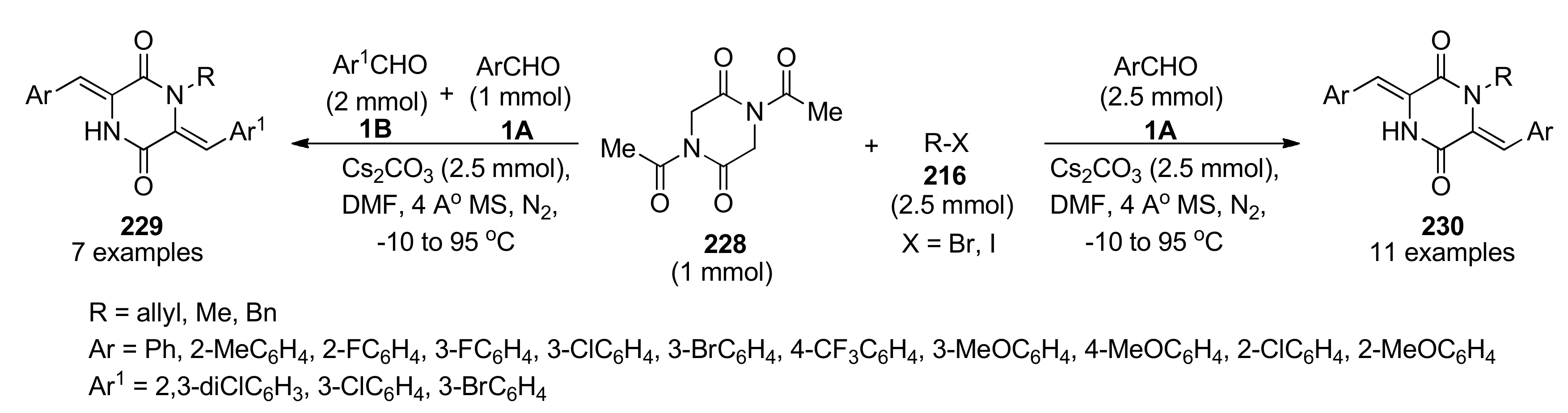{kind=link}
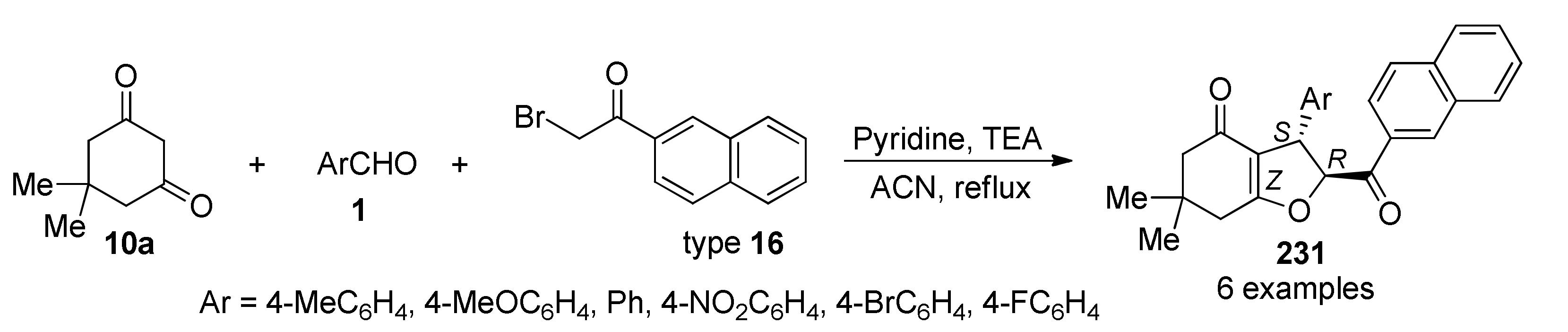{kind=link}
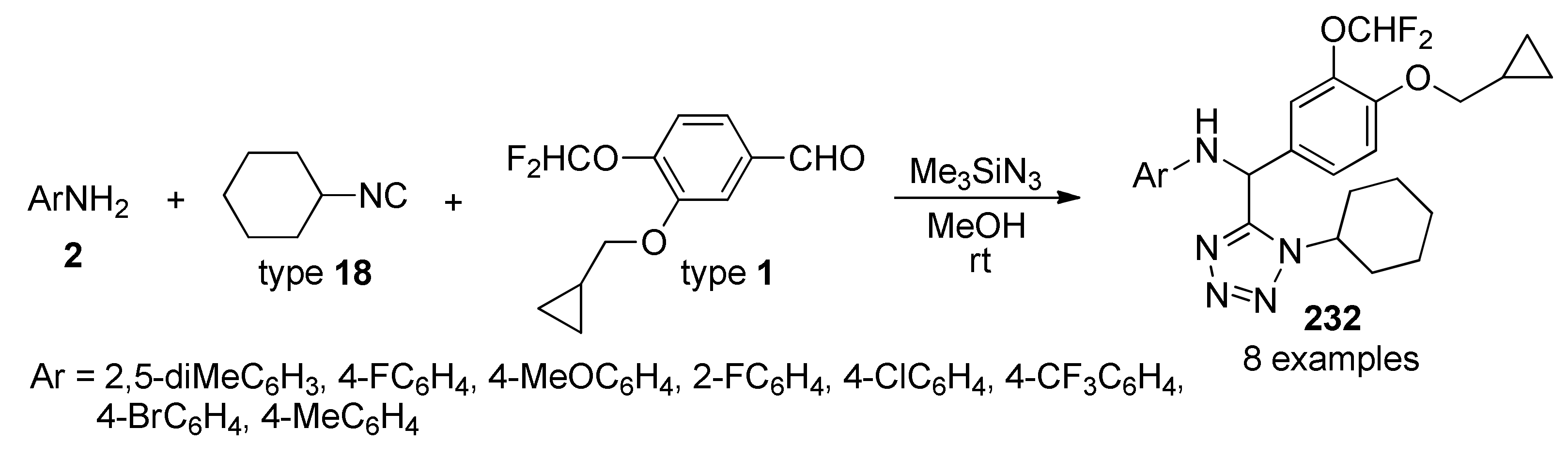{kind=link}
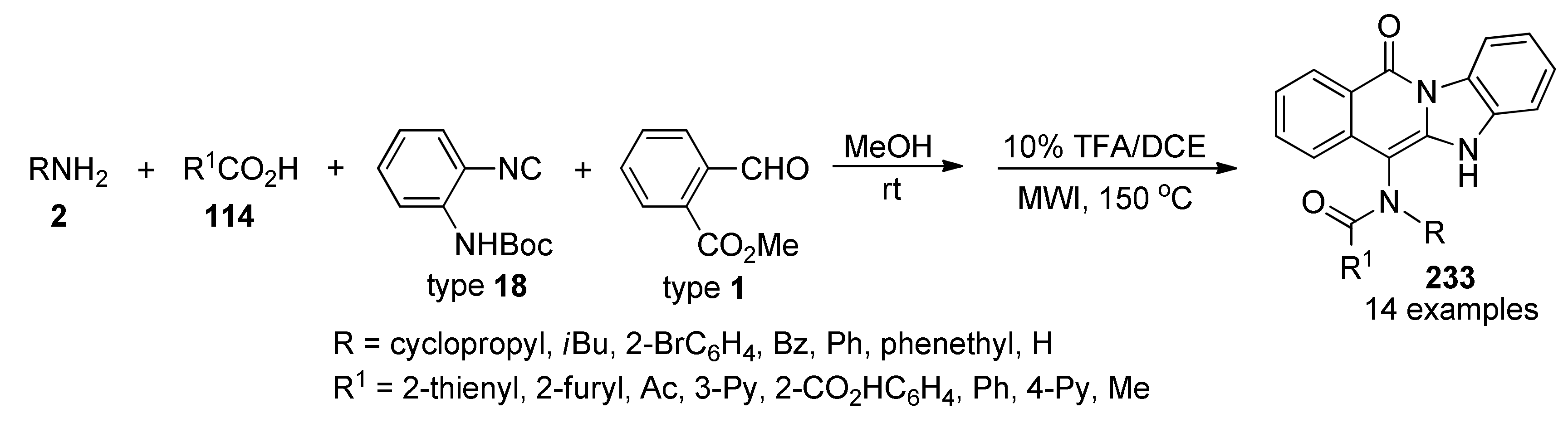{kind=link}
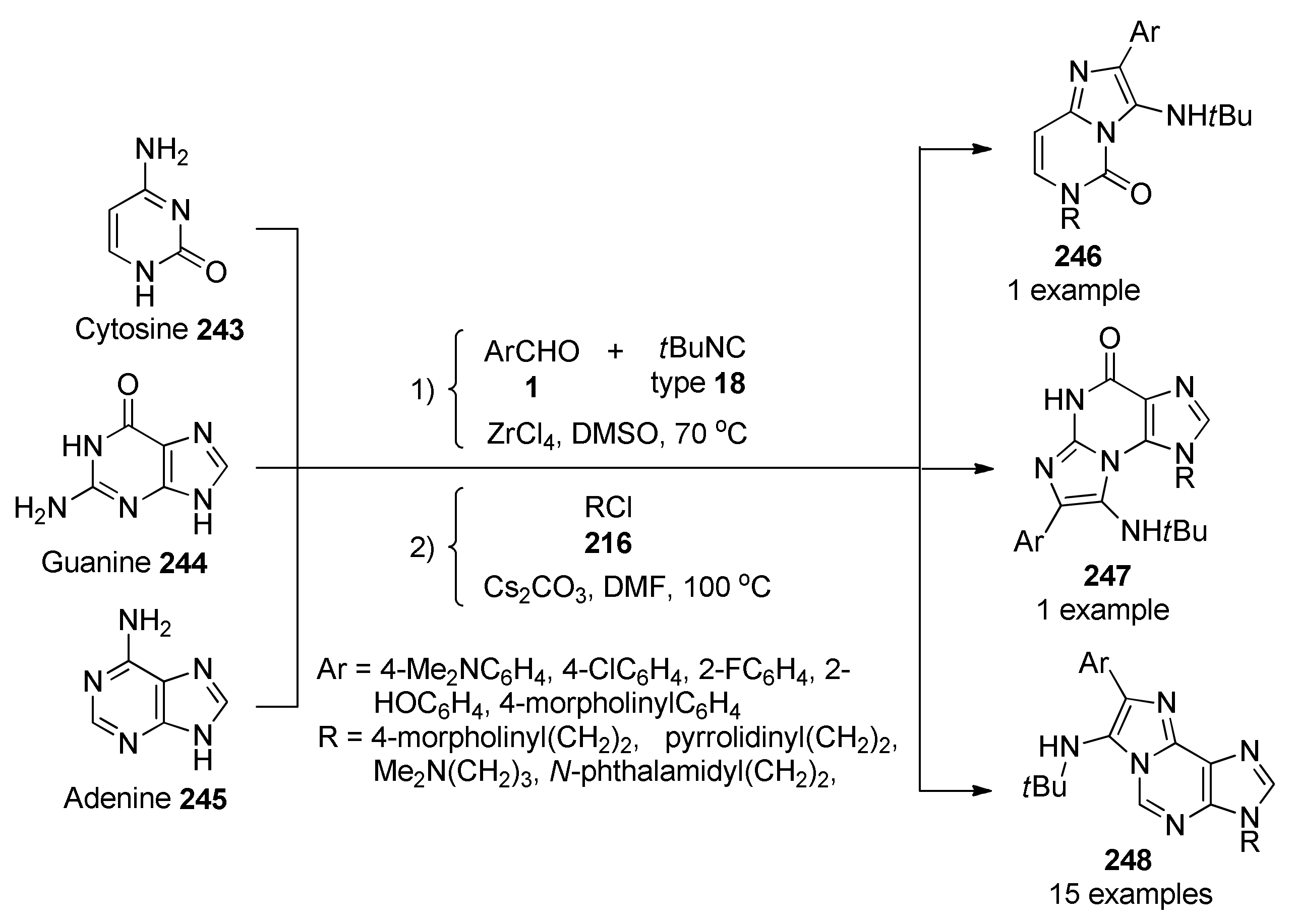{kind=link}
| Reagents | Conditions | Product | Reference | ||
|---|---|---|---|---|---|
 type 8 | RCHO type 1 |  6 | p-sulfonic acid calix[4]arene (0.5% mol), EtOH, reflux |  152a | [169] |
 type 27 |  type 1 | p-sulfonic acid, EtOH, reflux |  153b | [169] | |
 type 27 | ArCHO/R2CHO 1 | MAI·Fe2Cl7 (5 mol%), BMI·BF4 IL (1 mL), 80 °C |  154c | [170] | |
| Heterogeneous Zn- and Cd-based CPs catalysts (5 mol%), 70–100 °C, Continuous flow |  155d | [171] | |||
 type 27 |  type 6 | Bi(NO3)3.5H2O, 70 °C, solvent free |  156e | [172] | |
| Reagents | Conditions | Product | Reference | |||
