
PET Radiopharmaceuticals for Alzheimer’s Disease and Parkinson’s Disease Diagnosis, the Current and Future Landscape



Abstract
:1. Introduction
2. PET Imaging Agents for the Diagnosis AD and PD
2.1. PET-Tracers for the Imaging of Aβ Plaques
2.1.1. First Generation of Aβ PET Tracers
Benzothiazole (BTA) Derivatives
The Stilbene and Styrylpyridine Derivatives
2.1.2. Second Generation of Aβ PET Tracers
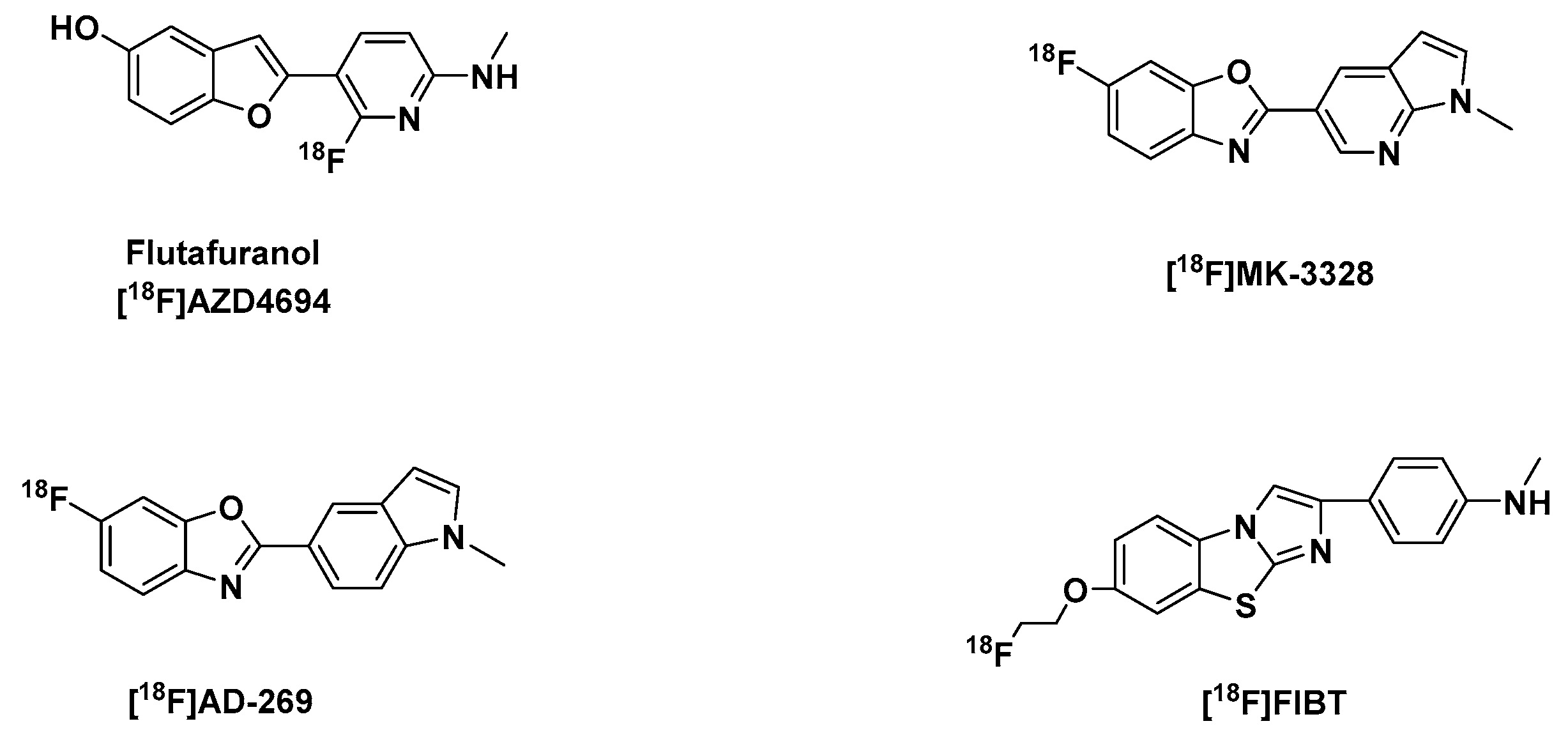
Benzofuran, Benzoxazole and Imidazobenzothiazole Derivatives
2.1.3. The Clinical Utility and Consequences of Clinically Approved PET-Aβ Radiotracers
2.2. PET-Tracers for the Imaging of Tau Aggregates
2.2.1. First Generation of Tau-PET Tracers
The Arylquinolines
The Phenylbutadienylbenzothiazoles (PBB)
The Carbazole and Benzimidazole Derivatives
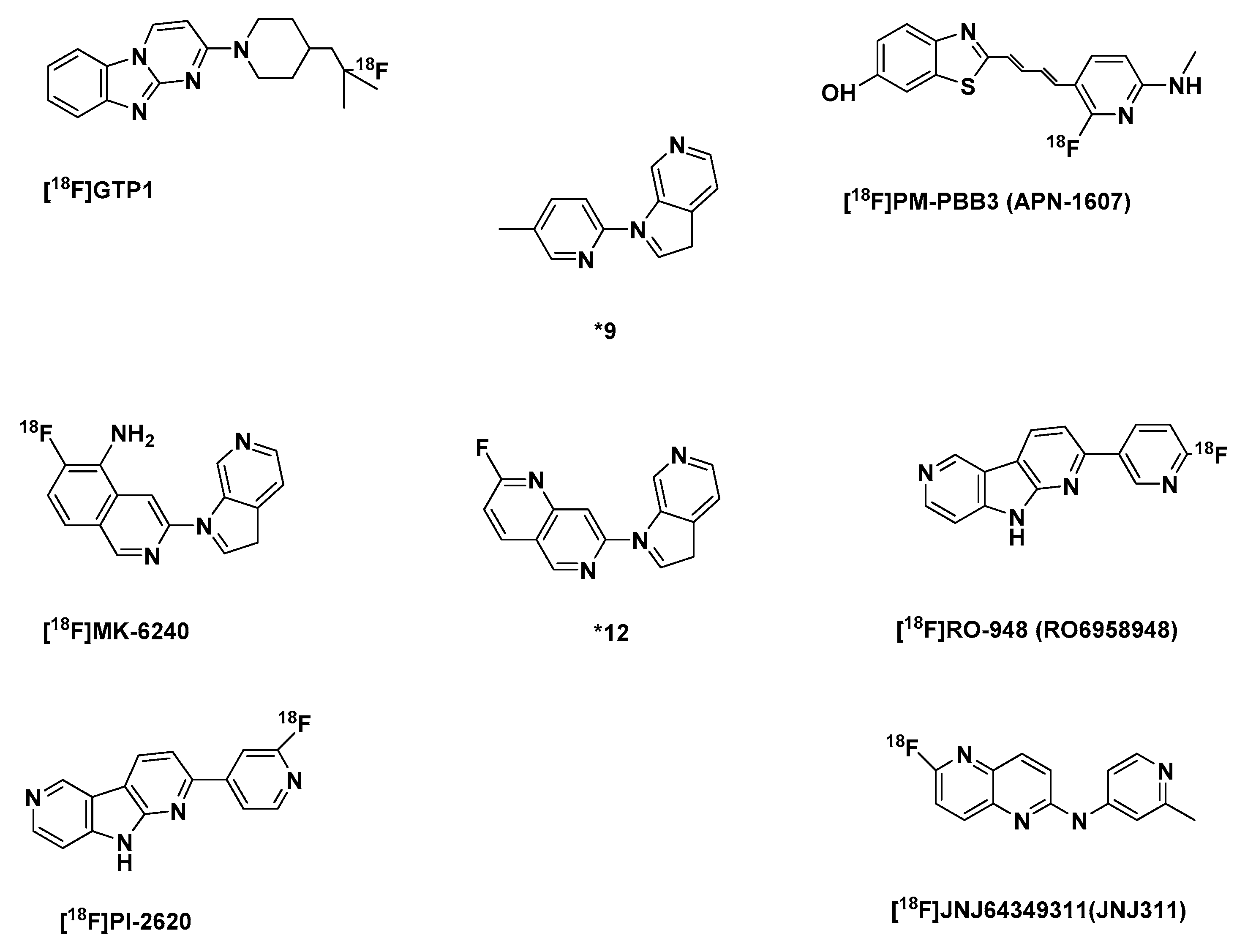
2.2.2. Second Generation of Selective Tau Tracers
Optimized First Generation Tau Tracers
The Azaindole-Isoquinoline and Naphthyridine Derivatives
2.3. Selective PET-Tracers for the Imaging of α-syn
2.3.1. The Phenothiazine Derivatives
2.3.2. The Indolinone and Indolinonediene Derivatives
2.3.3. Chalcone Derivatives and Structural Cogeners
2.3.4. Diarybisthiazole Compounds
3. Conclusions
Funding
Conflicts of Interest
References
- Alzheimer’s Disease International. The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends; World Alzheimer Report 2015; Alzheimer’s Disease International: London, UK, 2015. [Google Scholar]
- Alzheimer’s Disease International. World Alzheimer Report 2018—The State of the Art of Dementia Research: New Frontiers; Alzheimer’s Disease International: London, UK, 2018. [Google Scholar]
- World Health Organisation-Alzheimer’s Disease International. Dementia: A Public Health Priority; WHO: Geneva, Switzerland, 2012. [Google Scholar]
- Trevisan, K.; Cristina-Pereira, R.; Silva-Amaral, D.; Aversi-Ferreira, T.A. Theories of Aging and the Prevalence of Alzheimer’s Disease. Biomed. Res. Int. 2019, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Xie, A.; Gao, J.; Xu, L.; Meng, D. Shared mechanisms of neurodegeneration in Alzheimer’s disease and Parkinson’s disease. Biomed. Res. Int. 2014, 2014, 648740. [Google Scholar] [CrossRef]
- Murphy, M.P.; LeVine, H. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Association 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2018, 14, 367–429. [CrossRef]
- Zhang, Y.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, L.; Guo, Z. Alzheimer’s Aβ42 and Aβ40 peptides form interlaced amyloid fibrils. J. Neurochem. 2013, 126, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Lasagna-Reeves, C.A. Molecular mechanisms of amyloid oligomers toxicity. J. Alzheimers Dis. 2013, 33 (Suppl. 1), S67–78. [Google Scholar] [CrossRef] [Green Version]
- Trojanowski, J.Q.; Clark, C.M.; Schmidt, M.L.; Arnold, S.E.; Lee, V.M. Strategies for Improving the Postmortem Neuropathological Diagnosis of Alzheimer’s Disease. Neurobiol. Aging 1997, 18, S75–S79. [Google Scholar] [CrossRef]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef]
- Delacourte, A. Les diagnostics de la maladie d’Alzheimer. Ann. Biol. Clin. (Paris) 1998, 56, 133–142. [Google Scholar]
- Takizawa, C.; Thompson, P.L.; van Walsem, A.; Faure, C.; Maier, W.C. Epidemiological and economic burden of Alzheimer’s disease: A systematic literature review of data across Europe and the United States of America. J. Alzheimers Dis. 2015, 43, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.A.; Fox, N.C.; Sperling, R.A.; Klunk, W.E. Brain imaging in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006213. [Google Scholar] [CrossRef] [PubMed]
- Budson, A.E.; Solomon, P.R. New criteria for Alzheimer disease and mild cognitive impairment: implications for the practicing clinician. Neurologist 2012, 18, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Yu, C.-E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freudenberg-Hua, Y.; Li, W.; Davies, P. The Role of Genetics in Advancing Precision Medicine for Alzheimer’s Disease-A Narrative Review. Front. Med. (Lausanne) 2018, 5, 108. [Google Scholar] [CrossRef] [Green Version]
- Williamson, J.; Goldman, J.; Marder, K.S. Genetic aspects of Alzheimer disease. Neurologist 2009, 15, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Isaac, M.; Vamvakas, S.; Abadie, E.; Jonsson, B.; Gispen, C.; Pani, L. Qualification opinion of novel methodologies in the predementia stage of Alzheimer’s disease: Cerebro-spinal-fluid related biomarkers for drugs affecting amyloid burden—Regulatory considerations by European Medicines Agency focusing in improving benefit/risk in regulatory trials. Eur. Neuropsychopharmacol. 2011, 21, 781–788. [Google Scholar] [CrossRef]
- Blennow, K.; Hampel, H.; Zetterberg, H. Biomarkers in amyloid-β immunotherapy trials in Alzheimer’s disease. Neuropsychopharmacology 2014, 39, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Palmqvist, S.; Zetterberg, H.; Mattsson, N.; Johansson, P.; Minthon, L.; Blennow, K.; Olsson, M.; Hansson, O. Detailed comparison of amyloid PET and CSF biomarkers for identifying early Alzheimer disease. Neurology 2015, 85, 1240–1249. [Google Scholar] [CrossRef]
