Amyloid-Beta1-42 -Induced Increase in GABAergic Tonic Conductance in Mouse Hippocampal CA1 Pyramidal Cells


 and
and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
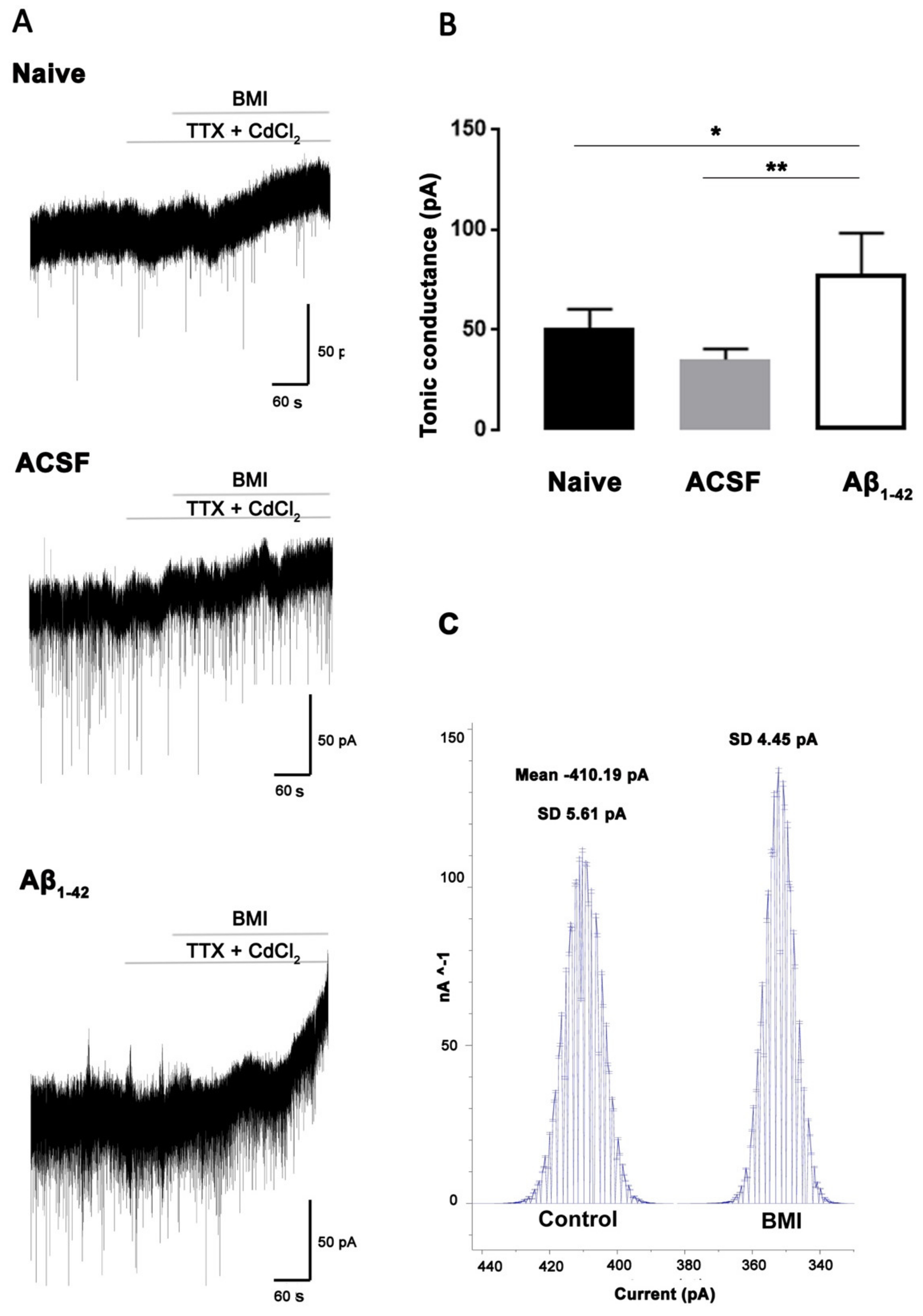
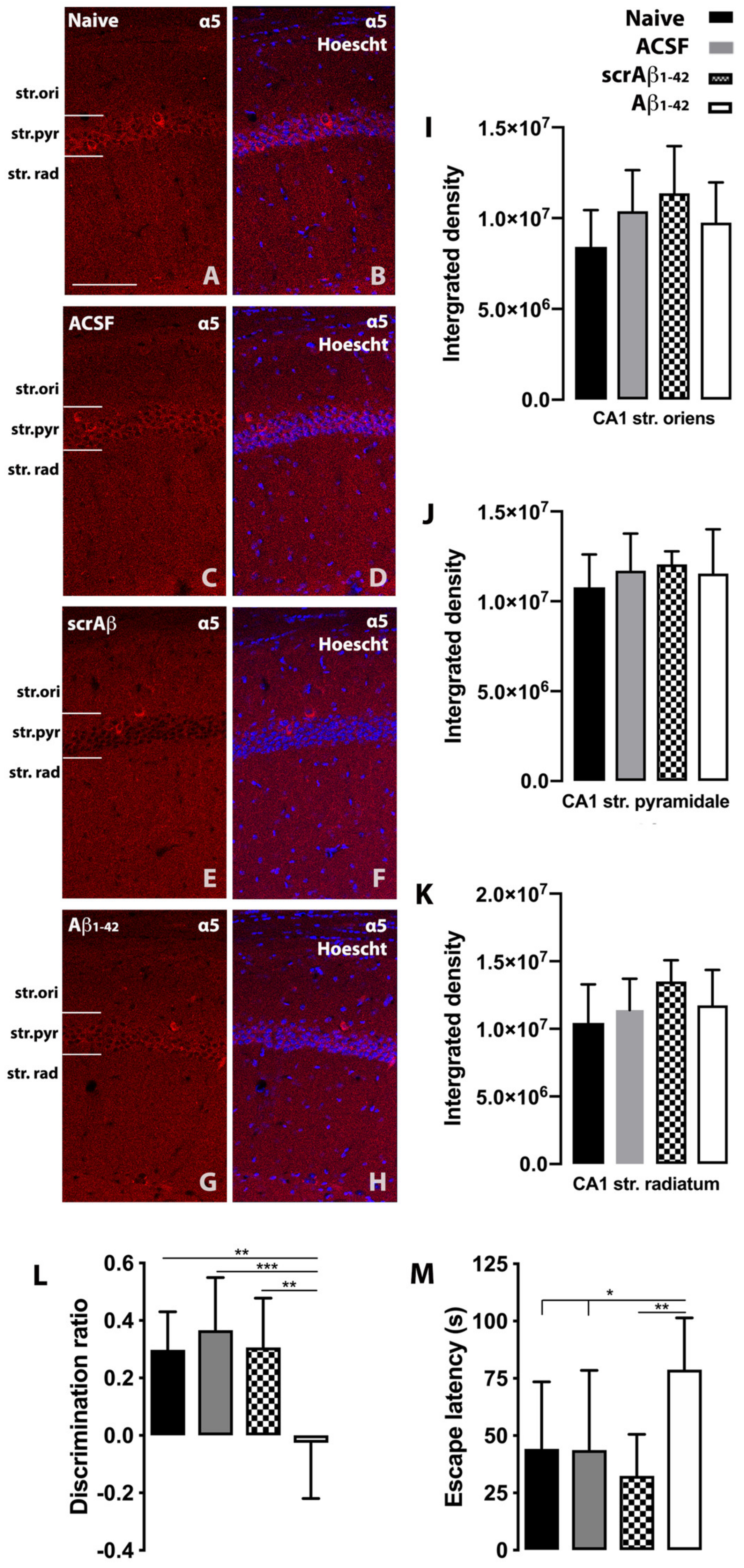
2. Results and Discussion
3. Materials and Methods
3.1. Animals
3.2. Aβ1-42 Preparation
3.3. Aβ1-42 Stereotaxic Injection
3.4. Whole-Cell Voltage-Clamp Recording
3.4.1. Slice Preparation
3.4.2. Whole-Cell Patch-Clamp Recording
3.4.3. Tonic Current Measurements in Hippocampal Slices
3.5. Behavioral Testing
3.5.1. Novel Object Recognition Test
3.5.2. Morris Water Maze Test
3.6. Tissue Processing and Immunohistochemistry
3.7. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pakaski, M.; Farkas, Z.; Kasa, P.; Forgon, M.; Papp, H.; Zarandi, M.; Penke, B.; Kasa, P. Vulnerability of small GABAergic neurons to human beta-amyloid pentapeptide. Brain Res. 1998, 796, 239–246. [Google Scholar] [CrossRef]
- Gao, R.; Penzes, P. Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr. Mol. Med. 2015, 15, 146–167. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.-Q.; Yao, W.; Yan, J.-Z.; Jin, C.; Yin, J.-J.; Yuan, J.; Yu, S.; Cheng, Z. Amyloid β causes excitation/inhibition imbalance through dopamine receptor 1-dependent disruption of fast-spiking GABAergic input in anterior cingulate cortex. Sci. Rep. 2018, 8, 302. [Google Scholar] [CrossRef]
- Li, Y.; Sun, H.; Chen, Z.; Xu, H.; Bu, G.; Zheng, H. Implications of GABAergic Neurotransmission in Alzheimer’s Disease. Front Aging Neurosci. 2016, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madl, J.E.; Royer, S.M. Glutamate dependence of GABA levels in neurons of hypoxic and hypoglycemic rat hippocampal slices. Neuroscience 2000, 96, 657–664. [Google Scholar] [CrossRef]
- Wu, Z.; Guo, Z.; Gearing, M.; Chen, G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s disease model. Nat. Commun. 2014, 5, 4159. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef]
