3.3. Synthesis
The general procedure for the synthesis of 8-amino-10-phenyl-5-hydroxy-2-(4-hydroxy-phenyl)-4-oxo-3,4-dihydro-2H,10H-pyrano[2,3-f]chromene-9-carbonitrile derivatives 2a–2g is as follows.
Aryl aldehyde (2.3 mmol) was added to a stirred solution that contained Compound 1 (2.3 mmol) in 5 ml of ethanol and malononitrile or ethyl cyanoacetate (2.3 mmol) with a few drops of piperidine. The reaction mixture was stirred at reflux. After the completion of the reaction (monitored by TLC), the mixture was kept at room temperature, and the formed solid product was collected by filtration and washed with ethanol and hexane to yield 2a–2g.
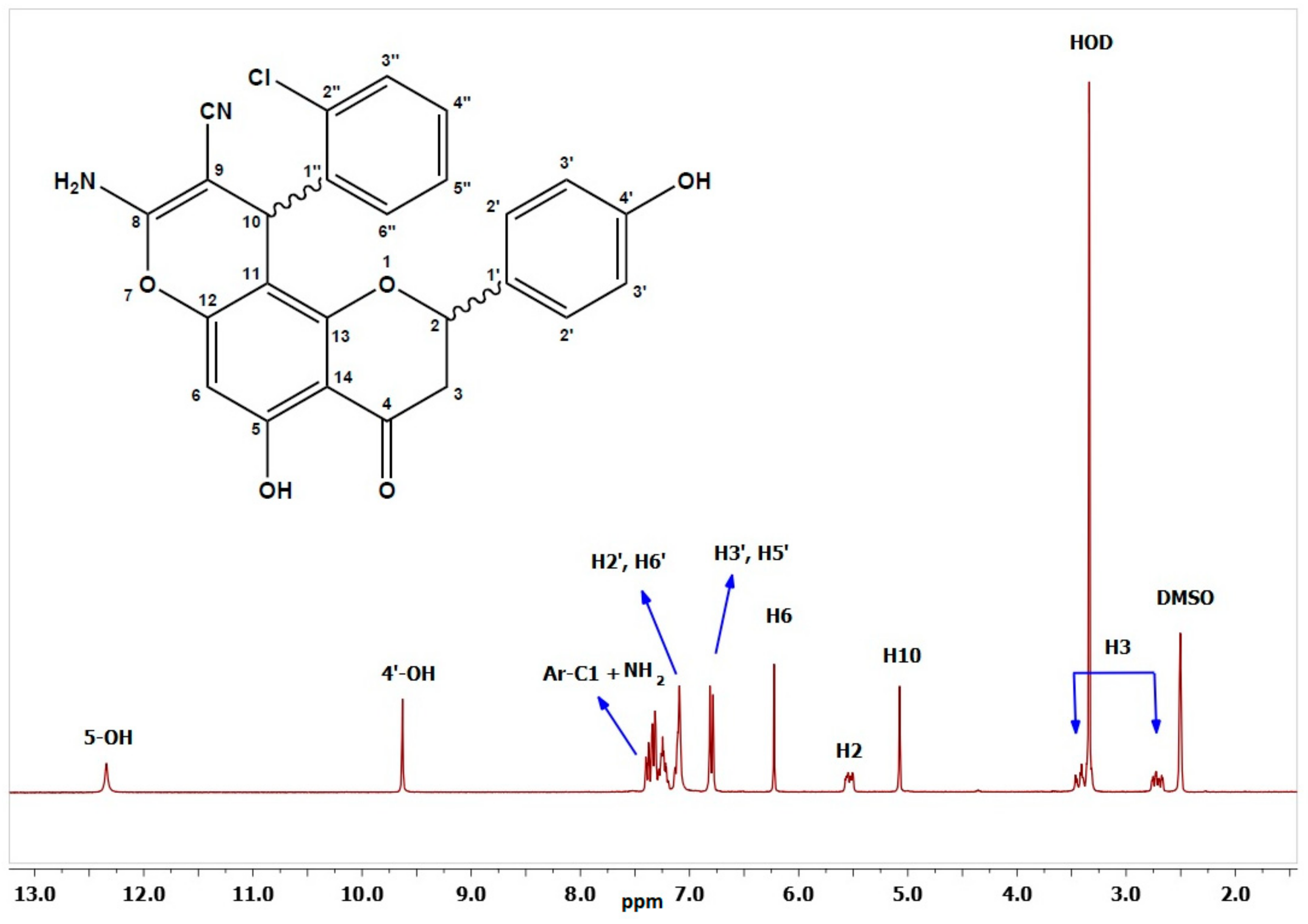
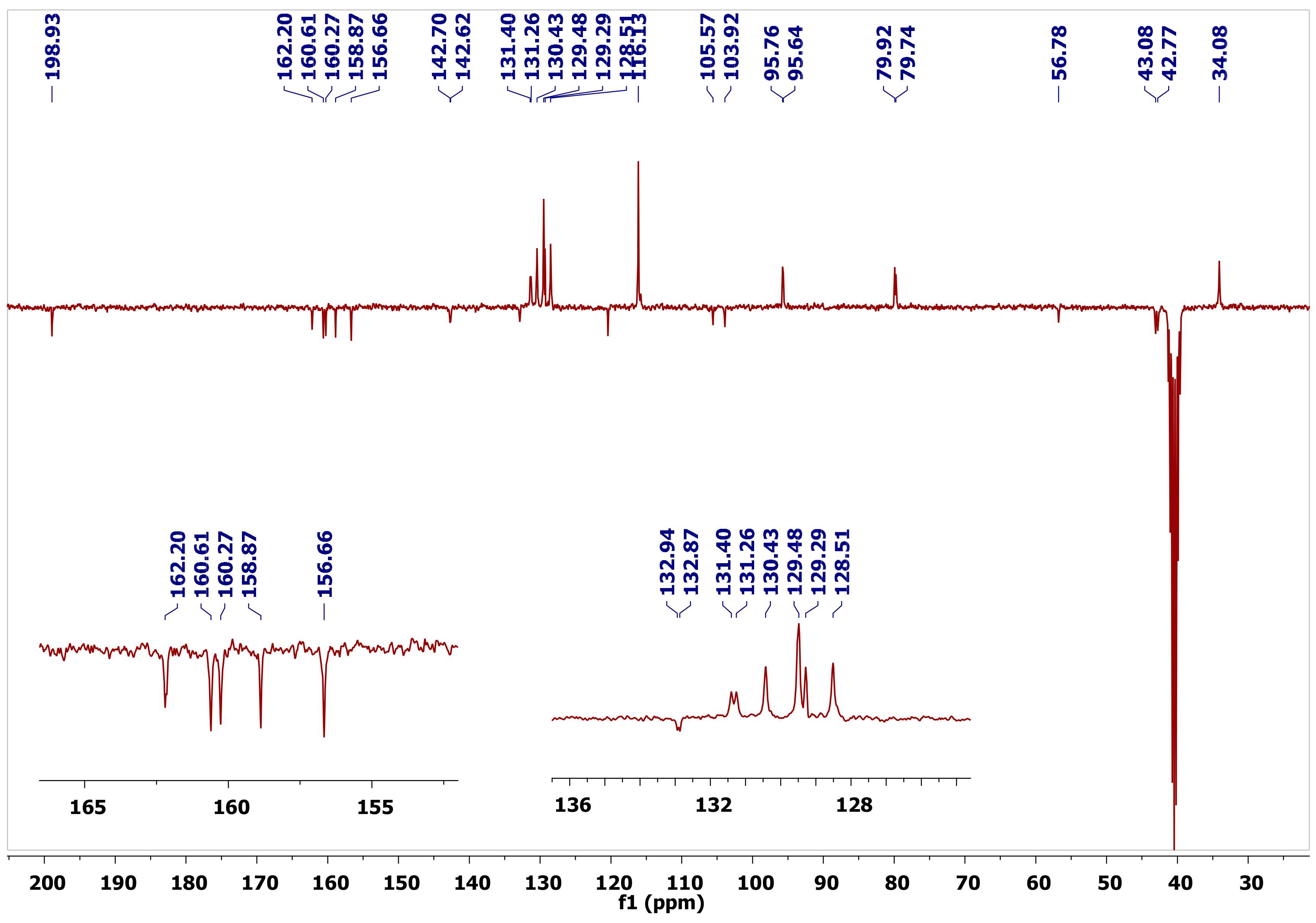
8-amino-10-(2-chlorophenyl)-5-hydroxy-2-(4-hydroxy-phenyl)-4-oxo-3,4-dihydro-2H,10H-pyrano[2,3-f] chromene-9-carbonitrile (2a). White solid (0.23 g, 27.45%), m.p. 262 °C; IR (KBr) cm−1: 3489 (OH), 3360 (NH2), 2178 (CN), and 1634 (C=O); 1H-NMR (DMSO, 300 MHz) δ 12.3 (s, 1H, C5–OH), 9.63 (s, 1H, C4′–OH), 7.39 (d, J = 7.5 Hz, 1H, C2″–H), 7.33 (d, J = 8.5 Hz, 2H, diastereomeric C2′–H), 7.32 (d, J = 8.5 Hz, 2H, diastereomeric C2′–H), 7.29–7.18 (m, 2H, C4″–H and C4″–H), 7.15–7.04 (m, 3H, overlapping Ar–C6″–H and NH2), 6.80 (d, J = 8.5 Hz, 2H, C3′–H), 6.23 (s, 1H, C6–H), 5.55 (dd, J = 12.8, 2.8 Hz, 1H, diastereomeric C2–H), 5.53 (dd, J = 12.8, 2.8 Hz, 1H, diastereomeric C2–H), 5.08 (s, 1H, C10–H), 3.40 (dd, J = 17.1, 13.0 Hz, 1H, diastereomeric trans C3–H), 3.41 (dd, J = 17.1, 13.0 Hz, 1H, diastereomeric trans C3–H), 2.73 (dd, J = 17.1, 2.7 Hz, 1H, diastereomeric cis C3–H), and 2.70 (dd, J = 17.1, 2.7 Hz, 1H, diastereomeric cis C3–H); 13C-NMR (DMSO, 75 MHz) δ 198.9 (C=O), 162.1 (C5–O), 160.5 (C13–O), 160.2 (C8–O), 158.8 (C4′–O), 156.6 (C12–O), 142.6 (diastereomeric C1″), 142.5 (diastereomeric C1″), 132.9 (C–Cl), 132.8 (C1′), 131.3 (diastereomeric CH), 131.2 (diastereomeric CH), 130.4 (C3″–H), 129.4 (C2′–H), 129.2 (CH), 128.4 (CH), 120.3 (CN), 116.1 (C3′–H), 105.9 (C14), 103.8 (C11), 95.7 (diastereomeric C6–H), 95.6 (diastereomeric C6–H), 79.8 (diastereomeric C2–H), 79.7 (diastereomeric C2–H), 56.7 (diastereomeric C9), 56.6 (diastereomeric C9), 43.0 (diastereomeric C3), 42.7 (diastereomeric C3), and 34.0 (C10–H); Anal. Calcd for C25H17N2O5Cl: C, 65.15; H, 3.72; N, 6.08. Found: C, 65.34; H, 3.63; N, 6.02.
8-amino-10-(2-fluorophenyl)-5-hydroxy-2-(4-hydroxy-phenyl)-4-oxo-3,4-dihydro-2H,10H-pyrano[2,3-f] chromene-9-carbonitrile (2b). Yellow solid (0.42 g, 57.76% ), m.p. 290 °C; IR (KBr) cm−1: 3497 (OH), 3403, 3192 (NH2), and 2198 (CN), 1647 (C=O); 1H-NMR (DMSO, 300 MHz) δ 12.4 (s, 1H, C5–OH), 9.63 (s, 1H, C4′–OH), 7.32 (d, J = 8.5 Hz, 1H, diastereomeric C2′–H), 7.31 (d, J = 8.5 Hz, 