- Hampel, H.; Bürger, K.; Teipel, S.J.; Bokde, A.L.W.; Zetterberg, H.; Blennow, K. Core candidate neurochemical and imaging biomarkers of Alzheimer’s disease. Alzheimers Dement. 2008, 4, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Tullhög, K.; Hansson, O.; Minthon, L.; Londos, E.; Blennow, K. Low incidence of post-lumbar puncture headache in 1,089 consecutive memory clinic patients. Eur. Neurol. 2010, 63, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Jagust, W.J. Brain imaging in the study of Alzheimer’s disease. Neuroimage 2012, 61, 505–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fawaz, M.V.; Brooks, A.F.; Rodnick, M.E.; Carpenter, G.M.; Shao, X.; Desmond, T.J.; Sherman, P.; Quesada, C.A.; Hockley, B.G.; Kilbourn, M.R.; et al. High Affinity Radiopharmaceuticals Based Upon Lansoprazole for PET Imaging of Aggregated Tau in Alzheimer’s Disease and Progressive Supranuclear Palsy: Synthesis, Preclinical Evaluation, and Lead Selection. ACS Chem. Neurosci. 2014, 5, 718–730. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Marek, K. Looking backward to move forward: early detection of neurodegenerative disorders. Science 2003, 302, 830–834. [Google Scholar] [CrossRef] [Green Version]
- Bailey, D.L.; Maisey, M.N.; Townsend, D.W.; Valk, P.E. Positron Emission Tomography. Basic Sciences; Springer-Verlag London Limited: London, UK, 2005; ISBN 9781852337988. [Google Scholar]
- van der Born, D.; Pees, A.; Poot, A.J.; Orru, R.V.A.; Windhorst, A.D.; Vugts, D.J. Fluorine-18 labelled building blocks for PET tracer synthesis. Chem. Soc. Rev. 2017, 46, 4709–4773. [Google Scholar] [CrossRef] [Green Version]
- Filippi, L.; Chiaravalloti, A.; Bagni, O.; Schillaci, O. 18F-labeled radiopharmaceuticals for the molecular neuroimaging of amyloid plaques in Alzheimer’s disease. Am. J. Nucl. Med. Mol. Imaging 2018, 8, 268–281. [Google Scholar]
- Mathis, C.A.; Wang, Y.; Klunk, W.E. Imaging beta-amyloid plaques and neurofibrillary tangles in the aging human brain. Curr. Pharm. Des. 2004, 10, 1469–1492. [Google Scholar] [CrossRef]
- Klunk, W.E.; Engler, H.; Nordberg, A.; Wang, Y.; Blomqvist, G.; Holt, D.P.; Bergström, M.; Savitcheva, I.; Huang, G.-f.; Estrada, S.; et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann. Neurol. 2004, 55, 306–319. [Google Scholar] [CrossRef]
- Jacobson, O.; Kiesewetter, D.O.; Chen, X. Fluorine-18 radiochemistry, labeling strategies and synthetic routes. Bioconjug. Chem. 2015, 26, 1–18. [Google Scholar] [CrossRef]
- Ariza, M.; Kolb, H.C.; Moechars, D.; Rombouts, F.; Andrés, J.I. Tau Positron Emission Tomography (PET) Imaging: Past, Present, and Future. J. Med. Chem. 2015, 58, 4365–4382. [Google Scholar] [CrossRef] [PubMed]
- Barthel, H.; Sabri, O. Florbetaben to trace amyloid-β in the Alzheimer brain by means of PET. J. Alzheimers Dis. 2011, 26 (Suppl. 3), 117–121. [Google Scholar] [CrossRef]
- Johnson, K.A.; Sperling, R.A.; Gidicsin, C.M.; Carmasin, J.S.; Maye, J.E.; Coleman, R.E.; Reiman, E.M.; Sabbagh, M.N.; Sadowsky, C.H.; Fleisher, A.S.; et al. Florbetapir (F18-AV-45) PET to assess amyloid burden in Alzheimer’s disease dementia, mild cognitive impairment, and normal aging. Alzheimers Dement. 2013, 9, S72–S83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatashita, S.; Yamasaki, H.; Suzuki, Y.; Tanaka, K.; Wakebe, D.; Hayakawa, H. 18FFlutemetamol amyloid-beta PET imaging compared with 11CPIB across the spectrum of Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Maccioni, R.B.; Farías, G.; Morales, I.; Navarrete, L. The revitalized tau hypothesis on Alzheimer’s disease. Arch. Med. Res. 2010, 41, 226–231. [Google Scholar] [CrossRef]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Villemagne, V.L.; Furumoto, S.; Fodero-Tavoletti, M.; Harada, R.; Mulligan, R.S.; Kudo, Y.; Masters, C.L.; Yanai, K.; Rowe, C.C.; Okamura, N. The challenges of tau imaging. Future Neurol. 2012, 7, 409–421. [Google Scholar] [CrossRef]
- Ke, Y.D.; Suchowerska, A.K.; van der Hoven, J.; de Silva, D.M.; Wu, C.W.; van Eersel, J.; Ittner, A.; Ittner, L.M. Lessons from tau-deficient mice. Int. J. Alzheimers Dis. 2012, 2012, 873270. [Google Scholar] [CrossRef] [Green Version]
- Ludolph, A.C.; Kassubek, J.; Landwehrmeyer, B.G.; Mandelkow, E.; Mandelkow, E.-M.; Burn, D.J.; Caparros-Lefebvre, D.; Frey, K.A.; de Yebenes, J.G.; Gasser, T.; et al. Tauopathies with parkinsonism: clinical spectrum, neuropathologic basis, biological markers, and treatment options. Eur. J. Neurol. 2009, 16, 297–309. [Google Scholar] [CrossRef]
- Medeiros, R.; Baglietto-Vargas, D.; LaFerla, F.M. The role of tau in Alzheimer’s disease and related disorders. CNS Neurosci. Ther. 2011, 17, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Winners. Available online: https://www.eanm.org/congresses-events/awards-grants/winners/ (accessed on 22 December 2019).
- Vanhaute, H.; Ceccarini, J.; Michiels, L.; Koole, M.; Emsell, L.; Lemmens, R.; Vandenbulcke, M.; van Laere, K. PET-MR Imaging of Tau and Synaptic Density in Prodromal Alzheimer’s Disease; Alzheimer’s Disease International: London, UK, 2019. [Google Scholar]
- U.S. Food and Drug Administration. Available online: https://www.fda.gov/ (accessed on 20 November 2019).
