3.2. Chemical Synthesis
3.2.1. 3-(3-Formyl-4-methoxyphenyl)-(E)-propenoic acid (9)
5-Iodo-2-methoxy-benzaldehyde (1.0 g, 3.82 mmol) was dissolved in 25 mL of acetonitrile and oxygen was removed by bubbling nitrogen through the solution. Triethylamine (3.71 mL, 26.7 mmol) and methyl acrylate (0.686, mL, 7.63 mmol) were added dropwise with stirring to the solution, followed by four portions of tri-o-tolylphosphine (0.0214 g, 0.095 mmol) and palladium (II) acetate (0.008 g, 0.047 mmol) at 1 h intervals. The mixture was then heated at 65–70 °C for 4 h, volatiles were removed by distillation, and the residue was dissolved in dichloromethane. The resulting solution was percolated through silica gel, eluting with a mixture of hexane–ethyl acetate (6:4). The percolate was evaporated to dryness, re-dissolved in 20 mL of methanol, and 20% aqueous potassium carbonate solution (10 mL) added. The mixture was stirred for 6 h at 20 °C and concentrated under reduced pressure to a third of its volume. The solution was acidified with conc. HCl (to pH 1) and the precipitate was filtered and recrystallized from isopropanol, to give 3-formyl-4-methoxycinnamic acid 9 as a crystalline white solid (0.472 g). A second harvest obtained by concentration of the mother liquor gave an additional 0.105 g (73%); m.p. 223–225 °C; 1H-NMR (DMSO-d6) δ: 12.35 (s, 1H, COOH), 10.33 (s, 1H, ArCHO), 8.03 (dd, J = 2.4, 8.8 Hz, 1H, 6′-H), 7.93 (d, J = 2.4 Hz, 1H, 2′-H), 7.59 (d, J = 16.0 Hz, 1H, 3-H), 7.28 (d, J = 8.8 Hz, 1H, 5′-H), 6.47 (d, J = 16.0 Hz, 1H, 2-H), 3.96 (s, 3H, CH3O); 13C-NMR (DMSO-d6) δ: 188.9 (ArCHO), 167.5 (1-C), 162.6 (4′-C), 142.6 (3-C), 135.6 (6′-C), 128.2 (2′-C), 126.9 (1′-C), 124.2 (3′-C), 118.3 (2-C), 113.4 (5′-C), 56.4 (CH3O). EIMS m/z (%): 206 (18, M+), 81 (33), 69 (100), 57 (25), 55 (33), 43 (39), 41 (58).
3.2.2. 3-[3-(difluoromethyl)-4-methoxyphenyl]-(E)-propenoyl fluoride (10)
A solution of Deoxofluor® 50% in toluene (0.107 mL, 0.435 mmol) was added dropwise to a suspension of 0.030 g of 3-formyl-4-methoxycinnamic acid (9, 0.030 g, 0.145 mmol) in 0.4 mL of dry dichloromethane under an argon atmosphere. The reaction mixture was stirred for 45 min at room temperature, diluted with 2 mL of dry dichloromethane and subsequently percolated through 2.5 g of silica gel under an argon atmosphere. The silica gel bed was rinsed with 8 mL of dry dichloromethane and the resulting solution was concentrated to dryness. The resulting solid was purified by flash column chromatography eluting with mixtures of ethyl acetate–hexane of increasing polarity to give 10 as a white solid (0.028 g, 84%). 1H-NMR (CDCl3, 200.13 MHz) δ: 7.80 (d, J = 16.0 Hz, 1H, 3-H), 7.79 (bs, 1H, 2′-H), 7.64 (m, 1H, 6′-H), 7.00 (m, 1H, 5′-H), 6.93 (t, J = 55.3 Hz, 1H, CF2H), 6.29 (dd, J = 7.1, 16.0 Hz, 1H, 2-H), 3.94 (s, 3H, CH3O); 13C-NMR (CDCl3, 50.32 MHz) δ: 157.2 (d, J = 338 Hz, 1-C), 159.9 (t, J = 6 Hz, 4′-C), 150.1 (d, J = 6 Hz, 3-C), 133.0 (s, 6′-C), 126.7 (t, J = 6.0 Hz, 2′-C), 125.9 (s, 1′-C), 123.7 (t, J = 22 Hz, 3′-C), 111.5 (s, 5′-C), 110.8 (t, J = 236 Hz, CF2H), 110.7 (d, J = 68 Hz, 2-C), 56.0 (s, CH3O).
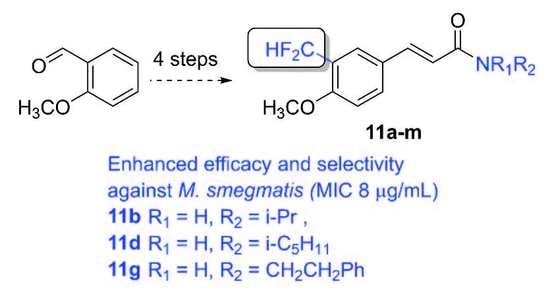
3.2.3. Representative Procedure. Preparation of N-(1-Methylethyl)-3-[3-(difluoromethyl)-4-methoxyphenyl]-(E)-propenamide (11b)
A solution of Deoxofluor® 50% in toluene (0.107 mL, 0.435 mmol) was added dropwise to a suspension of 0.030 g of 3-formyl-4-methoxycinnamic acid (9, 0.030 g, 0.145 mmol) in 0.4 mL of dry dichloromethane under an argon atmosphere. The reaction mixture was stirred for 45 min at room temperature, diluted with 2 mL of dry dichloromethane, and subsequently percolated through 2.5 g of silica gel under an argon atmosphere. The silica gel bed was rinsed with 8 mL of dry dichloromethane and the resulting solution was concentrated under a nitrogen flow to a final volume of 1 mL. Isopropylamine (0.037 mL, 0.435 mmol) and triethylamine (0.061 mL, 0.435 mmol) were added and the mixture was subsequently stirred for 1 h at room temperature. The solution was diluted with 10 mL of dichloromethane, washed twice with 1M HCl and once with water, dried with anhydrous sodium sulfate, and the solvent evaporated. The resulting solid was purified by flash column chromatography eluting with mixtures of ethyl acetate–hexane of increasing polarity to give 11b as a crystalline white solid (0.025 g, 64%). m.p. 113–114 °C; 1H-NMR (CDCl3) δ: 7.75–7.71 (m, 1H, 2′-H), 7.57 (d, J = 15.5 Hz, 1H, 3-H), 7.53–7.49 (m, 1H, 6′-H), 6.92 (t, J = 55.5 Hz, 1H, CF2H), 6.94–6.88 (m, 1H, 5′-H), 6.31 (d, J = 15.6 Hz, 1H, 2-H), 5.60–5.54 (m, NH), 4.29 – 4.16 (m, 1H, 1′′-H), 3.89 (s, 3H, CH3O), 1.22 (d, J = 6.5 Hz, 6H, 2′′-H); 13C-NMR (CDCl3) δ: 165.1 (s, 1-C), 158.3 (t, J = 6.3 Hz, 4′-C), 139.6 (s, 3-C), 132.4 (t, J = 2.1 Hz, 6′-C), 127.8 (s, 1′-C), 125.0 (t, J = 5.9 Hz, 2′-C), 123.2 (t, J = 22.2 Hz, 3′-C), 120.1 (s, 2-C), 111.3 (t, J = 236.3 Hz, CF2H), 111.3 (s, 5′-C), 56.0 (s, CH3O), 41.7 (s, 1′′-C), 23.0 (s, 2′′-C). EIMS m/z (%): 269 (57, M+), 211 (100), 183 (23), 132 (19), 58 (34). Analysis for C14H17F2NO2: Calcd: C, 62.42; H, 6.36; N, 5.20%. Found: C, 62.45; H, 6.32; N, 4.98%.
