Resistance of Gram-Negative Bacteria to Current Antibacterial Agents and Approaches to Resolve It




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
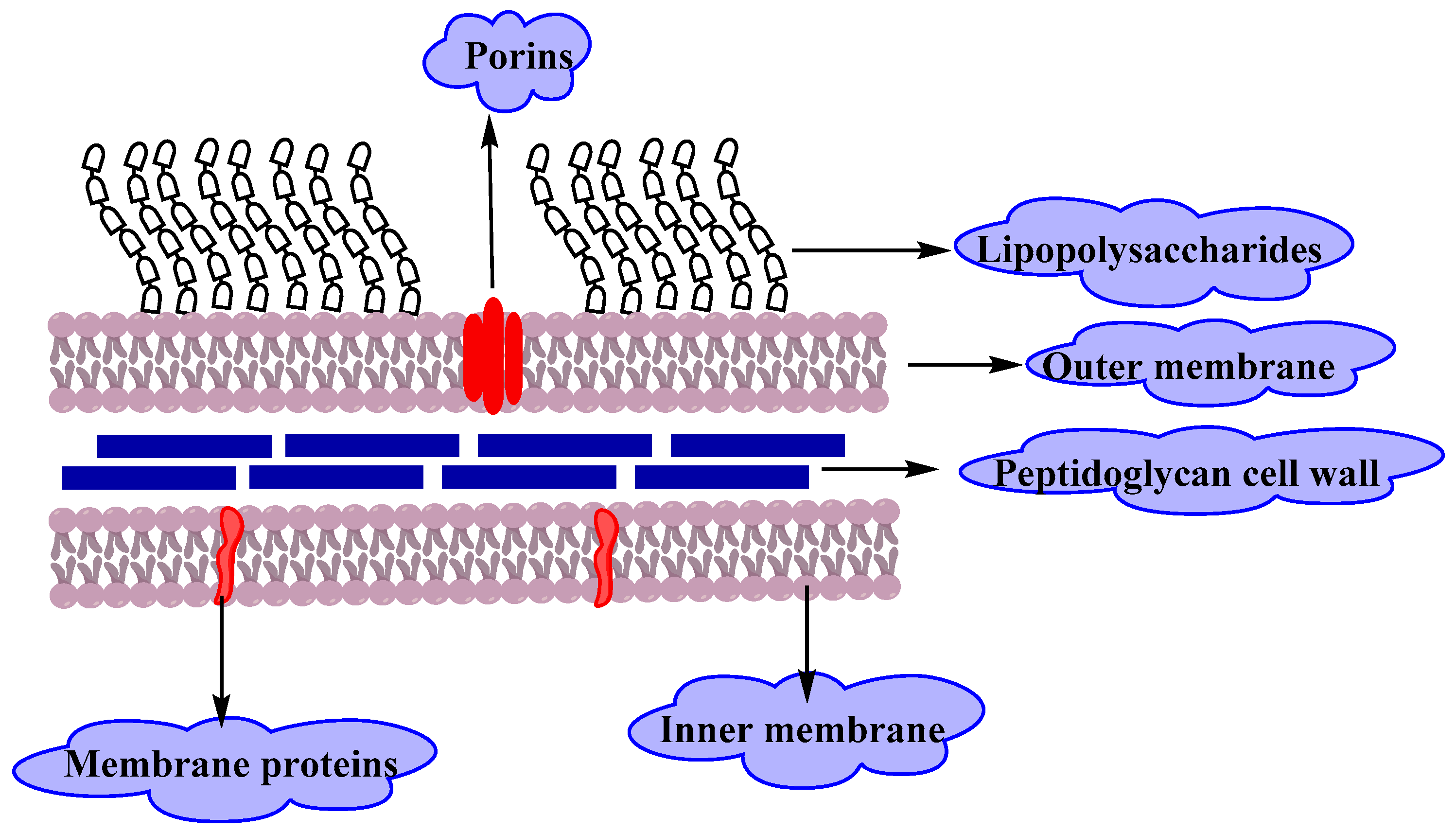
Gram Negative Bacteria
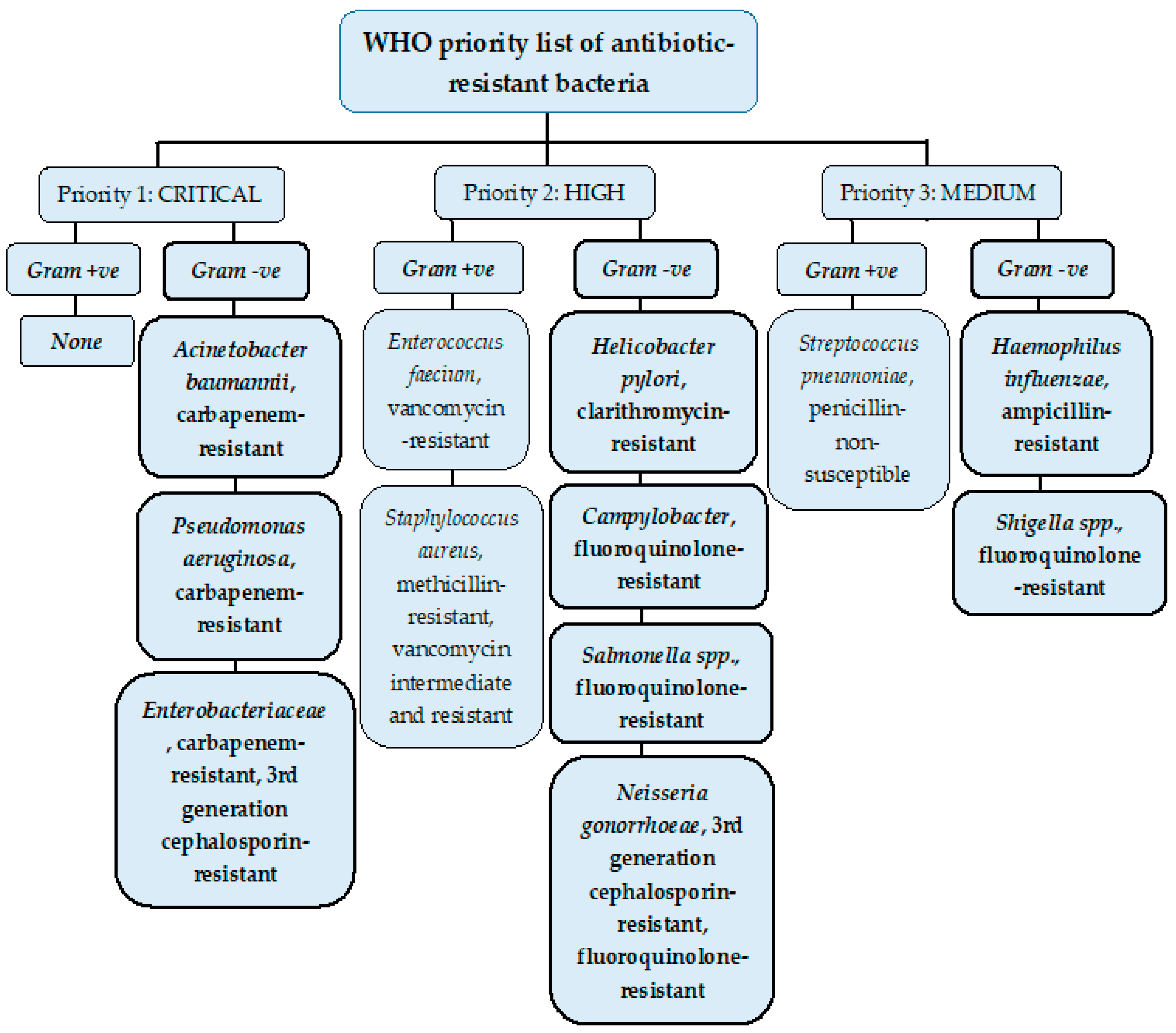
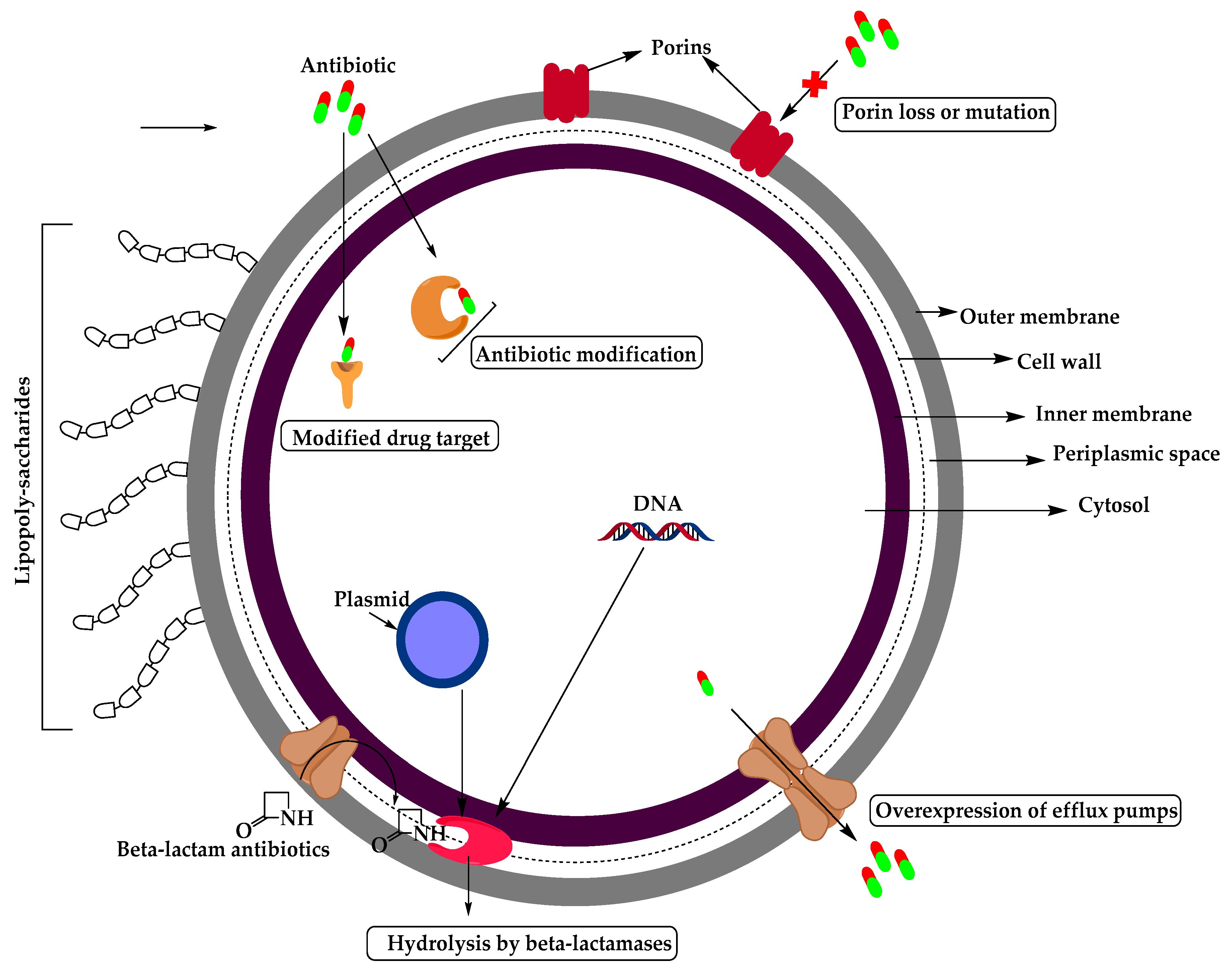
2. Resistant Gram-Negative Bacteria
2.1. Enterobacteriaceae
2.1.1. Enterobacteriaceae- 3rd Generation Cephalosporin-Resistant
2.1.2. Enterobacteriaceae- Carbapenem-Resistant
2.2. Acinetobacter baumannii
2.3. Pseudomonas aeruginosa
2.4. Helicobacter pylori- Clarithromycin-Resistant
2.5. Campylobacter- Fluoroquinolone-Resistant
2.6. Salmonella spp.- Fluoroquinolone-Resistant
2.6.1. Typhoidal Salmonella Resistance
2.6.2. Non-typhoidal Salmonella Resistance
2.7. Neisseria gonorrhoeae
2.7.1. Neisseria gonorrhoeae- 3rd Generation Cephalosporin-Resistant
2.7.2. Neisseria gonorrhoeae- Fluoroquinolone-Resistant
2.8. Haemophilus influenza- Ampicillin-resistant
2.9. Shigella spp.- Fluoroquinolone-Resistant
3. Treatment
3.1. Antibiotic Adjuvants
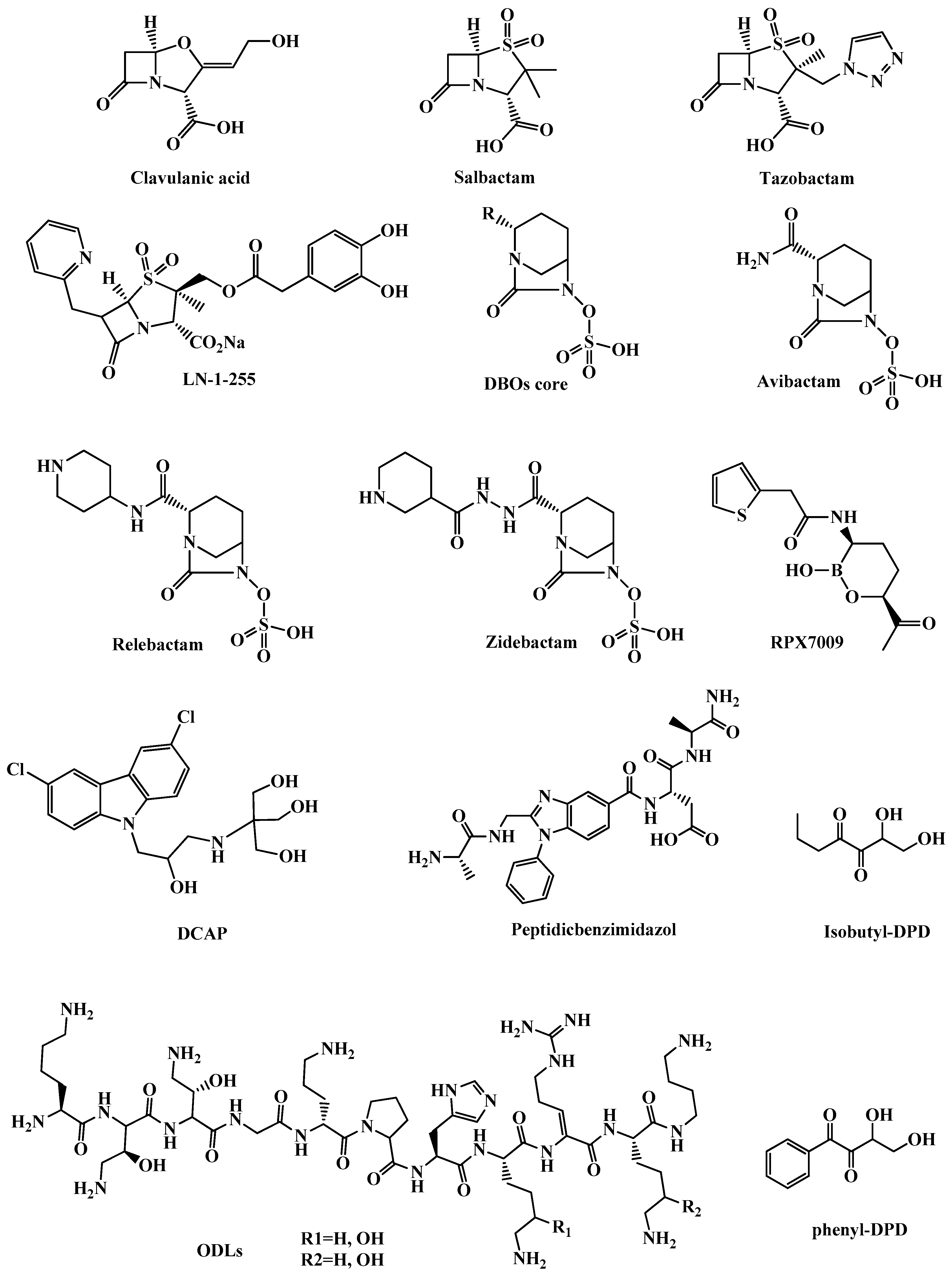
3.1.1. β-Lactamase Inhibitors
3.1.2. Clavulanic Acid and Penicillin-based Sulfones
3.1.3. Diazabicyclooctanes (DBOs)
3.1.4. Boronic Acids as Transition State Analogs
3.2. Antibiotic Alternatives
3.2.1. Bacteriophages
3.2.2. DCAP
3.2.3. Odilorhabdins (ODLs)