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villemagne, V.L.; Okamura, N. In vivo tau imaging: obstacles and progress. Alzheimers Dement. 2014, 10, S254–S264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamura, N.; Harada, R.; Ishiki, A.; Kikuchi, A.; Nakamura, T.; Kudo, Y. The development and validation of tau PET tracers: current status and future directions. Clin. Transl. Imaging 2018, 6, 305–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguero, C.; Dhaynaut, M.; Normandin, M.D.; Amaral, A.C.; Guehl, N.J.; Neelamegam, R.; Marquie, M.; Johnson, K.A.; El Fakhri, G.; Frosch, M.P.; et al. Autoradiography validation of novel tau PET tracer F-18-MK-6240 on human postmortem brain tissue. Acta Neuropathol. Commun. 2019, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Kroth, H.; Oden, F.; Molette, J.; Schieferstein, H.; Capotosti, F.; Mueller, A.; Berndt, M.; Schmitt-Willich, H.; Darmency, V.; Gabellieri, E.; et al. Discovery and preclinical characterization of 18FPI-2620, a next-generation tau PET tracer for the assessment of tau pathology in Alzheimer’s disease and other tauopathies. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2178–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillantini, M.G.; Goedert, M. The alpha-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Ann. N. Y. Acad. Sci. 2000, 920, 16–27. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Yoshimoto, M.; Tsuji, S.; Takahashi, H. α-Synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neuroscience Letters 1998, 249, 180–182. [Google Scholar] [CrossRef]
- Tu, P.H.; Galvin, J.E.; Baba, M.; Giasson, B.; Tomita, T.; Leight, S.; Nakajo, S.; Iwatsubo, T.; Trojanowski, J.Q.; Lee, V.M. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann. Neurol. 1998, 44, 415–422. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Vilar, M.; Chou, H.-T.; Lührs, T.; Maji, S.K.; Riek-Loher, D.; Verel, R.; Manning, G.; Stahlberg, H.; Riek, R. The fold of alpha-synuclein fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 8637–8642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uéda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, D.F.; George, J.M. Synucleins in synaptic plasticity and neurodegenerative disorders. J. Neurosci. Res. 1999, 58, 120–129. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chartier-Harlin, M.-C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [Green Version]
- Ibáñez, P.; Bonnet, A.-M.; Débarges, B.; Lohmann, E.; Tison, F.; Agid, Y.; Dürr, A.; Brice, A.; Pollak, P. Causal relation between α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1169–1171. [Google Scholar] [CrossRef]
- Billingsley, K.J.; Bandres-Ciga, S.; Saez-Atienzar, S.; Singleton, A.B. Genetic risk factors in Parkinson’s disease. Cell Tissue Res. 2018, 373, 9–20. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef]
- Nalls, M.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Parkinson’s disease genetics: Novel risk loci, genomic context, causal insights and heritable risk. BiorRxiv 2018. [CrossRef] [Green Version]
- Vernon, A.C.; Ballard, C.; Modo, M. Neuroimaging for Lewy body disease: is the in vivo molecular imaging of α-synuclein neuropathology required and feasible? Brain Res. Rev. 2010, 65, 28–55. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.H. The prodromal phase of sporadic Parkinson’s disease: does it exist and if so how long is it? Mov. Disord. 2008, 23, 1799–1807. [Google Scholar] [CrossRef] [PubMed]
- Ross, G.W.; Petrovitch, H.; Abbott, R.D.; Nelson, J.; Markesbery, W.; Davis, D.; Hardman, J.; Launer, L.; Masaki, K.; Tanner, C.M.; et al. Parkinsonian signs and substantia nigra neuron density in decendents elders without PD. Ann. Neurol. 2004, 56, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 1991, 114, 2283–2301. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Wuketich, S. Time course of nigrostriatal degeneration in parkinson’s disease. A detailed study of influential factors in human brain amine analysis. J. Neural Transm. 1976, 38, 277–301. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Tredici, K.D.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [Green Version]
- Deramecourt, V.; Bombois, S.; Maurage, C.-A.; Ghestem, A.; Drobecq, H.; Vanmechelen, E.; Lebert, F.; Pasquier, F.; Delacourte, A. Biochemical staging of synucleinopathy and amyloid deposition in dementia with Lewy bodies. J. Neuropathol. Exp. Neurol. 2006, 65, 278–288. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.; Seibyl, J.; Cartier, A.; Bhatt, R.; Catafau, A.M. Molecular imaging insights into neurodegeneration: focus on α-synuclein radiotracers. J. Nucl. Med. 2014, 55, 1397–1400. [Google Scholar] [CrossRef] [Green Version]
- Barrett, P.J.; Timothy Greenamyre, J. Post-translational modification of α-synuclein in Parkinson’s disease. Brain Res. 2015, 1628, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.J.; Lee, V.M.-Y.; Trojanowski, J.Q. Parkinson’s disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci. 2013, 14, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Kotzbauer, P.T.; Cairns, N.J.; Campbell, M.C.; Willis, A.W.; Racette, B.A.; Tabbal, S.D.; Perlmutter, J.S. Pathologic accumulation of α-synuclein and Aβ in Parkinson disease patients with dementia. Arch. Neurol. 2012, 69, 1326–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathis, C.A.; Lopresti, B.J.; Ikonomovic, M.D.; Klunk, W.E. Small-molecule PET Tracers for Imaging Proteinopathies. Semin. Nucl. Med. 2017, 47, 553–575. [Google Scholar] [CrossRef]
- Kotzbauer, P.T.; Tu, Z.; Mach, R.H. Current status of the development of PET radiotracers for imaging alpha synuclein aggregates in Lewy bodies and Lewy neurites. Clin. Transl. Imaging 2017, 5, 3–14. [Google Scholar] [CrossRef]
- Pike, V.W. Considerations in the Development of Reversibly Binding PET Radioligands for Brain Imaging. Curr. Med. Chem. 2016, 23, 1818–1869. [Google Scholar] [CrossRef]
- Laruelle, M.; Slifstein, M.; Huang, Y. Relationships between radiotracer properties and image quality in molecular imaging of the brain with positron emission tomography. Mol. Imaging Biol. 2003, 5, 363–375. [Google Scholar] [CrossRef]
- Zhang, L.; Villalobos, A. Strategies to facilitate the discovery of novel CNS PET ligands. EJNMMI Radiopharm. Chem. 2017, 1, 13. [Google Scholar] [CrossRef] [Green Version]
- van de Bittner, G.C.; Ricq, E.L.; Hooker, J.M. A philosophy for CNS radiotracer design. Acc. Chem. Res. 2014, 47, 3127–3134. [Google Scholar] [CrossRef] [Green Version]
- Elghetany, M.T.; Saleem, A. Methods for staining amyloid in tissues: A review. Stain Technol. 1988, 63, 201–212. [Google Scholar] [CrossRef]
- Vassar, P.S.; Culling, C.F. Fluorescent stains, with special reference to amyloid and connective tissues. Arch. Pathol. 1959, 68, 487–498. [Google Scholar] [PubMed]
- Watanabe, H.; Ono, M.; Ariyoshi, T.; Katayanagi, R.; Saji, H. Novel Benzothiazole Derivatives as Fluorescent Probes for Detection of β-Amyloid and α-Synuclein Aggregates. ACS Chem. Neurosci. 2017, 8, 1656–1662. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Goodman, M.M. Fluorine-18 radiolabeled heterocycles as PET tracers for imaging β-amyloid plaques in Alzheimer’s disease. Curr. Top. Med. Chem. 2013, 13, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Mathis, C.A.; Mason, N.S.; Lopresti, B.J.; Klunk, W.E. Development of positron emission tomography β-amyloid plaque imaging agents. Semin. Nucl. Med. 2012, 42, 423–432. [Google Scholar] [CrossRef] [Green Version]
- Klunk, W.E.; Wang, Y.; Huang, G.f.; Debnath, M.L.; Holt, D.P.; Mathis, C.A. Uncharged thioflavin-T derivatives bind to amyloid-beta protein with high affinity and readily enter the brain. Life Sciences 2001, 69, 1471–1484. [Google Scholar] [CrossRef]
- Klunk, W.E.; Wang, Y.; Huang, G.-f.; Debnath, M.L.; Holt, D.P.; Shao, L.; Hamilton, R.L.; Ikonomovic, M.D.; DeKosky, S.T.; Mathis, C.A. The Binding of 2-(4′-Methylaminophenyl)Benzothiazole to Postmortem Brain Homogenates Is Dominated by the Amyloid Component. J. Neurosci. 2003, 23, 2086–2092. [Google Scholar] [CrossRef] [Green Version]
- Mathis, C.A.; Wang, Y.; Holt, D.P.; Huang, G.F.; Debnath, M.L.; Klunk, W.E. Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J. Med. Chem. 2003, 46, 2740–2754. [Google Scholar] [CrossRef]
- Price, J.C.; Klunk, W.E.; Lopresti, B.J.; Lu, X.; Hoge, J.A.; Ziolko, S.K.; Holt, D.P.; Meltzer, C.C.; DeKosky, S.T.; Mathis, C.A. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh Compound-B. J. Cereb. Blood Flow Metab. 2005, 25, 1528–1547. [Google Scholar] [CrossRef] [Green Version]
- Mathis, C.A.; Bacskai, B.J.; Kajdasz, S.T.; McLellan, M.E.; Frosch, M.P.; Hyman, B.T.; Holt, D.P.; Wang, Y.; Huang, G.-f.; Debnath, M.L.; et al. A lipophilic thioflavin-T derivative for positron emission tomography (PET) imaging of amyloid in brain. Bioorganic & Medicinal Chemistry Letters 2002, 12, 295–298. [Google Scholar] [CrossRef]
- Klunk, W.E.; Mathis, C.A. Whatever happened to Pittsburgh Compound-A? Alzheimer Dis. Assoc. Disord. 2008, 22, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Mintun, M.A.; Larossa, G.N.; Sheline, Y.I.; Dence, C.S.; Lee, S.Y.; Mach, R.H.; Klunk, W.E.; Mathis, C.A.; DeKosky, S.T.; Morris, J.C. 11CPIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology 2006, 67, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Ikonomovic, M.D.; Klunk, W.E.; Abrahamson, E.E.; Mathis, C.A.; Price, J.C.; Tsopelas, N.D.; Lopresti, B.J.; Ziolko, S.; Bi, W.; Paljug, W.R.; et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain 2008, 131, 1630–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitneni, S.K.; Serdons, K.; Evens, N.; Fonge, H.; Celen, S.; Deroose, C.M.; Debyser, Z.; Mortelmans, L.; Verbruggen, A.M.; Bormans, G.M. Efficient purification and metabolite analysis of radiotracers using high-performance liquid chromatography and on-line solid-phase extraction. J. Chromatogr. A 2008, 1189, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Serdons, K.; Terwinghe, C.; Vermaelen, P.; van Laere, K.; Kung, H.; Mortelmans, L.; Bormans, G.; Verbruggen, A. Synthesis and evaluation of 18F-labeled 2-phenylbenzothiazoles as positron emission tomography imaging agents for amyloid plaques in Alzheimer’s disease. J. Med. Chem. 2009, 52, 1428–1437. [Google Scholar] [CrossRef] [PubMed]
- Mathis, C.A.; Holt, D.; Wang, Y.; Huang, G.-f.; Debnath, M.; Shao, L.; Klunk, W.E. P2-178 Species-dependent formation and identification of the brain metabolites of the amyloid imaging agent [11C]PIB. Neurobiol. Aging 2004, 25, S277–S278. [Google Scholar] [CrossRef]
- Juréus, A.; Swahn, B.-M.; Sandell, J.; Jeppsson, F.; Johnson, A.E.; Johnström, P.; Neelissen, J.A.M.; Sunnemark, D.; Farde, L.; Svensson, S.P.S. Characterization of AZD4694, a novel fluorinated Abeta plaque neuroimaging PET radioligand. J. Neurochem. 2010, 114, 784–794. [Google Scholar] [CrossRef]
- Mason, N.S.; Mathis, C.A.; Klunk, W.E. Positron emission tomography radioligands for in vivo imaging of Aβ plaques. J. Labelled Comp. Radiopharm. 2013, 56, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Snellman, A.; Rokka, J.; Lopez-Picon, F.R.; Eskola, O.; Wilson, I.; Farrar, G.; Scheinin, M.; Solin, O.; Rinne, J.O.; Haaparanta-Solin, M. Pharmacokinetics of ¹⁸Fflutemetamol in wild-type rodents and its binding to beta amyloid deposits in a mouse model of Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 1784–1795. [Google Scholar] [CrossRef]
- Rabinovici, G.D. The translational journey of brain β-amyloid imaging: from positron emission tomography to autopsy to clinic. JAMA Neurol. 2015, 72, 265–266. [Google Scholar] [CrossRef]
- Curtis, C.; Gamez, J.E.; Singh, U.; Sadowsky, C.H.; Villena, T.; Sabbagh, M.N.; Beach, T.G.; Duara, R.; Fleisher, A.S.; Frey, K.A.; et al. Phase 3 trial of flutemetamol labeled with radioactive fluorine 18 imaging and neuritic plaque density. JAMA Neurol. 2015, 72, 287–294. [Google Scholar] [CrossRef]
- GE Healthcare. Gehealthcare Vizamyl Prescribing Information. Available online: http://www3.gehealthcare.com/~/media/documents/us-global/products/nuclear-imaging-agents_non-gatekeeper/clinical%20product%20info/vizamyl/gehealthcare-vizamyl-prescribing-information.pdf (accessed on 15 September 2016).