- Crestani, F.; Keist, R.; Fritschy, J.M.; Benke, D.; Vogt, K.; Prut, L.; Bluthmann, H.; Mohler, H.; Rudolph, U. Trace fear conditioning involves hippocampal alpha5 GABA(A) receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 8980–8985. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.; Oh, G.; Orser, B. Etomidate targets alpha5 gamma-aminobutyric acid subtype A receptors to regulate synaptic plasticity and memory blockade. Anesthesiology 2009, 111, 1025–1035. [Google Scholar] [CrossRef] [Green Version]
- Limon, A.; Reyes-Ruiz, J.M.; Miledi, R. GABAergic drugs and Alzheimer’s disease. Future Med. Chem. 2011, 3, 149–153. [Google Scholar] [CrossRef]
- Louzada, P.; Lima, A.; Mendonca-Silva, D.; Noël, F.; de Mello, F.; Ferreira, S. Taurine prevents the neurotoxicity of β-amyloid and glutamate receptor agonists: Activation of GABA receptors and possible implications for Alzheimer’s disease and other neurological disorders. Faseb. J. 2004, 18, 511–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, P.E.; Zhu, L.; Verret, L.; Vossel, K.A.; Orr, A.G.; Cirrito, J.R.; Devidze, N.; Ho, K.; Yu, G.-Q.; Palop, J.J. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. P. Natl. Acad. Sci. 2012, 109, E2895–E2903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcade, M.; Bourdin, J.; Loiseau, N.; Peillon, H.; Rayer, A.; Drouin, D.; Schweighoffer, F.; Désiré, L. Etazolate, a neuroprotective drug linking GABAA receptor pharmacology to amyloid precursor protein processing. J. Neurochem. 2008, 106, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Vellas, B.; Sol, O.; Snyder, P.J.; Ousset, P.J.; Haddad, R.; Maurin, M.; Lemarie, J.C.; Desire, L.; Pando, M.P. EHT0202 in Alzheimer’s disease: A 3-month, randomized, placebo-controlled, double-blind study. Curr. Alzheimer Res. 2011, 8, 203–212. [Google Scholar] [CrossRef]
- Calvo-Flores Guzman, B.; Vinnakota, C.; Govindpani, K.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The GABAergic system as a therapeutic target for Alzheimer’s disease. J. Neurochem. 2018, 146, 649–669. [Google Scholar] [CrossRef] [Green Version]
- Glykys, J.; Mody, I. Hippocampal network hyperactivity after selective reduction of tonic inhibition in GABA A receptor alpha5 subunit-deficient mice. J. Neurophysiol. 2006, 95, 2796–2807. [Google Scholar] [CrossRef] [Green Version]