2H, diastereomeric C2′–H), 7.29–7.20 (m, 1H, C4″–H), 7.20–7.11 (m, 3H, C3″–H, C5″–H and C6″–H), 7.09 (s, NH2), 6.80 (d, J = 8.5 Hz, 2H, C3′–H), 6.22 (s, 1H, C6–H), 5.55 (dd, J = 12.2, 2.7 Hz, 1H, diastereomeric C2–H), 5.52 (dd, J = 12.6, 2.5 Hz, 1H, diastereomeric C2–H), 4.82 (s, 1H, C10–H), 3.51–3.37 (m, 1H, diastereomeric trans C3–H), 2.73 (dd, J = 17.1, 2.7 Hz, 1H, diastereomeric cis C3–H), and 2.70 (dd, J = 17.1, 2.7 Hz, 1H, diastereomeric cis C3–H); 13C-NMR (DMSO, 75 MHz) δ 198.0 (C=O), 162.6 (dd, J = 230 Hz, C–F), 162.2 (C5–O), 159.5 (C13–O), 159.3 (C8–O), 157.9 (C4′–O), 155.7 (C12–O), 129.9 (C1″), 128.5 (C2′–H), 128.3 (C1′), 128.2 (C4″–H), 124.5 (C5″–H), 119.7 (CN), 115.5 (d, J = 21.0 Hz, diastereomeric C3″), 115.2 (C3′–H), 115.1 (d, J = 21.0 Hz, diastereomeric C3″), 107.9 (diastereomeric C14), 104.7 (diastereomeric C14), 102.8 (diastereomeric C11), 100.1 (diastereomeric C11), 95.8 (minor atropic isomer C6–H), 95.6 (minor atropic isomer C6–H), 94.8 (diastereomeric C6–H), 94.8 (diastereomeric C6–H), 79.0 (diastereomeric C2–H), 78.8 (diastereomeric C2–H), 78.6 (minor atropic isomer C2–H), 56.0 (diastereomeric C9), 55.7 (diastereomeric C9), 42.2 (diastereomeric C3), 41.8 (diastereomeric C3), 30.3 (diastereomeric C10), and 30.3 (diastereomeric C10); Anal. Calcd for C25H17N2O5F: C, 67.57; H, 3.86; N, 6.30. Found: C, 66.85; H, 3.98; N, 6.09.