- Kung, M.-P.; Hou, C.; Zhuang, Z.-P.; Skovronsky, D.; Kung, H.F. Binding of two potential imaging agents targeting amyloid plaques in postmortem brain tissues of patients with Alzheimer’s disease. Brain Res. 2004, 1025, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Wilson, A.; Nobrega, J.; Westaway, D.; Verhoeff, P.; Zhuang, Z.-P.; Kung, M.-P.; Kung, H.F. 11C-labeled stilbene derivatives as Aβ-aggregate-specific PET imaging agents for Alzheimer’s disease. Nucl. Med. Biol. 2003, 30, 565–571. [Google Scholar] [CrossRef]
- Verhoeff, N.P.L.G.; Wilson, A.A.; Takeshita, S.; Trop, L.; Hussey, D.; Singh, K.; Kung, H.F.; Kung, M.-P.; Houle, S. In-vivo imaging of Alzheimer disease beta-amyloid with 11CSB-13 PET. Am. J. Geriatr. Psychiatry 2004, 12, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Oya, S.; Kung, M.-P.; Hou, C.; Maier, D.L.; Kung, H.F. F-18 stilbenes as PET imaging agents for detecting beta-amyloid plaques in the brain. J. Med. Chem. 2005, 48, 5980–5988. [Google Scholar] [CrossRef] [Green Version]
- Stephenson, K.A.; Chandra, R.; Zhuang, Z.-P.; Hou, C.; Oya, S.; Kung, M.-P.; Kung, H.F. Fluoro-pegylated (FPEG) imaging agents targeting Abeta aggregates. Bioconjug. Chem. 2007, 18, 238–246. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Oya, S.; Kung, M.-P.; Hou, C.; Maier, D.L.; Kung, H.F. F-18 Polyethyleneglycol stilbenes as PET imaging agents targeting Abeta aggregates in the brain. Nucl. Med. Biol. 2005, 32, 799–809. [Google Scholar] [CrossRef]
- Kung, H.F.; Choi, S.R.; Qu, W.; Zhang, W.; Skovronsky, D. 18F stilbenes and styrylpyridines for PET imaging of A beta plaques in Alzheimer’s disease: A miniperspective. J. Med. Chem. 2010, 53, 933–941. [Google Scholar] [CrossRef] [Green Version]
- Rowe, C.C.; Ackerman, U.; Browne, W.; Mulligan, R.; Pike, K.L.; O’Keefe, G.; Tochon-Danguy, H.; Chan, G.; Berlangieri, S.U.; Jones, G.; et al. Imaging of amyloid β in Alzheimer’s disease with 18F-BAY94-9172, a novel PET tracer: proof of mechanism. Lancet Neurol. 2008, 7, 129–135. [Google Scholar] [CrossRef]
- Frequently Asked Questions about Beta-Amyloid Imaging. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/204677s000lbl.pdf (accessed on 20 February 2020).
- Zhang, W.; Kung, M.-P.; Oya, S.; Hou, C.; Kung, H.F. 18F-labeled styrylpyridines as PET agents for amyloid plaque imaging. Nucl. Med. Biol. 2007, 34, 89–97. [Google Scholar] [CrossRef]
- Choi, S.R.; Golding, G.; Zhuang, Z.; Zhang, W.; Lim, N.; Hefti, F.; Benedum, T.E.; Kilbourn, M.R.; Skovronsky, D.; Kung, H.F. Preclinical properties of 18F-AV-45: A PET agent for Abeta plaques in the brain. J. Nucl. Med. 2009, 50, 1887–1894. [Google Scholar] [CrossRef] [Green Version]
- Wong, D.F.; Rosenberg, P.B.; Zhou, Y.; Kumar, A.; Raymont, V.; Ravert, H.T.; Dannals, R.F.; Nandi, A.; Brasić, J.R.; Ye, W.; et al. In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18F-AV-45 (florbetapir corrected F 18). J. Nucl. Med. 2010, 51, 913–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamura, N.; Yanai, K. Florbetapir (18F), a PET imaging agent that binds to amyloid plaques for the potential detection of Alzheimer’s disease. IDrugs 2010, 13, 890–899. [Google Scholar] [PubMed]
- Kolata, G. Promise Seen for Detection of Alzheimer’s. Available online: https://www.nytimes.com/2010/06/24/health/research/24scans.html (accessed on 5 December 2019).
- Johnson, A.E.; Jeppsson, F.; Sandell, J.; Wensbo, D.; Neelissen, J.A.M.; Juréus, A.; Ström, P.; Norman, H.; Farde, L.; Svensson, S.P.S. AZD2184: A radioligand for sensitive detection of beta-amyloid deposits. J. Neurochem. 2009, 108, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Swahn, B.-M.; Sandell, J.; Pyring, D.; Bergh, M.; Jeppsson, F.; Juréus, A.; Neelissen, J.; Johnström, P.; Schou, M.; Svensson, S. Synthesis and evaluation of pyridylbenzofuran, pyridylbenzothiazole and pyridylbenzoxazole derivatives as ¹⁸F-PET imaging agents for β-amyloid plaques. Bioorg. Med. Chem. Lett. 2012, 22, 4332–4337. [Google Scholar] [CrossRef] [PubMed]
- Nelissen, N.; van Laere, K.; Thurfjell, L.; Owenius, R.; Vandenbulcke, M.; Koole, M.; Bormans, G.; Brooks, D.J.; Vandenberghe, R. Phase 1 study of the Pittsburgh compound B derivative 18F-flutemetamol in healthy volunteers and patients with probable Alzheimer disease. J. Nucl. Med. 2009, 50, 1251–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathis, C.; Lopresti, B.; Mason, N.; Price, J.; Flatt, N.; Bi, W.; Ziolko, S.; DeKosky, S.; Klunk, W. Comparison of the Amyloid Imaging Agents [F-18]3′-F-PIB and [C-11]PIB in Alzheimer’s Disease and Control Subjects. Available online: http://jnm.snmjournals.org/content/48/supplement_2/56P.3.short (accessed on 6 December 2019).
- Lopresti, B.J.; Klunk, W.E.; Mathis, C.A.; Hoge, J.A.; Ziolko, S.K.; Lu, X.; Meltzer, C.C.; Schimmel, K.; Tsopelas, N.D.; DeKosky, S.T.; et al. Simplified quantification of Pittsburgh Compound B amyloid imaging PET studies: A comparative analysis. J. Nucl. Med. 2005, 46, 1959–1972. [Google Scholar]
- Cselényi, Z.; Jönhagen, M.E.; Forsberg, A.; Halldin, C.; Julin, P.; Schou, M.; Johnström, P.; Varnäs, K.; Svensson, S.; Farde, L. Clinical validation of 18F-AZD4694, an amyloid-β-specific PET radioligand. J. Nucl. Med. 2012, 53, 415–424. [Google Scholar] [CrossRef] [Green Version]
- A Phase 3 Clinical Trial to Evaluate the Efficacy and Safety of [18F]NAV4694 PET for Detection of Cerebral Beta-Amyloid When Compared With Postmortem Histopathology-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01886820 (accessed on 6 December 2019).
- Flutafuranol F 18-Navidea Biopharmaceuticals-AdisInsight. Available online: https://adisinsight.springer.com/drugs/800031066 (accessed on 6 December 2019).
- Hostetler, E.D.; Sanabria-Bohórquez, S.; Fan, H.; Zeng, Z.; Gammage, L.; Miller, P.; O’Malley, S.; Connolly, B.; Mulhearn, J.; Harrison, S.T.; et al. 18FFluoroazabenzoxazoles as potential amyloid plaque PET tracers: synthesis and in vivo evaluation in rhesus monkey. Nucl. Med. Biol. 2011, 38, 1193–1203. [Google Scholar] [CrossRef]
- Merck Clinical Trials. Available online: https://www.merck.com/clinical-trials/study.html?id=3328-002&kw=alzheimer%27s&tab=eligibility (accessed on 6 December 2019).