|---|---|---|---|---|---|---|
| Ar1CHO 1 | ArCOMe 16 |  38 | AcONH4 | EtOH, reflux |  157a | [176] |
 12 |  158a | |||||
 8 |  8 | ArNH2 2 | PEG-400, 200 W, 80 °C |  159b | [177] | |
 27 |  27 | (NH4)2CO3 |  160b | |||
 47 |  12 | ArNH2 2 | Montmorillonite-K10, H2O/EtOH (2:1), 60 °C |  161c | [178] | |
| Reagents | Conditions | Product | Reference | ||
|---|---|---|---|---|---|
 27 | ArCHO 1 |  2 | AcOH, MWI, 120 °C |  162a | [145] |
 94 | RCHO 1 |  2 | L-Proline, EtOH, reflux |  163b | [181] |
 27 | ArCHO 1 |  2 | EtOH, reflux |  164c | [182] |
 10a | EtOH, MWI, 150 °C |  165d | [183] | ||
| Reagents | Conditions | Product | Reference | |||
|---|---|---|---|---|---|---|
| ArCHO 1 |  119 |  12 | HCONH2 | K2CO3, 90 °C → 150 °C |  166a | [187] |
 38 |  167b | |||||
 38 | HC(OEt)3 | EtOH, K2CO3, 90 °C → 150 °C |  168c | |||
 119 | HCO2H | EtOH, K2CO3, 90 °C → 100 °C |  169d | |||
| Reagents | Conditions | Product | Reference | |||
|---|---|---|---|---|---|---|
 12/38 X = CN, CO2Et, CONH2 |  16 | ArCHO 1 | TEA | EtOH, reflux |  234a | [231] |
 1 |  235b | |||||
| ArCHO 1 | AcONH4 |  236a | ||||
 1 |  237b | |||||
 119 | PhCHO 1 | CH2(CN)2 12 | ----- | 1,4-dioxane, TEA, reflux |  238 | |
| Reagents | Conditions | Product | Reference | ||
|---|---|---|---|---|---|
| S8 |  type 16 | PhNCS type 54 | EtOH, TEA, reflux |  239 | [231] |
 type 119 | 1,4-dioxane, TEA, reflux |  240 | |||
| CH2(CN)2 12 | 241 | ||||
 type 200 |  type 12/38 X = CN, CO2Et | EtOH, TEA, reflux |  242a | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Insuasty, D.; Castillo, J.; Becerra, D.; Rojas, H.; Abonia, R. Synthesis of Biologically Active Molecules through Multicomponent Reactions. Molecules 2020, 25, 505. https://doi.org/10.3390/molecules25030505
Insuasty D, Castillo J, Becerra D, Rojas H, Abonia R. Synthesis of Biologically Active Molecules through Multicomponent Reactions. Molecules. 2020; 25(3):505. https://doi.org/10.3390/molecules25030505
Chicago/Turabian StyleInsuasty, Daniel, Juan Castillo, Diana Becerra, Hugo Rojas, and Rodrigo Abonia. 2020. "Synthesis of Biologically Active Molecules through Multicomponent Reactions" Molecules 25, no. 3: 505. https://doi.org/10.3390/molecules25030505
APA StyleInsuasty, D., Castillo, J., Becerra, D., Rojas, H., & Abonia, R. (2020). Synthesis of Biologically Active Molecules through Multicomponent Reactions. Molecules, 25(3), 505. https://doi.org/10.3390/molecules25030505