1. Introduction
The famous
Vinca alkaloid family was isolated first in the 1950s from the leaves of
Catharanthus roseus. Some of these natural compounds, along with their semisynthetic analogues, are still being used as chemotherapeutic agents in anticancer therapy (especially in the case of lymphomas and leukemia) [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
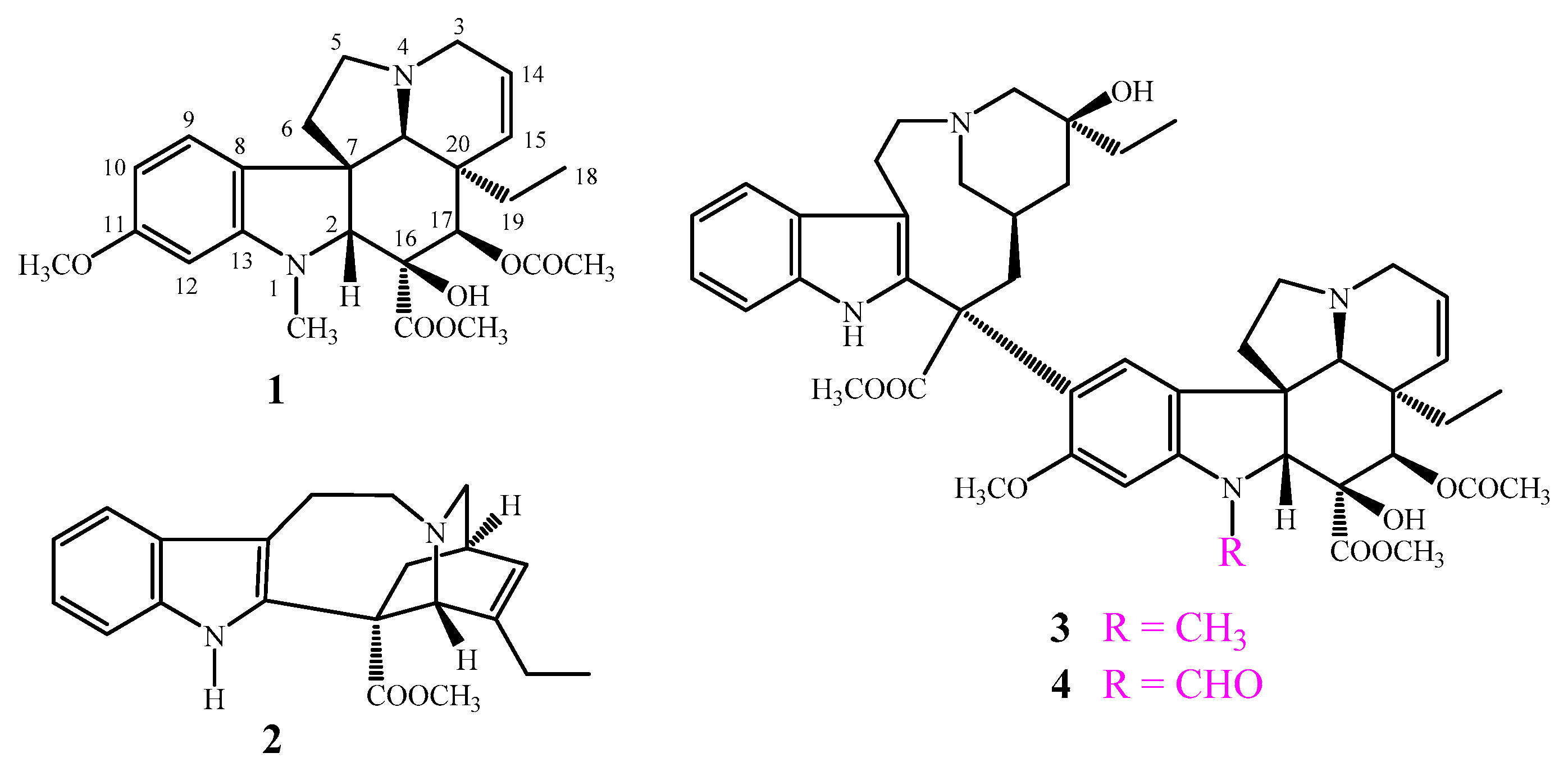
15]. Vindoline (
1) and catharanthine (
2) are the two subunits of the “dimeric” alkaloids vinblastine (
3) and vincristine (
4), which are remarkable representatives of the
Vinca family with a significant cytotoxic activity (
Figure 1). Vindoline (
1) is present in the plant in a much larger quantity than the dimers, however, it does not have any anticancer effect.
Recently, a development program has been started aiming at the synthesis of special complex molecules by incorporating two drug pharmacophores into one single molecule so as to obtain selective anticancer drugs. The objective of this project is to make available new derivatives of Vinca alkaloids that could serve as alternatives to vinblastine (3) and vincristine (4), which have a favorable antitumor effect, however, also have a high molecular weight, low absorption and several unwanted effects (e.g., toxicity and multidrug-resistance). Therefore, we have attempted to test vindoline (1) as a potential antitumor agent by connecting it to different natural and synthetic pharmacophores.
Accordingly, our main purpose was to test the idea that not only vinblastine (3) and vincristine (4), but a “monomer” Vinca alkaloid unit could also have anticancer activity, when it is combined with certain structural units.
Vinca alkaloids are usually used in the form of sulfate salts and are administered via intravenous injection in clinical therapy since their absorption is poor from the gastrointestinal tract. Their most significant adverse effects are neurotoxicity (peripheral neuropathy) and myelosuppression. Besides their significant toxicity, multidrug-resistance (MDR) is another problem that restricts the applicability of these pharmaceutical molecules in clinical therapy. Therefore, the basic aim of our research project was to synthesize new Vinca alkaloid derivatives in order to increase their effectiveness and/or reduce their serious side effects.
There are several pharmaceuticals on the market, which include the pharmacophores that we wished to introduce. 1,2,3-triazole is a widely used moiety in modern drug discovery due to its advantageous structural properties (e.g., moderate dipole character, rigidity, in vivo metabolic stability and the ability to forma hydrogen bond, which increases water solubility) as a potential connecting unit. Furthermore, this particular azaheterocycle has several beneficial biological activities, such as anticancer, antifungal and antibacterial effects. There are also HIV protease and histone deacetylase inhibitors on the market, which include 1,2,3-triazole. These compounds can be easily synthesized via click chemistry, which is an increasingly used method in medicinal chemistry [
16,
17]. Other nitrogen-containing heterocycles, such as piperazine and morpholine could also have significant therapeutic value [
18]. Piperazine analogues show diverse biological activities (e.g., antimalarial, antipsychotic, and antidepressant), too. Finally, morpholine derivatives also have outstanding pharmaceutical applications as anti-inflammatory, analgesic, neuroprotective, or antitumor agents, just to mention a few examples. The wide spectrum of biological utilities that these molecules offer made it clear that it is worthwhile to try these pharmacophores within the
Vinca alkaloid family.
Recently, several experiments were performed to conjugate
Vinca alkaloids with different types of amino acid esters, steroids and triphenylphosphine [
19,
20,
21,
22,
23,
24]. The advantage of linking with amino acids is that the given products, bound to carrier peptides (e.g., octaarginine), are able to enter directly into the cancer cells, thereby enabling more targeted therapy, and thus reducing the mentioned serious side effects. On the other hand, the steroid vector can facilitate the internalization of the drug into the cell. Finally, a triphenylphospine unit could help
Vinca compounds to fight against multidrug resistance and promote the accumulation of the drug inside cancer cells. Moreover, it has antitumor activity on its own [
25]. The products obtained showed promising half maximal inhibitory concentration (IC
50) values [
19,
20,
21], or were measured across the entire NCI cell panel and had promising in vitro cytotoxic activities (in terms of growth percent rates (GPR) and growth inhibition of 50% of cells (GI
50) values) [
22,
23,
24], according to the National Institutes of Health (NIH), US [
26,
27,
28,
29].
3. Experimental Section
3.1. General
All the reagents were purchased from Sigma-Aldrich (Budapest, Hungary) and used as received. Melting points were measured on a VEB Analytik Dresden PHMK-77/1328 apparatus (Dresden, Germany) and are uncorrected. IR spectra were recorded on Zeiss IR 75 and 80 instruments (Thornwood, NY, USA). NMR measurements were performed on a Bruker Avance III HDX 400 MHz NMR spectrometer equipped with a
15N-
31P{
1H-
19F} 5 mm CryoProbe Prodigy (
31P: 161.8 MHz), a Bruker Avance III HDX 500 MHz NMR spectrometer equipped with a
1H{
13C/
15N} 5 mm TCI CryoProbe (
1H: 499.9 MHz,
13C: 125.7 MHz), and a Bruker Avance III HDX 800 MHz NMR spectrometer equipped with a
1H-
19F{
13C/
15N} 5 mm TCI CryoProbe (
1H: 799.7 MHz,
13C: 201.1 MHz) (Bruker Corporation, Billerica, MA, USA).
1H-
1H, direct
1H-
13C, and long-range
1H-
13C scalar spin-spin connectivity were established from two-dimensional
1H-
1H correlation spectroscopy (2D COSY), HSQC, and
1H-
13C heteronuclear single quantum coherence (HMBC) experiments.
1H-
1H spatial proximities were determined using two-dimensional
1H-
1H nuclear Overhauser effect spectroscopy (NOESY) or
1H-
1H rotating frame nuclear Overhauser effect spectroscopy (ROESY) experiments.
1H and
13C chemical shifts are given on the delta scale relative to tetramethylsilane.
15N chemical shifts were determined on the basis of the
1H-
15N HMBC data and are given on the delta scale relative to nitromethane. All pulse sequences were applied by using the standard spectrometer software package. All experiments were performed at 298 K. NMR spectra were processed using Bruker TopSpin 3.5 pl 7 (Bruker Corporation, Billerica, MA, USA) and ACD/Spectrus Processor version 2017.2.2 (Advanced Chemistry Development, Inc., Toronto, ON, Canada). ESI-HRMS and MS-MS analyses were performed on a Thermo Velos Pro Orbitrap Elite (Thermo Fisher Scientific, Bremen, Germany) system. The ionization method was ESI, operated in positive ion mode. The protonated molecular ion peaks were fragmented by CID (collision-induced dissociation) at a normalized collision energy of 35–45%. For the CID experiment helium was used as the collision gas. The samples were dissolved in methanol. Data acquisition and analysis were accomplished with Xcalibur software version 2.0 (Thermo Fisher Scientific, Bremen, Germany). EI-HRMS analyses were performed on a Thermo Q Exactive GC Orbitrap system. The ionization method was EI and was operated in positive ion mode. The electron energy was 70 eV and the source temperature was set to 250 °C. Data acquisition and analysis were accomplished with Xcalibur software version 4.0 (Thermo Fisher Scientific, Bremen, Germany). TLC was carried out using DC-Alufolien Kieselgel 60 F
254 (Merck, Budapest, Hungary) plates. Preparative TLC analyses were performed on silica gel 60 PF
254+366 (Merck, Budapest, Hungary) glass plates.
1H and
13C NMR data of all the new compounds are available online as a
Supplementary Materials.
