
Design and Construction of a Focused DNA-Encoded Library for Multivalent Chromatin Reader Proteins

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
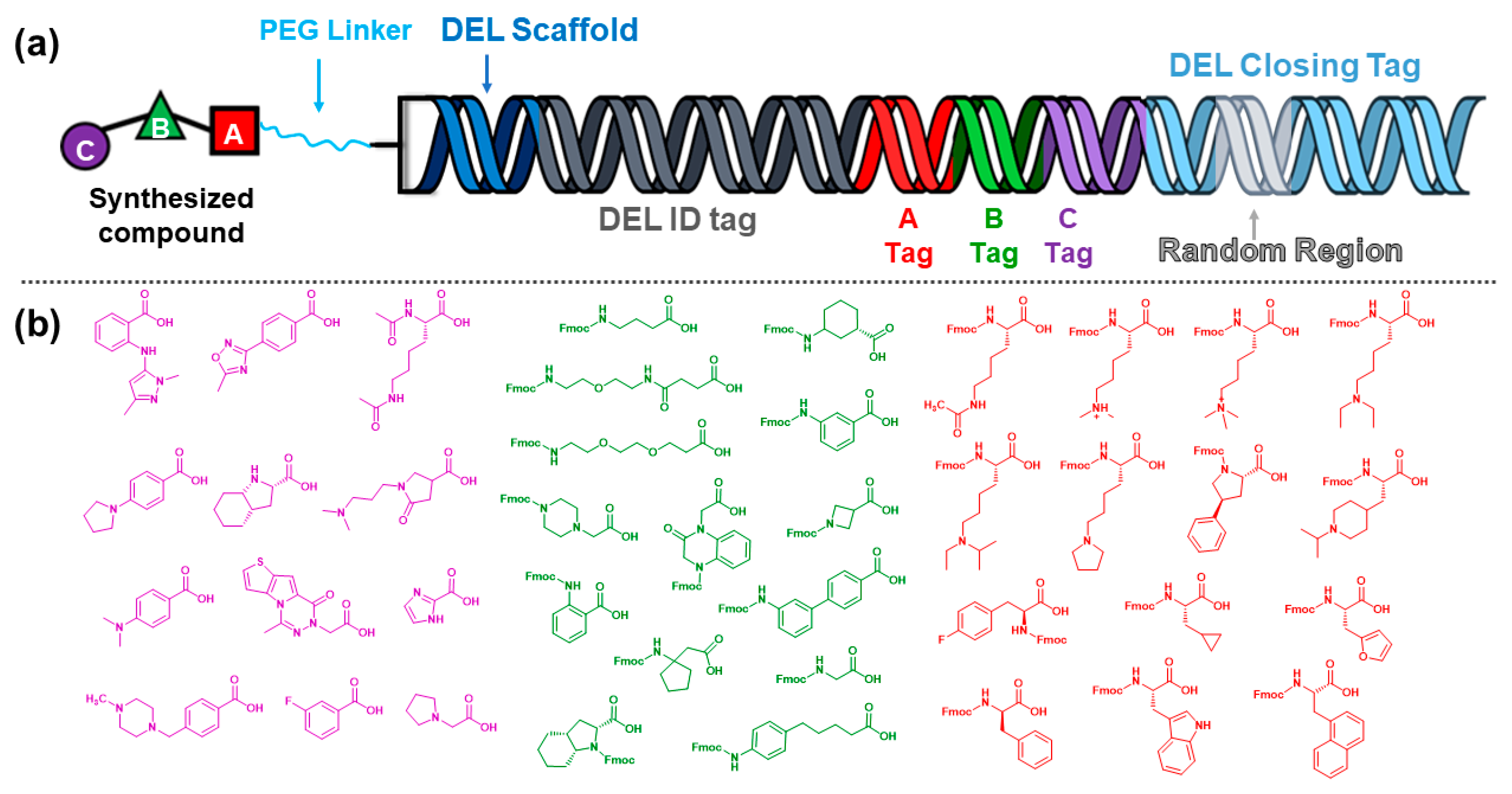
2.1. Computational Selection of the Library Building Blocks
2.2. Summary of DEL Production Strategy
2.3. Production of Barcoded Control Compounds to Inform DEL Selection Conditions
2.4. Evidence of Selection Power Using Control Compounds
3. Materials and Methods
3.1. Materials
3.2. Methods
4. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Krishnamurthy, V.M.; Estroff, L.; Whitesides, G.M. Multivalency in Ligand Design; Erlanson, D.D.A., Wolfgang, D.J., Eds.; WILEY_VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006. [Google Scholar] [CrossRef]
- Ruthenburg, A.J.; Li, H.; Patel, D.J.; Allis, C.D. Multivalent Engagement of Chromatin Modifications by Linked Binding Modules. Nat. Rev. Mol. Cell Biol. 2007, 8, 983–994. [Google Scholar] [CrossRef] [Green Version]
- Jurkowska, R.Z.; Qin, S.; Kungulovski, G.; Tempel, W.; Liu, Y.; Bashtrykov, P.; Stiefelmaier, J.; Jurkowski, T.P.; Kudithipudi, S.; Weirich, S.; et al. H3K14ac Is Linked to Methylation of H3K9 by the Triple Tudor Domain of SETDB1. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Yang, Y.; Fang, J.; Xiao, J.; Zhu, T.; Chen, F.; Wang, P.; Li, Z.; Yang, H.; Xu, Y. Structural Insight into Coordinated Recognition of Trimethylated Histone H3 Lysine 9 (H3K9me3) by the Plant Homeodomain (PHD) and Tandem Tudor Domain (TTD) of UHRF1 (Ubiquitin-like, Containing PHD and RING Finger Domains, 1) Protein. J. Biol. Chem. 2013, 288, 1329–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, W.-W.; Wang, Z.; Yiu, T.T.; Akdemir, K.C.; Xia, W.; Winter, S.; Tsai, C.-Y.; Shi, X.; Schwarzer, D.; Plunkett, W.; et al. TRIM24 Links a Non-Canonical Histone Signature to Breast Cancer. Nature 2010, 468, 927–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, Q.; Wang, Z.; Zaromytidou, A.I.; Zhang, X.H.F.; Chow-Tsang, L.F.; Liu, J.X.; Kim, H.; Barlas, A.; Manova-Todorova, K.; Kaartinen, V.; et al. A Poised Chromatin Platform for TGF-β Access to Master Regulators. Cell 2011, 147, 1511–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, J.R.; Upadhyay, A.K.; Qi, H.H.; Zhang, X.; Shi, Y.; Cheng, X. Enzymatic and Structural Insights for Substrate Specificity of a Family of Jumonji Histone Lysine Demethylases. Nat. Struct. Mol. Biol. 2009, 17, 38–43. [Google Scholar] [CrossRef]
- Ramón-Maiques, S.; Kuo, A.J.; Carney, D.; Matthews, A.G.W.; Oettinger, M.A.; Gozani, O.; Yang, W. The Plant Homeodomain Finger of RAG2 Recognizes Histone H3 Methylated at Both Lysine-4 and Arginine-2. Proc. Natl. Acad. Sci. USA 2007, 104, 18993–18998. [Google Scholar]