- Yousefi, B.H.; Drzezga, A.; von Reutern, B.; Manook, A.; Schwaiger, M.; Wester, H.-J.; Henriksen, G. A Novel (18)F-Labeled Imidazo2,1-bbenzothiazole (IBT) for High-Contrast PET Imaging of β-Amyloid Plaques. ACS Med. Chem. Lett. 2011, 2, 673–677. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, B.H.; von Reutern, B.; Scherübl, D.; Manook, A.; Schwaiger, M.; Grimmer, T.; Henriksen, G.; Förster, S.; Drzezga, A.; Wester, H.-J. FIBT versus florbetaben and PiB: A preclinical comparison study with amyloid-PET in transgenic mice. EJNMMI Res. 2015, 5, 6519. [Google Scholar] [CrossRef] [Green Version]
- Westwell, A.D. Fluorinated Pharmaceuticals. Development of 18F-labeled compounds for imaging of Aβ plaques by means of PET.; Future Science Ltd.: London, UK, 2015; ISBN 9781910419007. [Google Scholar]
- Yousefi, B.H.; Manook, A.; Grimmer, T.; Arzberger, T.; von Reutern, B.; Henriksen, G.; Drzezga, A.; Förster, S.; Schwaiger, M.; Wester, H.-J. Characterization and first human investigation of FIBT, a novel fluorinated Aβ plaque neuroimaging PET radioligand. ACS Chem. Neurosci. 2015, 6, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Rosenberg, P.; Oh, E. A Review of Diagnostic Impact of Amyloid Positron Emission Tomography Imaging in Clinical Practice. Dement. Geriatr. Cogn. Disord. 2018, 46, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Barthel, H.; Sabri, O. Clinical Use and Utility of Amyloid Imaging. J. Nucl. Med. 2017, 58, 1711–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiotis, K.; Saint-Aubert, L.; Boccardi, M.; Gietl, A.; Picco, A.; Varrone, A.; Garibotto, V.; Herholz, K.; Nobili, F.; Nordberg, A. Clinical validity of increased cortical uptake of amyloid ligands on PET as a biomarker for Alzheimer’s disease in the context of a structured 5-phase development framework. Neurobiol. Aging 2017, 52, 214–227. [Google Scholar] [CrossRef] [Green Version]
- Ceccaldi, M.; Jonveaux, T.; Verger, A.; Salmon, P.K.; Houzard, C.; Godefroy, O.; Shields, T.; Perrotin, A.; Gismondi, R.; Bullich, S.; et al. Impact of Florbetaben PET Imaging on Diagnosis and Management of Patients with Suspected Alzheimer’s Disease Eligible for CSF Analysis in France. Available online: http://jnm.snmjournals.org/content/58/supplement_1/561?related-urls=yes&legid=jnumed;58/supplement_1/561 (accessed on 8 February 2020).
- Hattori, N.; Ono, S.; UDO, N.; Yamamoto, S.; Ogawa, M.; Sugie, H. Clinical Impact of F-18 Flutemetamol (FMM) PET to Assess Cerebral Aß Pathology in Patients with Various Cognitive Disorders. Available online: http://jnm.snmjournals.org/content/59/supplement_1/480 (accessed on 8 February 2020).
- Leuzy, A.; Savitcheva, I.; Chiotis, K.; Lilja, J.; Andersen, P.; Bogdanovic, N.; Jelic, V.; Nordberg, A. Clinical impact of 18Fflutemetamol PET among memory clinic patients with an unclear diagnosis. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1276–1286. [Google Scholar] [CrossRef] [Green Version]
- Sabri, O.; Seibyl, J.; Rowe, C.; Barthel, H. Beta-amyloid imaging with florbetaben. Clin. Transl. Imaging 2015, 3, 13–26. [Google Scholar] [CrossRef] [Green Version]
- de Wilde, A.; van der Flier, W.M.; Pelkmans, W.; Bouwman, F.; Verwer, J.; Groot, C.; van Buchem, M.M.; Zwan, M.; Ossenkoppele, R.; Yaqub, M.; et al. Association of Amyloid Positron Emission Tomography With Changes in Diagnosis and Patient Treatment in an Unselected Memory Clinic Cohort: The ABIDE Project. JAMA Neurol. 2018, 75, 1062–1070. [Google Scholar] [CrossRef]
- Zannas, A.S.; Doraiswamy, P.M.; Shpanskaya, K.S.; Murphy, K.R.; Petrella, J.R.; Burke, J.R.; Wong, T.Z. Impact of ¹⁸F-florbetapir PET imaging of β-amyloid neuritic plaque density on clinical decision-making. Neurocase 2014, 20, 466–473. [Google Scholar] [CrossRef]
- University of Zurich. Investigating the Clinical Consequences of Flutemetamol-PET-Scanning-ICH GCP-Clinical Trials Registry. Available online: https://ichgcp.net/clinical-trials-registry/NCT02353949 (accessed on 8 February 2020).
- Buée, L.; Bussière, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders11These authors contributed equally to this work. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Fichou, Y.; Al-Hilaly, Y.K.; Devred, F.; Smet-Nocca, C.; Tsvetkov, P.O.; Verelst, J.; Winderickx, J.; Geukens, N.; Vanmechelen, E.; Perrotin, A.; et al. The elusive tau molecular structures: can we translate the recent breakthroughs into new targets for intervention? Acta Neuropathol. Commun. 2019, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- Harada, R.; Okamura, N.; Furumoto, S.; Tago, T.; Yanai, K.; Arai, H.; Kudo, Y. Characteristics of Tau and Its Ligands in PET Imaging. Biomolecules 2016, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Mott, R.T.; Dickson, D.W.; Trojanowski, J.Q.; Zhukareva, V.; Lee, V.M.; Forman, M.; van Deerlin, V.; Ervin, J.F.; Wang, D.-S.; Schmechel, D.E.; et al. Neuropathologic, biochemical, and molecular characterization of the frontotemporal dementias. J. Neuropathol. Exp. Neurol. 2005, 64, 420–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, D.W.; Kouri, N.; Murray, M.E.; Josephs, K.A. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J. Mol. Neurosci. 2011, 45, 384–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, R.; Okamura, N.; Furumoto, S.; Yanai, K. Imaging Protein Misfolding in the Brain Using β-Sheet Ligands. Front. Neurosci. 2018, 12, 585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinton, L.K.; Blurton-Jones, M.; Myczek, K.; Trojanowski, J.Q.; LaFerla, F.M. Synergistic Interactions between Abeta, tau, and alpha-synuclein: Acceleration of neuropathology and cognitive decline. J. Neurosci. 2010, 30, 7281–7289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Näslund, J.; Haroutunian, V.; Mohs, R.; Davis, K.L.; Davies, P.; Greengard, P.; Buxbaum, J.D. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA 2000, 283, 1571–1577. [Google Scholar] [CrossRef]
- Okamura, N.; Suemoto, T.; Furumoto, S.; Suzuki, M.; Shimadzu, H.; Akatsu, H.; Yamamoto, T.; Fujiwara, H.; Nemoto, M.; Maruyama, M.; et al. Quinoline and benzimidazole derivatives: candidate probes for in vivo imaging of tau pathology in Alzheimer’s disease. J. Neurosci. 2005, 25, 10857–10862. [Google Scholar] [CrossRef]
- Fodero-Tavoletti, M.T.; Okamura, N.; Furumoto, S.; Mulligan, R.S.; Connor, A.R.; McLean, C.A.; Cao, D.; Rigopoulos, A.; Cartwright, G.A.; O’Keefe, G.; et al. 18F-THK523: A novel in vivo tau imaging ligand for Alzheimer’s disease. Brain 2011, 134, 1089–1100. [Google Scholar] [CrossRef] [Green Version]
- Okamura, N.; Furumoto, S.; Harada, R.; Tago, T.; Yoshikawa, T.; Fodero-Tavoletti, M.; Mulligan, R.S.; Villemagne, V.L.; Akatsu, H.; Yamamoto, T.; et al. Novel 18F-labeled arylquinoline derivatives for noninvasive imaging of tau pathology in Alzheimer disease. J. Nucl. Med. 2013, 54, 1420–1427. [Google Scholar] [CrossRef] [Green Version]
- Villemagne, V.L.; Furumoto, S.; Fodero-Tavoletti, M.T.; Mulligan, R.S.; Hodges, J.; Harada, R.; Yates, P.; Piguet, O.; Pejoska, S.; Doré, V.; et al. In vivo evaluation of a novel tau imaging tracer for Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 816–826. [Google Scholar] [CrossRef]
- Tago, T.; Furumoto, S.; Okamura, N.; Harada, R.; Adachi, H.; Ishikawa, Y.; Yanai, K.; Iwata, R.; Kudo, Y. Preclinical Evaluation of (18)FTHK-5105 Enantiomers: Effects of Chirality on Its Effectiveness as a Tau Imaging Radiotracer. Mol. Imaging Biol. 2016, 18, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Okamura, N.; Furumoto, S.; Fodero-Tavoletti, M.T.; Mulligan, R.S.; Harada, R.; Yates, P.; Pejoska, S.; Kudo, Y.; Masters, C.L.; Yanai, K.; et al. Non-invasive assessment of Alzheimer’s disease neurofibrillary pathology using 18F-THK5105 PET. Brain 2014, 137, 1762–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saint-Aubert, L.; Almkvist, O.; Chiotis, K.; Almeida, R.; Wall, A.; Nordberg, A. Regional tau deposition measured by 18FTHK5317 positron emission tomography is associated to cognition via glucose metabolism in Alzheimer’s disease. Alzheimers Res. Ther. 2016, 8, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiotis, K.; Saint-Aubert, L.; Savitcheva, I.; Jelic, V.; Andersen, P.; Jonasson, M.; Eriksson, J.; Lubberink, M.; Almkvist, O.; Wall, A.; et al. Imaging in-vivo tau pathology in Alzheimer’s disease with THK5317 PET in a multimodal paradigm. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1686–1699. [Google Scholar] [CrossRef] [PubMed]
- Tago, T.; Furumoto, S.; Okamura, N.; Harada, R.; Adachi, H.; Ishikawa, Y.; Yanai, K.; Iwata, R.; Kudo, Y. Structure-Activity Relationship of 2-Arylquinolines as PET Imaging Tracers for Tau Pathology in Alzheimer Disease. J. Nucl. Med. 2016, 57, 608–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, R.; Okamura, N.; Furumoto, S.; Furukawa, K.; Ishiki, A.; Tomita, N.; Tago, T.; Hiraoka, K.; Watanuki, S.; Shidahara, M.; et al. 18F-THK5351: A Novel PET Radiotracer for Imaging Neurofibrillary Pathology in Alzheimer Disease. J. Nucl. Med. 2016, 57, 208–214. [Google Scholar] [CrossRef] [Green Version]