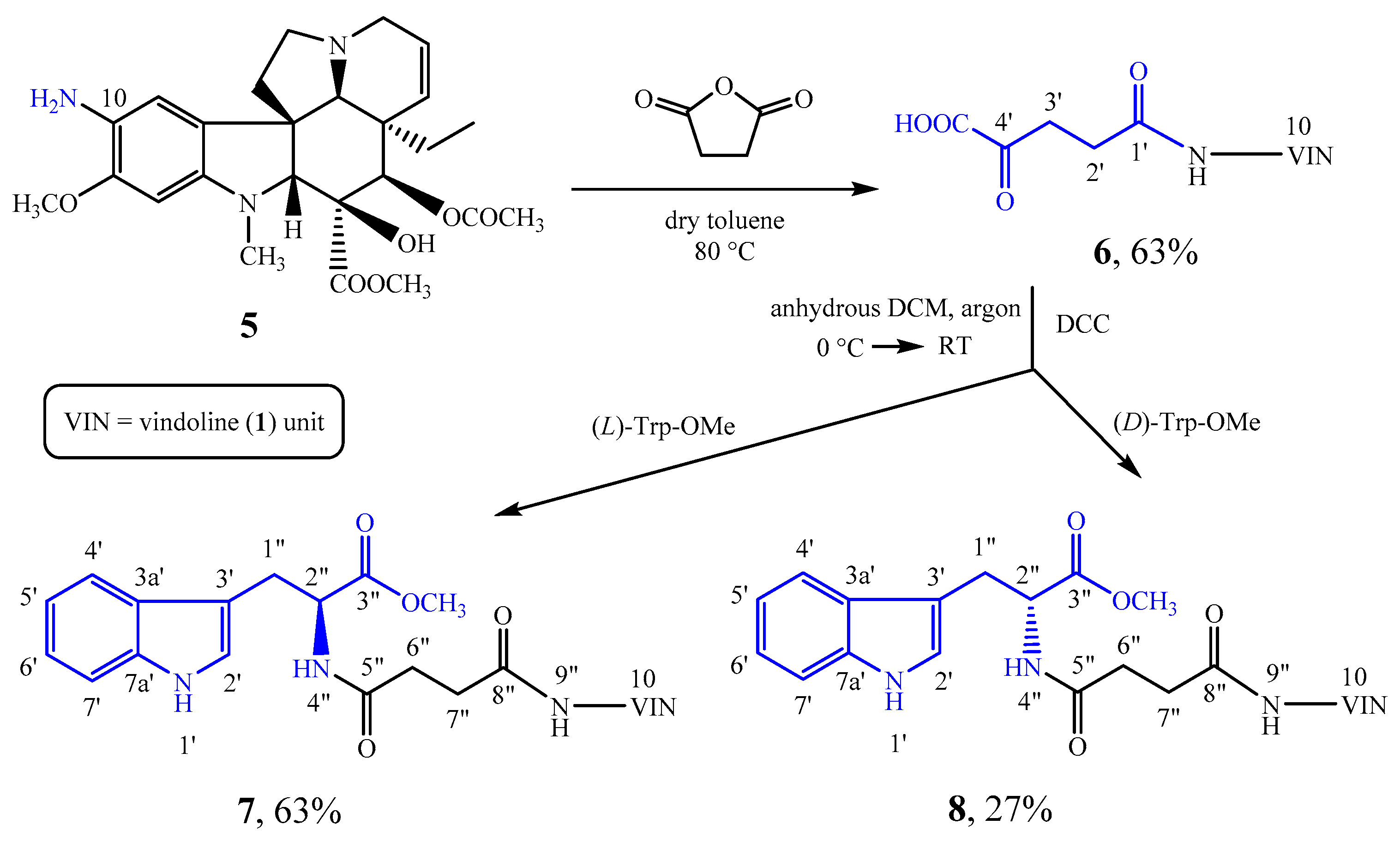
3.2. Synthesis of 10-(3-Carboxypropanamido)vindoline (6)
10-Aminovindoline (
5) [
30] (450 mg, 0.95 mmol) and succinic anhydride (144 mg, 1.4 mmol) were dissolved in 20 mL of dry toluene. MS 4 Å molecular sieve was added to the mixture under argon. The reaction mixture was heated to 80 °C and worked up after 32 h. The molecular sieve was filtered and the precipitate remaining on the filter was washed with EtOH. The mixture was evaporated under reduced pressure, then 20 mL of distilled water was added, and the pH was adjusted to 7 with ammonia solution. The aqueous phase was extracted with chloroform (4 × 40 mL). The organic phase was dried over MgSO
4, then the solvent evaporated under reduced pressure. After preparative TLC (chloroform-methanol = 10:1) 342 mg (63%) of a yellow solid (
6) was obtained. Mp 147–149 °C. TLC (chloroform-methanol = 10:1); R
f = 0.43. IR (KBr): 3425, 2964, 1742, 1527, 1245, 1039 cm
−1.
1H NMR (399.8 MHz; DMSO-d6) δ (ppm) 0.43 (t; J = 7.3 Hz; 3H; H3-18); 0.95 (dq; J = 13.9; 7.3 Hz; 1H; Hx-19); 1.47 (dq; J = 13.9; 7.3 Hz; 1H; Hy-19); 1.94 (s; 3H; C(17)-OC(O)CH3); 2.17–2.25 (m; 2H; H2-6); 2.41–2.48 (br m; 2H; H2-3′); 2.50–2.57 (m; 3H; Hx-5. H2-2′); 2.58 (s; 3H; N(1)-CH3); 2.59 (s; 1H; H-21); 2.78–2.86 (m; 1H; Hx-3); 3.24–3.32 (m; 1H; Hy-5); 3.37–3.44 (m; 1H; Hy-3); 3.51 (s; 1H; H-2); 3.66 (s; 3H; C(16)-COOCH3); 3.80 (s; 3H; C(11)-OCH3); 5.06–5.10 (m; 1H; H-15); 5.21 (s; 1H; H-17); 5.82 (ddd; J = 10.1; 4.8; 1.1 Hz; 1H; H-14); 6.39 (s; 1H; H-12); 7.57 (s; 1H; H-9); 8.80 (br; 1H; C(16)-OH); 8.92 (s; 1H; C(10)-NH).
13C NMR (100.5 MHz; DMSO-d6) δ (ppm) 7.5 (C-18); 20.6 (C(17)-OC(O)CH3); 29.1 (C-3′); 30.4 (C-19); 30.7 (C-2′); 38.7 (N(1)-CH3); 42.4 (C-20); 43.6 (C-6); 50.3 (C-3); 51.2 (C-5); 51.6 (C(16)-COOCH3); 52.3 (C-7); 55.6 (C(11)-OCH3); 66.2 (C-21); 75.8 (C-17); 78.6 (C-16); 82.9 (C-2); 93.9 (C-12); 117.5 (C-9); 119.3 (C-10); 123.5 (C-8); 124.4 (C-14); 129.8 (C-15); 149.2 (C-13); 151.1 (C-11); 169.6 (C-1′); 170.0 (C(17)-OC(O)CH3); 171.6 (C(16)-COOCH3); 173.9 (br; C-4′).
ESI-HRMS: M + H = 572.26097 (delta = 1.3 ppm; C29H38O9N3). HR-ESI-MS-MS (CID = 35%; rel. int. %): 554(12); 512(100); 494(3); 480(2); 303(9).
3.3. Amidation of 10-(3-Carboxypropanamido)vindoline (6) and (l)-Tryptophan Methyl Ester
77 mg (0.35 mmol) of (l)-tryptophan methyl ester was liberated from its hydrochloric acid salt, and dissolved in 10 mL of anhydrous DCM. Under argon at 0 °C, 200 mg (0.35 mmol) of compound 6 was added. Then, 110 mg (0.53 mmol) of DCC was dissolved in 6 mL of anhydrous DCM and added dropwise into the reaction mixture. Subsequently, the mixture was allowed to reach room temperature. After 19 h, the reaction mixture was filtered, and the filtrate was evaporated under reduced pressure. After preparative TLC (dichloromethane-methanol = 10:1) 169 mg (63%) of a pale yellow solid (7) was obtained. Mp 140–141 °C. TLC (dichloromethane-methanol = 10:1); Rf = 0.35. IR (KBr): 3370, 2952, 1742, 1525, 1245, 1039, 743 cm−1.
1H NMR (799.7 MHz; DMSO-d6) δ (ppm) 0.42 (t; J = 7.3 Hz; 3H; H3-18); 0.94 (dq; J = 14.1; 7.3 Hz; 1H; Hx-19); 1.47 (dq; J = 14.1; 7.3 Hz; 1H; Hy-19); 1.94 (s; 3H; C(17)-OC(O)CH3); 2.17–2.23 (m; 2H; H2-6); 2.35–2.44 (m; 2H; H2-6′’); 2.47–2.53 (m; 3H; H2-7′’; Hx-5); 2.58 (~s; 4H; N(1)-CH3; H-21); 2.75–2.79 (m; 1H; Hx-3); 3.03 (dd; J = 14.5; 8.1 Hz; 1H; Hx-1′’); 3.11 (dd; J = 14.5; 6.0 Hz; 1H; Hy-1′’); 3.25–3.28 (m; 1H; Hy-5); 3.37–3.41 (m; 1H; Hy-3); 3.50 (s; 1H; H-2); 3.55 (s; 3H; C(3′’)-OCH3); 3.65 (s; 3H; C(16)-COOCH3); 3.78 (s; 3H; C(11)-OCH3); 4.47–4.51 (m; 1H; H-2′’); 5.06–5.09 (m; 1H; H-15); 5.20 (s; 1H; H-17); 5.81 (ddd; J = 10.1; 4.9; 1.1 Hz; 1H; H-14); 6.38 (s; 1H; H-12); 6.97–7.00 (m; 1H; H-5′); 7.05–7.08 (m; 1H; H-6′); 7.16 (d; J = 1.6 Hz; 1H; H-2′); 7.33 (d; J = 8.1 Hz; 1H; H-7′); 7.48 (d; J = 7.8 Hz; 1H; H-4′); 7.53 (s; 1H; H-9); 8.33 (d; J = 7.5 Hz; 1H; NH-4′’); 8.78 (s; 1H; C(16)-OH); 8.89 (s; 1H; NH-9′’); 10.85–10.87 (m; 1H; NH-1′).
13C NMR (201.1 MHz; DMSO-d6) δ (ppm) 7.5 (C-18); 20.6 (C(17)-OC(O)CH3); 27.1 (C-1′’); 30.2 (C-6′’); 30.4 (C-19); 31.0 (C-7′’); 38.7 (N(1)-CH3); 42.4 (C-20); 43.6 (C-6); 50.3 (C-3); 51.1 (C-5); 51.6 (C(16)-COOCH3; C(3′’)-OCH3); 52.3 (C-7); 53.1 (C-2′’); 55.6 (C(11)-OCH3); 66.2 (C-21); 75.8 (C-17); 78.6 (C-16); 82.9 (C-2); 93.9 (C-12); 109.3 (C-3′); 111.3 (C-7′); 117.8 (C-9); 117.9 (C-4′); 118.3 (C-5′); 119.1 (C-10); 120.8 (C-6′); 123.4 (C-8); 123.6 (C-2′); 124.3 (C-14); 126.9 (C-3a’); 129.8 (C-15); 135.9 (C-7a’); 149.3 (C-13); 151.3 (C-11); 169.8 (C-8′’); 169.9 (C(17)-OC(O)CH3); 171.4 (C-5′’); 171.5 (C(16)-COOCH3); 172.3 (C-3′’).
ESI-HRMS: M + H = 772.35419 (delta = −1.3 ppm; C41H50O10N5). HR-ESI-MS-MS (CID=35%; rel. int. %): 754(13); 712(100); 694(3); 680(3); 610(2); 554(3); 503(11); 494(3); 472(3); 443(4); 412(4).
3.4. Amidation of 10-(3-Carboxypropanamido)vindoline (6) and (d)-Tryptophan Methyl Ester
39 mg (0.18 mmol) of (d)-tryptophan methyl ester was liberated from its hydrochloric acid salt, and dissolved in 5 mL of anhydrous DCM. Under argon at 0 °C, 100 mg (0.18 mmol) of compound 6 was added. Then, 55 mg (0.26 mmol) of N,N’-dicyclohexylcarbodiimide (DCC) was dissolved in 3 mL of anhydrous DCM and added dropwise into the reaction mixture. Subsequently, the mixture was allowed to reach room temperature. After 8 h, the reaction mixture was filtered, and the filtrate was evaporated under reduced pressure. After preparative TLC (dichloromethane-methanol = 10:1) 36 mg (27%) of a pale yellow solid (8) was obtained. Mp 152–153 °C. TLC (dichloromethane-methanol = 10:1); Rf = 0.41. IR (KBr): 3308, 2952, 1742, 1525, 1225, 1039, 743 cm−1.