- Tong, Q.; Mazur, S.J.; Rincon-Arano, H.; Rothbart, S.B.; Kuznetsov, D.M.; Cui, G.; Liu, W.H.; Gete, Y.; Klein, B.J.; Jenkins, L.; et al. An Acetyl-Methyl Switch Drives a Conformational Change in P53. Structure 2015, 23, 322–331. [Google Scholar] [CrossRef] [Green Version]
- Teske, K.A.; Hadden, M.K. Methyllysine Binding Domains: Structural Insight and Small Molecule Probe Development. Eur. J. Med. Chem. 2017, 136, 14–35. [Google Scholar] [CrossRef]
- Wagner, T.; Robaa, D.; Sippl, W.; Jung, M. Mind the Methyl: Methyllysine Binding Proteins in Epigenetic Regulation. ChemMedChem 2014, 9, 466–483. [Google Scholar] [CrossRef]
- Dougherty, D.A. The Cation-π Interaction. Acc. Chem. Res. 2013, 46, 885–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnash, K.D.; James, L.I.; Frye, S.V. Target Class Drug Discovery. Nat. Chem. Biol. 2017, 13, 1053–1056. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic Modifications and Human Disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Mayr, L.M.; Bojanic, D. Novel Trends in High-Throughput Screening. Curr. Opin. Pharmacol. 2009, 9, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Macarron, R.; Banks, M.N.; Bojanic, D.; Burns, D.J.; Cirovic, D.A.; Garyantes, T.; Green, D.V.S.; Hertzberg, R.P.; Janzen, W.P.; Paslay, J.W.; et al. Impact of High-Throughput Screening in Biomedical Research. Nat. Rev. Drug Discov. 2011, 10, 188–195. [Google Scholar] [CrossRef]
- Goodnow, R.A., Jr.; Dumelin, C.E.; Keefe, A.D. DNA-Encoded Chemistry: Enabling the Deeper Sampling of Chemical Space. Nat. Rev. Drug Discov. 2016, 16, 131–147. [Google Scholar] [CrossRef]
- Yuen, L.H.; Dana, S.; Liu, Y.; Bloom, S.I.; Thorsell, A.; Neri, D.; Donato, A.J.; Kireev, D.; Schu, H.; Franzini, R.M. A Focused DNA-Encoded Chemical Library for the Discovery of Inhibitors of NAD + -Dependent Enzymes. J. Am. Chem. Soc. 2019, 141, 5169–5181. [Google Scholar] [CrossRef]
- Eidam, O.; Satz, A. Analysis of the Productivity of DNA Encoded Libraries. Med. Chem. Commun. 2016, 7, 1323–1331. [Google Scholar] [CrossRef]
- Satz, A.L.; Hochstrasser, R.; Petersen, A.C. Analysis of Current DNA Encoded Library Screening Data Indicates Higher False Negative Rates for Numerically Larger Libraries. ACS Comb. Sci. 2017, 19, 234–238. [Google Scholar] [CrossRef]
- Hutti, J.E.; Porter, M.A.; Cheely, A.W.; Cantley, L.C.; Wang, X.; Kireev, D.; Baldwin, A.S.; Janzen, W.P. Development of a High-Throughput Assay for Identifying Inhibitors of TBK1 and IKKε. PLoS ONE 2012. [Google Scholar] [CrossRef] [Green Version]
- Elkins, J.M.; Fedele, V.; Szklarz, M.; Abdul Azeez, K.R.; Salah, E.; Mikolajczyk, J.; Romanov, S.; Sepetov, N.; Huang, X.P.; Roth, B.L.; et al. Comprehensive Characterization of the Published Kinase Inhibitor Set. Nat. Biotechnol. 2016, 34, 95–103. [Google Scholar] [CrossRef]
- Neri, D.; Franzini, R.M.; Nauer, A.; Neri, D. Interrogating Target-Specificity by Parallel Screening of a DNA-Encoded Chemical Library against Closely Related Proteins. Chem. Commun. 2015, 51, 38–41. [Google Scholar] [CrossRef]