3.2.4. N-Methyl-3-[3-(difluoromethyl)-4-methoxyphenyl]-(E)-propenamide (11a)
Compound 11a was prepared from acid 9 (30.0 mg, 0.145 mmol), Deoxofluor® 50% in toluene (0.107 mL, 0.435 mmol), methylamine hydrochloride (0.030 g, 0.435 mmol), and triethylamine (0.082 mL, 0.580 mmol) following the procedure described for 11b. Compound 11a was obtained as a crystalline white solid (0.021 g, 60 %); m.p. 169–171 °C; 1H-NMR (CDCl3-CD3OD 9:1) δ: 7.76–7.71 (m, 1H, 2′-H), 7.57–7.54 (m, 1H, 6′-H), 7.54 (d, J = 15.5 Hz, 1H, 3-H), 6.97–6.93 (m, 1H, 5′-H), 6.93 (t, J = 55.5 Hz, 1H, CF2H), 6.40 (d, J = 15.7 Hz, 1H, 2-H), 3.90 (s, 3H, CH3O), 2.89 (s, 3H, NCH3); 13C-NMR (CDCl3-CD3OD 9:1) δ: 167.5 (1-C), 158.2 (t, JCF = 5.7 Hz, 4′-C), 139.4 (3-C), 132.2 (6′-C), 127.6 (1′-C), 125.0 (t, JCF = 5.9 Hz, 2′-C), 123.0 (t, JCF = 22.2 Hz, 3′-C), 119.3 (2-C), 111.2 (t, JCF = 236.1 Hz, CF2H), 111.2 (5′-C), 55.8 (CH3O), 26.2 (NCH3). EIMS m/z (%): 241 (55, M+), 240 (26), 211 (100), 183 (25), 132 (22). Analysis for C12H13F2NO2: Calcd: C, 59.75; H, 5.43; N, 5.81 %. Found: C, 59.27; H, 5.28; N, 5.66 %.
3.2.5. N-(2-Methylpropyl)-3-[3-(difluoromethyl)-4-methoxyphenyl]-(E)-propenamide (11c)
Compound 11c was prepared from acid 9 (30.0 mg, 0.145 mmol), Deoxofluor® 50% in toluene (0.107 mL, 0.435 mmol), isobutylamine (0.044 mL, 0.435 mmol), and triethylamine (0.061 mL, 0.435 mmol) following the procedure described for 11b. Compound 11c was obtained as a crystalline white solid (0.027 g, 66 %); m.p. 145–146 °C; 1H-NMR (CDCl3) δ: 7.77–7.72 (m, 1H, 2′-H), 7.59 (d, J = 15.5 Hz, 1H, 3-H), 7.56–7.49 (m, 1H, 6′-H), 6.93 (t, J = 55.5 Hz, 1H, CF2H), 6.94–6.90 (m, 1H, 5′-H), 6.35 (d, J = 15.5 Hz, 1H, 2-H), 5.74 (t, J = 5.4 Hz, 1H, NH), 3.89 (s, 3H, CH3O), 3.23 (dd, J = 6.2, 6.7 Hz, 2H, 1′′-H), 1.92–1.77 (1 H, m, 2′′-H), 0.96 (d, J = 6.7 Hz, 6H, 3′′-H); 13C-NMR (CDCl3) δ: 166.0 (1-C), 158.3 (t, JCF = 5.7 Hz, 4′-C), 139.8 (3-C), 132.4 (6′-C), 127.8 (1′-C), 125.0 (t, JCF = 5.9 Hz, 2′-C), 123.3 (t, JCF = 22.3 Hz, 3′-C), 119.8 (2-C), 111.3 (t, JCF = 236.2 Hz, CF2H), 111.3 (5′-C), 56.0 (CH3O), 47.2 (1′′-C), 28.8 (2′′-C), 20.3 (3′′-C). EIMS m/z (%): 283 (30, M+), 226 (53), 211 (100), 183 (22), 132 (20), 43 (18). Analysis for C15H19F2NO2·0.5H2O: Calcd: C, 61.63; H, 6.90; N, 4.79%. Found: C, 61.51; H, 6.57; N, 4.76%.
3.2.6. N-(3-Methylbutyl)-3-[3-(difluoromethyl)-4-methoxyphenyl]-(E)-propenamide (11d)
Compound 11d was prepared from acid 9 (30.0 mg, 0.145 mmol), Deoxofluor® 50% in toluene (0.107 mL, 0.435 mmol), isopentylamine (0.051 mL, 0.435 mmol), and triethylamine (0.061 mL, 0.435 mmol) following the procedure described for 11b. Compound 11d was obtained as a crystalline white solid (0.025 g, 58%); m.p. 110–111 °C; 1H-NMR (CDCl3) δ: 7.80–7.68 (m, 1H, 2′-H), 7.58 (d, J = 15.5 Hz, 1H, 3-H), 7.55–7.49 (m, 1H, 6′-H), 6.92 (t, J = 55.5 Hz, 1H, CF2H), 6.97–6.85 (m, 1H, 5′-H), 6.33 (d, J = 15.5 Hz, 1H, 2-H), 5.71 (t, J = 5.6 Hz, 1H, NH), 3.89 (s, 3H, CH3O), 3.50–3.33 (m, 2H, 1′′-H), 1.72–1.61 (1 H, m, 2′′-H), 1.46 (dt, J = 7.0, 8.5 Hz, 2H, 3′′-H), 0.94 (d, J = 6.6 Hz, 6H, 4′′-H); 13C-NMR (CDCl3) δ: 165.9 (1-C), 158.3 (t, JCF = 5.7 Hz, 4′-C), 139.7 (3-C), 132.4 (t, JCF = 1.9 Hz, 6′-C), 127.8 (1′-C), 125.0 (t, JCF = 5.9 Hz, 2′-C), 123.2 (t, JCF = 22.2 Hz, 3′-C), 119.9 (2-C), 111.3 (t, JCF = 236.2 Hz, CF2H), 111.3 (5′-C), 56.0 (CH3O), 38.7 (2′′-C), 38.2 (1′′-C), 26.0 (3′′-C), 22.6 (4′′-C). EIMS m/z (%): 297 (30, M+), 241 (44), 240 (32), 226 (22), 211 (100), 183 (27), 132 (25). Analysis for. C16H21F2NO2: Calcd: C, 64.63; H, 7.12; N, 4.71%. Found: C, 64.48; H, 7.05; N, 4.74%.
3.2.7. N-Cyclohexyl-3-[3-(difluoromethyl)-4-methoxyphenyl]-(E)-propenamide (11e)
Compound 11e was prepared from acid 9 (30.0 mg, 0.145 mmol), Deoxofluor® 50% in toluene (0.107 mL, 0.435 mmol), cyclohexylamine (0.050 mL, 0.435 mmol), and triethylamine (0.061 mL, 0.435 mmol) Following the procedure used for 11b. Compound 11e was obtained as a crystalline white solid (0.034 g, 76%); m.p. 181–183 °C; 1H-NMR (CDCl3) δ: 7.75–7.73 (m, 1H, 2′-H), 7.58 (d, J = 15.5 Hz, 1H, 3-H), 7.52 (m, 1H, 6′-H), 6.92 (t, J = 55.5 Hz, 1H, CF2H), 6.93–6.90 (m, 1H, 5′-H), 6.32 (d, J = 15.5 Hz, 1H, 2-H), 5.61 (d, J = 6.2 Hz, 1H, NH), 3.97–3.90 (m, 1H, 1′′-H), 3.89 (s, 3H, CH3O), 2.04–1.92 (m, 2H, 2′′-Heq), 1.79–1.69 (m, 2H, 3′′-Heq), 1.68–1.59 (m, 1H, 4′′-Heq), 1.48–1.34 (m, 2H, 3′′-Hax), 1.29–1.11 (m, 3H, 2′′-Hax and 4′′-Hax); 13C-NMR (CDCl3) δ: 165.0 (1-C), 158.3 (t, JCF = 5.6 Hz, 4′-C), 139.6 (3-C), 132.4 (6′-C), 127.9 (1′-C), 125.0 (t, JCF = 5.8 Hz, 2′-C), 123.3 (t, JCF = 22.3 Hz, 3′-C), 120.2 (2-C), 111.3 (t, JCF = 236.2 Hz, CF2H), 111.3 (5′-C), 56.0 (CH3O), 48.5 (1′′-C), 33.4 (2′′-C), 25.7 (4′′-C), 25.0 (3′′-C). EIMS m/z (%): 309 (62, M+), 226 (64), 211 (100), 183 (33), 132 (25), 98 (39). Analysis for C17H21F2NO2: Calcd C, 66.00; H, 6.84; N, 4.53%. Found: C, 65.85; H, 6.86; N, 4.36%.