1H NMR (499.9 MHz; DMSO-d6) δ (ppm) 0.42 (t; J = 7.3 Hz; 3H; H3-18); 0.95 (dq; J = 14.2; 7.3 Hz; 1H; Hx-19); 1.46 (dq; J = 14.2; 7.3 Hz; 1H; Hy-19); 1.93 (s; 3H; C(17)-OC(O)CH3); 2.15–2.24 (m; 2H; H2-6); 2.37–2.43 (m; 2H; H2-6′’); 2.46–2.54 (m; 3H; H2-7′’; Hx-5); 2.58 (br s; 1H; H-21. s; 3H; N(1)-CH3); 2.74–2.79 (m; 1H; Hx-3); 3.00–3.06 (m; 1H; Hx-1′’); 3.10–3.15 (m; 1H; Hy-1′’); 3.24–3.29 (m; 1H; Hy-5); 3.35–3.42 (m; 1H; Hy-3); 3.51 (s; 1H; H-2); 3.55 (s; 3H; C(3′’)-OCH3); 3.65 (s; 3H; C(16)-COOCH3); 3.78 (s; 3H; C(11)-OCH3); 4.47–4.53 (m; 1H; H-2′’); 5.05–5.09 (m; 1H; H-15); 5.20 (s; 1H; H-17); 5.81 (ddd; J = 10.1; 4.9; 1.5 Hz; 1H; H-14); 6.38 (s; 1H; H-12); 6.96–7.00 (m; 1H; H-5′); 7.04–7.08 (m; 1H; H-6′); 7.16 (d; J = 2.2 Hz; 1H; H-2′); 7.31–7.35 (m; 1H; H-7′); 7.46–7.50 (m; 1H; H-4′); 7.54 (s; 1H; H-9); 8.32 (d; J = 7.5 Hz; 1H; NH-4′’); 8.78 (br s; 1H; C(16)-OH); 8.88 (s; 1H; NH-9′’); 10.86 (br d; J = 2.2 Hz; 1H; NH-1′).
13C NMR (125.7 MHz; DMSO-d6) δ (ppm) 7.5 (C-18); 20.6 (C(17)-OC(O)CH3); 27.1 (C-1′’); 30.2 (C-6′’); 30.4 (C-19); 31.1 (C-7′’); 38.7 (N(1)-CH3); 42.4 (C-20); 43.6 (C-6); 50.3 (C-3); 51.1 (C-5); 51.6 (C(3′’)-OCH3. C(16)-COOCH3); 52.3 (C-7); 53.1 (C-2′’); 55.6 (C(11)-OCH3); 66.2 (C-21); 75.8 (C-17); 78.6 (C-16); 82.9 (C-2); 93.9 (C-12); 109.3 (C-3′); 111.3 (C-7′); 117.8 (C-9); 117.9 (C-4′); 118.3 (C-5′); 119.2 (C-10); 120.8 (C-6′); 123.5 (C-8); 123.6 (C-2′); 124.4 (C-14); 127.0 (C-3a’); 129.8 (C-15); 136.0 (C-7a’); 149.3 (C-13); 151.3 (C-11); 169.8 (C-8′’); 170.0 (C(17)-OC(O)CH3); 171.4 (C-5′’); 171.5 (C(16)-COOCH3); 172.3 (C-3′’).
ESI-HRMS: M + H = 772.35387 (delta = −1.75 ppm; C41H50O10N5). HR-ESI-MS-MS (CID=35%; rel. int. %): 754(15); 712(100); 694(3); 680(3); 610(2); 554(3); 503(13); 494(4); 472(4); 443(6); 412(5).
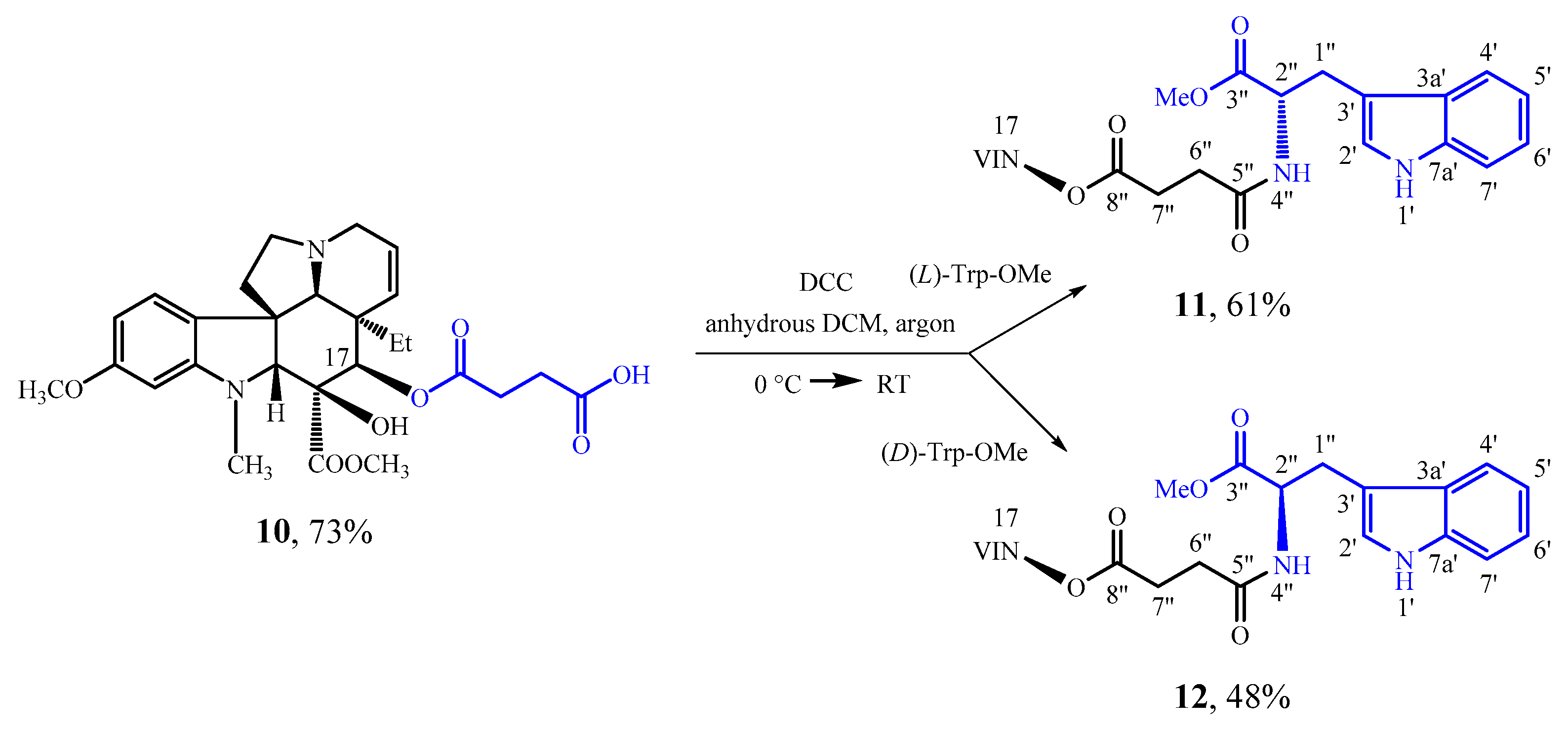
3.5. Amidation of 17-(3-Carboxypropanoyloxy)vindoline (10) and (l)-Tryptophan Methyl Ester
85 mg (0.39 mmol) of (
l)-tryptophan methyl ester was liberated from its hydrochloric acid salt, and dissolved in 10 mL of anhydrous DCM. Under argon the mixture was cooled to 0 °C, and 200 mg (0,39 mmol) of compound
10 [
31] was added. Then, 82 mg (0.39 mmol) of DCC dissolved in 5 mL anhydrous DCM was added dropwise into the reaction mixture. Then, the mixture was allowed to reach room temperature. After stirring for 22 h, the precipitate was filtered, and the filtrate was evaporated under reduced pressure. After preparative TLC (dichloromethane-methanol = 10:1) 170 mg (61%) of a pale yellow solid (
11) was obtained. Mp 131–133 °C. TLC (dichloromethane-methanol = 20:1); R
f = 0.34. IR (KBr): 3369, 2952, 1741, 1502, 1225, 1168, 1027, 744 cm
−1.
1H NMR (499.9 MHz; DMSO-d6) δ (ppm) 0.41 (t; J = 7.3 Hz; 3H; H3-18); 0.92 (dq; J = 14.1; 7.3 Hz; 1H; Hx-19); 1.46 (dq; J = 14.1; 7.3 Hz; 1H; Hy-19); 2.14–2.24 (m. 2H; H2-6); 2.27–2.45 (m; 4H; H2-6′’. H2-7′’); 2.52–2.59 (m; 1H; Hx-5); 2.56 (s; 3H; N(1)-CH3); 2.65 (s; 1H; H-21); 2.74–2.80 (m; 1H; Hx-3); 2.99–3.06 (m; 1H; Hx-1′’); 3.10–3.15 (m; 1H; Hy-1′’); 3.24–3.30 (m; 1H; Hy-5); 3.35–3.41 (m; 1H; Hy-3); 3.54 (~s; 4H; H-2. C(3′’)-OCH3); 3.63 (s; 3H; C(16)-COOCH3); 3.70 (s; 3H; C(11)-OCH3); 4.45–4.50 (m; 1H; H-2′’); 5.11–5.15 (m; 1H; H-15); 5.18 (s; 1H; H-17); 5.75 (ddd; J = 10.2; 4.8; 1.3 Hz; 1H; H-14); 6.19 (d; J = 2.2 Hz; 1H; H-12); 6.28 (dd; J = 8.2; 2.2 Hz; 1H; H-10); 6.96–7.00 (m; 1H; H-5′); 7.04 (d; J = 8.2 Hz; 1H; H-9); 7.04–7.08 (m; 1H; H-6′); 7.14 (d; J = 2.3 Hz; 1H; H-2′); 7.32–7.35 (m; 1H; H-7′); 7.46–7.49 (m; 1H; H-4′); 8.36 (d; J = 7.5 Hz; 1H; NH-4′’); 8.80 (br s; 1H; C(16)-OH); 10.86 (br d; J = 2.3 Hz; 1H; NH-1′).
13C NMR (125.7 MHz; DMSO-d6) δ (ppm) 7.5 (C-18); 27.0 (C-1′’); 28.9. 29.6 (C-6′’. C-7′’); 30.3 (C-19); 38.0 (N(1)-CH3); 42.4 (C-20); 43.7 (C-6); 50.3 (C-3); 51.0 (C-5); 51.6 (C(3′’)-OCH3); 51.7 (C(16)-COOCH3); 52.0 (C-7); 53.2 (C-2′’); 55.0 (C(11)-OCH3); 66.0 (C-21); 75.8 (C-17); 78.6 (C-16); 82.8 (C-2); 95.4 (C-12); 104.4 (C-10); 109.3 (C-3′); 111.3 (C-7′); 117.9 (C-4′); 118.3 (C-5′); 120.9 (C-6′); 123.0 (C-9); 123.6 (C-2′); 124.1 (C-14); 125.3 (C-8); 127.0 (C-3a’); 129.9 (C-15); 136.0 (C-7a’); 153.4 (C-13); 160.4 (C-11); 170.7 (C-5′’); 171.5 (C(16)-COOCH3); 171.8 (C-8′’); 172.3 (C-3′’).
15N NMR (50.7 MHz; DMSO-d6) δ (ppm) −323.2 (N-4); -315.0 (N-1); −262.2 (N-4′’); −250.8 (N-1′).
ESI-HRMS: M + H = 715.33393 (delta = 0.24 ppm; C39H47O9N4). HR-ESI-MS-MS (CID = 35%; rel. int. %): 697(100); 415(6); 397(9).