- Sone, D.; Imabayashi, E.; Maikusa, N.; Okamura, N.; Furumoto, S.; Kudo, Y.; Ogawa, M.; Takano, H.; Yokoi, Y.; Sakata, M.; et al. Regional tau deposition and subregion atrophy of medial temporal structures in early Alzheimer’s disease: A combined positron emission tomography/magnetic resonance imaging study. Alzheimers Dement. 2017, 9, 35–40. [Google Scholar] [CrossRef]
- Betthauser, T.J.; Lao, P.J.; Murali, D.; Barnhart, T.E.; Furumoto, S.; Okamura, N.; Stone, C.K.; Johnson, S.C.; Christian, B.T. In Vivo Comparison of Tau Radioligands 18F-THK-5351 and 18F-THK-5317. J. Nucl. Med. 2017, 58, 996–1002. [Google Scholar] [CrossRef] [Green Version]
- Edmondson, D.E.; Mattevi, A.; Binda, C.; Li, M.; Hubálek, F. Structure and mechanism of monoamine oxidase. Curr. Med. Chem. 2004, 11, 1983–1993. [Google Scholar] [CrossRef]
- Tipton, K.F.; Boyce, S.; O’Sullivan, J.; Davey, G.P.; Healy, J. Monoamine oxidases: certainties and uncertainties. Curr. Med. Chem. 2004, 11, 1965–1982. [Google Scholar] [CrossRef]
- Ishiki, A.; Harada, R.; Kai, H.; Sato, N.; Totsune, T.; Tomita, N.; Watanuki, S.; Hiraoka, K.; Ishikawa, Y.; Funaki, Y.; et al. Neuroimaging-pathological correlations of 18FTHK5351 PET in progressive supranuclear palsy. Acta Neuropathol. Commun. 2018, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Harada, R.; Ishiki, A.; Kai, H.; Sato, N.; Furukawa, K.; Furumoto, S.; Tago, T.; Tomita, N.; Watanuki, S.; Hiraoka, K.; et al. Correlations of 18F-THK5351 PET with Postmortem Burden of Tau and Astrogliosis in Alzheimer Disease. J. Nucl. Med. 2018, 59, 671–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebs, M.R.H.; Bromley, E.H.C.; Donald, A.M. The binding of thioflavin-T to amyloid fibrils: localisation and implications. J. Struct. Biol. 2005, 149, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, M.; Shimada, H.; Suhara, T.; Shinotoh, H.; Ji, B.; Maeda, J.; Zhang, M.-R.; Trojanowski, J.Q.; Lee, V.M.-Y.; Ono, M.; et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron 2013, 79, 1094–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, H.; Kawamura, K.; Igarashi, N.; Takei, M.; Fujishiro, T.; Aihara, Y.; Shiomi, S.; Muto, M.; Ito, T.; Furutsuka, K.; et al. Radiosynthesis, photoisomerization, biodistribution, and metabolite analysis of 11C-PBB3 as a clinically useful PET probe for imaging of tau pathology. J. Nucl. Med. 2014, 55, 1532–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, S.; Ono, M.; Sahara, N.; Higuchi, M.; Dickson, D.W. Fluorescence and autoradiographic evaluation of tau PET ligand PBB3 to α-synuclein pathology. Mov. Disord. 2017, 32, 884–892. [Google Scholar] [CrossRef]
- Perez-Soriano, A.; Arena, J.E.; Dinelle, K.; Miao, Q.; McKenzie, J.; Neilson, N.; Puschmann, A.; Schaffer, P.; Shinotoh, H.; Smith-Forrester, J.; et al. PBB3 imaging in Parkinsonian disorders: Evidence for binding to tau and other proteins. Mov. Disord. 2017, 32, 1016–1024. [Google Scholar] [CrossRef]
- Hsieh, C.-J.; Mach, R.H.; Zhude, T.; Kotzbauer, P.T. Imaging of Aggregated Alpha-Synuclein in Parkinson’s Disease: A Work in Progress. The newsletter of the SNMI. Centrer for Molecular Imaging Innovation and Translation. 2018, 12. Available online: http://s3.amazonaws.com/rdcmssnmmi/ files/production/public/FileDownloads/MiGateway_2_2018_final.pdf (accessed on 8 December 2019).
- Cashion, D.K.; Chen, G.; Kasi, D.; Kolb, H.C.; Liu, C.; Sinha, A.; Szardenings, A.K.; Wang, E.; Yu, C.; Zhang, W.; et al. Imaging Agents for Detecting Neurological Disorders. Available online: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011119565 (accessed on 8 December 2019).
- Xia, C.-F.; Arteaga, J.; Chen, G.; Gangadharmath, U.; Gomez, L.F.; Kasi, D.; Lam, C.; Liang, Q.; Liu, C.; Mocharla, V.P.; et al. (18)FT807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimers Dement. 2013, 9, 666–676. [Google Scholar] [CrossRef]
- Zhang, W.; Arteaga, J.; Cashion, D.K.; Chen, G.; Gangadharmath, U.; Gomez, L.F.; Kasi, D.; Lam, C.; Liang, Q.; Liu, C.; et al. A highly selective and specific PET tracer for imaging of tau pathologies. J. Alzheimers Dis. 2012, 31, 601–612. [Google Scholar] [CrossRef]
- Chien, D.T.; Bahri, S.; Szardenings, A.K.; Walsh, J.C.; Mu, F.; Su, M.-Y.; Shankle, W.R.; Elizarov, A.; Kolb, H.C. Early clinical PET imaging results with the novel PHF-tau radioligand F-18-T807. J. Alzheimers Dis. 2013, 34, 457–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mintun, M.; Schwarz, A.; Joshi, A.; Shcherbinin, S.; Chien, D.; Elizarov, A.; Su, M.-Y.; Shankle, W.; Pontecorvo, M.; Tauscher, J.; et al. Exploratory analyses of regional human brain distribution of the PET tau tracer F18-labeled T807 (AV-1541) in subjects with normal cognitive function or cognitive impairment thought to be due to Alzheimer’s disease. Alzheimer’s Dement. 2013, 9, P842. [Google Scholar] [CrossRef]
- Ono, M.; Sahara, N.; Kumata, K.; Ji, B.; Ni, R.; Koga, S.; Dickson, D.W.; Trojanowski, J.Q.; Lee, V.M.-Y.; Yoshida, M.; et al. Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain 2017, 140, 764–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Choi, J.Y.; Lee, S.H.; Ryu, Y.H.; Lee, M.S.; Lyoo, C.H. 18 F-AV-1451 binds to putamen in multiple system atrophy. Mov. Disord. 2017, 32, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Leuzy, A.; Chiotis, K.; Lemoine, L.; Gillberg, P.-G.; Almkvist, O.; Rodriguez-Vieitez, E.; Nordberg, A. Tau PET imaging in neurodegenerative tauopathies-still a challenge. Mol. Psychiatry 2019, 24, 1112–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, H.C.; Andrés, J.I. Tau Positron Emission Tomography Imaging. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Lowe, V.J.; Curran, G.; Fang, P.; Liesinger, A.M.; Josephs, K.A.; Parisi, J.E.; Kantarci, K.; Boeve, B.F.; Pandey, M.K.; Bruinsma, T.; et al. An autoradiographic evaluation of AV-1451 Tau PET in dementia. Acta Neuropathol. Commun. 2016, 4, 58. [Google Scholar] [CrossRef] [Green Version]
- Hostetler, E.D.; Walji, A.M.; Zeng, Z.; Miller, P.; Bennacef, I.; Salinas, C.; Connolly, B.; Gantert, L.; Haley, H.; Holahan, M.; et al. Preclinical Characterization of 18F-MK-6240, a Promising PET Tracer for In Vivo Quantification of Human Neurofibrillary Tangles. J. Nucl. Med. 2016, 57, 1599–1606. [Google Scholar] [CrossRef] [Green Version]
- Vermeiren, C.; Motte, P.; Viot, D.; Mairet-Coello, G.; Courade, J.-P.; Citron, M.; Mercier, J.; Hannestad, J.; Gillard, M. The tau positron-emission tomography tracer AV-1451 binds with similar affinities to tau fibrils and monoamine oxidases. Mov. Disord. 2018, 33, 273–281. [Google Scholar] [CrossRef]
- Wooten, D.W.; Guehl, N.J.; Verwer, E.E.; Shoup, T.M.; Yokell, D.L.; Zubcevik, N.; Vasdev, N.; Zafonte, R.D.; Johnson, K.A.; El Fakhri, G.; et al. Pharmacokinetic Evaluation of the Tau PET Radiotracer 18F-T807 (18F-AV-1451) in Human Subjects. J. Nucl. Med. 2017, 58, 484–491. [Google Scholar] [CrossRef] [Green Version]
- Shcherbinin, S.; Schwarz, A.J.; Joshi, A.; Navitsky, M.; Flitter, M.; Shankle, W.R.; Devous, M.D.; Mintun, M.A. Kinetics of the Tau PET Tracer 18F-AV-1451 (T807) in Subjects with Normal Cognitive Function, Mild Cognitive Impairment, and Alzheimer Disease. J. Nucl. Med. 2016, 57, 1535–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Declercq, L.; Celen, S.; Lecina, J.; Ahamed, M.; Tousseyn, T.; Moechars, D.; Alcazar, J.; Ariza, M.; Fierens, K.; Bottelbergs, A.; et al. Comparison of New Tau PET-Tracer Candidates With 18FT808 and 18FT807. Mol. Imaging 2016, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.T.; Edison, P. Tau Imaging in Neurodegenerative Diseases Using Positron Emission Tomography. Curr. Neurol. Neurosci. Rep. 2019, 19, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanabria Bohórquez, S.; Marik, J.; Ogasawara, A.; Tinianow, J.N.; Gill, H.S.; Barret, O.; Tamagnan, G.; Alagille, D.; Ayalon, G.; Manser, P.; et al. 18FGTP1 (Genentech Tau Probe 1), a radioligand for detecting neurofibrillary tangle tau pathology in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2077–2089. [Google Scholar] [CrossRef]
- Barret, O.; Alagille, D.; Sanabria, S.; Comley, R.A.; Weimer, R.M.; Borroni, E.; Mintun, M.; Seneca, N.; Papin, C.; Morley, T.; et al. Kinetic Modeling of the Tau PET Tracer 18F-AV-1451 in Human Healthy Volunteers and Alzheimer Disease Subjects. J. Nucl. Med. 2017, 58, 1124–1131. [Google Scholar] [CrossRef] [Green Version]
- Seki, C.; Tagai, K.; Shimada, H.; Takahata, K.; Kubota, M.; Takado, Y.; Shinitoh, H.; Kimura, Y.; Ichise, M.; Okada, M. Establishment of a Simplified Method to Quantify [18F]PM-PBB3 ([18F]APN-1607) Binding in the Brains of Living Human Subjects. 2019. Available online: https://repo.qst.go.jp/?action=pages_view_main&active_action=repository_view_main_item_detail&item_id=78239&item_no=1&page_id=13&block_id=21 (accessed on 18 February 2020).