8-amino-10-(3-bromophenyl)-5-hydroxy-2-(4-hydroxy-phenyl)-4-oxo-3,4-dihydro-2H,10H-pyrano[2,3-f] chromene-9-carbonitrile (2c). White solid (0.46 g, 50.07%), m.p. 182 °C; IR (KBr) cm−1: 3634(OH), 3424 (NH2), 2207 (CN), and 1653 (C=O); 1H-NMR (DMSO, 300 MHz) δ 12.4 (s, 1H, C5–OH), 9.63 (s, 1H, C4′–OH), 7.45–7.37 (m, 2H, H2′ and H6′), 7.36–7.23 (m, 3H, H2″, H4″ and H5″), 7.21–7.10 (m, 3H, H6″ and NH2), 6.80 (d, J = 8.5 Hz, 2H, H3′ and H5′), 6.24 (s, 1H, H6), 5.56 (dd, J = 12.5, 2.8 Hz, 1H, diastereomeric H2), 5.53 (dd, J = 13.1, 2.8 Hz, 1H, diastereomeric H2), 4.62 (s, 1H, diastereomeric H10), 4.61 (s, 1H, diastereomeric H10), 3.42 (dd, J = 17.2, 13.5 Hz, 1H, diastereomeric trans H3ax), 3.41 (dd, J = 17.6, 13.3 Hz, 1H, diastereomeric trans H3ax), 2.75 (dd, J = 17.4, 3.0 Hz, 1H, diastereomeric cis H3eq), and 2.71 (dd, J = 17.1, 2.7 Hz, 1H, diastereomeric cis H3eq); 13C-NMR (DMSO, 75 MHz) δ 198.9 (C=O), 162.9 (C-5), 160.4 (C-13), 160.3 (C-8), 158.8 (C-4′), 156.2 (C-12), 148.5 (C-1′’), 131.7 (C-2′’), 130.6 (C-5′’), 129.4 (C-2′ and C-6′), 129.2 (C-1′), 128.5 (C-4′’), 127.2 (C-6′’), 122.5 (C-3′’), 120.7 (CN), 116.1 (C-3′ and C-5′), 105.7 (C-14), 104.3 (C-11), 96.0 (C6–H), 79.8 (C-2), 57.7 (C-9), 43.0 (diastereomeric C-3), and 42.9 (diastereomeric C-3), 36.2 (C-10); Anal. Calcd for C25H17N2O5Br: C, 59.42; H, 3.39; N, 5.54. Found: C, 59.82; H, 3.28; N, 5.31.