3.6. Amidation of 17-(3-Carboxypropanoyloxy)vindoline (10) and (d)-Tryptophan Methyl Ester
85 mg (0.39 mmol) of (
d)-tryptophan methyl ester was liberated from its hydrochloric acid salt, and dissolved in 10 mL of anhydrous DCM. Under argon the mixture was cooled to 0 °C, and 200 mg (0,39 mmol) of compound
10 [
31] was added. Then, 82 mg (0.39 mmol) of DCC dissolved in 5 mL of anhydrous DCM was added dropwise into the reaction mixture. Then, the mixture was allowed to reach room temperature. After stirring for 22 h, the precipitate was filtered, and the filtrate was evaporated under reduced pressure. After preparative TLC (dichloromethane-methanol = 15:1) 133 mg (48%) of a pale yellow solid (
12) was obtained. Mp 141–143 °C. TLC (dichloromethane-methanol = 15:1); R
f = 0.54. IR (KBr): 3380, 2952, 1741, 1502, 1225, 1168, 1028, 744 cm
−1.
1H NMR (499.9 MHz; DMSO-d6) δ (ppm) 0.40 (t; J = 7.4 Hz; 3H; H3-18); 0.92 (dq; J = 14.3; 7.4 Hz; 1H; Hx-19); 1.46 (dq; J = 14.3; 7.4 Hz; 1H; Hy-19); 2.14–2.24 (m. 2H; H2-6); 2.27–2.45 (m; 4H; H2-6′’. H2-7′’); 2.52–2.59 (m; 1H; Hx-5); 2.56 (s; 3H; N(1)-CH3); 2.65 (s; 1H; H-21); 2.75–2.82 (m; 1H; Hx-3); 2.98–3.05 (m; 1H; Hx-1′’); 3.08–3.14 (m; 1H; Hy-1′’); 3.24–3.30 (m; 1H; Hy-5); 3.38–3.44 (m; 1H; Hy-3); 3.54 (s; 1H; H-2); 3.55 (s; 3H; C(3′’)-OCH3); 3.63 (s; 3H; C(16)-COOCH3); 3.70 (s; 3H; C(11)-OCH3); 4.46–4.52 (m; 1H; H-2′’); 5.12–5.16 (m; 1H; H-15); 5.19 (s; 1H; H-17); 5.81 (ddd; J = 10.3; 4.8; 1.3 Hz; 1H; H-14); 6.18 (d; J = 2.3 Hz; 1H; H-12); 6.28 (dd; J = 8.2; 2.3 Hz; 1H; H-10); 6.94–6.98 (m; 1H; H-5′); 7.03–7.07 (m; 1H; H-6′); 7.04 (d; J = 8.2 Hz; 1H; H-9); 7.13 (d; J = 2.3 Hz; 1H; H-2′); 7.31–7.34 (m; 1H; H-7′); 7.46–7.48 (m; 1H; H-4′); 8.35 (d; J = 7.5 Hz; 1H; NH-4′’); 8.79 (br s; 1H; C(16)-OH); 10.85 (br d; J = 2.3 Hz; 1H; NH-1′).
13C NMR (125.7 MHz; DMSO-d6) δ (ppm) 7.5 (C-18); 27.0 (C-1′’); 28.9. 29.6 (C-6′’. C-7′’); 30.3 (C-19); 38.0 (N(1)-CH3); 42.4 (C-20); 43.7 (C-6); 50.3 (C-3); 51.0 (C-5); 51.6 (C(3′’)-OCH3); 51.7 (C(16)-COOCH3); 52.0 (C-7); 53.1 (C-2′’); 55.0 (C(11)-OCH3); 66.1 (C-21); 75.8 (C-17); 78.6 (C-16); 82.8 (C-2); 95.4 (C-12); 104.4 (C-10); 109.3 (C-3′); 111.3 (C-7′); 117.9 (C-4′); 118.3 (C-5′); 120.8 (C-6′); 123.0 (C-9); 123.5 (C-2′); 124.1 (C-14); 125.3 (C-8); 127.0 (C-3a’); 130.0 (C-15); 136.0 (C-7a’); 153.4 (C-13); 160.4 (C-11); 170.7 (C-5′’); 171.6 (C(16)-COOCH3); 171.8 (C-8′’); 172.3 (C-3′’).
ESI-HRMS: M + H = 715.33370 (delta = −0.08 ppm; C39H47O9N4). HR-ESI-MS-MS (CID = 45%; rel. int. %): 697(100); 415(5); 397(9).
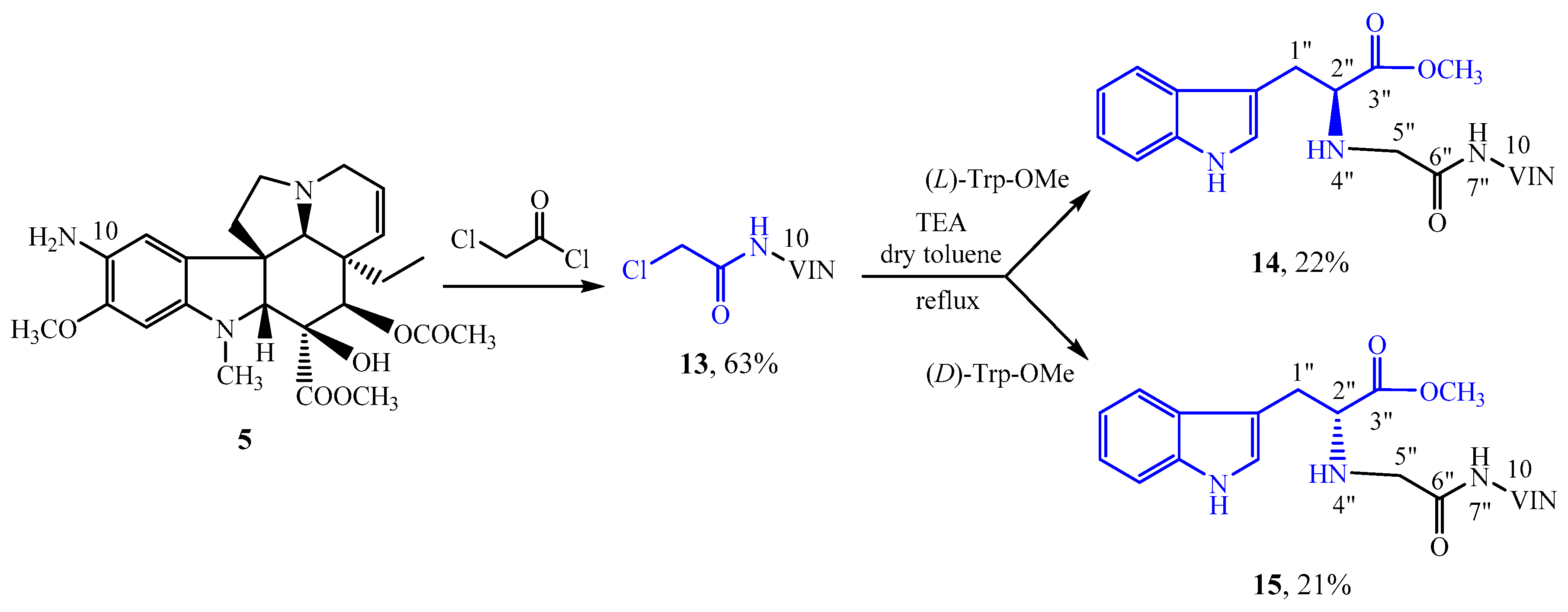
3.7. N-Acylation of 10-Aminovindoline (5) with Chloroacetyl Chloride
978 mg (2.1 mmol) of 10-aminovindoline (
5) [
30] was dissolved in 20 mL of anhydrous DCM under argon, then 321 mg (2.3 mmol) of anhydrous K
2CO
3 was added. The reaction mixture was cooled to 0 °C, and 1.71 mL (21.5 mmol) of chloroacetyl chloride was added dropwise. The mixture was allowed to reach room temperature. After stirring for 6 h, the reaction mixture was filtered, and the filtrate was washed with 50 mL of 10% NaHCO
3 solution. The aqueous phase was extracted with 2 × 50 mL DCM. The organic phase was dried over MgSO
4, then the solvent evaporated under reduced pressure. After preparative TLC (ethyl acetate: methanol = 15:1) 712 mg (63%) of a white crystalline pure product (
13) was obtained. Mp 168 °C. TLC (ethyl acetate: methanol = 20:1); R
f = 0.30. IR (KBr): 3392, 2964, 1743,1533, 1245, 1039 cm
−1.
1H NMR (499.9 MHz; CDCl3) δ (ppm) 0.53 (3H; t; J = 7.3 Hz; H3-18); 1.08 (1H; dq; J = 14.3; 7.3 Hz; Hx-19); 1.62 (1H; dq; J = 14.3; 7.3 Hz; Hy-19); 2.08 (3H; s; C(17)-OC(O)CH3); 2.25–2.40 (2H; m; H2-6); 2.54–2.63 (1H; m; Hx-5); 2.69 (3H; s; N(1)-CH3); 2.77 (1H; s; H-21); 2.81–2.89 (1H; m; Hx-3); 3.36–3.45 (1H; m; Hy-5); 3.45–3.54 (1H; m; Hy-3); 3.73 (1H; s; H-2); 3.79 (3H; s; C(16)-COOCH3); 3.91 (3H; s; C(11)-OCH3); 4.16 (2H; ~s; C(10)-NH-C(O)CH2Cl); 5.24 (1H; br d; J = 10.4 Hz; H-15); 5.43 (1H; s; H-17); 5.86 (1H; ddd; J = 10.4; 5.0; 1.4 Hz; H-14); 6.13 (1H; s; H-12); 8.04 (1H; s; H-9); 8.72 (1H; br; C(10)-NH); 9.60 (1H; br; C(16)-OH).
13C NMR (201.1 MHz; CDCl3) δ (ppm) 7.6 (C-18); 21.1 (C(17)-OC(O)CH3); 31.0 (C-19); 38.9 (N(1)-CH3); 42.9 (C-20); 43.1 (C(10)-NH-C(O)CH2Cl); 43.9 (br; C-6); 50.9 (C-3); 51.5 (br; C-5); 52.3 (C(16)-COOCH3); 53.3 (br; C-7); 56.1 (C(11)-OCH3); 66.5 (br; C-21); 76.4 (C-17); 79.7 (C-16); 83.4 (br; C-2); 93.2 (C-12); 114.5 (C-9); 119.0 (C-10); 123.6 (br; C-8); 124.2 (br; C-14); 130.4 (C-15); 149.5 (C-13); 149.9 (C-11); 163.0 (C(10)-NH-C(O)CH2Cl); 170.8 (C(17)-OC(O)CH3); 171.8 (C(16)-COOCH3).
ESI-HRMS: M + H = 548.21557 (delta = −0.4 ppm; C27H35O7N3Cl). HR-ESI-MS-MS (CID = 35%. rel. int. %): 530(11); 488(100); 470(2);456(3); 428(2); 386(1); 279(7).
3.8. N-Alkylation of (l)-Tryptophan Methyl Ester with 10-Chloroacetamido-Vindoline (13)
10-Chloroacetamido-vindoline (13) (200 mg, 0.37 mmol) and (l)-tryptophan methyl ester (88 mg, 0.40 mmol; previously liberated from its hydrochloric acid salt) were dissolved in 5 mL of dry toluene in a sealed glass. Then, 0.05 mL (0.37 mmol) of TEA was added. The sealed glass was placed in an oil bath at 150 °C. After stirring for 15 h, the precipitate was filtered, and the filtrate was evaporated under reduced pressure. After preparative TLC (dichloromethane-methanol = 10:1) 59 mg (22%) of a pale green solid (14) was obtained. Mp 150–152 °C. TLC (dichloromethane-methanol = 10:1); Rf = 0.54. IR (KBr): 3319, 2950, 1740, 1528, 1459, 1243, 1039, 743 cm−1.