3.2.8. N-Phenyl-3-[3-(difluoromethyl)-4-methoxyphenyl]-(E)-propenamide (11f)
Compound 11f was prepared from acid 9 (30.0 mg, 0.145 mmol), Deoxofluor® 50% in toluene (0.107 mL, 0.435 mmol), aniline (0.040 mL, 0.435 mmol), and triethylamine (0.061 mL, 0.435 mmol) following the procedure used for 11b. Compound 11f was obtained as a crystalline white solid (0.025 g, 57%); m.p. 138–139 °C; 1H-NMR (CDCl3) δ: 7.76 (m, 1H, 2′-H), 7.70 (d, J = 15.5 Hz, 1H, 3-H), 7.63 (d, J = 7.2 Hz, 2H, 2′′-H), 7.60 (bs, 1H, NH), 7.54–7.50 (m, 1H, 6′-H), 7.37–7.31 (m, 2H, 3′′-H), 7.14–7.10 (m, 1H, 4′′-H), 6.92 (t, J = 55.5 Hz, 1H, CF2H), 6.92–6.88 (m, 1H, 5′-H), 6.50 (d, J = 15.5 Hz, 1H, 2-H), 3.89 (s, 3H, CH3O); 13C-NMR (CDCl3) δ: 164.0 (1-C), 158.5 (t, JCF = 5.6 Hz, 4′-C), 141.2 (3-C), 138.1 (1′′-C), 132.5 (6′-C), 129.1 (3′′-C), 127.4 (1′-C), 125.2 (t, JCF = 5.7 Hz, 2′-C), 124.4 (4′′-C), 123.2 (t, JCF = 22.2 Hz, 3′-C), 120.0 (2′′-C), 119.7 (2-C), 111.1 (5′-C), 111.2 (t, JCF = 236.4 Hz, CF2H), 55.9 (CH3O). EIMS m/z (%): 303 (25, M+), 211 (100), 183 (17), 132 (15), 93 (19). Analysis for C17H15F2NO2: Calcd C, 67.32; H, 4.98; N, 4.62%. Found: C, 67.31; H, 5.00; N, 4.37%.
3.2.9. N-(2-Phenylethyl)-3-[3-(difluoromethyl)-4-methoxyphenyl]-(E)-propenamide (11g)
Compound 11g was prepared from acid 9 (30.0 mg, 0.145 mmol), Deoxofluor® 50% in toluene (0.107 mL, 0.435 mmol), 2-phenylethylamine (0.055 mL, 0.435 mmol), and triethylamine (0.061 mL, 0.435 mmol) following the procedure used for 11b. Compound 11g was obtained as a crystalline white solid (0.029 g, 60%); m.p. 104–105 °C; 1H-NMR (CDCl3) δ: 7.72–7.71 (m, 1H, 2′-H), 7.57 (d, J = 15.6 Hz, 1H, 3-H), 7.52–7.49 (m, 1H, 6′-H), 7.35–7.30 (m, 2H, 3′′′-H), 7.26–7.21 (m, 3H, 2′′′-H and 4′′′-H), 6.91 (t, J = 55.5 Hz, 1H, CF2H), 6.92–6.88 (m, 1H, 5′-H), 6.26 (d, J = 15.5 Hz, 1H, 2-H), 5.74 (t, J = 5.3 Hz, 1H, NH), 3.88 (s, 3H, CH3O), 3.66 (td, J = 6.9, 5.4 Hz, 2H, 1′′-H), 2.89 (t, J = 6.9 Hz, 2H, 2′′-H); 13C-NMR (CDCl3) δ: 166.0 (1-C), 158.3 (t, JCF = 5.6 Hz, 4′-C), 139.9 (3-C), 139.0 (1′′′-C), 132.4 (6′-C), 128.9 (2′′′-C), 128.8 (3′′′-C), 127.7 (1′-C), 126.7 (4′′′-C), 125.1 (t, JCF = 5.9 Hz, 2′-C), 123.2 (t, JCF = 22.2 Hz, 3′-C), 119.6 (2-C), 111.3 (t, JCF = 236.3 Hz, CF2H), 111.3 (5′-C), 56.0 (CH3O), 40.9 (1′′-C), 35.8 (2′′-C). EIMS m/z (%): 331 (34, M+), 226 (32), 211 (100), 183 (19), 132 (16), 104 (17), 91 (47). Analysis for C19H19F2NO2: Calcd C, 68.87; H, 5.78; N, 4.23%. Found: C, 68.86; H, 5.83; N, 4.10%.
3.2.10. N-Methoxy-N-methyl-3-[3-(difluoromethyl)-4-methoxyphenyl]-(E)-propenamide (11h)
Compound 11h was prepared from acid 9 (30.0 mg, 0.145 mmol), Deoxofluor® 50% in toluene (0.107 mL, 0.435 mmol), N,O-dimethylhydroxylamine hydrochloride (0.043 g, 0.435 mmol), and triethylamine (0.082 mL, 0.580 mmol) following the procedure used for 11b. Compound 11h was obtained as a crystalline white solid (0.026 g, 66%); m.p. 101 °C; 1H-NMR (CDCl3) δ: 7.83–7.80 (m, 1H, 2′-H), 7.69 (d, J = 15.8 Hz, 1H, 3-H), 7.62–7.58 (m, 1H, 6′-H), 6.96 (d, J = 15.8 Hz, 1H, 2-H), 6.96–6.93 (m, 1H, 5′-H), 6.94 (t, J = 55.5 Hz, 1H, CF2H), 3.91 (s, 3H, CH3OAr), 3.78 (s, 3H, N(CH3)OCH3), 3.31 (s, 3H, N(CH3)OCH3); 13C-NMR (CDCl3) δ: 167.1 (1-C), 158.5 (t, JCF = 5.7 Hz, 4′-C), 142.4 (3-C), 132.7 (6′-C), 128.1 (1′-C), 125.5 (t, JCF = 5.9 Hz, 2′-C), 123.3 (t, JCF = 22.1 Hz, 3′-C), 114.8 (2-C), 111.4 (t, JCF = 236.3 Hz, CF2H), 111.3 (5′-C), 62.1 (N(CH3)OCH3), 56.0 (CH3OAr), 32.7 (N(CH3)OCH3). EIMS m/z (%): 271 (3.4, M+), 211 (100), 183 (18), 132 (12). Analysis for C13H15F2NO3: Calcd C, 57.56; H, 5.57; N, 5.16%. Found: C, 57.84; H, 5.65; N, 5.24%.
3.2.11. N,N-Bis(2-methoxyethyl)-3-[3-(difluoromethyl)-4-methoxyphenyl]-(E)-propenamide (11i)
A solution of Deoxofluor® 50% in toluene (0.107 mL, 0.435 mmol) was added dropwise to a suspension of 0.030 g of 3-formyl-4-methoxycinnamic acid (9, 0.030 g, 0.145 mmol) in 0.4 mL of dry dichloromethane under an argon atmosphere. The reaction mixture was stirred for 45 min at room temperature. Triethylamine (0.061 mL, 0.435 mmol) was added and the mixture was subsequently stirred for 1 h at room temperature. The solution was diluted with 10 mL of dichloromethane, washed twice with 1M HCl and once with water, dried with anhydrous sodium sulfate, and the solvent evaporated. The resulting solid was purified by flash column chromatography eluting with mixtures of ethyl acetate–hexane of increasing polarity to give 11i as a crystalline white solid (0.042 g, 84%); m.p. 60–61 °C; 1H-NMR (CDCl3) δ: 7.76–7.71 (m, 1H, 2′-H), 7.64 (d, J = 15.4 Hz, 1H, 3-H), 7.58–7.52 (m, 1H, 6′-H), 6.93 (t, J = 55.5 Hz, 1H, CF2H), 6.95–6.91 (m, 1H, 5′-H), 6.89 (d, J = 15.4 Hz, 1H, 2-H), 3.90 (s, 3H, CH3OAr), 3.72 (t, J = 5.8 Hz, 2H, NCH2), 3.68 (t, J = 5.3 Hz, 2H, NCH2), 3.59 (t, J = 5.4 Hz, 2H, CH3OCH2), 3.56 (t, J = 5.8 Hz, 2H, CH3OCH2), 3.35 (s, 3H, CH3OCH2), 3.34 (s, 3H, CH3OCH2); 13C-NMR (CDCl3) δ: 167.0 (1-C), 158.3 (t, JCF = 5.60 Hz, 4′-C), 141.3 (3-C), 132.3 (t, JCF = 2.05 Hz, 6′-C), 128.4 (1′-C), 125.3 (t, JCF = 5.8 Hz, 2′-C), 123.2 (t, JCF = 22.1 Hz, 3′-C), 117.0 (2-C), 111.4 (t, JCF = 236.2 Hz, CF2H), 111.3 (5′-C), 71.4 (×2, CH3OCH2), 59.3 and 59.0 (CH3OCH2), 56.0 (CH3OAr), 49.2 and 47.5 (NCH2). EIMS m/z (%): 344 (5.4, M + 1), 343 (3.2, M+), 211 (100), 183 (9), 132 (3). Analysis for C17H23F2NO4: Calcd C, 59.46; H, 6.75; N, 4.08%. Found: C, 59.08; H, 6.55; N, 4.10%.