8-amino-10-(4-tert-butylphenyl)-5-hydroxy-2-(4-hydroxy-phenyl)-4-oxo-3,4-dihyd-ro-2H,10H-pyrano[2,3-f] chromene-9-carbonitrile (2d). Yellowish white solid (0.20 g, 22.80%), m.p. 264 °C; IR (KBr) cm−1: 3488 (OH), 3381 (NH2), 2961– 2869 (CH), 2185 (CN), and 1643 (C=O); Diastereomeric ratio: 1:1; atropic isomeric ratio 1:4.3 due to locked conformation and hindered rotation. Major atropic isomer: 1H-NMR (DMSO, 300 MHz) δ 11.9 (s, 1H, C5–OH), 9.62 (s, 1H, C4′–OH), 7.30 (d, J = 8.3 Hz, 2H, C3″–H), 7.03 (br s, 2H, NH2), 6.94 (d, J = 8.2 Hz, 2H, C2″–H), 6.91 (d, J = 8.0 Hz, 2H, C2′–H), 6.74 (d, J = 8.1 Hz, 2H, C3′–H), 6.21 (s, 1H, C6–H), 5.07 (dd, J = 12.8, 2.5 Hz, 1H, C2–H), 4.46 (s, 1H, C10–H), 3.34 (dd, J = 17.1, 13.2 Hz, 1H, trans C3–H), and 2.75 (dd, J = 17.1, 2.7 Hz, 1H, cis C3–H); minor atropic isomer: 1H–NMR δ 9.56 (s, 1H, C4′–OH), 6.98 (d, J = 8.4 Hz, 2H, C3′–H), 6.20 (s, 1H, C6–H), 5.61 (dd, J = 12.0, 3.1 Hz, 1H, C2–H), 4.49 (s, 1H, C10–H), 3.08 (dd, J = 17.3, 12.1 Hz, 1H, trans C3–H), and 2.83 (dd, J = 17.1, 3.3 Hz, 1H, cis C3–H); major atropic isomer: 13C-NMR (DMSO, 75 MHz) δ 198.7 (C=O), 161.9 (C5–O), 160.2 (C13–O), 160.0 (C8–O), 158.7 (C4′–O), 156.3 (C12–O), 149.7 (C4″), 142.9 (C1″), 129.4 (C2′–H), 127.7 (C1′), 127.4 (C2″–H), 126.0 (C3″–H), 120.9 (CN), 115.9 (C3′–H), 106.2 (C14), 104.4 (C11), 96.8 (C6–H), 79.9 (C2–H), 58.0 (C9), 41.7 (C3–H2), 36.2 (C10–H), and 35.0 (t-Bu quaternary carbon), 32.0 (CH3); minor atropic isomer: 13C-NMR δ 198.1 (C=O), 161.7 (C5–O), 159.7 (C8–O), 158.2 (C4′–O), 142.8 (C1″), 108.8 (C14), 104.6 (C11), 96.5 (C6–H), 79.4 (C2–H), 58.4 (C9), 42.9 (C3–H2), and 36.5 (C10–H); Anal. Calcd for C29H26N2O5: C, 72.18; H, 5.43; N, 5.81. Found: C, 71.99; H, 5.20; N, 5.73.
8-amino-10-(thiophene)-5-hydroxy-2-(4-hydroxy-phenyl)-4-oxo-3,4-dihydro-2H-10H-pyrano[2,3-f] chromene-9-carbonitrile (2e). White solid (0.23 g, 29.25 %), m.p. 293 °C; IR (KBr) cm−1: 3501 (OH), 3402, 3205 (NH2), 2206 (CN), and 1653 (C=O); 1H-NMR (DMSO, 300 MHz) δ 12.5 (s, 1H, C5–OH), 9.63 (s, 1H, C4′–OH), 7.46–7.37 (m, 1H, H4″), 7.32 (d, J = 8.3 Hz, 2H, H2′ and H6′), 7.20–7.14 (m, 1H, H2″), 7.11 (br s, 2H, NH2), 6.92–6.85 (m, 1H, H5″), 6.81 (d, J = 8.3 Hz, 2H, H3′ and H5′), 6.21 (s, 1H, H6), 5.59–5.43 (m, 1H, H2), 4.73 (s, 1H, diastereomeric H10), 4.71 (s, 1H, diastereomeric H10), 3.40 (dd, J = 17.3, 13.2 Hz, 1H, trans H3ax), and 2.83–2.68 (m, 1H, cis H3eq); 13C-NMR (DMSO, 75 MHz) δ 198.9 (C=O), 161.9 (C-8), 160.7 (C-13), 160.2 (C-5), 158.8 (C-4′), 156.3 (C-12), 146.2 (C-1″), 129.4 (C-2′ and C-6′), 129.1 (C-1′), 127.5 (diastereomeric C-5″), 127.4 (diastereomeric C-5″), 127.3 (C-4″), 121.8 (diastereomeric C-2″), 121.7 (diastereomeric C-2″), 121.0 (CN), 116.1 (C-3′ and C-5′), 106.7 (diastereomeric C-14), 106.6 (diastereomeric C-14), 105.3 (diastereomeric C-11), 105.2 (diastereomeric C-11), 96.0 (diastereomeric C-6), 95.9 (diastereomeric C-6), 79.8 (diastereomeric C-2), 79.7 (diastereomeric C-2), 57.6 (diastereomeric C-9), 57.5 (diastereomeric C-9), 43.1 (diastereomeric C-3), 42.8 (diastereomeric C-3), and 31.5 (C-10); Anal. Calcd for C23H16N2O5S: C, 63.88; H, 3.73; N, 6.48. Found: C, 63.83; H, 3.60; N, 6.16.