1H NMR (499.9 MHz; DMSO-d6) δ (ppm) 0.42 (t; J = 7.3 Hz; 3H; H3-18); 0.96 (dq; J = 13.8; 7.3 Hz; 1H; Hx-19); 1.49 (dq; J = 13.8; 7.3 Hz; 1H; Hy-19); 1.95 (s; 3H; C(17)-OC(O)CH3); 2.17–2.28 (m; 2H; H2-6); 2.52–2.60 (m; 1H; Hx-5); 2.60 (s; 3H; N(1)-CH3); 2.65 (br s; 1H; H-21); 2.81–2.90 (m; 1H; Hx-3); 3.08–3.19 (m; 3H; H2-1′’. Hx-5′’); 3.31–3.41 (m; 2H; Hy-5. Hy-5′’); 3.41–3.48 (m; 1H; Hy-3); 3.53 (s; 1H; H-2); 3.55 (s; 3H; C(3′’)-OCH3); 3.58–3.62 (m; 1H; H-2′’); 3.66 (s; 3H; C(16)-COOCH3); 3.82 (s; 3H; C(11)-OCH3); 5.08–5.12 (m; 1H; H-15); 5.21 (s; 1H; H-17); 5.83 (ddd; J = 10.1; 4.8; 1.5 Hz; 1H; H-14); 6.45 (s; 1H; H-12); 6.93–6.97 (m; 1H; H-5′); 7.04–7.07 (m; 1H; H-6′); 7.14 (d; J = 2.2 Hz; 1H; H-2′); 7.32–7.35 (m; 1H; H-7′); 7.47–7.50 (m; 1H; H-4′); 7.91 (s; 1H; H-9); 8.79 (br s; 1H; C(16)-OH); 9.49 (s; 1H; NH-7′’); 10.89 (br d; J = 2.2 Hz; 1H; NH-1′).
13C NMR (125.7 MHz; DMSO-d6) δ (ppm): 7.4 (C-18). 20.6 (C(17)-OC(O)CH3). 28.5 (C-1′’). 30.4 (C-19). 38.8 (N(1)-CH3). 42.4 (C-20). 43.5 (C-6). 50.3 (C-3). 50.8 (C-5′’). 51.1 (C-5). 51.4 (C(3′’)-OCH3). 51.7 (C(16)-COOCH3). 52.4 (C-7). 55.9 (C(11)-OCH3). 61.7 (C-2′’). 66.2 (C-21). 75.7 (C-17). 78.6 (C-16). 82.6 (C-2). 94.0 (C-12). 109.2 (C-3′). 111.3 (C-7′). 114.5 (C-9). 117.9 (C-4′). 118.2 (C-5′). 119.3 (C-10). 120.8 (C-6′). 123.4 (C-8). 123.5 (C-2′). 124.3 (C-14). 127.1 (C-3a’). 129.8 (C-15). 136.0 (C-7a’). 148.7 (C-13). 149.8 (C-11). 168.3 (br; C-6′’). 170.0 (C(17)-OC(O)CH3). 171.5 (C(16)-COOCH3). 173.8 (br; C-3′’).
ESI-HRMS: M + H = 730.34419 (delta = −0.64 ppm; C39H48O9N5). HR-ESI-MS-MS (CID = 35%; rel. int. %): 712(12); 670(100); 652(3); 568(4); 461(5); 433(3); 346(3).
3.9. N-Alkylation of (d)-Tryptophan Methyl Ester with 10-Chloroacetamido-Vindoline (13)
10-Chloroacetamido-vindoline (13) (200 mg, 0.37 mmol) and (d)-tryptophan methyl ester (88 mg, 0.40 mmol; previously liberated from its hydrochloric acid salt) were dissolved in 5 mL of dry toluene in a sealed glass. Then, 0.05 mL (0.37 mmol) of TEA was added. The sealed glass was placed in an oil bath at 150 °C. After stirring for 12 h, the precipitate was filtered, and the filtrate was evaporated under reduced pressure. After preparative TLC (dichloromethane-methanol = 10: 1) 56 mg (21%) of a pale green solid (15) was obtained. Mp 157–159 °C. TLC (dichloromethane-methanol = 10:1); Rf = 0.58. IR (KBr): 3325, 2950, 1740, 1528, 1459, 1244, 1039, 743 cm−1.
1H NMR (499.9 MHz; DMSO-d6) δ (ppm) 0.42 (t; J = 7.3 Hz; 3H; H3-18); 0.94 (dq; J =14.1; 7.3 Hz; 1H; Hx-19); 1.47 (dq; J =14.1; 7.3 Hz; 1H; Hy-19); 1.94 (s; 3H; C(17)-OC(O)CH3); 2.16–2.27 (m; 2H; H2-6); 2.50–2.56 (m; 1H; Hx-5); 2.60 (~s; 4H; N(1)-CH3. H-21); 2.79–2.85 (m; 1H; Hx-3); 2.92–3.11 (br m; 1H; NH-4′’); 3.07–3.12 (m; 3H; Hx-5′’. H2-1′’); 3.28–3.38 (m; 2H; Hy-5. Hy-5′’); 3.39–3.45 (m; 1H; Hy-3); 3.52 (s; 1H; H-2); 3.55 (s; 3H; C(3′’)-OCH3); 3.55–3.60 (m; 1H; H-2′’); 3.66 (s; 3H; C(16)-COOCH3); 3.82 (s; 3H; C(11)-OCH3); 5.07–5.11 (m; 1H; H-15); 5.20 (s; 1H; H-17); 5.83 (ddd; J = 10.4; 4.2; 1.1 Hz; 1H; H-14); 6.45 (s; 1H; H-12); 6.94–6.98 (m; 1H; H-5′); 7.03–7.07 (m; 1H; H-6′); 7.14 (d; J = 2.0 Hz; 1H; H-2′); 7.31–7.35 (m; 1H; H-7′); 7.47–7.51 (m; 1H; H-4′); 7.91 (s; 1H; H-9); 8.77 (br s; 1H; C(16)-OH); 9.48 (s; 1H; NH-7′’); 10.88 (br d; J = 2.0 Hz; 1H; NH-1′).
13C NMR (125.7 MHz; DMSO-d6) δ (ppm) 7.5 (C-18); 20.7 (C(17)-OC(O)CH3); 28.6 (C-1′’); 30.4 (C-19); 38.8 (N(1)-CH3); 42.4 (C-20); 43.6 (C-6); 50.4 (C-3); 51.0 (C-5′’); 51.1 (C-5); 51.4 (C(3′’)-OCH3); 51.6 (C(16)-COOCH3); 52.5 (C-7); 55.9 (C(11)-OCH3); 61.8 (C-2′’); 66.1 (C-21); 75.8 (C-17); 78.7 (C-16); 82.8 (C-2); 94.0 (C-12); 109.3 (C-3′); 111.3 (C-7′); 114.3 (C-9); 117.9 (C-4′); 118.3 (C-5′); 119.4 (C-10); 120.8 (C-6′); 123.4 (C-8); 123.5 (C-2′); 124.4 (C-14); 127.1 (C-3a’); 129.8 (C-15); 136.0 (C-7a’); 148.6 (C-13); 149.7 (C-11); 168.4 (C-6′’); 170.0 (C(17)-OC(O)CH3); 171.5 (C(16)-COOCH3); 174.0 (C-3′’).
ESI-HRMS: M+H=730.34353 (delta=-1.54 ppm; C39H48O9N5). HR-ESI-MS-MS (CID=35%; rel. int. %): 712(13); 670(100); 652(3); 568(4); 461(7); 433(4); 346(4).
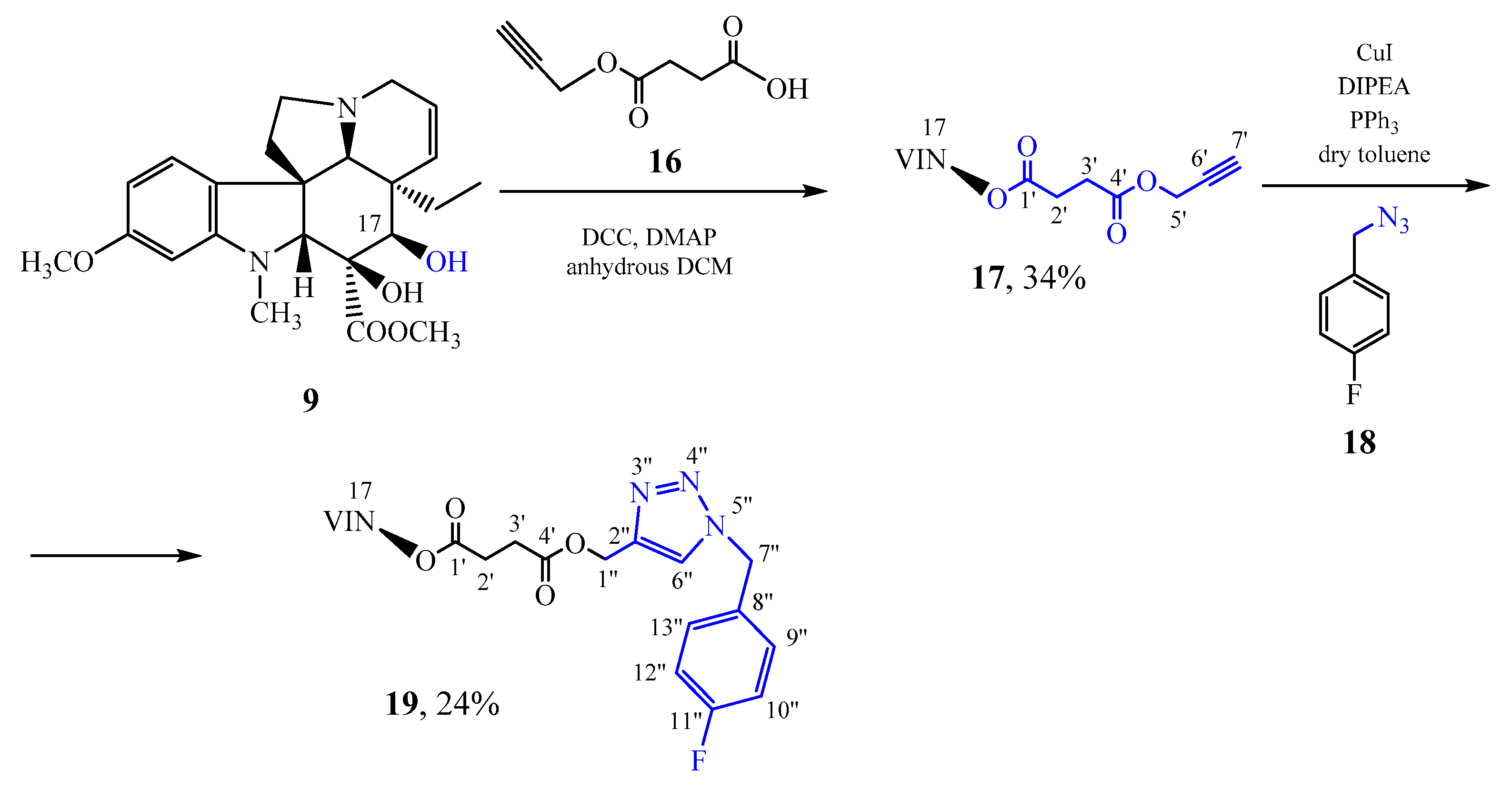
3.10. Synthesis of 17-[4-Oxo-4-(prop-2-ynyloxy)butanoyloxy]vindoline (17)
17-Desacetylvindoline (
9) [
31] (600 mg, 1.5 mmol) and 4-oxo-4- (prop-2-ynyloxy) butanoic acid (
16) (240 mg, 1.5 mmol) were dissolved in 15 mL of anhydrous DCM followed by a dropwise addition of 330 mg (0.53 mmol) of DCC and 15 mg (0.039 mmol) of 4-dimethylaminopyridine (DMAP) dissolved in another 15 mL of anhydrous DCM. The reaction mixture was refluxed for 4h, then further 250 mg of DCC and 30 mg of DMAP dissolved in 20 mL of anhydrous DCM were added in two portions. After refluxing for 46 h, the reaction mixture was filtered, and the filtrate was evaporated under reduced pressure. After preparative TLC (dichloromethane: methanol = 10: 1) 269 mg (34%) of a pale brown crystalline product (
17) was obtained. Mp 123–130 °C. TLC (dichloromethane: methanol = 10: 1); R
f = 0.75. IR (KBr): 1156; 1223; 1262; 1619; 1732; 2944; 3258 cm
−1.