3.2.12. (E)-3-[3-(Difluoromethyl)-4-methoxyphenyl]-1-(morpholin-4-yl)propenone (11j)
Compound 11j was prepared from acid 9 (30.0 mg, 0.145 mmol), Deoxofluor® 50% in toluene (0.107 mL, 0.435 mmol), morpholine (0.0376 mL, 0.435 mmol), and triethylamine (0.061 mL, 0.435 mmol) following the procedure used for 11b. Compound 11j was obtained as a crystalline white solid (0.024 g, 56%); m.p. 120–121 °C; 1H-NMR (CDCl3) δ: 7.82–7.75 (m, 1H, 2′-H), 7.68 (d, J = 15.4 Hz, 1H, 3-H), 7.56–7.54 (m, 1H, 6′-H), 6.95 (t, J = 55.5 Hz, 1H, CF2H), 6.95–6.93 (m, 1H, 5′-H), 6.78 (d, J = 15.4 Hz, 1H, 2-H), 3.91 (s, 3H, CH3O), 3.79–3.66 (m, 8H, 2′′-H, 3′′-H, 5′′-H and 6′′-H); 13C-NMR (CDCl3) δ: 165.7 (1-C), 158.4 (t, JCF = 5.7 Hz, 4′-C), 142.2 (3-C), 132.7 (6′-C), 128.0 (1′-C), 124.9 (t, JCF = 5.8 Hz, 2′-C), 123.3 (t, JCF = 22.2 Hz, 3′-C), 115.4 (2-C), 111.3 (t, JCF = 237.0 Hz, CF2H), 111.3 (5′-C), 67.0 (2′′-C and 6′′-C), 56.0 (CH3O), 46.4 and 42.6 (br s, 3′′-C and 5′′-C). EIMS m/z (%): 297 (53, M+), 211 (100), 183 (22), 132 (15), 86 (12). Analysis for C15H17F2NO3: Calcd C, 60.60; H, 5.76; N, 4.71%. Found: C, 61.02; H, 5.74; N, 4.42%.
3.2.13. N-(2,5-Bis(trifluoromethyl)phenyl)-3-[3-(difluoromethyl)-4-methoxyphenyl]-(E)-propenamide (11k)
A solution of Deoxofluor® 50% in toluene (0.107 mL, 0.435 mmol) was added dropwise to a suspension of 3-formyl-4-methoxycinnamic acid (9, 0.030 g, 0.145 mmol) in 0.4 mL of dry dichloromethane under an argon atmosphere. The reaction mixture was stirred for 45 min at room temperature, diluted with 2 mL of dry dichloromethane, and percolated through 2.5 g of silica gel under an argon atmosphere. The silica gel bed was rinsed with 8 mL of dry dichloromethane and the resulting solution was dried under a nitrogen flow to give a white solid. Anhydrous THF (1.0 mL), 2,5-bis(trifluoromethyl)aniline (0.068 mL, 0.435 mmol) and 2.0 M lithium diisopropylamide in THF-heptane-ethylbenzene (0.145 mL, 0.29 mmol) were added and the mixture was stirred for 2 h at 25 °C under an argon atmosphere. The solution was diluted with 10 mL of dichloromethane, quenched with ice-water, washed twice with 1M HCl and once with water, dried with anhydrous sodium sulfate, and the solvent evaporated. The resulting oil was purified by flash column chromatography eluting with mixtures hexane–ethyl acetate of increasing polarity to give 11k as a white solid (0.030 g, 47%); m.p. 168–169 °C; 1H-NMR (CDCl3) δ: 8.83 (bs, 1H, 6′′-H), 7.85–7.82 (m, 1H, 2′-H), 7.78 (d, J = 15.5 Hz, 1H, 3-H), 7.76 (d, J = 8.3 Hz, 1H, 3′′-H), 7.64 (s, 1H, NH), 7.66–7.59 (m, 1H, 6′-H), 7.49 (d, J = 8.2 Hz, 1H, 4′′-H), 7.00–6.96 (m, 1H, 5′-H), 6.96 (t, J = 55.41 Hz, 1H, CF2H), 6.47 (d, J = 15.4 Hz, 1H, 2-H), 3.93 (s, 3H, CH3O); 13C-NMR (CDCl3) δ: 164.1 (1-C), 159.1 (t, JCF = 5.57 Hz, 4′-C), 143.3 (3-C), 136.6 (1′′-C), 135.2 (q, JCF = 33.4 Hz, 5′′-C), 133.0 (6′-C), 127.0 (q, JCF = 5.40 Hz, 3′′-C), 127.0 (1′-C), 125.8 (t, JCF = 5.78 Hz, 2′-C), 123.6 (q, JCF = 273.1 Hz, 2′′-CF3), 123.6 (t, JCF = 22.2 Hz, 3′-C), 123.3 (q, JCF = 273.2 Hz, 5′′-CF3), 122.3 (q, JCF = 30.1 Hz, 2”-C), 121.0 (q, JCF = 3.8 Hz, 6”-C), 120.9 (q, JCF = 3.7 Hz, 4”-C), 118.5 (2-C), 111.5 (5′-C), 111.2 (t, JCF = 236.6 Hz, CF2H), 56.1 (CH3O). EIMS m/z (%): 439 (5, M+), 420 (2), 212 (12), 211 (100), 183 (15), 132 (12). Analysis for C19H13F8NO2·0.5H2O: Calcd C, 50.90; H, 3.15; N, 3.12%. Found: C, 51.20; H, 3.10; N, 3.50%.
3.2.14. N,N’-Bis [3-[3-(difluoromethyl)-4-methoxyphenyl]-(E)-propenoyl]-1,3-propanediamine (11l)
Compound 11l was prepared from acid 9 (30.0 mg, 0.145 mmol), Deoxofluor® 50% in toluene (0.107 mL, 0.435 mmol), 1,3-diaminopropane (0.0055 mL, 0.065 mmol), and triethylamine (0.061 mL, 0.435 mmol) following the procedure used for 11b. Compound 11l was obtained as a white solid (0.019 g, 59%); m.p. 174–175 °C; 1H-NMR (CDCl3-CD3OD 9:1) δ: 7.74 (m, 2H, 2”-H), 7.56 (d, J = 15.8 Hz, 2H, 3′-H), 7.57–7.53 (m, 2H, 6”-H), 6.94–6.90 (m, 2H, 5”-H), 6.92 (t, J = 55.5 Hz, 2H, CF2H), 6.43 (d, J = 15.7 Hz, 2H, 2′-H), 3.90 (s, 6H, CH3O), 3.45–3.35 (m, 4H, 1-H and 3-H), 1.80–1.74 (m, 2H, 2-H); 13C-NMR (CDCl3-CD3OD 9:1) δ: 167.2 (1′-C), 158.3 (t, JCF = 5.6 Hz, 4”-C), 139.8 (3′-C), 132.1 (6”-C), 127.6 (1”-C), 125.4 (t, JCF = 5.8 Hz, 2”-C), 123.1 (t, JCF = 22.1 Hz, 3”-C), 119.6 (2′-C), 111.3 (t, JCF = 236.2 Hz, CF2H), 111.3 (5”-C), 55.9 (CH3O), 36.5 (1-C and 3-C), 29.3 (2-C). HRMS: calcd for C25H27F4N2O4+ (M + H)+: 495.1902, found: 495.1893; HRMS/MS (33 eV) from (M + H)+ m/z (%): 268.1155 (4), 211.0570 (100), 183.0614 (13). Analysis for C25H26F4N2O4: Calcd C, 60.72; H, 5.30; N, 5.67%. Found: C, 60.46; H, 5.41; N, 5.23%.
3.2.15. 1,4-Bis[3-[3-(difluoromethyl)-4-methoxyphenyl]-(E)-propenoyl]-piperazine (11m)
Compound 11m was prepared from acid 9 (30.0 mg, 0.145 mmol), Deoxofluor® 50% in toluene (0.107 mL, 0.435 mmol), piperazinium diacetate (0.015 g, 0.073 mmol), and triethylamine (0.102 mL, 0.730 mmol) following the procedure used for 11b. Compound 11m was obtained as a white solid (0.017 g, 46%); m.p. 259–261 °C; 1H-NMR (CDCl3-CD3OD 9:1) δ: 7.80 (m, 2H, 2′-H), 7.66 (d, J = 15.3 Hz, 2H, 3-H), 7.65–7.61 (m, 2H, 6′-H), 7.03–6.99 (m, 2H, 5′-H), 6.96 (t, J = 55.5 Hz, 2H, CF2H), 6.88 (d, J = 15.4 Hz, 2H, 2-H), 3.93 (s, 6H, CH3O), 3.86–3.76 (m, 8H, N(CH2CH2)2N); 13C-NMR (CDCl3-CD3OD 9:1) δ: 166.3 (1-C), 158.5 (t, JCF = 5.6 Hz, 4′-C), 142.8 (3-C), 132.3 (br s, 6′-C), 127.4 (1′-C), 125.2 (br s, 2′-C), 123.1 (t, JCF = 22.2 Hz, 3′-C), 114.8 (2-C), 111.2 (5′-C), 111.1 (t, JCF = 236.1 Hz, CF2H), 55.7 (CH3O), 45.4 and 42.2 (br s, N(CH2CH2)2N). HRMS calcd for C26H27F4N2O4+ (M + H)+: 507.1902, found: 507.1892; HRMS/MS (33 eV) from (M + H)+ m/z (%): 211.0558 (100), 183.0610 (31), 160.0515 (4). Analysis for C26H26F4N2O4·0.5H2O: Calcd C, 60.58; H, 5.28; N, 5.43%. Found: C, 60.13; H, 5.15; N, 5.19%.