8-amino-10-(4-fluorophenyl)-5-hydroxy-2-(4-hydroxy-phenyl)-4-oxo-3,4-dihydro-2H,10H-pyrano[2,3-f]chromene-9-carbonitrile (2f). White solid (0.26 g, 27.31%), m.p. 230 °C; IR (KBr) cm−1: 3477 (OH), 3411, 3294 (NH2), 2914–2880 (CH), and 1686 (C=O); 1528, 1349 (NO); 1H-NMR (DMSO, 300 MHz) δ 12.4 (s, 1H, C5–OH), 9.63 (s, 1H, C4′–OH), 7.32 (d, J = 8.5 Hz, 1H, diastereomeric H2′ and H6′), 7.31 (d, J = 8.5 Hz, 2H, diastereomeric H2′ and H6′), 7.23–7.11 (m, 4H, H2″, H6″, H5″ and H3″), 7.10 (s, NH2), 6.80 (d, J = 8.5 Hz, 2H, H3′ and H5′), 6.23 (s, 1H, H6), 5.54 (dd, J = 12.8, 3.0 Hz, 1H, diastereomeric H2), 5.53 (dd, J = 13.1, 3.0 Hz, 1H, diastereomeric H2), 4.61 (s, 1H, diastereomeric H10), 4.59 (s, 1H, diastereomeric H10), 3.41 (dd, J = 17.4, 13.2 Hz, 1H, diastereomeric trans H3ax), 2.74 (dd, J = 17.1, 2.9 Hz, 1H, diastereomeric cis H3eq), and 2.71 (dd, J = 17.3, 2.9 Hz, 1H, diastereomeric cis H3eq); 13C-NMR (75 MHz, DMSO-d6) δ 198.82 (C=O), 163.37 (C-4″), 161.93 (C-5), 159.92 (C-13), 159.36 (C-8), 157.56 (C-4′), 154.95 (C-12), 129.91 (C-1″), 131.39.81 (C-2′ and C-6′), 130.07 (C-1′), 129.72 ( C-2″ and 6″), 128.47 (C-3″ and C-5″), 120.74 (CN), 115.99 (diastereomeric C-3′ and C-5′),116.85 (diastereomeric C-3′ and C-5′) 105.64 (C-14), 104.97 (C-11), 95.88 (diastereomeric C-6), 95.76 (diastereomeric C-6), 79.78 (diastereomeric C-2), 79.67 (diastereomeric C-2), 53.58 (C-9), and 42.75 (C-3), 36.09 (C-10); Anal. Calcd for C25H17N2O5F: C, 67.57; H, 3.86; N, 6.30. Found: C, 66.85; H, 3.98; N, 6.0.