1H NMR (499.9 MHz; DMSO-d6) δ (ppm) 0.42 (t; J = 7.4 Hz; 3H; H3-18); 0.94 (dq; J = 14.0. 7.4 Hz; 1H; Hx-19); 1.46 (dq; J = 14.0. 7.4 Hz; 1H; Hy-19); 2.17–2.25 (m; 2H; H2-6); 2.41–2.60 (m; 8H; H2-2′. H2-3′. N(1)-CH3. Hx-5); 2.66 (s; 1H; H-21); 2.79 (ddd; J = 16.2. 2.7. 1.5 Hz; 1H; Hx-3); 3.25–3.31 (m; 1H; Hy-5); 3.42 (ddd; J = 16.2. 4.9. 1.2 Hz; 1H; Hy-3); 3.54 (t; J = 2.5 Hz; 1H; H-7′); 3.55 (s; 1H; H-2); 3.65 (s; 3H; C(16)-COOCH3); 3.70 (s; 3H; C(11)-OCH3); 4.64–4.72 (m; 2H; H2-5′); 5.13 (ddd; J = 10.2. 2.7. 1.2 Hz; 1H; H-15); 5.20 (s; 1H; H-17); 5.83 (ddd; J = 10.2. 4.9. 1.5 Hz; 1H; H-14); 6.19 (d; J = 2.3 Hz; 1H; H-12); 6.28 (dd; J = 8.2. 2.3 Hz; 1H; H-10); 7.05 (d; J = 8.2 Hz; 1H; H-9); 8.79 (s; 1H; C(16)-OH).
13C NMR (125.7 MHz; DMSO-d6) δ (ppm) 7.5 (C-18); 28.3 (C-3′); 28.5 (C-2′); 30.3 (C-19); 38.0 (N(1)-CH3); 42.4 (C-20); 43.6 (C-6); 50.3 (C-3); 51.0 (C-5); 51.70 (C(16)-COOCH3); 51.73 (C-5′); 52.0 (C-7); 55.0 (C(11)-OCH3); 66.1 (C-21); 76.1 (C-17); 77.6 (C-7′); 78.2 (C-6′); 78.6 (C-16); 82.8 (C-2); 95.4 (C-12); 104.5 (C-10); 123.0 (C-9); 124.3 (C-14); 125.3 (C-8); 129.8 (C-15); 153.4 (C-13); 160.4 (C-11); 171.1 (C-4′); 171.3 (C-1′); 171.5 (C(16)-COOCH3).
15N NMR (50.7 MHz; DMSO-d6) δ (ppm) −322.4 (N-4); −314.2 (N-1).
ESI-HRMS: M + H = 553.25395 (delta = −0.9ppm; C30H37O8N2). HR-ESI-MS-MS (CID = 35%; rel. int. %): 535(11); 493(13); 397(100); 365(4); 188(25).
3.11. The Click Reaction of 17-[4-Oxo-4-(prop-2-ynyloxy)butanoyloxy]vindoline (17) and 4-Fluorobenzyl-Azide (18)
299 mg (0.54 mmol) of 17-[4-oxo-4-(prop-2-ynyloxy)butanoyloxy]vindoline (17), 86 mg (0.57 mmol) of 4-fluorobenzyl-azide (18), 29 mg (0.11 mmol) of triphenylphosphine and 11 mg (0.054 mmol) of copper (I) iodide were dissolved in 12 mL of dry toluene, then 0.29 mL (1.6 mmol) of N,N-diisopropylethylamine (DIPEA) was added, and the reaction mixture was refluxed for 4 h. Then, 75 mL of toluene was added and the mixture was washed with 90 mL of distilled water, finally the aqueous phase was washed with another 30 mL of toluene. The organic phase was dried over MgSO4, then the solvent evaporated under reduced pressure. After preparative TLC (dichloromethane-methanol = 30:1 then dichloromethane-methanol = 15:1) 90 mg (24%) of a white crystalline product (19) was obtained. Mp 80–87 °C. TLC (dichloromethane-methanol = 15:1); Rf = 0,72. IR (KBr): 1158; 1225; 1503; 1738; 2838; 2962 cm−1.
1H NMR (499.9 MHz; DMSO-d6) δ (ppm) 0.41 (t; J = 7.4 Hz; 3H; H3-18); 0.93 (dq; J = 13.9. 7.4 Hz; 1H; Hx-19); 1.44 (dq; J = 13.9. 7.4 Hz; 1H; Hy-19); 2.17–2.25 (m; 2H; H2-6); 2.39–2.61 (m; 8H; H2-3′. H2-2′. N(1)-CH3. Hx-5); 2.66 (s; 1H; H-21); 2.79 (br d; J = 16.1 Hz; 1H; Hx-3); 3.25–3.31 (m; 1H; Hy-5); 3.40 (br dd; J = 16.1. 4.9 Hz; 1H; Hy-3); 3.55 (s; 1H; H-2); 3.63 (s; 3H; C(16)-COOCH3); 3.70 (s; 3H; C(11)-OCH3); 5.08–5.12 (m; 1H; H-15), 5.11 (s; 2H; H2-1′’); 5.20 (s; 1H; H-17); 5.58 (s; 2H; H2-7′’); 5.79 (ddd; J = 10.2. 4.9. 1.4 Hz; 1H; H-14); 6.19 (d; J = 2.2 Hz; 1H; H-12); 6.28 (dd; J = 8.2. 2.2 Hz; 1H; H-10); 7.05 (d; J = 8.2 Hz; 1H; H-9); 7.17–7.23 (m; 2H; H-10′’. H-12′’); 7.36–7.42 (m; 2H; H-9′’. H-13′’); 8.19 (s; 1H; H-6′’); 8.80 (s; 1H; C(16)-OH).
13C NMR (125.7 MHz; DMSO-d6) δ (ppm) 7.5 (C-18); 28.4 (C-2′); 28.5 (C-3′); 30.4 (C-19); 38.0 (N(1)-CH3); 42.4 (C-20); 43.6 (C-6); 50.3 (C-3); 51.1 (C-5); 51.7 (C(16)-COOCH3); 51.9 (C-7′’); 52.0 (C-7); 55.0 (C(11)-OCH3); 57.3 (C-1′’); 66.1 (C-21); 76.1 (C-17); 78.6 (C-16); 82.7 (C-2); 95.4 (C-12); 104.5 (C-10); 115.5 (d; 2JFC = 21.6 Hz; C-10′’. C-12′’); 123.0 (C-9); 124.2 (C-14); 124.6 (C-6′’); 125.3 (C-8); 129.8 (C-15); 130.3 (d; 3JFC = 8.4 Hz; C-9′’. C-13′’); 132.1 (d; 4JFC = 3.1 Hz; C-8′’); 142.0 (C-2′’); 153.4 (C-13); 160.5 (C-11); 161.8 (d; 1JFC = 244.4 Hz; C-11′’); 171.4 (C-1′); 171.6 (C-4′. C(16)-COOCH3).
15N NMR (50.7 MHz; DMSO-d6) δ (ppm) −322.8 (N-4); −314.4 (N-1); −130.8 (N-5′’); −27.7 (N-3′’); −18.7 (N-4′’).
ESI-HRMS: M + H = 704.30669 (delta = −3.3ppm; C37H43O8N5F). HR-ESI-MS-MS (CID = 35%; rel. int. %): 686(100); 644(5); 397(22).
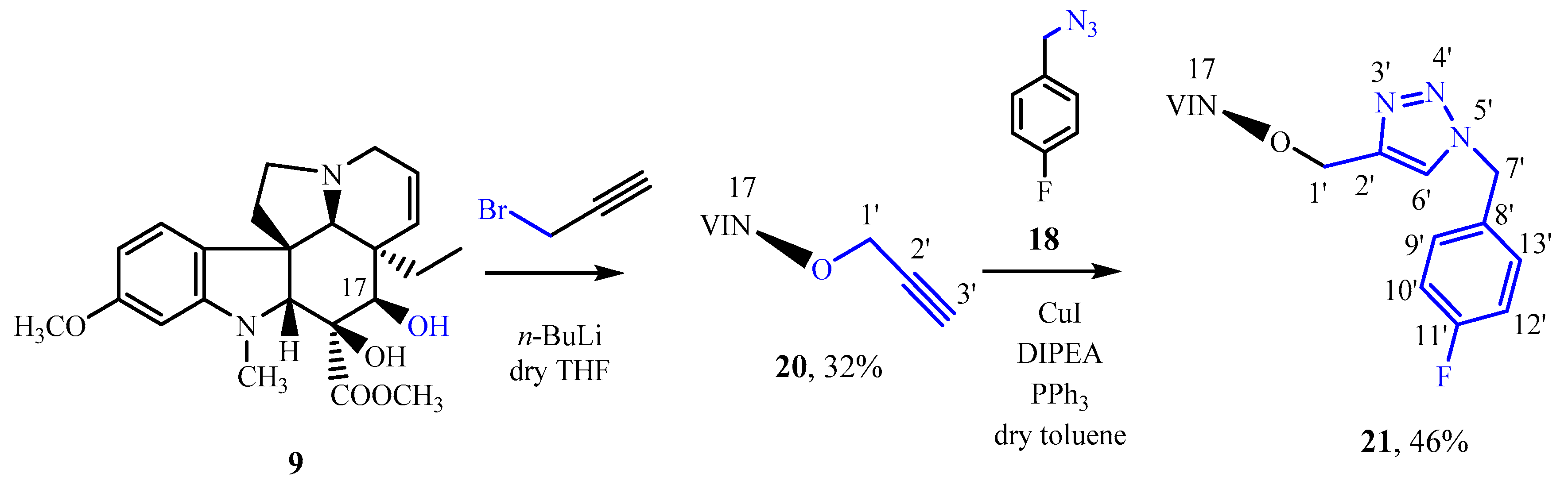
3.12. Synthesis of 17-Propargylvindoline (20)
17-Desacetylvindoline (
9) [
31] (600 mg, 1.4 mmol) and hexamethylphosphoramide (0.25 mL, 1.5 mmol) were dissolved in 18 mL of dry tetrahydrofuran under argon. The reaction mixture was cooled to 0 °C, and a solution of 0.60 mL (1.6 mmol) 2.5 M n-butyllithium in hexane was added dropwise via a syringe. After stirring for 5 min at 0 °C, propargyl bromide (0.156 mL, 1.4 mmol) was added, while maintaining the temperature at 0 ° C. After stirring for 16 h at room temperature, the mixture was diluted with 30 mL of distilled water and 60 mL of 10% Na
2CO
3 solution, then it was extracted with dichloromethane (3 × 60 mL). The organic phase was dried over MgSO
4, then the solvent evaporated under reduced pressure. After preparative TLC (dichloromethane-methanol = 20:1) 185 mg (32%) of a brown crystalline product (
20) was obtained. Mp 80–88 °C. TLC (dichloromethane-methanol = 20:1); R
f = 0,33. IR (KBr): 1080; 1245; 1502; 1735; 2931; 3276 cm
−1.