3.2.16. N-(1-Methylethyl)-3-(4-methoxyphenyl)-(E)-propenamide (13a)
To a solution of 4-methoxycinnamic acid (12a, 50 mg, 0.28 mmol) in dry DMF (0.6 mL), triethylamine (0.059 mL, 0.42 mmol), isopropylamine (0.048 mL, 0.56 mmol) and a solution of (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (0.186 g, 0.42 mmol) in dry dichloromethane (0.6 mL) were added at 0 °C. The reaction mixture was stirred at 0 °C for 30 min and then at 25 °C for 2 h. Water (15 mL) was added and the mixture was extracted with dichloromethane (30 mL). The extract was washed with 1M HCl and water, dried over anhydrous sodium sulfate, and the solvent evaporated. The residue was recrystallized from hexane to give 13a as a crystalline white solid (0.052 g, 85%); m.p. 131 °C; 1H-NMR (CDCl3) δ: 7.57 (d, J = 15.6 Hz, 1H, 3-H), 7.44 (d, J = 8.6 Hz, 2H, 2′-H and 6′-H), 6.88 (d, J = 8.7 Hz, 2H, 3′-H and 5′-H), 6.24 (d, J = 15.6 Hz, 1H, 2-H), 5.49 (d, J = 8.2 Hz, 1 H, NH), 4.28–4.17 (m, 1H, 1”-H), 3.82 (s, 3 H, CH3O), 1.22 (d, J = 6.6 Hz, 6 H, 2”-H); 13C-NMR (CDCl3) δ: 165.5 (1-C), 160.9 (4′-C), 140.5 (3-C), 129.4 (2′-C and 6′-C), 127.8 (1′-C), 118.8 (2-C), 114.3 (3′-C and 5′-C), 55.5 (CH3O), 41.7 (1”-C), 23.0 (2”-C). EIMS m/z (%): 220 (18, M + 1), 219 (51, M+), 161 (100), 134 (28), 133 (40), 118 (14). Analysis for C13H17NO2: C, 71.21; H, 7.81; N, 6.39%. Found: C, 70.93; H, 7.82; N, 6.36%.
3.2.17. N-(1-Methylethyl)-3-(4-methoxy-3-methylphenyl)-(E)-propenamide (13b)
Compound 13b was prepared from acid 12b (50 mg, 0.26 mmol), triethylamine (0.054 mL, 0.39 mmol), isopropylamine (0.045 mL, 0.52 mmol), and (benzotriazol-1-yloxy)-tris(dimethylamino)-phosphonium hexafluorophosphate (0.173 g, 0.39 mmol) following the procedure used for 13a. The reaction product was purified by flash column chromatography eluting with mixtures of hexane–ethyl acetate of increasing polarity to give 13c as a pale yellow solid (0.049 g, 80%); m.p. 124–125 °C; 1H-NMR (CDCl3) δ: 7.54 (d, J = 15.5 Hz, 1H, 3-H), 7.33–7.29 (m, 2H, 2′-H and 6′-H), 6.80 (d, J = 9.0 Hz, 1H, 5′-H), 6.22 (d, J = 15.5 Hz, 1H, 2-H), 5.38 (d, J = 6.6 Hz, 1H, NH), 4.30–4.16 (m, 1H, 1”-H), 3.85 (s, 3H, CH3O), 2.21 (s, 3H, CH3Ar), 1.22 (d, J = 6.5 Hz, 6H, 2”-H); 13C-NMR (CDCl3) δ: 165.6 (1-C), 159.2 (4′-C), 140.8 (3-C), 129.8 (2′-C), 127.5 (6′-C), 127.2 (1′-C and 3′-C), 118.4 (2-C), 110.0 (5′-C), 55.5 (CH3O), 41.6 (1”-C), 23.1 (2”-C), 16.4 (CH3Ar). EIMS m/z (%): 234 (13, M + 1), 233 (36, M+), 175 (100), 148 (32), 147 (26), 115 (17). Analysis for C14H19NO2·0.25H2O: Calcd: C, 70.71; H, 8.26; N, 5.89%. Found: C, 70.77; H, 8.15; N, 5.86%.
3.2.18. N-(1-Methylethyl)-3-(3-hydroxy-4-methoxyphenyl)-(E)-propenamide (13c)
Compound 13c was prepared from isoferulic acid (8, 54 mg, 0.28 mmol), triethylamine (0.059 mL, 0.42 mmol), isopropylamine (0.048 mL, 0.56 mmol), and (benzotriazol-1-yloxy)-tris(dimethylamino)-phosphonium hexafluorophosphate (0.186 g, 0.42 mmol) following the procedure used for 13a. The reaction product was purified by flash column chromatography eluting with mixtures of hexane–ethyl acetate of increasing polarity to give 13b as a white solid (0.048 g, 73%); m.p. 164–165 °C; 1H-NMR (CDCl3-CD3OD 9:1) δ: 7.46 (d, J = 15.6 Hz, 1H, 3-H), 7.08 (d, J = 2.1 Hz, 1H, 2′-H), 6.98 (dd, J = 2.0, 8.6 Hz, 1H, 6′-H), 6.83 (d, J = 8.3 Hz, 1H, 5′-H), 6.25 (d, J = 15.6 Hz,1H, 2-H), 4.21–4.12 (m, 1H, 1”-H), 3.90 (s, 3H, CH3O), 1.21 (d, J = 6.6 Hz, 6H, 2”-H); 13C-NMR (CDCl3-CD3OD 9:1) δ: 166.1 (1-C), 148.7 (4′-C), 146.0 (3′-C), 140.6 (3-C), 128.4 (1′-C), 121.4 (6′-C), 118.8 (2-C), 112.9 (2′-C), 111.0 (5′-C), 55.9 (CH3O), 41.5 (1”-C), 22.6 (2”-C). EIMS m/z (%): 235 (43, M+), 178 (35), 177 (98),150 (27), 145 (28), 134 (27), 117 (43), 89 (69), 58 (100). Analysis for C13H17NO3: Calcd C, 66.36; H, 7.28; N, 5.95%. Found: C, 66.28; H, 7.17; N, 5.69%.
3.2.19. N-(1-Methylethyl)-3-[4-(difluoromethyl)-3-methoxyphenyl]-(E)-propenamide (15)
Methyl-3-[4-formyl-3-methoxyphenyl]-(E)-propenoate (14, 33 mg, 0.150 mmol) was dissolved in a mixture of methanol (2 mL) and 20% aqueous potassium carbonate solution (1 mL). The reaction mixture was stirred for 4 h at 20 °C and concentrated under reduced pressure to a third of its volume. The solution was acidified with conc. HCl (to pH 1) and the precipitate was filtered and used without further purification. The crude product was treated with Deoxofluor® 50% in toluene (0.107 mL, 0.435 mmol), isopropylamine (0.044 mL, 0.435 mmol) and triethylamine (0.061 mL, 0.435 mmol) following the procedure described for 11b. Compound 15 was obtained as a crystalline white solid (0.025 g, 62% 2 steps); m.p. 128–130 °C; 1H-NMR (CDCl3) δ: 7.62 (d, J = 15.5 Hz, 1H, 3-H), 7.59–7.56 (m, 1H, 5′-H), 7.22–7.18 (m, 1H, 6′-H), 7.05–7.01 (m, 1H, 2′-H), 6.95 (t, J = 55.5 Hz, 1H, CF2H), 6.41 (d, J = 15.5 Hz, 1H, 2-H), 5.48 (d, J = 6.5 Hz, 1H NH), 4.32–4.20 (m, 1H, 1′′-H), 3.92 (s, 3H, CH3O), 1.26 (d, J = 6.6 Hz, 6H, 2”-H); 13C-NMR (CDCl3) δ: 164.6 (s, 1-C), 157.6 (t, J = 6.0 Hz, 3′-C), 140.0 (s, 3-C), 138.9 (t, J = 1.8 Hz, 1′-C), 126.8 (t, J = 5.8 Hz, 5′-C), 123.8 (t, J = 22.3 Hz, 4′-C), 122.9 (s, 2-C), 120.0 (s, 6′-C), 111.4 (t, J = 236.4 Hz, CF2H), 110.2 (s, 2′-C), 55.9 (s, CH3O), 41.9 (s, 1”-C), 23.0 (s, 2”-C). EIMS m/z (%): 269 (55, M+), 211 (100), 183 (24), 132 (18), 58 (33). Analysis for C14H17F2NO2: Calcd C, 62.42; H, 6.36; N, 5.20%. Found: C, 62.44; H, 6.35; N, 4.99%.