- Franzini, R.M.; Ekblad, T.; Zhong, N.; Wichert, M.; Decurtins, W.; Nauer, A.; Zimmermann, M.; Samain, F.; Scheuermann, J.; Brown, P.J.; et al. Identification of Structure-Activity Relationships from Screening a Structurally Compact DNA-Encoded Chemical Library. Angew. Chemie Int. Ed. 2015, 54, 3927–3931. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Lee, J.; Hagerty, S.W.; Soh, B.Y.; Mcalpin, S.E.; Cormier, K.A.; Smith, K.M.; Ferrante, R.J. ESET/SETDB1 Gene Expression and Histone H3 (K9) Trimethylation in Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2006, 103, 19176–19181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batham, J.; Lim, P.S.; Rao, S. SETDB-1: A Potential Epigenetic Regulator in Breast Cancer Metastasis. Cancers (Basel) 2019, 11, 1143. [Google Scholar] [CrossRef] [Green Version]
- Fei, Q.; Shang, K.; Zhang, J.; Chuai, S.; Kong, D.; Zhou, T.; Fu, S.; Liang, Y.; Li, C.; Chen, Z.; et al. Histone Methyltransferase SETDB1 Regulates Liver Cancer Cell Growth through Methylation of P53. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.M.; Wei, L.; Law, C.T.; Ho, D.W.H.; Tsang, F.H.C.; Au, S.L.K.; Sze, K.M.F.; Lee, J.M.F.; Wong, C.C.L.; Ng, I.O.L. Up-Regulation of Histone Methyltransferase SETDB1 by Multiple Mechanisms in Hepatocellular Carcinoma Promotes Cancer Metastasis. Hepatology 2016, 63, 474–487. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Wei, M.; Ren, S.C.; Chen, R.; Xu, W.D.; Wang, F.B.; Lu, J.; Shen, J.; Yu, Y.W.; Hou, J.G.; et al. Histone Methyltransferase SETDB1 Is Required for Prostate Cancer Cell Proliferation, Migration and Invasion. Asian J. Androl. 2014, 16, 319–324. [Google Scholar] [CrossRef]
- Kong, X.; Chen, J.; Xie, W.; Brown, S.M.; Cai, Y.; Wu, K.; Fan, D.; Nie, Y.; Yegnasubramanian, S.; Tiedemann, R.L.; et al. Defining UHRF1 Domains That Support Maintenance of Human Colon Cancer DNA Methylation and Oncogenic Properties. Cancer Cell 2019, 35, 633–648. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.P.; Sun, H.F.; Li, L.D.; Fu, W.Y.; Jin, W. UHRF1 Promotes Breast Cancer Progression by Suppressing KLF17 Expression by Hypermethylating Its Promoter. Am. J. Cancer Res. 2017, 7, 1554–1565. [Google Scholar]
- Jin, W.; Chen, L.; Chen, Y.; Xu, S.G.; Di, G.H.; Yin, W.J.; Wu, J.; Shao, Z.M. UHRF1 Is Associated with Epigenetic Silencing of BRCA1 in Sporadic Breast Cancer. Breast Cancer Res. Treat. 2010, 123, 359–373. [Google Scholar] [CrossRef] [PubMed]