1H NMR (499.9 MHz; DMSO-d6) δ (ppm) 0.49 (t; J = 7.3 Hz; 3H; H3-18); 0.93 (dq; J = 13.7. 7.3 Hz; 1H; Hx-19); 1.43 (dq; J = 13.7. 7.3 Hz; 1H; Hy-19); 2.11–2.19 (m; 2H; H2-6); 2.50–2.56 (m; 1H; Hx-5); 2.57 (s; 3H; N(1)-CH3); 2.60 (s; 1H; H-21); 2.77 (ddd; J = 16.1. 2.5. 1.5 Hz; 1H; Hx-3); 3.22–3.28 (m; 1H; Hy-5); 3.37 (ddd; J = 16.1. 4.9. 1.2 Hz; 1H; Hy-3); 3.38 (t; J = 2.4 Hz; 1H; H-3′); 3.44 (s; 1H; H-2); 3.68 (s; 1H; H-17); 3.69 (s; 6H; C(16)-COOCH3. C(11)-OCH3); 4.20–4.27 (m; 2H; H2-1′); 5.46 (ddd; J = 10.2. 2.5. 1.2 Hz; 1H; H-15); 5.78 (ddd; J = 10.2. 4.9. 1.5 Hz; 1H; H-14); 6.16 (d; J = 2.3 Hz; 1H; H-12); 6.26 (d; J = 8.2. 2.3 Hz; 1H; H-10); 7.02 (d; J = 8.2 Hz; 1H; H-9); 8.47 (s; 1H; C(16)-OH).
13C NMR (125.7 MHz; DMSO-d6) δ (ppm) 7.7 (C-18); 32.1 (C-19); 38.4 (N(1)-CH3); 43.2 (C-20); 44.0 (C-6); 50.5 (C-3); 51.1 (C-5); 51.6 (C(16)-COOCH3); 51.9 (C-7); 55.0 (C(11)-OCH3); 60.7 (C-1′); 66.6 (C-21); 76.5 (C-3′); 79.9 (C-16); 81.1 (C-2′); 83.4 (C-2); 84.3 (C-17); 95.3 (C-12); 104.3 (C-10); 122.9 (C-9); 123.0 (C-14); 125.5 (C-8); 131.2 (C-15); 153.4 (C-13); 160.4 (C-11); 172.6 (C(16)-COOCH3).
ESI-HRMS: M + H = 453.23785 (delta = −1.2ppm; C26H33O5N2). HR-ESI-MS-MS (CID = 35%; rel. int. %): 435(21); 397(45); 393(100); 188(32).
3.13. The Click Reaction of 17-Propargylvindoline (20) and 4-Fluorobenzyl-Azide (18)
205 mg (0.45 mmol) of 17-propargylvindoline (20), 72 mg (0.48 mmol) of 4-fluorobenzyl-azide, 24 mg (0.09 mmol) of triphenylphosphine and 9 mg (0.05 mmol) of copper (I) iodide were dissolved in 10 mL of dry toluene, then 0 24 mL (1.4 mmol) of DIPEA was added, and the reaction mixture was stirred at reflux for 5 h. Then, 60 mL of toluene was added, and the mixture was washed with 75 mL of distilled water, finally the aqueous phase was washed with another 25 mL of toluene. The organic phase was dried over MgSO4, then the solvent evaporated under reduced pressure. After preparative TLC (dichloromethane-methanol = 30:1) 126 mg (46%) of a yellow crystalline product (21) was obtained. Mp 92–95 °C. TLC (dichloromethane-methanol = 30:1); Rf = 0,24. IR (KBr): 1085, 1224; 1245; 1502; 1736; 2837; 2946 cm−1.
1H NMR (799.7 MHz; DMSO-d6) δ (ppm) 0.50 (t; J = 7.3 Hz; 3H; H3-18); 0.98 (dq; J = 13.7. 7.3 Hz; 1H; Hx-19); 1.38 (dq; J = 13.7. 7.3 Hz; 1H; Hy-19); 2.13–2.17 (m; 2H; H2-6); 2.50–2.54 (m; 1H; Hx-5); 2.59 (s; 3H; N(1)-CH3); 2.61 (s; 1H; H-21); 2.74–2.78 (m; 1H; Hx-3); 3.21–3.25 (m; 1H; Hy-5); 3.32–3.36 (m; 1H; Hy-3); 3.44 (s; 1H; H-2); 3.66 (s; 3H; C(16)-COOCH3); 3.70 (s; 3H; C(11)-OCH3); 3.85 (s; 1H; H-17); 4.62.–4.68 (AB; J = 11.2 Hz; 2H; H2-1′); 5.42–5.45 (m; 1H; H-15); 5.56 (~s; 2H; H2-7′); 5.73 (ddd; J = 10.2. 5.0. 1.5 Hz; 1H; H-14); 6.17 (d; J = 2.3 Hz; 1H; H-12); 6.27 (dd; J = 8.2. 2.3 Hz; 1H; H-10); 7.02 (d; J = 8.2 Hz; 1H; H-9); 7.18–7.21 (m; 2H; H-10′. H-12′); 7.37–7.40 (m; 2H; H-9′. H-13′); 7.97 (s; 1H; H-6′); 8.43 (br s; 1H; C(16)-OH).
13C NMR (201.1 MHz; DMSO-d6) δ (ppm) 7.7 (C-18); 32.5 (C-19); 38.4 (N(1)-CH3); 43.4 (C-20); 44.1 (C-6); 50.5 (C-3); 51.0 (C-5); 51.5 (C(16)-COOCH3); 51.7 (C-7′); 51.9 (C-7); 54.9 (C(11)-OCH3); 66.56 (C-1′); 66.60 (C-21); 80.0 (C-16); 83.4 (C-2); 83.6 (C-17); 95.3 (C-12); 104.2 (C-10); 115.5 (d; 2JFC = 21.6 Hz; C-10′. C-12′); 122.9 (C-9); 123.0 (C-14); 123.6 (C-6′); 125.6 (C-8); 130.2 (d; 3JFC = 8.4 Hz; C-9′. C-13′); 131.1 (C-15); 132.3 (d; 4JFC = 3.0 Hz; C-8′); 144.7 (C-2′); 153.4 (C-13); 160.4 (C-11); 161.7 (d; 1JFC = 244.4 Hz; C-11′); 172.6 (C(16)-COOCH3).
15N NMR (81.0 MHz; DMSO-d6) δ (ppm) −322.7 (N-4); −314.1 (N-1); −131.5 (N-5′); −28.6 (N-3′); -20.5 (N-4′).
ESI-HRMS: M + H = 604.29147 (delta = −2.5ppm; C33H39O5N5F). HR-ESI-MS-MS (CID = 35%; rel. int. %): 586(18); 554(19); 544(82); 484(9); 415(100); 397(43); 377(27); 365(17); 355(28); 188(22).
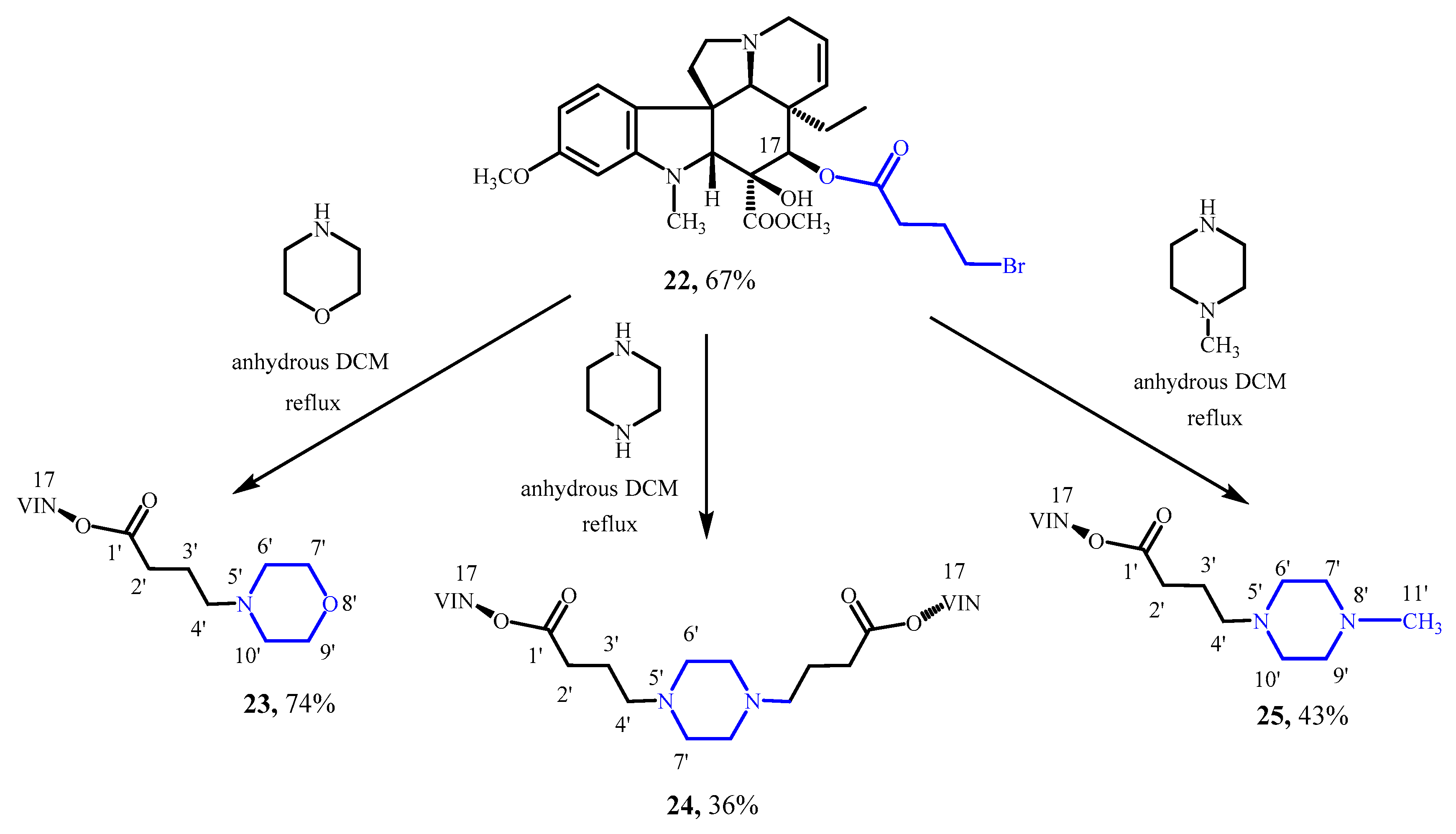
3.14. N-Alkylation of Morpholine with 17-(4-Bromobutanoyloxy)vindoline (22)
17-(4-Bromobutanoyloxy)vindoline (
22) [
22,
23] (120 mg, 0.21 mmol) was dissolved in 6 mL of anhydrous DCM, and the stirring was started under argon. Then, 0,04 mL (0.43 mmol) of morpholine was added. The reaction mixture was stirred at room temperature for 2 h, then at reflux for 11 h. The mixture was then allowed to cool to room temperature. After the addition of 4 mL of DCM, the mixture was extracted with 6 mL of 10% Na
2CO
3 solution. The aqueous phase was washed with 6 mL of DCM, then the organic phase was washed with 6 mL of distilled water. The organic phase was dried over MgSO
4, then the solvent evaporated under reduced pressure. After preparative TLC (dichloromethane-methanol = 15:1) 90 mg (74%) of a yellow oleic product (
23) was obtained. TLC (dichloromethane-methanol = 20:1); R
f = 0.27. IR (KBr): 2954, 2808, 1735, 1613, 1500, 1223, 1115, 1024 cm
−1.