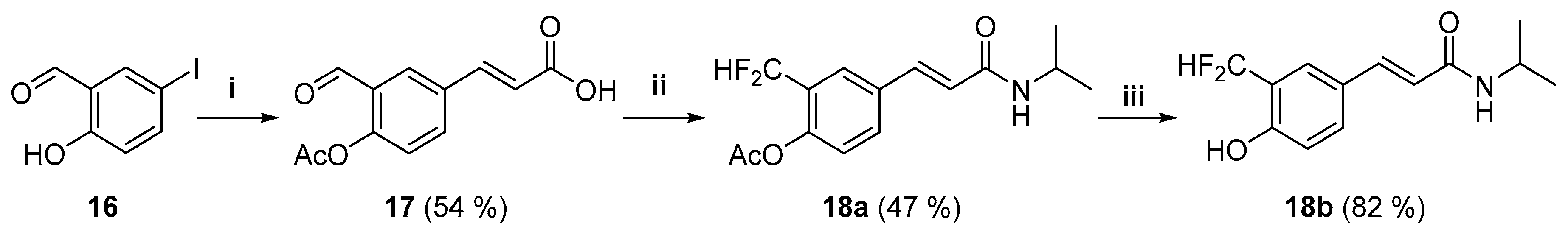
3.2.20. 3-(4-Acetyloxy-3-formylphenyl)-(E)-propenoic acid (17)
To a solution of 5-iodosalicylaldehyde 16 (2.68 g, 10.8 mmol) in dry acetone (50 mL) was added K2CO3 (3.0 g, 21.6 mmol) and then acetic anhydride (2.15 mL, 21.6 mmol) was added dropwise to the suspension with vigorous stirring. Stirring was continued at room temperature for 2.5 h and the reaction mixture was percolated through a silica gel pad using acetone as eluent. The percolate was evaporated to dryness and the residue recrystallized from n-hexane to give 5-iodo-2-acetyloxy-benzaldehyde as pale yellow needles (2.9 g, 92%); m.p. 89–90 °C; 1H-NMR (CDCl3) δ: 10.01 (s, 1H, ArCHO), 8.16 (d, J = 2.2 Hz, 1H, 2-H), 7.91 (dd, J = 2.3, 8.5 Hz, 1H, 6-H), 6.96 (d, J = 8.5 Hz, 1H, 5-H), 2.38 (s, 3H, CH3C(O)Ar); 13C-NMR (CDCl3) δ: 187.0 (ArCHO), 168.7 (CH3C(O)Ar), 151.3 (2-C), 143.8 (4-C), 139.5 (6-C), 129.5 (1-C), 125.5 (3-C), 90.2 (5-C), 20.8 (CH3C(O)Ar).
The 5-iodo-2-acetyloxybenzaldehyde obtained above (0.2 g, 0.69 mmol) was dissolved in 5 mL of acetonitrile and oxygen was removed by bubbling nitrogen through the solution. Triethylamine (0.673 mL, 4.82 mmol) and acrylic acid (0.095 mL, 1.38 mmol) were added dropwise with stirring to the solution, followed by tri-o-tolylphosphine (16 mg, 0.069 mmol) and palladium (II) acetate (8.0 mg, 0.047 mmol). The mixture was then heated at 65–70 °C for 4 h, volatiles were removed by distillation, and the residue was dissolved in dichloromethane. The resulting solution was percolated through silica gel eluting with a mixture of hexane–ethyl acetate (6:4). The percolate was evaporated to dryness and purified by flash column chromatography eluting with mixtures of hexane–ethyl acetate of increasing polarity to give compound 17 as a white solid (0.095 g, 54%); m.p. 205–207 °C; 1H-NMR (DMSO-d6) δ: 12.53 (s, 1H, COOH), 10.07 (s, 1H, ArCHO), 8.22 (d, J = 2.3 Hz, 1H, 2′-H), 8.08 (dd, J = 8.6, 2.3 Hz, 1H, 6′-H), 7.67 (d, J = 16.0 Hz, 1H, 3-H), 7.36 (d, J = 8.5 Hz, 1H, 5′-H), 6.63 (d, J =16.1 Hz, 1H, 2-H), 2.35 (s, 3H, CH3CO); 13C-NMR (DMSO-d6) δ: 189.9 (ArCHO), 169.1 (CH3CO), 167.3 (1-C), 151.7 (4′-C), 141.8 (3-C), 134.7 (6′-C), 132.8 (1′-C), 131.2 (2′-C), 128.2 (3′-C), 124.5 (5′-C), 120.9 (2-C), 20.7 (CH3CO). EIMS m/z (%): 234 (1, M+), 192 (23), 146 (9), 91 (20), 89 (45), 63 (19), 43 (100).
3.2.21. N-(1-Methylethyl)-3-[4-acetyloxy-3-(difluoromethyl)phenyl]-(E)-propenamide (18a)
Compound 18a was prepared from acid 17 (30.0 mg, 0.128 mmol), Deoxofluor® 50% in toluene (0.095 mL, 0.386 mmol), isopropylamine (0.033 mL, 0.386 mmol), and triethylamine (0.054 mL, 0.386 mmol) following the procedure used for 11b. Compound 18a was obtained as a white solid (0.018 g, 47%); m.p. 131–133 °C; 1H-NMR (CDCl3) δ: 7.76–7.71 (m, 1 H, 2′-H), 7.60 (d, J = 15.5 Hz, 1H, 3-H), 7.61–7.55 (m, 1H, 6′-H), 7.23–7.18 (m, 1H, 5′-H), 6.74 (t, J = 55.2 Hz, 1H, CF2H), 6.36 (d, J = 15.6 Hz, 1H, 2-H), 5.50 (d, J = 7.8 Hz, 1 H, NH), 4.28–4.17 (m, 1H, 1”-H), 2.34 (s, 3 H, CH3CO), 1.23 (d, J = 6.6 Hz, 6 H, 2”-H); 13C-NMR (CDCl3) δ: 168.7 (CH3CO), 164.5 (1-C), 149.1 (t, JCF = 5.2 Hz, 4′-C), 138.9 (3-C), 133.4 (1′-C), 131.2 (br s, 6′-C), 127.0 (t, JCF = 22.6 Hz, 3′-C), 125.5 (t, JCF = 6.4 Hz, 2′-C), 123.8 (s, 5′-C), 122.6 (s, 2-C), 111.6 (t, J = 238.9 Hz, CF2H), 41.9 (s, 1′′-C), 23.0 (s, 2”-C), 21.0 (CH3CO). EIMS m/z (%): 298 (23, M + 1), 297 (18, M+), 255 (88), 197 (68), 177 (44), 101 (30), 58 (100), 43 (60). Analysis for C15H17F2NO3: Calcd C, 60.60; H, 5.76; N, 4.71%. Found: C, 60.21; H, 5.76; N, 4.64%.
3.2.22. N-(1-Methylethyl)-3-[3-(difluoromethyl)-4-hydroxyphenyl]-(E)-propenamide (18b)
To a solution of 18a (10.0 mg, 0.034 mmol) in MeOH (1.0 mL) was added conc. H2SO4 (50 µL) and the mixture stirred at 60 °C for 2 h. The reaction mixture was diluted with water (15 mL) and concentrated under reduced pressure. The milky suspension was extracted with ethyl acetate (30 mL), the organic layer was dried with anhydrous sodium sulfate, and the solvent evaporated. The residue was purified by flash column chromatography eluting with mixtures of hexane–ethyl acetate of increasing polarity to give compound 18b as a white amorphous solid (0.007 g, 82%); m.p. 153–154 °C; 1H-NMR (CDCl3-CD3OD 9:1) δ: 7.70–7.65 (m, 1H, 2′-H), 7.50 (d, J 15.6, 1H, 3-H), 7.41–7.35 (m, 1H, 6′-H), 6.95 (t, 1H, J = 55.7 Hz, CF2H), 6.87–6.82 (m, 1H, 5′-H), 6.29 (d, J = 15.6 Hz, 1H, 2-H), 4.23–4.13 (m, 1H, 1”-H), 1.21 (d, J = 6.5 Hz, 6H, 2”-H); 13C-NMR (CDCl3-CD3OD 9:1) δ: 165.9 (1-C), 156.6 (t, JCF = 5.6 Hz, 4′-C), 140.0 (3-C), 132.0 (6′-C), 126.6 (1′-C), 125.3 (t, JCF = 5.6 Hz, 2′-C), 121.5 (t, JCF = 22.3 Hz, 3′-C), 118.9 (2-C), 116.1 (5′-C), 111.8 (t, JCF = 235.5 Hz, CF2H), 41.6 (1”-C), 22.7 (2”-C). EIMS m/z (%): 255 (13, M+), 197 (36), 177 (45), 121 (23), 101 (77), 58 (100), 43 (33). HRMS: calcd for C13H16F2NO2+ (M + H)+: 256.1144, found: 256.1145.