- Daskalos, A.; Oleksiewicz, U.; Filia, A.; Nikolaidis, G.; Xinarianos, G.; Gosney, J.R.; Malliri, A.; Field, J.K.; Liloglou, T. UHRF1-Mediated Tumor Suppressor Gene Inactivation in Nonsmall Cell Lung Cancer. Cancer 2011, 117, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Babbio, F.; Pistore, C.; Curti, L.; Castiglioni, I.; Kunderfranco, P.; Brino, L.; Oudet, P.; Seiler, R.; Thalman, G.N.; Roggero, E.; et al. The SRA Protein UHRF1 Promotes Epigenetic Crosstalks and Is Involved in Prostate Cancer Progression. Oncogene 2012, 31, 4878–4887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, A.; Trippel, F.; Wagner, A.; Joppien, S.; Felle, M.; Vokuhl, C.; Schwarzmayr, T.; Strom, T.M.; von Schweinitz, D.; Längst, G.; et al. Overexpression of UHRF1 Promotes Silencing of Tumor Suppressor Genes and Predicts Outcome in Hepatoblastoma. Clin. Epigenetics 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambon, M.; Orsetti, B.; Berthe, M.L.; Bascoul-Mollevi, C.; Rodriguez, C.; Duong, V.; Gleizes, M.; Thénot, S.; Bibeau, F.; Theillet, C.; et al. Prognostic Significance of TRIM24/TIF-1α Gene Expression in Breast Cancer. Am. J. Pathol. 2011, 178, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Herquel, B.; Ouararhni, K.; Khetchoumian, K.; Ignat, M.; Teletin, M.; Mark, M.; Béchade, G.; Van Dorsselaer, A.; Sanglier-Cianférani, S.; Hamiche, A.; et al. Transcription Cofactors TRIM24, TRIM28, and TRIM33 Associate to Form Regulatory Complexes That Suppress Murine Hepatocellular Carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 8212–8217. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Sun, L.; Tang, Z.; Fu, L.; Xu, Y.; Li, Z.; Luo, W.; Qiu, X.; Wang, E. Overexpression of TRIM24 Correlates with Tumor Progression in Non-Small Cell Lung Cancer. PLoS ONE 2012, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Appikonda, S.; Thakkar, K.N.; Barton, M.C. Regulation of Gene Expression in Human Cancers by TRIM24. Drug Discov. Today Technol. 2016, 19, 57–63. [Google Scholar] [CrossRef]
- Vincent, D.F.; Yan, K.P.; Treilleux, I.; Gay, F.; Arfi, V.; Kaniewsky, B.; Marie, J.C.; Lepinasse, F.; Martel, S.; Goddard-Leon, S.; et al. Inactivation of TIF1γ Cooperates with KrasG12D to Induce Cystic Tumors of the Pancreas. PLoS Genet. 2009, 5, 1–9. [Google Scholar] [CrossRef]
- Aucagne, R.; Droin, N.; Paggetti, J.; Lagrange, B.; Largeot, A.; Hammann, A.; Bataille, A.; Martin, L.; Yan, K.P.; Fenaux, P.; et al. Transcription Intermediary Factor 1γ Is a Tumor Suppressor in Mouse and Human Chronic Myelomonocytic Leukemia. J. Clin. Invest. 2011, 121, 2361–2370. [Google Scholar] [CrossRef] [Green Version]
- Tong, D.; Liu, Q.; Liu, G.; Yuan, W.; Wang, L.; Guo, Y.; Lan, W.; Zhang, D.; Dong, S.; Wang, Y.; et al. The HIF/PHF8/AR Axis Promotes Prostate Cancer Progression. Oncogenesis 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maina, P.K.; Shao, P.; Liu, Q.; Fazli, L.; Tyler, S.; Nasir, M.; Dong, X.; Qi, H.H. C-MYC Drives Histone Demethylase PHF8 during Neuroendocrine Differentiation and in Castration-Resistant Prostate Cancer. Oncotarget 2016, 7, 75585–75602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Ma, S.; Song, N.; Li, X.; Liu, L.; Yang, S.; Ding, X.; Shan, L.; Zhou, X.; Su, D.; et al. Stabilization of Histone Demethylase PHF8 by USP7 Promotes Breast Carcinogenesis. J. Clin. Invest. 2016, 126, 2205–2220. [Google Scholar] [CrossRef] [Green Version]