- Marczynski, T.J. GABAergic deafferentation hypothesis of brain aging and Alzheimer’s disease revisited. Brain Res. Bull. 1998, 45, 341–379. [Google Scholar] [CrossRef]
- Fuhrer, T.E.; Palpagama, T.H.; Waldvogel, H.J.; Synek, B.J.L.; Turner, C.; Faull, R.L.; Kwakowsky, A.L. Impaired expression of GABA transporters in the human Alzheimer’s disease hippocampus, subiculum, entorhinal cortex and superior temporal gyrus. Neuroscience 2017, 351, 108–118. [Google Scholar] [CrossRef]
- Govindpani, K.; Calvo-Flores Guzman, B.; Vinnakota, C.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. Towards a Better Understanding of GABAergic Remodeling in Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1813. [Google Scholar] [CrossRef] [Green Version]
- Kwakowsky, A.; Calvo-Flores Guzmán, B.; Govindpani, K.; Waldvogel, H.J.; Faull, R.L. Gamma-aminobutyric acid A receptors in Alzheimer’s disease: Highly localized remodeling of a complex and diverse signaling pathway. Neural Regen. Res. 2018, 8, 1362–1363. [Google Scholar] [CrossRef]
- Kwakowsky, A.; Calvo-Flores Guzman, B.; Pandya, M.; Turner, C.; Waldvogel, H.J.; Faull, R.L. GABAA receptor subunit expression changes in the human Alzheimer’s disease hippocampus, subiculum, entorhinal cortex and superior temporal gyrus. J. Neurochem. 2018, 145, 374–392. [Google Scholar] [CrossRef] [PubMed]
- Masurkar, A.V. Towards a circuit-level understanding of hippocampal CA1 dysfunction in Alzheimer’s disease across anatomical axes. J. Alzheimers Dis. Parkinsonism 2018, 8, 412. [Google Scholar] [CrossRef] [PubMed]
- Maruki, K.; Izaki, Y.; Nomura, M.; Yamauchi, T. Differences in paired-pulse facilitation and long-term potentiation between dorsal and ventral CA1 regions in anesthetized rats. Hippocampus 2001, 11, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Collinson, N.; Kuenzi, F.M.; Jarolimek, W.; Maubach, K.A.; Cothliff, R.; Sur, C.; Smith, A.; Otu, F.M.; Howell, O.; Atack, J.R.; et al. Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the alpha 5 subunit of the GABAA receptor. J. Neurosci. 2002, 22, 5572–5580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semyanov, A.; Walker, M.C.; Kullmann, D.M. GABA uptake regulates cortical excitability via cell type–specific tonic inhibition. Nat. neurosci. 2003, 6, 484. [Google Scholar] [CrossRef]