3.2.23. N-(1-Methylethyl)-trans-2-[3-(difluoromethyl)-4-methoxyphenyl]-cyclopropanecarboxamide (19)
Compound
S2 (see the
Supplementary Materials) (0.018 g, 0.066 mmol) was dissolved in diethyl ether (0.1 mL) and water (0.003 mL) and potassium
t-butoxide (40 mg, 0.036 mmol) were added. The mixture was stirred at room temperature for 3 h. After this, the reaction mixture was diluted with 2M HCl (2 mL) and extracted with dichloromethane, the organic layer was dried with anhydrous sodium sulfate, and the solvent evaporated. The resulting crude product was treated with BOP and isopropylamine following the same procedure used for compound
13a. The residue was purified by flash column chromatography eluting with mixtures of hexane–ethyl acetate of increasing polarity to give compound
19 as a white solid (0.015 g, 80%); m.p. 166–167 °C;
1H-NMR (CDCl
3) δ: 7.22 (bs, 1H, 2′-H), 7.24–7.19 (m, 1H, 6′-H), 6.91 (t,
J = 55.7 Hz, 1H, CF
2H), 6.86 (m, 1H, 5′-H), 5.47 (d,
J = 7.2 Hz, 1H, N
H), 4.20–4.04 (m, 1H, 1′′-H), 3.84 (s, 3H, C
H3OAr), 2.47 (ddd,
J = 4.1, 6.3, 9.1 Hz, 1H, 2-H), 1.58 (ddd,
J = 4.1, 5.2, 9.1 Hz, 1H, 3a-H), 1.49 (ddd,
J = 4.1, 5.2, 8.2 Hz, 1H, 1-H), 1.17 (d,
J = 6.6 Hz, 3H, 2a’’-H), 1.19–1.14 (m, 1H, 3b-H), 1.17 (d,
J = 6.6 Hz, 3H, 2b’’-H);
13C-NMR (CDCl
3) δ: 170.8 (
C(O)N), 155.9 (t,
JCF = 6.0 Hz, 4′-C), 133.4 (1′-C), 130.4 (t,
JCF = 2.1 Hz, 6′-C), 123.3 (t,
JCF = 5.7 Hz, 2′-C), 122.8 (t,
JCF = 22.0 Hz, 3′-C), 111.6 (t,
JCF = 235.6 Hz,
CF
2H), 111.2 (5′-C), 55.9 (
CH
3OAr), 41.8 (1′′-C), 26.6 (1-C), 24.1 (1-C), 23.1 (2b’’-C), 23.0 (2a’’-C), 15.8 (3-C). EIMS
m/
z (%): 284 (39, M + 1), 283 (100, M
+), 264 (29), 224 (21), 197 (30), 178 (21), 146 (42), 43 (15). Analysis for C
15H
19F
2NO
2: Calcd C, 63.59; H, 6.76; N, 4.94%. Found: C, 63.29; H, 6.50; N, 4.82%.
3.2.24. N-(1-Methylethyl)-3-[3-(difluoromethyl)-4-methoxyphenyl]-propanamide (20)
Compound 11b (0.030 g, 0.111 mmol) was dissolved in ethyl acetate (10 mL), 10% wt. Pd/C (0.003 g) added and the mixture hydrogenated at 3 bar and room temperature for 8 h. The catalyst was filtered and the residue crystallized from n-hexane to give compound 20 as a crystalline white solid (0.028 g, 93%); m.p. 84 °C; 1H-NMR (CDCl3) δ: 7.38 (m, 1H, 2′-H), 7.30 – 7.23 (m, 1H, 6′-H), 6.92 (t, J = 55.7 Hz, 1H, CF2H), 6.86–6.83 (m, 1H, 5′-H), 5.16 (d, J = 6.4 Hz, 1H, NH), 4.05 (d heptet, J = 6.6, 7.9 Hz, 1H, 1′′-H), 3.84 (s, 3H, CH3OAr), 2.94 (t, J = 7.6 Hz, 2H, 3-H), 2.40 (t, J = 7.6 Hz, 2H, 2-H), 1.08 (d, J = 6.6 Hz, 6H, 2′′-H); 13C-NMR (CDCl3) δ: 171.1 (1-C), 155.9 (t, JCF = 6.1 Hz, 4′-C), 133.3 (1′-C), 132.1 (6′-C), 126.0 (t, JCF = 5.7 Hz, 2′-C), 122.7 (t, JCF = 22.2 Hz, 3′-C), 111.7 (t, JCF = 235.3 Hz, CF2H), 111.2 (5′-C), 55.9 (CH3OAr), 41.5 (1′′-C), 38.9 (2-C), 31.0 (3-C), 22.9 (2′′-C). EIMS m/z (%): 272 (70, M + 1), 271 (100, M+), 252 (21), 184 (46), 171 (46), 100 (19). Analysis for C14H19F2NO2: Calcd C, 61.98; H, 7.06; N, 5.16%. Found: C, 62.04; H, 7.02; N, 5.11%.
3.2.25. N-(1-Methylethyl)-2-methyl-3-[3-(difluoromethyl)-4-methoxyphenyl]-(E)-propenamide (21)
Compound
21 was prepared from 2-Methyl-3-(3-formyl-4-methoxyphenyl)-(
E)-propenoic acid (
S3, see the
Supplementary Materials) (24.0 mg, 0.109 mmol), Deoxofluor
® 50% in toluene (0.160 mL, 0.327 mmol), isopropylamine (0.028 mL, 0.327 mmol), and triethylamine (0.046 mL, 0.327 mmol) following the procedure used for
11b. Compound
21 was obtained as a crystalline white solid (0.020 g, 65%); m.p. 96–97 °C;
1H-NMR (CDCl
3) δ: 7.57–7.53 (m, 1H, 2′-H), 7.47–7.37 (m, 1H, 6′-H), 7.25 (bs, 1H, 3-H), 6.97 – 6.93 (m, 1H, 5′-H), 6.95 (t,
J = 55.6 Hz, 1H, CF
2H), 5.66 (d,
J = 7.3 Hz, 1H, N
H), 4.20 (dhept,
J = 6.6, 7.7 Hz, 1′′-H), 3.90 (s, 3H, C
H3O), 2.09 (d,
J = 1.4 Hz, 3H, C=CC
H3), 1.24 (d,
J = 6.6 Hz, 6H, 2”-H);
13C-NMR (CDCl
3) δ: 168.8 (s, 1-C), 156.8 (t,
J = 5.9 Hz, 4′-C), 133.2 (t,
J = 2.1 Hz, 6′-C), 132.4 (s, 3-C), 131.9 (s, 2-C), 128.9 (s, 1′-C), 127.4 (t,
J = 5.8 Hz, 2′-C), 122.7 (t,
J = 22.2 Hz, 3′-C), 111.4 (t,
J = 236.1 Hz,
CF
2H), 111.0 (s, 5′-C), 55.9 (s,
CH
3O), 41.9 (s, 1”-C), 23.0 (s, 2”-C), 14.4 (s, C=C
CH
3). EIMS
m/
z (%): 284 (55, M + 1), 283 (100, M
+), 225 (36), 197 (30), 146 (97), 131 (16), 58 (11). Analysis for C
15H
19F
2NO
2: Calcd C, 63.59; H, 6.76; N, 4.94%. Found: C, 63.45; H, 6.76; N, 4.94%.