- Björkman, M.; Östling, P.; Härmä, V.; Virtanen, J.; Mpindi, J.P.; Rantala, J.; Mirtti, T.; Vesterinen, T.; Lundin, M.; Sankila, A.; et al. Systematic Knockdown of Epigenetic Enzymes Identifies a Novel Histone Demethylase PHF8 Overexpressed in Prostate Cancer with an Impact on Cell Proliferation, Migration and Invasion. Oncogene 2012, 31, 3444–3456. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Sun, A.; Liang, X.; Ma, L.; Shen, L.; Li, T.; Zheng, L.; Shang, W.; Zhao, W.; Jia, J. Histone Demethylase PHF8 Promotes Progression and Metastasis of Gastric Cancer. Am. J. Cancer Res. 2017, 7, 448–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, I.H.; Melek, M.; Jayaratne, N.D.; Gellert, M. Inverse Transposition by the RAG1 and RAG2 Proteins: Role Reversal of Donor and Target DNA. EMBO J. 2002, 21, 6625–6633. [Google Scholar] [CrossRef] [Green Version]
- O’Driscoll, M.; Jeggo, P. Immunological Disorders and DNA Repair. Mutat. Res.Fundam. Mol. Mech. Mutagen. 2002, 509, 109–126. [Google Scholar] [CrossRef]
- Li, X.; Kong, X.; Wang, Y.; Yang, Q. 53BP1 Is a Novel Regulator of Angiogenesis in Breast Cancer. Cancer Sci. 2013, 104, 1420–1426. [Google Scholar] [CrossRef]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; Van Der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 Loss Rescues BRCA1 Deficiency and Is Associated with Triple-Negative and BRCA-Mutated Breast Cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Li, X.; Zhao, Y.; Yang, Q.; Kong, B. 53BP1 Suppresses Tumor Growth and Promotes Susceptibility to Apoptosis of Ovarian Cancer Cells through Modulation of the Akt Pathway. Oncol. Rep. 2012, 27, 1251–1257. [Google Scholar] [CrossRef] [Green Version]
- Bi, J.; Huang, A.; Liu, T.; Zhang, T.; Ma, H. Expression of DNA Damage Checkpoint 53BP1 Is Correlated with Prognosis, Cell Proliferation and Apoptosis in Colorectal Cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 6070–6082. [Google Scholar] [PubMed]
- James, L.I.; Barsyte-Lovejoy, D.; Zhong, N.; Krichevsky, L.; Korboukh, V.K.; Herold, J.M.; MacNevin, C.J.; Norris, J.L.; Sagum, C.A.; Tempel, W.; et al. Discovery of a Chemical Probe for the L3MBTL3 Methyllysine Reader Domain. Nat. Chem. Biol. 2013, 9, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Herold, J.M.; Wigle, T.J.; Norris, J.L.; Lam, R.; Korboukh, V.K.; Gao, C.; Ingerman, L.A.; Kireev, D.B.; Senisterra, G.; Vedadi, M.; et al. Small-Molecule Ligands of Methyl-Lysine Binding Proteins. J. Med. Chem. 2011, 54, 2504–2511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnash, K.D.; The, J.; Norris-drouin, J.L.; Cholensky, S.H.; Worley, B.M.; Li, F.; Stuckey, J.I.; Brown, P.J.; Vedadi, M.; Arrowsmith, C.H.; et al. Discovery of Peptidomimetic Ligands of EED as Allosteric Inhibitors of PRC2. ACS Chem. Biol. 2017, 19, 161–172. [Google Scholar] [CrossRef]