1H NMR (499.9 MHz; CDCl3) δ (ppm) 0.48 (t; J = 7.4 Hz; 3H; H3-18); 1.14 (dq; J = 14.4. 7.4 Hz; 1H; Hx-19); 1.64 (dq; J = 14.4. 7.4 Hz; 1H; Hy-19); 1.78–1.84 (m; 2H; H2-3′); 2.27–2.41 (m; 6H; H2-6. H2-4′. H2-2′); 2.41–2.48 (br m; 4H; H2-6′. H2-10′); 2.49–2.55 (m; 1H; Hx-5); 2.66 (s; 1H; H-21); 2.67 (s; 3H; N(1)-CH3); 2.82 (ddd; J = 16.0. 2.6. 1.6 Hz; 1H; Hx-3); 3.39–3.45 (m; 1H; Hy-5); 3.49 (ddd; J = 16.0. 4.9. 1.3 Hz; 1H; Hy-3); 3.67–3.73 (br m; 4H; H2-7′. H2-9′); 3.74 (s; 1H; H-2); 3.779 (s; 3H). 3.780 (s; 3H): C(16)-COOCH3. C(11)-OCH3; 5.23 (ddd; J = 10.2. 2.6. 1.3 Hz; 1H; H-15); 5.46 (s; 1H; H-17); 5.84 (ddd; J = 10.2. 4.9. 1.6 Hz; 1H; H-14); 6.07 (d; J = 2.3 Hz; 1H; H-12); 6.30 (dd; J = 8.2. 2.3 Hz; 1H; H-10); 6.89 (d; J = 8.2 Hz; 1H; H-9); 9.52 (br; 1H; C(16)-OH).
13C NMR (125.7 MHz; CDCl3) δ (ppm) 7.7 (C-18); 21.7 (br; C-3′); 30.8 (C-19); 31.8 (C-2′); 38.3 (N(1)-CH3); 42.9 (C-20); 44.1 (C-6); 51.1 (C-3); 52.0 (C-5); 52.2 (C(16)-COOCH3); 52.8 (C-7); 53.5 (br; C-6′. C-10′); 55.4 (C(11)-OCH3); 57.9 (C-4′); 66.9 (br; C-7′. C-9′); 67.1 (C-21); 76.3 (C-17); 79.6 (C-16); 83.4 (C-2); 95.8 (C-12); 104.6 (C-10); 122.7 (C-9); 124.1 (C-14); 125.1 (C-8); 130.6 (C-15); 153.7 (C-13); 161.2 (C-11); 171.9 (C(16)-COOCH3); 173.2 (C-1′).
ESI-HRMS: M + H = 570.31692 (delta = −0.8 ppm; C31H44O7N3). HR-ESI-MS-MS (CID = 35%; rel. int. %): 552(100); 415(2); 397(25); 188(1).
3.15. N-Alkylation of Piperazine with 17-(4-Bromobutanoyloxy)vindoline (22)
17-(4-Bromobutanoyloxy)vindoline (
22) [
22,
23] (140 mg, 0.25 mmol) was dissolved in 6 mL of anhydrous DCM, and the stirring was started under argon. Then, 43 mg (0.50 mmol) of piperazine was added. The reaction mixture was stirred at reflux for 7 h. The mixture was then allowed to cool to room temperature, and after the addition of a few DCM, the mixture was extracted with 6 mL of 10% Na
2CO
3 solution. The aqueous phase was washed with 6 mL of DCM, then the organic phase was washed with 6 mL of distilled water. The organic phase was dried over MgSO
4, then the solvent evaporated under reduced pressure. After preparative TLC (dichloromethane-methanol = 15:1) 47 mg (36%) of a pale yellow crystalline product (
24) was obtained. Mp 118–124 °C. TLC (dichloromethane-methanol = 15:1); R
f = 0,05. IR (KBr): 2947, 2808, 1740, 1616 1502, 1246, 1170 cm
−1.
The molecule is symmetric, it has two equivalent halves. The NMR assignment below applies to the other half of the molecule as well.
1H NMR (499.9 MHz; DMSO-d6) δ (ppm) 0.42 (t; J = 7.4 Hz; 6H; H3-18); 0.95 (dq; J = 13.9. 7.4 Hz; 2H; Hx-19); 1.48 (dq; J = 13.9. 7.4 Hz; 2H; Hy-19); 1.54–1.68 (m; 4H; H2-3′); 2.16–2.28 (m; 12H; H2-6. H2-2′. H2-4′); 2.22–2.43 (br; 8H; H2-6′. H2-7′); 2.53–2.60 (m; 8H; N(1)-CH3. Hx-5); 2.64 (s; 2H; H-21); 2.79 (br d; J = 16.4 Hz; 2H; Hx-3); 3.25–3.31 (m; 2H; Hy-5); 3.41 (br dd; J = 16.4. 4.8 Hz; 2H; Hy-3); 3.54 (s; 2H; H-2); 3.63 (s; 6H; C(16)-COOCH3); 3.70 (s; 6H; C(11)-OCH3); 5.09 (br d; J = 10.2 Hz; 2H; H-15); 5.19 (s; 2H; H-17); 5.81 (ddd; J = 10.2. 4.8. 1.2 Hz; 2H; H-14); 6.19 (d; J = 2.2 Hz; 2H; H-12); 6.28 (dd; J = 8.2. 2.2 Hz; 2H; H-10); 7.05 (d; J = 8.2 Hz; 2H; H-9); 8.75 (s; 2H; C(16)-OH).
13C NMR (125.7 MHz; DMSO-d6) δ (ppm) 7.5 (C-18); 21.5 (C-3′); 30.3 (C-19); 31.3 (C-2′); 38.0 (N(1)-CH3); 42.4 (C-20); 43.6 (C-6); 50.4 (C-3); 51.1 (C-5); 51.6 (C(16)-COOCH3); 52.0 (C-7); 52.6 (C-6′. C-7′); 55.0 (C(11)-OCH3); 56.6 (C-4′); 66.1 (C-21); 75.7 (C-17); 78.6 (C-16); 82.8 (C-2); 95.4 (C-12); 104.5 (C-10); 123.0 (C-9); 124.3 (C-14); 125.3 (C-8); 129.9 (C-15); 153.4 (C-13); 160.4 (C-11); 171.6 (C(16)-COOCH3); 172.4 (C-1′).
ESI-HRMS: M + H = 1051.57418 (delta = −0.8 ppm; C58H79O12N6). HR-ESI-MS-MS (CID = 35%; rel. int. %): 1034(100); 1016(10); 655(2); 637(22); 619(11); 397(12).
3.16. N-Alkylation of N-Methylpiperazine with 17-(4-Bromobutanoyloxy)vindoline (22)
17-(4-Bromobutanoyloxy)vindoline (
22) [
22,
23] (180 mg, 0.32 mmol) was dissolved in 8 mL of anhydrous DCM, and the stirring was started under argon. Then, 0,072 mL (0.64 mmol) of
N-methylpiperazine was added. The reaction mixture was stirred at reflux for 12 h. The mixture was then allowed to cool to room temperature, and after the addition of a few DCM, the mixture was extracted with 6 mL of 10% Na
2CO
3 solution. The aqueous phase was washed with 6 mL of DCM, then the organic phase was washed with 6 mL of distilled water. The organic phase was dried over MgSO
4, then the solvent evaporated under reduced pressure. After preparative TLC (dichloromethane-methanol = 15:1) 80 mg (43%) of a white crystalline product (
25) was obtained. Mp 65–70 °C. TLC (dichloromethane-methanol = 20:1); R
f = 0.06. IR (KBr): 2937, 2794, 1740, 1616, 1502, 1244, 1166 cm
−1.
1H NMR (499.9 MHz; DMSO-d6) δ (ppm) 0.42 (t; J = 7.4 Hz; 3H; H3-18); 0.95 (dq; J = 14.0. 7.4 Hz; 1H; Hx-19); 1.49 (dq; J = 14.0. 7.4 Hz; 1H; Hy-19); 1.55–1.68 (m; 2H; H2-3′); 2.13 (s; 3H; H3-11′); 2.16–2.28 (m; 6H; H2-6. H2-2′. H2-4′); 2.17–2.44 (br; 8H; H2-7′. H2-9′. H2-6′. H2-10′); 2.53–2.61 (m; 4H; N(1)-CH3. Hx-5); 2.66 (s; 1H; H-21); 2.79 (br d; J = 16.3 Hz; 1H; Hx-3); 3.25–3.31 (m; 1H; Hy-5); 3.41 (br dd; J = 16.3. 4.9 Hz; 1H; Hy-3); 3.54 (s; 1H; H-2); 3.64 (s; 3H; C(16)-COOCH3); 3.71 (s; 3H; C(11)-OCH3); 5.10 (br d; J = 10.2 Hz; 1H; H-15); 5.19 (s; 1H; H-17); 5.81 (ddd; J = 10.2. 4.9. 1.2 Hz; 1H; H-14); 6.19 (d; J = 2.2 Hz; 1H; H-12); 6.28 (dd; J = 8.2. 2.2 Hz; 1H; H-10); 7.05 (d; J = 8.2 Hz; 1H; H-9); 8.76 (s; 1H; C(16)-OH).
13C NMR (125.7 MHz; DMSO-d6) δ (ppm) 7.5 (C-18); 21.5 (C-3′); 30.3 (C-19); 31.2 (C-2′); 38.0 (N(1)-CH3); 42.4 (C-20); 43.6 (C-6); 45.6 (C-11′); 50.4 (C-3); 51.1 (C-5); 51.6 (C(16)-COOCH3); 52.0 (C-7); 52.5 (C-6′. C-10′); 54.6 (C-7′. C-9′); 55.0 (C(11)-OCH3); 56.6 (C-4′); 66.1 (C-21); 75.7 (C-17); 78.7 (C-16); 82.8 (C-2); 95.4 (C-12); 104.5 (C-10); 123.0 (C-9); 124.3 (C-14); 125.3 (C-8); 129.9 (C-15); 153.4 (C-13); 160.5 (C-11); 171.6 (C(16)-COOCH3); 172.4 (C-1′).
EI-HRMS: M = 582.34230 (delta = 1.9 ppm; C32H46O6N4).
3.17. Cell Lines and Reagents
Human breast cancer cell lines MCF-7 and MDA-MB-231, and human cervical cancer HeLa cells were purchased from ECCAC - European Collection of Cell Cultures, Salisbury, UK. SiHa cervical cancer cells were obtained from the American Type Culture Collection (ATCC), Manassas, VA, USA. Eagle’s minimum essential medium (EMEM), fetal bovine serum (FBS), penicillin–amphotericin B, non-essential amino acids, trypsin, were purchased from Lonza Group Ltd. (Basel, Switzerland) while Trypan blue solution, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and dimethyl-sulfoxide (DMSO) were from Sigma-Aldrich Ltd. (Budapest, Hungary). All the cancer cell lines were cultured in 75 cm
2 flask (Orange Scientific, Hungary) in EMEM media according to the distributor’s instructions, supplemented with 10% fetal bovine serum, 1% non-essential amino acids and an antibiotic–antimycotic mixture. The cells were incubated in cultured conditions at 37 °C under humidified atmosphere with 5% carbon dioxide CO
2 [
33].
3.18. Antiproliferative Assay
The antiproliferative activity of the prepared compounds was determined by colorimetric MTT assay on a panel of human cancer cell lines isolated from breast (MCF-7 and MDA-MB-231) and cervical (HeLa and SiHa) cancers as published before [
34]. Briefly, the cells were seeded in 96-well microplates with an initial cell density of 5 × 10
3 cells per well and incubated overnight to allow cells’ attachment in the bottom of wells. Afterwards the cells were treated with mediums containing six different concentrations (0.1, 0.3, 1, 3, 10, 30 µM) of the tested compounds and incubated for 72 h. We note that, under the cell culture conditions, the highest DMSO content of the medium (0.3%) did not have any substantial effect on cell proliferation. Subsequently, the cells were incubated with 44 µL of 5 mg/mL MTT solution for 4 h. Then, the medium was discarded and the precipitated formazan crystals were solubilized in 100 µL of DMSO by gently shaking for 60 min at 37 °C. Finally, absorbance was measured at 545 nm wavelength by using a microplate reader. Antiproliferative activity was determined as a percentage of inhibition compared to the untreated cell control. IC
50 values, representing the 50% inhibitory concentrations, were obtained from nonlinear regression using Graph Pad Prism 5.01 (Graph Pad Software; San Diego, CA, USA). All in vitro tests were carried out in two separate experiments, n = 5 each.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}