3.2.4. Peptidic benzimidazoles
3.2.5. Quorum Sensing (QS) Inhibitors
3.2.6. Metal-Based Antibacterial Agents
4. Patient Education
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Coico, R. Gram staining. Curr. Protoc. Microbiol. 2006, 1, A.3C.1–A.3C.2. [Google Scholar]
- Moyes, R.B.; Reynolds, J.; Breakwell, D.P. Differential Staining of Bacteria: Gram Stain. Curr. Protoc. Microbiol. 2009, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Silhavy, T.J.; Kahne, D.; Walker, S. The Bacterial Cell Envelope. Cold Spring Harb. Perspect. Boil. 2010, 2, a000414. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.R.; Rosenthal, K.S.; Pfaller, M.A. Medical Microbiology; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Miller, S.I. Antibiotic Resistance and Regulation of the Gram-Negative Bacterial Outer Membrane Barrier by Host Innate Immune Molecules. mBio 2016, 7, e01541-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, P.; Gupta, V. Next-generation strategy for treating drug resistant bacteria: Antibiotic hybrids. Indian J. Med. Res. 2019, 149, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Exner, M.; Bhattacharya, S.; Christiansen, B.; Gebel, J.; Goroncy-Bermes, P.; Hartemann, P.; Heeg, P.; Ilschner, C.; Kramer, A.; Larson, E.; et al. Antibiotic resistance: What is so special about multidrug-resistant Gram-negative bacteria? GMS Hyg. Infect. Control. 2017, 12. [Google Scholar] [CrossRef]