- Stuckey, J.I.; Dickson, B.M.; Cheng, N.; Liu, Y.; Norris-Drouin, J.L.; Cholensky, S.H.; Tempel, W.; Qin, S.; Huber, K.G.; Sagum, C.; et al. A Cellular Chemical Probe Targeting the Chromodomains of Polycomb Repressive Complex 1. Nat. Chem. Biol. 2016, 12, 180–187. [Google Scholar] [CrossRef] [Green Version]
- Lingel, A.; Sendzik, M.; Huang, Y.; Shultz, M.D.; Cantwell, J.; Dillon, M.P.; Fu, X.; Fuller, J.; Gabriel, T.; Gu, X.J.; et al. Structure-Guided Design of EED Binders Allosterically Inhibiting the Epigenetic Polycomb Repressive Complex 2 (PRC2) Methyltransferase. J. Med. Chem. 2017, 60, 415–427. [Google Scholar] [CrossRef]
- Wagner, T.; Greschik, H.; Burgahn, T.; Schmidtkunz, K.; Xiong, Y.; Fedorov, O.; Schott, A.; Mcmillan, J.; Baranauskien, L.; Jin, J.; et al. Identification of a Small-Molecule Ligand of the Epigenetic Reader Protein Spindlin1 via a Versatile Screening Platform. Nucleic Acids Res. 2016, 44, e88. [Google Scholar] [CrossRef]
- Senisterra, G.; Zhu, H.Y.; Luo, X.; Zhang, H.; Xun, G.; Lu, C.; Xiao, W.; Hajian, T.; Loppnau, P.; Chau, I.; et al. Discovery of Small-Molecule Antagonists of the H3K9me3 Binding to UHRF1 Tandem Tudor Domain. SLAS Discov. Adv. Life Sci. R&D 2018, 23, 930–940. [Google Scholar] [CrossRef] [Green Version]
- Ren, C.; Morohashi, K.; Plotnikov, A.N.; Jakoncic, J.; Steven, G.; Li, J.; Zeng, L.; Rodriguez, Y.; Stojanoff, V.; Walsh, M. Small Molecule Modulators of Methyl-Lysine Binding for the CBX7 Chromodomain. Chem. Biol. 2015, 22, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Liu, Y.; Fan, M.; Zhu, G.; Jin, H.; Liang, J.; Liu, Z.; Huang, Z.; Zhang, L. Identification and Characterization of Benzo[d]Oxazol-2(3H)-One Derivatives as the First Potent and Selective Small-Molecule Inhibitors of Chromodomain Protein CDYL. Eur. J. Med. Chem. 2019, 182. [Google Scholar] [CrossRef]
- Barnash, K.D.; Lamb, K.N.; Stuckey, J.I.; Norris-Drouin, J.L.; Cholensky, S.H.; Kireev, D.B.; Frye, S.V.; James, L.I. Chromodomain Ligand Optimization via Target-Class Directed Combinatorial Repurposing. ACS Chem. Biol. 2016, 11, 2475–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wigle, T.J.; Herold, J.M.; Senisterra, G.A.; Vedadi, M.; Kireev, D.B.; Arrowsmith, C.H.; Frye, S.V.; Janzen, W.P. Screening for Inhibitors of Low-Affinity Epigenetic Peptide-Protein Interactions: An AlphaScreenTM-Based Assay for Antagonists of Methyl-Lysine Binding Proteins. J. Biomol. Screen. 2010, 15, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Rectenwald, J.M.; Hardy, P.B.; Norris-Drouin, J.L.; Cholensky, S.H.; James, L.I.; Frye, S.V.; Pearce, K.H. A General TR-FRET Assay Platform for High-Throughput Screening and Characterizing Inhibitors of Methyl-Lysine Reader Proteins. SLAS Discov. Adv. Life Sci. R&D 2019, 24, 693–700. [Google Scholar] [CrossRef]