- Bartos, M.; Vida, I.; Jonas, P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat. rev. neurosci. 2007, 8, 45. [Google Scholar] [CrossRef]
- Mann, E.O.; Paulsen, O. Role of GABAergic inhibition in hippocampal network oscillations. Trends Neurosci. 2007, 30, 343–349. [Google Scholar] [CrossRef]
- Song, J.; Zhong, C.; Bonaguidi, M.A.; Sun, G.J.; Hsu, D.; Gu, Y.; Meletis, K.; Huang, Z.J.; Ge, S.; Enikolopov, G.; et al. Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature 2012, 489, 150–154. [Google Scholar] [CrossRef]
- Lee, V.; Maguire, J. The impact of tonic GABAA receptor-mediated inhibition on neuronal excitability varies across brain region and cell type. Front Neural Circuits 2014, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Fox, N.C.; Scahill, R.I.; Crum, W.R.; Rossor, M.N. Correlation between rates of brain atrophy and cognitive decline in AD. Neurology 1999, 52, 1687–1689. [Google Scholar] [CrossRef]
- Lauren, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revett, T.; Baker, G.; Jhamandas, J.; Kar, S. Glutamate system, amyloid β peptides and tau protein: Functional interrelationships and relevance to Alzheimer disease pathology. J. Psychiatry Neurosci. 2013, 38, 6–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Miguel, A.; Hercher, C.; Beasley, C.L.; Barr, A.M.; Bayer, T.A.; Falkai, P.; Leurgans, S.E.; Schneider, J.A.; Bennett, D.A.; Honer, W.G. Loss of Munc18-1 long splice variant in GABAergic terminals is associated with cognitive decline and increased risk of dementia in a community sample. Mol. Neurodegener. 2015, 10, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwakowsky, A.; Potapov, K.; Kim, S.; Peppercorn, K.; Tate, W.; Ábrahám, I. Treatment of beta amyloid 1-42(Aβ1-42)-induced basal forebrain cholinergic damage by a non-classical estrogen signaling activator in vivo. Sci. Rep. 2016, 6, 21101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, J.; Tate, W.; Palpagama, T.; Peppercorn, K.; Waldvogel, H.; Faull, R.L.; Kwakowsky, A. The Acute Effects of Amyloid-Beta1−42 on Glutamatergic Receptor and Transporter Expression in the Mouse Hippocampus. Front. Neurosci. 2020, 13, 1427. [Google Scholar] [CrossRef] [Green Version]