- Oliveira, J.; Reygaert, W.C. Gram Negative Bacteria; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Ruppé, E.; Woerther, P.-L.; Barbier, F. Mechanisms of antimicrobial resistance in Gram-negative bacilli. Ann. Intensiv. Care 2015, 5, 61. [Google Scholar] [CrossRef] [Green Version]
- Paterson, D.L. Resistance in gram-negative bacteria: Enterobacteriaceae. Am. J. Med. 2006, 34, S20–S28. [Google Scholar] [CrossRef]
- Van Duin, D. Carbapenem-resistant Enterobacteriaceae: What we know and what we need to know. Virulence 2017, 8, 379–382. [Google Scholar] [CrossRef] [Green Version]
- Lutgring, J.D. Carbapenem-resistant Enterobacteriaceae: An emerging bacterial threat. Semin. Diagn. Pathol. 2019, 36, 182–186. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, J.H.; Park, M.; Park, K.S.; Bae, I.K.; Kim, Y.B.; Cha, C.-J.; Jeong, B.C.; Lee, S.H. Biology of Acinetobacter baumannii: Pathogenesis, Antibiotic Resistance Mechanisms, and Prospective Treatment Options. Front. Microbiol. 2017, 7, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.-F.; Lan, C.-Y. Antimicrobial resistance inAcinetobacter baumannii: From bench to bedside. World J. Clin. Cases 2014, 2, 787–814. [Google Scholar] [CrossRef] [PubMed]