- Clark, M.A.; Acharya, R.A.; Arico-Muendel, C.C.; Belyanskaya, S.L.; Benjamin, D.R.; Carlson, N.R.; Centrella, P.A.; Chiu, C.H.; Creaser, S.P.; Cuozzo, J.W.; et al. Design, Synthesis and Selection of DNA-Encoded Small-Molecule Libraries. Nat. Chem. Biol. 2009, 5, 647–654. [Google Scholar] [CrossRef]
- Deng, H.; O’Keefe, H.; Davie, C.P.; Lind, K.E.; Acharya, R.A.; Franklin, G.J.; Larkin, J.; Matico, R.; Neeb, M.; Thompson, M.M.; et al. Discovery of Highly Potent and Selective Small Molecule ADAMTS-5 Inhibitors That Inhibit Human Cartilage Degradation via Encoded Library Technology (ELT). J. Med. Chem. 2012, 55, 7061–7079. [Google Scholar] [CrossRef]
- Li, Y.; Gabriele, E.; Samain, F.; Favalli, N.; Sladojevich, F.; Scheuermann, J.; Neri, D. Optimized Reaction Conditions for Amide Bond Formation in DNA-Encoded Combinatorial Libraries. ACS Comb. Sci. 2016, 18, 438–443. [Google Scholar] [CrossRef] [Green Version]
- Lamb, K.N.; Bsteh, D.; Dishman, S.N.; James, L.I.; Bell, O.; Frye Correspondence, S.V. Discovery and Characterization of a Cellular Potent Positive Allosteric Modulator of the Polycomb Repressive Complex 1 Chromodomain, CBX7. Cell Chem. Biol. 2019, 26, 1365–1379.e22. [Google Scholar] [CrossRef]
- Connelly, K.E.; Weaver, T.M.; Alpsoy, A.; Gu, B.X.; Musselman, C.A.; Dykhuizen, E.C. Engagement of DNA and H3K27me3 by the CBX8 Chromodomain Drives Chromatin Association. Nucleic Acids Res. 2019, 47, 2289–2305. [Google Scholar] [CrossRef] [Green Version]
- Bostick, M.; Kim, J.K.; Estève, P.-O.; Clark, A.; Pradhan, S.; Jacobsen, S.E. UHRF1 Plays a Role in Maintaining DNA Methylation in Mammalian Cells. Science 2007, 317, 1760–1765. [Google Scholar] [CrossRef] [Green Version]
- Denton, K.E.; Wang, S.; Gignac, M.C.; Milosevich, N.; Hof, F.; Dykhuizen, E.C.; Krusemark, C.J. Robustness of In Vitro Selection Assays of DNA-Encoded Peptidomimetic Ligands to CBX7 and CBX8. SLAS Discov. Adv. Life Sci. R&D 2018, 23, 417–428. [Google Scholar] [CrossRef]
- Wang, S.; Denton, K.E.; Hobbs, K.F.; Weaver, T.; Mcfarlane, J.M.B.; Connelly, K.E.; Gignac, M.C.; Milosevich, N.; Hof, F.; Paci, I.; et al. Optimization of Ligands Using Focused DNA-Encoded Libraries To Develop a Selective, Cell-Permeable CBX8 Chromodomain Inhibitor. ACS Chem. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.L.; Watts, B.; Stuckey, J.I.; Norris-Drouin, J.L.; Cholensky, S.H.; Dickson, B.M.; An, Y.; Mathea, S.; Salah, E.; Knapp, S.; et al. Quantitative Characterization of Bivalent Probes for a Dual Bromodomain Protein, Transcription Initiation Factor TFIID Subunit 1. Biochemistry 2018, 57, 2140–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Protein | UniProt Accession | Domains | PTM Recognition | PDB ID | Disease Relevance | References |