Ethyl-8-amino-10-(3-nitrophenyl)-5-hydroxy-2-(4-hydroxy-phenyl)-4-oxo-3,4-dih-ydro-2H,10H-pyrano[2,3-f] chromene-9-carboxlate (2g). White solid (0.26 g, 27.31%), m.p. 230 °C; IR (KBr) cm−1: 3477 (OH), 3411, 3294 (NH2), 2914–2880 (CH), and 1686 (C=O); 1528, 1349 (NO); 1H-NMR (DMSO, 300 MHz) δ 11.9 (s, 1H, C5–OH), 9.71 (s, 1H, minor atropic isomer C4′–OH), 9.70 (s, 1H, diastereomeric C4′–OH), 9.64 (s, 1H, minor atropic isomer C4′–OH), 9.62 (s, 1H, diastereomeric C4′–OH), 8.08–7.98 (app t (two overlapped diastereomeric C4″–H doublets), J = 7.3 Hz, 1H, C4″–H), 7.88 (s, 1H, diastereomeric C2″–H), 7.79 (br s, 2H, NH2 and diastereomeric C2″–H), 7.62–7.35 (m, 2H, C5″–H and C6″–H), 7.17 (d, J = 9.2 Hz, 2H, minor atropic isomer C2′–H), 7.11 (d, J = 8.7 Hz, 2H, diastereomeric C2′–H), 7.06 (d, J = 8.7 Hz, 2H, diastereomeric C2′–H), 6.79 (d, J = 8.4 Hz, 2H, diastereomeric C3′–H), 6.72 (d, J = 8.6 Hz, 2H, diastereomeric C3′–H), 6.27 (s, 1H, diastereomeric C6–H), 6.26 (s, 1H, diastereomeric C6–H), 5.64 (dd, J = 12.5, 2.9 Hz, 1H, diastereomeric C2–H), 5.07 (dd, J = 13.6, 2.5 Hz, 1H, diastereomeric C2–H), 5.14 (dd, J = 13.6, 2.7 Hz, 1H, minor atropic isomer C2–H), 4.91 (s, 1H, minor atropic isomer C10–H), 4.90 (s, 1H, diastereomeric C10–H), 4.85 (s, 1H, minor atropic isomer C10–H), 4.83 (s, 1H, diastereomeric C10–H), 4.03–3.81 (m, 2H, OCH2CH3) 3.45 (dd, J = 17.2, 13.4 Hz, 1H, diastereomeric trans C3–H), 3.11 (dd, J = 17.4, 12.9 Hz, 1H, diastereomeric trans C3–H), 2.72 (dd, J = 17.6, 3.1 Hz, 1H, diastereomeric cis C3–H), and 2.66 (dd, J = 17.2, 2.7 Hz, 1H, diastereomeric cis C3–H); 13C-NMR (DMSO, 75 MHz) δ 198.7 (diastereomeric C=O), 198.3 (diastereomeric C=O), 168.7 (Minor atropic isomer ester C=O), 168.7 (Ester C=O), 162.1 (diastereomeric C5–O), 162.0 (diastereomeric C5–O),160.8 (diastereomeric C13–O), 160.7 (diastereomeric C13–O), 160.0 (diastereomeric C8–O), 159.9 (diastereomeric C8–O), 158.9 (diastereomeric C4′–O), 158.5 (diastereomeric C4′–O), 156.1 (C12–O), 149.6 (diastereomeric C–NO2), 149.6 (diastereomeric C–NO2), 148.3 (Minor atropic isomer C1″), 148.0 (C1″), 135.2 (diastereomeric C6″–H), 135.0 (diastereomeric C6″–H), 130.5 (diastereomeric C5″), 130.4 (diastereomeric C5″), 129.5 (diastereomeric C2′–H), 128.7 (Minor atropic isomer C2′–H), 128.5 (diastereomeric C2′–H), 128.4 (C1′), 123.4 (diastereomeric C4″), 123.3 (diastereomeric C4″), 122.9 (Minor atropic isomer C4″), 122.7 (Minor atropic isomer C4″), 122.0 (diastereomeric C2″), 121.9 (diastereomeric C2″), 116.9 (Minor atropic isomer C3′–H), 116.0 (diastereomeric C3′–H), 115.9 (diastereomeric C3′–H), 106.6 (diastereomeric C14), 106.7 (diastereomeric C14), 106.6 (Minor atropic isomer C14), 104.3 (C11), 96.7 (diastereomeric C6–H), 96.6 (diastereomeric C6–H), 80.2 (diastereomeric C2–H), 80.0 (diastereomeric C2–H), 76.8 (diastereomeric C9), 76.6 (diastereomeric C9), 59.8 (OCH2CH3), 43.4 (diastereomeric C3–H2), 43.0 (diastereomeric C3–H2), 35.3 (diastereomeric C10–H), 35.3 (diastereomeric C10–H), and 15.0 (OCH2CH3); Anal. Calcd for C27H22N2O9: C, 62.55; H, 4.28; N, 5.40. Found: C, 61.99; H, 3.79; N, 5.32.


 ,
, 


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}