- Pachori, P.; Gothalwal, R.; Gandhi, P. Emergence of antibiotic resistance Pseudomonas aeruginosa in intensive care unit; a critical review. Gene Funct. Dis. 2019, 6, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Savoldi, A.; Carrara, E.; Graham, D.Y.; Conti, M.; Tacconelli, E. Prevalence of Antibiotic Resistance in Helicobacter pylori: A Systematic Review and Meta-analysis in World Health Organization Regions. Gastroenterology 2018, 155, 1372–1382.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Dunbar, K.B.; Mitui, M.; Arnold, C.A.; Lam-Himlin, R.M.; Valasek, M.A.; Thung, I.; Okwara, C.; Coss, E.; Cryer, B.; et al. Helicobacter pylori Clarithromycin Resistance and Treatment Failure Are Common in the USA. Dig. Dis. Sci. 2016, 61, 2373–2380. [Google Scholar] [CrossRef]
- Alba, C.; Blanco, A.; Alarcón, T. Antibiotic resistance in Helicobacter pylori. Curr. Opin. Infect. Dis. 2017, 30, 489–497. [Google Scholar] [CrossRef]
- Smith, J.L.; Fratamico, P.M. Fluoroquinolone Resistance in Campylobacter. J. Food Prot. 2010, 73, 1141–1152. [Google Scholar] [CrossRef]
- Sproston, E.L.; Wimalarathna, H.M.L.; Sheppard, S.K. Trends in fluoroquinolone resistance in Campylobacter. Microb. Genom. 2018, 4. [Google Scholar] [CrossRef]
- Cuypers, W.; Jacobs, J.; Wong, V.; Klemm, E.J.; Deborggraeve, S.; Van Puyvelde, S. Fluoroquinolone resistance in Salmonella: Insights by whole-genome sequencing. Microb. Genom. 2018, 4. [Google Scholar] [CrossRef]