|---|---|---|---|---|---|---|
| SETDB1 | Q15047 | Triple Tudor | H3K9me2/3K14ac | 6BHD, 6BHE, 6BHI | Huntington’s, Cancer – Breast, Liver, Prostate | [3,25,26,27,28,29] |
| UHRF1 | Q96T88 | PHD, Tandem Tudor | H3(N-terminus)K9me3 | 4GY5 | Cancer – Breast, Lung, Prostate, Liver | [4,30,31,32,33,34,35] |
| TRIM24 | O15164 | PHD, Bromo | H3K9K23ac | 3O34 | Cancer – Breast, Liver, Lung, Colon | [5,36,37,38,39] |
| TRIM33 | Q9UPN9 | PHD, Bromo | H3K9me3K14ac, H3K9me3K14acK18ac | 3U5N, 3U5O | Cancer – Liver, Pancreatic, Leukemia | [6,37,40,41] |
| PHF8 | Q9UPP1 | PHD, Jumonji | H3K4me3K9me2 | 3KV4 | Cancer – Prostate, Lung, Breast, Gastric | [7,42,43,44,45,46] |
| RAG2 | P55895 | PHD | H3R2meK4me3 | 2V85 | Lymphoid tumors and immunological disorders | [8,47,48] |
| 53BP1 | Q12888 | Tandem Tudor | *p53K381acK382me2 | 4X34 | Cancer – Breast, Ovarian, Colon | [9,49,50,51,52] |
| Input Library/ Experiment Sequenced | Number of Compound Barcodes | Total Reads | Positive Control Frequency | Negative Control Frequency |
|---|---|---|---|---|
| UNCDEL003 with controls, Prep 1 | 45,202 | 120,038 | 6 | 0 |
| CBX7 with DEL Prep 1 | 935 | 10,570 | 8570 | 0 |
| CBX7(short) with DEL Prep 1 | 47,933 | 152,754 | 117 | 0 |
| MyOne Dynabeads with DEL Prep 1 | 1,145 | 5847 | 35 | 0 |
| UNCDEL003 with controls, Prep 2 | 30,989 | 53,239 | 1 | 0 |
| CBX7 with DEL Prep 2 | 1236 | 7460 | 5308 | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rectenwald, J.M.; Guduru, S.K.R.; Dang, Z.; Collins, L.B.; Liao, Y.-E.; Norris-Drouin, J.L.; Cholensky, S.H.; Kaufmann, K.W.; Hammond, S.M.; Kireev, D.B.; et al. Design and Construction of a Focused DNA-Encoded Library for Multivalent Chromatin Reader Proteins. Molecules 2020, 25, 979. https://doi.org/10.3390/molecules25040979
Rectenwald JM, Guduru SKR, Dang Z, Collins LB, Liao Y-E, Norris-Drouin JL, Cholensky SH, Kaufmann KW, Hammond SM, Kireev DB, et al. Design and Construction of a Focused DNA-Encoded Library for Multivalent Chromatin Reader Proteins. Molecules. 2020; 25(4):979. https://doi.org/10.3390/molecules25040979
Chicago/Turabian StyleRectenwald, Justin M., Shiva Krishna Reddy Guduru, Zhao Dang, Leonard B. Collins, Yi-En Liao, Jacqueline L. Norris-Drouin, Stephanie H. Cholensky, Kyle W. Kaufmann, Scott M. Hammond, Dmitri B. Kireev, and et al. 2020. "Design and Construction of a Focused DNA-Encoded Library for Multivalent Chromatin Reader Proteins" Molecules 25, no. 4: 979. https://doi.org/10.3390/molecules25040979
APA StyleRectenwald, J. M., Guduru, S. K. R., Dang, Z., Collins, L. B., Liao, Y. -E., Norris-Drouin, J. L., Cholensky, S. H., Kaufmann, K. W., Hammond, S. M., Kireev, D. B., Frye, S. V., & Pearce, K. H. (2020). Design and Construction of a Focused DNA-Encoded Library for Multivalent Chromatin Reader Proteins. Molecules, 25(4), 979. https://doi.org/10.3390/molecules25040979