- 18F-PM-PBB3 PET Study in Tauopathy Including Alzheimer’s Disease, Other Dementias and Normal Controls-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/study/NCT03625128 (accessed on 10 December 2019).
- Brendel, M.; Yousefi, B.H.; Blume, T.; Herz, M.; Focke, C.; Deussing, M.; Peters, F.; Lindner, S.; von Ungern-Sternberg, B.; Drzezga, A.; et al. Comparison of 18F-T807 and 18F-THK5117 PET in a Mouse Model of Tau Pathology. Front. Aging Neurosci. 2018, 10, 174. [Google Scholar] [CrossRef] [Green Version]
- Wong, D.F.; Comley, R.A.; Kuwabara, H.; Rosenberg, P.B.; Resnick, S.M.; Ostrowitzki, S.; Vozzi, C.; Boess, F.; Oh, E.; Lyketsos, C.G.; et al. Characterization of 3 Novel Tau Radiopharmaceuticals, 11C-RO-963, 11C-RO-643, and 18F-RO-948, in Healthy Controls and in Alzheimer Subjects. J. Nucl. Med. 2018, 59, 1869–1876. [Google Scholar] [CrossRef] [Green Version]
- Gobbi, L.C.; Knust, H.; Körner, M.; Honer, M.; Czech, C.; Belli, S.; Muri, D.; Edelmann, M.R.; Hartung, T.; Erbsmehl, I.; et al. Identification of Three Novel Radiotracers for Imaging Aggregated Tau in Alzheimer’s Disease with Positron Emission Tomography. J. Med. Chem. 2017, 60, 7350–7370. [Google Scholar] [CrossRef]
- Honer, M.; Gobbi, L.; Knust, H.; Kuwabara, H.; Muri, D.; Koerner, M.; Valentine, H.; Dannals, R.F.; Wong, D.F.; Borroni, E. Preclinical Evaluation of 18F-RO6958948, 11C-RO6931643, and 11C-RO6924963 as Novel PET Radiotracers for Imaging Tau Aggregates in Alzheimer Disease. J. Nucl. Med. 2018, 59, 675–681. [Google Scholar] [CrossRef] [Green Version]
- Marquié, M.; Normandin, M.D.; Vanderburg, C.R.; Costantino, I.M.; Bien, E.A.; Rycyna, L.G.; Klunk, W.E.; Mathis, C.A.; Ikonomovic, M.D.; Debnath, M.L.; et al. Validating novel tau positron emission tomography tracer F-18-AV-1451 (T807) on postmortem brain tissue. Ann. Neurol. 2015, 78, 787–800. [Google Scholar] [CrossRef] [Green Version]
- Walji, A.M.; Hostetler, E.D.; Selnick, H.; Zeng, Z.; Miller, P.; Bennacef, I.; Salinas, C.; Connolly, B.; Gantert, L.; Holahan, M.; et al. Discovery of 6-(Fluoro-(18)F)-3-(1H-pyrrolo2,3-cpyridin-1-yl)isoquinolin-5-amine ((18)F-MK-6240): A Positron Emission Tomography (PET) Imaging Agent for Quantification of Neurofibrillary Tangles (NFTs). J. Med. Chem. 2016, 59, 4778–4789. [Google Scholar] [CrossRef] [PubMed]
- Pascoal, T.A.; Shin, M.; Kang, M.S.; Chamoun, M.; Chartrand, D.; Mathotaarachchi, S.; Bennacef, I.; Therriault, J.; Ng, K.P.; Hopewell, R.; et al. In vivo quantification of neurofibrillary tangles with 18FMK-6240. Alzheimers Res. Ther. 2018, 10, 74. [Google Scholar] [CrossRef] [PubMed]
- Betthauser, T.J.; Cody, K.A.; Zammit, M.D.; Murali, D.; Converse, A.K.; Barnhart, T.E.; Stone, C.K.; Rowley, H.A.; Johnson, S.C.; Christian, B.T. In Vivo Characterization and Quantification of Neurofibrillary Tau PET Radioligand 18F-MK-6240 in Humans from Alzheimer Disease Dementia to Young Controls. J. Nucl. Med. 2019, 60, 93–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- [18F]MK-6240 Positron Emission Tomography (PET) Tracer First-in-Human Validation Study (MK-6240-001)-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02562989 (accessed on 8 December 2019).
- Declercq, L.; Rombouts, F.; Koole, M.; Fierens, K.; Mariën, J.; Langlois, X.; Andrés, J.I.; Schmidt, M.; Macdonald, G.; Moechars, D.; et al. Preclinical Evaluation of 18F-JNJ64349311, a Novel PET Tracer for Tau Imaging. J. Nucl. Med. 2017, 58, 975–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Politis, M. Neuroimaging in Parkinson disease: from research setting to clinical practice. Nat. Rev. Neurol. 2014, 10, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W.; Braak, H.; Duda, J.E.; Duyckaerts, C.; Gasser, T.; Halliday, G.M.; Hardy, J.; Leverenz, J.B.; Del Tredici, K.; Wszolek, Z.K.; et al. Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol. 2009, 8, 1150–1157. [Google Scholar] [CrossRef]
- Yu, L.; Cui, J.; Padakanti, P.K.; Engel, L.; Bagchi, D.P.; Kotzbauer, P.T.; Tu, Z. Synthesis and in vitro evaluation of α-synuclein ligands. Bioorg. Med. Chem. 2012, 20, 4625–4634. [Google Scholar] [CrossRef] [Green Version]
- Bagchi, D.P.; Yu, L.; Perlmutter, J.S.; Xu, J.; Mach, R.H.; Tu, Z.; Kotzbauer, P.T. Binding of the radioligand SIL23 to α-synuclein fibrils in Parkinson disease brain tissue establishes feasibility and screening approaches for developing a Parkinson disease imaging agent. PLoS ONE 2013, 8, e55031. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Jin, H.; Padakanti, P.K.; Li, J.; Yang, H.; Fan, J.; Mach, R.H.; Kotzbauer, P.; Tu, Z. Radiosynthesis and in Vivo Evaluation of Two PET Radioligands for Imaging α-Synuclein. Appl. Sci. 2014, 4, 66–78. [Google Scholar] [CrossRef] [Green Version]
- Honson, N.S.; Johnson, R.L.; Huang, W.; Inglese, J.; Austin, C.P.; Kuret, J. Differentiating Alzheimer disease-associated aggregates with small molecules. Neurobiol. Dis. 2007, 28, 251–260. [Google Scholar] [CrossRef] [Green Version]
- Chu, W.; Zhou, D.; Gaba, V.; Liu, J.; Li, S.; Peng, X.; Xu, J.; Dhavale, D.; Bagchi, D.P.; d’Avignon, A.; et al. Design, Synthesis, and Characterization of 3-(Benzylidene)indolin-2-one Derivatives as Ligands for α-Synuclein Fibrils. J. Med. Chem. 2015, 58, 6002–6017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, C.-J.; Xu, K.; Lee, I.; Graham, T.J.A.; Tu, Z.; Dhavale, D.; Kotzbauer, P.; Mach, R.H. Chalcones and Five-Membered Heterocyclic Isosteres Bind to Alpha Synuclein Fibrils in Vitro. ACS Omega 2018, 3, 4486–4493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, M.; Maya, Y.; Haratake, M.; Ito, K.; Mori, H.; Nakayama, M. Aurones serve as probes of beta-amyloid plaques in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2007, 361, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Haratake, M.; Mori, H.; Nakayama, M. Novel chalcones as probes for in vivo imaging of beta-amyloid plaques in Alzheimer’s brains. Bioorg. Med. Chem. 2007, 15, 6802–6809. [Google Scholar] [CrossRef]
- Ono, M.; Yoshida, N.; Ishibashi, K.; Haratake, M.; Arano, Y.; Mori, H.; Nakayama, M. Radioiodinated flavones for in vivo imaging of beta-amyloid plaques in the brain. J. Med. Chem. 2005, 48, 7253–7260. [Google Scholar] [CrossRef]
- Meng, X.; Munishkina, L.A.; Fink, A.L.; Uversky, V.N. Effects of Various Flavonoids on the α-Synuclein Fibrillation Process. Parkinsons. Dis. 2010, 2010, 650794. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Han, S.; Fink, A.L. Oxidized quercetin inhibits α-synuclein fibrillization. Biochim. Biophys. Acta 2013, 1830, 2872–2881. [Google Scholar] [CrossRef]