- A Crump, J.; Sjölund-Karlsson, M.; A Gordon, M.; Parry, C.M. Epidemiology, Clinical Presentation, Laboratory Diagnosis, Antimicrobial Resistance, and Antimicrobial Management of Invasive Salmonella Infections. Clin. Microbiol. Rev. 2015, 28, 901–937. [Google Scholar] [CrossRef] [Green Version]
- A Hill, S.; Masters, T.L.; Wachter, J. Gonorrhea - an evolving disease of the new millennium. Microb. Cell 2016, 3, 371–389. [Google Scholar] [CrossRef] [PubMed]
- Bala, M.; Sood, S. Cephalosporin Resistance in Neisseria gonorrhoeae. J. Glob. Infect. Dis. 2010, 2, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Barry, P.M.; Klausner, J.D. The use of cephalosporins for gonorrhea: The impending problem of resistance. Expert Opin. Pharmacother. 2009, 10, 555–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanem, K.G.; Giles, J.A.; Zenilman, J.M. Fluoroquinolone-resistant Neisseria gonorrhoeae: The Inevitable Epidemic. Infect. Dis. Clin. North Am. 2005, 19, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, A.P.R.D.C.; Dos Santos, K.T.B.; Moreira, B.M.; Fracalanzza, S.E.L.; Bonelli, R.R. Antimicrobial resistance in Neisseria gonorrhoeae: History, molecular mechanisms and epidemiological aspects of an emerging global threat. Braz. J. Microbiol. 2017, 48, 617–628. [Google Scholar] [CrossRef]