3.2.26. (1-Methylethyl)-3-[3-(difluoromethyl)-4-methoxyphenyl]-(E)-propenoate (22)
Compound 22 was prepared from acid 9 (30.0 mg, 0.145 mmol), Deoxofluor® 50% in toluene (0.107 mL, 0.435 mmol), isopropanol (0.034 mL, 0.435 mmol), and triethylamine (0.061 mL, 0.435 mmol) following the procedure used for 11b. Compound 22 was obtained as a white solid (0.033 g, 84%); m.p. 62 °C; 1H-NMR (CDCl3) δ: 7.77–7.74 (m, 1H, 2′-H), 7.63 (d, J = 16.0 Hz, 1H, 3-H), 7.60–7.56 (m, 1H, 6′-H), 6.97–6.93 (m, 1H, 5′-H), 6.92 (t, J = 55.5 Hz, 1H, CF2H), 6.35 (d, J = 16.0 Hz, 1H, 2-H), 5.13 (hept, J = 6.3 Hz, 1′′-H), 3.91 (s, 3H, CH3O), 1.31 (d, J = 6.3 Hz, 6H, 2”-H); 13C-NMR (CDCl3) δ: 166.6 (s, 1-C), 158.7 (t, J = 5.7 Hz, 4′-C), 143.1 (s, 3-C), 132.1 (t, J = 1.9 Hz, 6′-C), 127.5 (s, 1′-C), 126.0 (t, J = 5.9 Hz, 2′-C), 123.4 (t, J = 22.3 Hz, 3′-C), 117.8 (s, 2-C), 111.4 (s, 5′-C), 111.2 (t, J = 236.3 Hz, CF2H), 67.9 (s, 1”-C), 56.0 (s, CH3O), 22.1 (s, 2”-C). HRMS: calcd for C14H17F2O3+ (M + H)+ requires m/z 271.1140, found m/z 271.1141.
3.2.27. N-(1-Methylethyl)-3-(difluoromethyl)-4-methoxybenzamide (23)
p-Methoxybenzoic acid (91 mg, 0.60 mmol) was dissolved in anhydrous dichloromethane (0.90 mL) and cooled to −40 °C. Then, dichloromethylmethyl ether (0.49 mL, 0.54 mmol) and a solution of TiCl4 (0.144 mL, 1.30 mmol) in anhydrous dichloromethane (0.3 mL) were added dropwise under continuous stirring. The deep red solution was stirred at −40 °C for 1.5 h and then 1M HCl (3 mL) added. The resulting emulsion was stirred at room temperature for 0.5 h and extracted with dichloromethane. The organic layer was dried with anhydrous sodium sulfate and the solvent evaporated. The resulting product was treated with Deoxofluor® 50% in toluene (0.107 mL, 0.435 mmol), isopropyl amine (0.037 mL, 0.435 mmol), and triethylamine (0.061 mL, 0.435 mmol) following the procedure used for 11b. Compound 23 was obtained as a white solid (0.025 g, 52% for 2 steps); m.p. 99–100 °C; 1H-NMR (CDCl3) δ: 7.97–7.93 (m, 1H, 6-H), 7.90 – 7.87 (m, 1H, 2-H), 7.00–6.95 (m, 1H, 5-H), 6.94 (t, J = 55.4 Hz, 1H, CF2H), 5.91 (d, J = 6.7 Hz, 1H, NH), 4.28 (dhept, J = 6.6, 7.6 Hz, 1′-H), 3.92 (s, 3H, CH3O), 1.28 (d, J = 6.6 Hz, 6H, 2′-H); 13C-NMR (CDCl3) δ: 165.5 (s, C(O)N), 159.6 (t, J = 5.6 Hz, 4-C), 131.8 (s, 6-C), 127.4 (s, 1-C), 124.7 (t, J = 5.9 Hz, 2-C), 122.5 (t, J = 22.3 Hz, 3-C), 111.3 (t, J = 236.4 Hz, CF2H), 111.0 (s, 5-C), 56.1 (s, CH3O), 42.1 (s, 1′-C), 23.0 (s, 2′-C). EIMS m/z (%): 243 (13, M+), 185 (100), 157 (4), 127 (4), 109 (2). Analysis for C12H15F2NO2: Calcd C, 59.25; H, 6.22; N, 5.76%. Found: C, 59.14; H, 6.23; N, 5.77%.
3.2.28. N-(1-Methylethyl)-7-(difluoromethyl)-6-methoxy-3,4-dihydronaphthalene-2-carboxamide (24)
6-Methoxy-3,4-dihydronaphthalene-2-carboxylic acid (50 mg, 0.25 mmol) was dissolved in anhydrous dichloromethane (0.38 mL) and cooled to −40 °C. Then, dichloromethylmethyl ether (0.49 mL, 0.54 mmol) and a solution of TiCl4 (60 µL, 0.54 mmol) in anhydrous dichloromethane (0.12 mL) were added dropwise under continuous stirring. The deep red solution was stirred at −40 °C for 1.5 h and then 1M HCl (3 mL) was added. The resulting emulsion was stirred at room temperature for 0.5 h and then extracted with dichloromethane. The organic layer was dried with anhydrous sodium sulfate and the solvent evaporated. The resulting product was treated with Deoxofluor® 50% in toluene (0.192 mL, 0.779 mmol), isopropylamine (0.066 mL, 0.779 mmol), and triethylamine (0.109 mL, 0.779 mmol) following the procedure used for 11b. Compound 24 was obtained as a white solid (0.027 g, 35% for 2 steps); mp 150–152 °C; 1H-NMR (CDCl3) δ: 7.36 (m, 1H, 8-H), 7.10–7.06 (m, 1H, 1-H), 6.90 (t, J = 55.7 Hz, 1H, CF2H), 6.75 (s, 1H, 5-H), 5.65 (d, J = 7.8 Hz, 1H, NH), 4.20 (dhept, J = 6.5, 7.1 Hz, 1′-H), 3.88 (s, 3H, CH3O), 2.89 (t, J = 8.2 Hz, 2H, 4-H), 2.57 (td, J = 1.5, 8.2 Hz, 2H, 3-H), 1.23 (d, J = 6.5 Hz, 6H, 2′-H); 13C-NMR (CDCl3) δ: 167.2 (s, C(O)N), 157.6 (t, J = 6.0 Hz, 6-C), 141.0 (t, J = 1.7 Hz, 10-C), 132.0 (s, 2-C), 129.6 (s, 1-C), 125.8 (s, 9-C), 125.8 (t, J = 5.7 Hz, 8-C), 121.1 (t, J = 22.4 Hz, 7-C), 111.5 (t, J = 235.5 Hz, CF2H), 110.7 (s, 5-C), 56.0 (s, CH3O), 41.7 (s, 1′-C), 28.5 (s, 4-C), 23.0 (s, 2′-C), 22.7 (s, 3-C). HRMS: calcd for C16H20F2NO2+ (M + H)+ requires m/z 296.1456, found m/z 296.1457.
3.2.29. N-(1-Methylethyl)-7-(difluoromethyl)-6-methoxy-naphthalene-2-carboxamide (25)
Compound 24 (10 mg, 0.034 mmol) and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (11.5 mg, 0.05 mmol) were dissolved in dry toluene (0.8 mL) and the mixture was heated under reflux in an argon atmosphere for 1 h. Then, 5% NaHCO3 was added and the mixture extracted with dichloromethane, the organic layer was dried with anhydrous sodium sulfate, and the solvent evaporated. The residue was purified by flash column chromatography eluting with mixtures of hexane–ethyl acetate of increasing polarity to give compound 25 as a white solid (0.0085 g, 86%); m.p. 173–174 °C; 1H-NMR (CDCl3) δ: 8.22 (bs, 1H, 1-H), 8.11 (m, 1H, 8-H), 7.88 (dd, J = 1.8, 8.5 Hz, 1H, 3-H), 7.79 (d, J = 8.5 Hz, 1H, 4-H), 7.19 (s, 1H, 5-H), 7.02 (t, J = 55.3 Hz, 1H, CF2H), 6.06 (d, J = 7.6 Hz, 1H, NH), 4.35 (dhept, J = 6.6, 7.7 Hz, 1′-H), 4.00 (s, 3H, CH3O), 1.31 (d, J = 6.6 Hz, 6H, 2′-H); 13C-NMR (CDCl3) δ: 166.6 (s, C(O)N), 156.2 (t, J = 4.7 Hz, 6-C), 137.0 (s, 10-C), 131.0 (s, 2-C), 127.9 (t, J = 6.9 Hz, 8-C), 127.7 (s, 1-C), 127.1 (s, 4-C), 127.1 (s, 9-C), 125.9 (s, 3-C), 125.0 (t, J = 21.6 Hz, 7-C), 111.6 (t, J = 236.9 Hz, CF2H), 105.9 (s, 5-C), 55.9 (s, CH3O), 42.2 (s, 1′-C), 23.1 (s, 2′-C). HRMS: calcd for C16H18F2NO2+ (M + H)+ requires m/z 294.1307, found m/z 294.1300.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