- Hillen, H. The Beta Amyloid Dysfunction (BAD) Hypothesis for Alzheimer’s Disease. Front. Neurosci. 2019, 13, 1154. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.-Q.; Mobley, W.C. lzheimer Disease Pathogenesis: Insights From Molecular and Cellular Biology Studies of Oligomeric Aβ and Tau Species. Front Neurosci. 2019. [Google Scholar] [CrossRef]
- Paxinos, G.; Franklin, K. The Mouse Brain in Stereotaxic Coordinates, 2nd ed.; Academic Press: Cambridge, MA, USA, 2000. [Google Scholar]
- Palpagama, T.; Sagniez, M.; Kim, S.H.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. GABAA receptors are well preserved in the hippocampus of aged mice. eNeuro 2019. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are available from the authors. |


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calvo-Flores Guzmán, B.; Kim, S.; Chawdhary, B.; Peppercorn, K.; Tate, W.P.; Waldvogel, H.J.; Faull, R.L.; Montgomery, J.; Kwakowsky, A. Amyloid-Beta1-42 -Induced Increase in GABAergic Tonic Conductance in Mouse Hippocampal CA1 Pyramidal Cells. Molecules 2020, 25, 693. https://doi.org/10.3390/molecules25030693
Calvo-Flores Guzmán B, Kim S, Chawdhary B, Peppercorn K, Tate WP, Waldvogel HJ, Faull RL, Montgomery J, Kwakowsky A. Amyloid-Beta1-42 -Induced Increase in GABAergic Tonic Conductance in Mouse Hippocampal CA1 Pyramidal Cells. Molecules. 2020; 25(3):693. https://doi.org/10.3390/molecules25030693
Chicago/Turabian StyleCalvo-Flores Guzmán, Beatriz, SooHyun Kim, Bhavya Chawdhary, Katie Peppercorn, Warren P Tate, Henry J Waldvogel, Richard LM Faull, Johanna Montgomery, and Andrea Kwakowsky. 2020. "Amyloid-Beta1-42 -Induced Increase in GABAergic Tonic Conductance in Mouse Hippocampal CA1 Pyramidal Cells" Molecules 25, no. 3: 693. https://doi.org/10.3390/molecules25030693
APA StyleCalvo-Flores Guzmán, B., Kim, S., Chawdhary, B., Peppercorn, K., Tate, W. P., Waldvogel, H. J., Faull, R. L., Montgomery, J., & Kwakowsky, A. (2020). Amyloid-Beta1-42 -Induced Increase in GABAergic Tonic Conductance in Mouse Hippocampal CA1 Pyramidal Cells. Molecules, 25(3), 693. https://doi.org/10.3390/molecules25030693