- Heinz, E. The return of Pfeiffer’s bacillus: Rising incidence of ampicillin resistance in Haemophilus influenzae. Microb. Genom. 2018, 4, e000214. [Google Scholar] [CrossRef]
- Tristram, S.; Jacobs, M.R.; Appelbaum, P.C. Antimicrobial Resistance in Haemophilus influenzae. Clin. Microbiol. Rev. 2007, 20, 368–389. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.; Lee, J.; Lee, J.; Kim, E.; Lee, S.; Yu, J.; Kang, Y. Antimicrobial Resistance in Haemophilus influenzae Respiratory Tract Isolates in Korea: Results of a Nationwide Acute Respiratory Infections Surveillance▿. Antimicrob. Agents Chemother. 2009, 54, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Qin, T.; Qian, H.; Fan, W.; Ma, P.; Zhou, L.; Dong, C.; Gu, B.; Huo, X. Newest data on fluoroquinolone resistance mechanism of Shigella flexneri isolates in Jiangsu Province of China. Antimicrob. Resist. Infect. Control. 2017, 6, 97. [Google Scholar] [CrossRef] [Green Version]
- Nüesch-Inderbinen, M.; Heini, N.; Zurfluh, K.; Althaus, D.; Hächler, H.; Stephan, R. Shigella Antimicrobial Drug Resistance Mechanisms, 2004–2014. Emerg. Infect. Dis. 2016, 22, 1083–1085. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.-C.; Silverman, M.H.; Yu, X.Y.; Wright, G.; Brown, N. Discovery and development of DNA polymerase IIIC inhibitors to treat Gram-positive infections. Bioorganic Med. Chem. 2019, 27, 3209–3217. [Google Scholar] [CrossRef] [PubMed]