- Masuda, M.; Suzuki, N.; Taniguchi, S.; Oikawa, T.; Nonaka, T.; Iwatsubo, T.; Hisanaga, S.-i.; Goedert, M.; Hasegawa, M. Small molecule inhibitors of alpha-synuclein filament assembly. Biochemistry 2006, 45, 6085–6094. [Google Scholar] [CrossRef]
- Cui, M.; Ono, M.; Watanabe, H.; Kimura, H.; Liu, B.; Saji, H. Smart near-infrared fluorescence probes with donor-acceptor structure for in vivo detection of β-amyloid deposits. J. Am. Chem. Soc. 2014, 136, 3388–3394. [Google Scholar] [CrossRef]
- Ono, M.; Doi, Y.; Watanabe, H.; Ihara, M.; Ozaki, A.; Saji, H. Structure–activity relationships of radioiodinated diphenyl derivatives with different conjugated double bonds as ligands for α-synuclein aggregates. RSC Adv. 2016, 6, 44305–44312. [Google Scholar] [CrossRef]
- Fanti, S.; Bonfiglioli, R.; Decristoforo, C. Highlights of the 30th Annual Congress of the EANM, Vienna 2017: “Yes we can-make nuclear medicine great again”. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1781–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wester, H.-J.; Yousefi, B.H. US20170157274A1-Compounds Binding to Neuropathological Aggregates-Google Patents. Available online: https://patents.google.com/patent/US20170157274A1/en (accessed on 24 December 2019).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tracer | Log P | Aβ(1-40) fibrils, [nM] | Aβ(1-42) Fibrils, [nM] | Aβ plaques in Brain Homogenates, [nM] | Brain Uptake [%ID/g] (2 min p.i.) | Brain Clearance [%ID/g] (30 min p.i.) | |||
|---|---|---|---|---|---|---|---|---|---|
| Ki | Kd | Ki | Kd | Ki | Kd | ||||
| Th-T [92,96] | 0.57 | 890 580 | NA | NA | NA | NA | NA | NA | NA |
| [11C]PiB [91,94] [105,131] | 1.2 2.23 | 4.3 | 4.7 | NA | NA | IC50: 2.3 | 1.4 | 0.21%ID-kg/g 1 1.50 (5 min p.i.) | 0.018%ID-kg/g 1 0.31 |
| [18F]Florbetaben [114,119] | 2.41 | NA | NA | NA | NA | 6.7 2.22 | NA | 7.77 | 1.59 |
| [18F]Florbetapir [119] | NA | NA | NA | NA | NA | 2.87 | 3.72 | 7.33 | 1.88 (60 min p.i.) |
| [18F]Flutemetamol [103,105] | 3.2 2 | 15.3 | 1.6 | NA | NA | NA | NA | 3505 nM | 980 nM |
| NA | NA | NA | NA | NA | NA | NA | 3.67 (5 min p.i.) | 0.42 | |
| [11C]6-Me-BTA-1 [92,94,96] | 3.36 | 20.2 10 | NA | NA | NA | NA | NA | 7.61 0.223%ID-kg/g 1 | 2.76 0.083%ID-kg/g 1 |
| [11C]SB-13 [110,112] | 2.36 | 6.0 | NA | NA | NA | 1.2 | NA | 1.15 (cortex) 1.15 (cerebellum) | 0.42 (cortex) 0.41 (cerebellum) |
| [18F]FMAPO [112,114] | 2.95 | NA | NA | NA | NA | 5.0 | NA | 9.75 | 1.70 |
| [18F]Flutafuranol [18F]AZD4694 [103] | 2.8 2 | 18.5 | 2.3 | NA | NA | NA | NA | 1550 nM | 154 nM |
| [18F]MK-3328 [131] | 2.91 | NA | NA | NA | NA | IC50: 10.5 | NA | NA | NA |
| [18F]AD-269 [131] | 3.42 | NA | NA | NA | NA | IC50: 8.0 | NA | NA | NA |
| [18F]FIBT [133,134] | 1.92 | 2.1 | NA | NA | NA | NA | 0.7 | ~7.3 3 | ~1.25 3 |
| Tracer | Log P | Tau Affinity [nM] | Selectivity tau/Aβ | Aβ Affinity (nM) | Brain Uptake [%ID/g] | Brain Clearance [%ID/g] | |
|---|---|---|---|---|---|---|---|
| HITP | AD-PHF, Kd [nM] | 2 min p.i. | 30 min p.i. | ||||
| BF-158 [155] | 1.67 | EC50: 399 | NA | 1.60 1 | Ki: > 5000 | 11.3 | 3.1 |
| BF-170 [155] | 1.85 | EC50: 221 | NA | 3.50 1 | Ki: > 5000 | 9.1 | 0.25 |
| [18F]THK-523 [156,157] | 2.40 | Kd1:1.67 Kd2:21.74 Ki: 59.30 | 86.50 | 10 2 | Kd1 (Aβ fibrils): 20.7 | 2.75 | 1.47 |
| [18F]THK-5105 [157] | 3.03 | Kd1:1.45 Kd2:7.40 Ki: 7.80 | 2.63 | 25 2 | Kd1 (Aβ fibrils): 35.9 | 9.20 | 3.61 |
| [18F]THK-5117 [157] | 2.32 | 10.50 | 5.19 | 30 2 | NA | 6.06 | 0.59 |
| [18F]THK-5351 [164] | 1.5 | NA | 2.9 | NA | NA | NA | NA |
| [11C]PBB3 [172,173] | 3.3 | NA | Kd: 2.55 3 | 48 2 | Kd: 114 3 | 1.92 (1 min p.i.) | 0.11 |
| [18F]Flortaucipir (AV-1451,[18F]T807) [178,191] | 1.67 | NA | 14.6 3 | 25 2 | NA | 4.43 (5 min p.i.)7.5 | 0.620.8 |
| [18F]T808 [179,191] | NA | NA | 22 | 27 2 | NA | 4.9 | 0.4 |
| Tracer | Log P | Tau affinity [nM] | Selectivity, tau/Aβ | Ki, Aβ fibrils (nM) | Brain Uptake [%ID/g] | Brain Clearance [%ID/g] | |
|---|---|---|---|---|---|---|---|
| HITP | AD-PHF | 2 min p.i. | 30 min p.i. | ||||
| [18F]GTP1 [193] | NA | NA | Kd:10.8 | NA | NA | NA | NA |
| [18F]PM-PBB3 (APN-1607) | NA | NA | NA | NA | NA | NA | NA |
| (*9) [202] | NA | NA | Ki: 8.8 | >11361 | >10000 | NA | NA |
| [18F]MK-6240 [202] | 3.32 | NA | Ki: 0.36 | >277771 | >10000 | NA | NA |
| (*12) [202] | 2.90 | NA | Ki: 52.6 | >190 | >10000 | NA | NA |
| [18F]RO-948 (RO6958948)[53] [199] | 3.22 | NA | 44% 2 pIC50: 3 8.4 | NA | pIC50: < 6 4 | 5.7 5 | 10.9 6 |
| [18F]PI-2620 [53] | NA | NA | pIC50: 8.5 7 | NA | pIC50: < 6 4 | 5.9 5 | 16.6 6 |
| [18F]JNJ64349311 (JNJ311) [206] | 2.2 | NA | Ki: 8 8 | >500 | >4398 9 IC50: < 5 9 | 1.9 10 | 0.3 10 |
| Tracer | Log D | α-syn Affinity [nM] | Aβ Fibrils Affinity [nM] | Tau Fibrils Affinity [nM] | Brain Uptake [%ID/g] | Brain Clearance [%ID/g] | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ki | Kd | Ki | Kd | Ki | Kd | 5 min p.i. | 60 min p.i. | |||
| α-syn Fibrils | Human PD Homogenate | |||||||||
| [11C]SIL5 [209,210,211] | 3.79 | 32.1. 66.2. | 83.1 | NA | 110 | NA | 136 | NA | 0.953 | 0.158 |
| [125I]SIL23 [209,210] | 5.72 | 57.9 | NA | 148 | NA | 635 | NA | 230 | NA | NA |
| [18F]SIL26 [209,210,211] | 4.02 | 49.0 15.5. | 33.5 | NA | 103 | NA | 125 | NA | 0.758 | 0.410 |
| *14 [[213] | 4.2 1 | 79.5 | NA | NA | 113.3 | NA | 853.5 | NA | NA | NA |
| *20 [213] | 3.5 2 | 40.7 | NA | NA | 27.6 | NA | 53.7 | NA | NA | NA |
| [18F]46a [213] | 4.18 | 2.1 | NA | 8.9 | 142.4 | NA | 80.1 | NA | NA | NA |
| *11a,b [214] | 3.54 3 | 18.5 | NA | NA | 91.5 | NA | >1000 | NA | NA | NA |
| [125I]IDP-4 [222] | NA | NA | NA | 5.4 | NA | 16.24 | NA | NA | 0.45 (2 min p.i.) | 0.42 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uzuegbunam, B.C.; Librizzi, D.; Hooshyar Yousefi, B. PET Radiopharmaceuticals for Alzheimer’s Disease and Parkinson’s Disease Diagnosis, the Current and Future Landscape. Molecules 2020, 25, 977. https://doi.org/10.3390/molecules25040977
Uzuegbunam BC, Librizzi D, Hooshyar Yousefi B. PET Radiopharmaceuticals for Alzheimer’s Disease and Parkinson’s Disease Diagnosis, the Current and Future Landscape. Molecules. 2020; 25(4):977. https://doi.org/10.3390/molecules25040977
Chicago/Turabian StyleUzuegbunam, Bright Chukwunwike, Damiano Librizzi, and Behrooz Hooshyar Yousefi. 2020. "PET Radiopharmaceuticals for Alzheimer’s Disease and Parkinson’s Disease Diagnosis, the Current and Future Landscape" Molecules 25, no. 4: 977. https://doi.org/10.3390/molecules25040977
APA StyleUzuegbunam, B. C., Librizzi, D., & Hooshyar Yousefi, B. (2020). PET Radiopharmaceuticals for Alzheimer’s Disease and Parkinson’s Disease Diagnosis, the Current and Future Landscape. Molecules, 25(4), 977. https://doi.org/10.3390/molecules25040977