- Bernal, P.; Molina-Santiago, C.; Daddaoua, A.; Llamas, M.A. Antibiotic adjuvants: Identification and clinical use. Microb. Biotechnol. 2013, 6, 445–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, E.E.; Franco, O.L.; Hancock, R.E.W. Antibiotic Adjuvants: Diverse Strategies for Controlling Drug-Resistant Pathogens. Chem. Boil. Drug Des. 2014, 85, 56–78. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C. Molecular mechanisms that confer antibacterial drug resistance. Nature 2000, 406, 775–781. [Google Scholar] [CrossRef]
- Gonzalez-Bello, C. Antibiotic adjuvants – A strategy to unlock bacterial resistance to antibiotics. Bioorganic Med. Chem. Lett. 2017, 27, 4221–4228. [Google Scholar] [CrossRef]
- Drawz, S.M.; Papp-Wallace, K.M.; Bonomo, R.A. New β-Lactamase Inhibitors: A Therapeutic Renaissance in an MDR World. Antimicrob. Agents Chemother. 2013, 58, 1835–1846. [Google Scholar] [CrossRef] [Green Version]
- Leonard, D.A.; Bonomo, R.A.; Powers, R.A. Class D β-Lactamases: A Reappraisal after Five Decades. Accounts Chem. Res. 2013, 46, 2407–2415. [Google Scholar] [CrossRef] [Green Version]
- Klingler, F.-M.; Wichelhaus, T.A.; Frank, D.; Cuesta-Bernal, J.; El-Delik, J.; Müller, H.F.; Sjuts, H.; Göttig, S.; Koenigs, A.; Pos, K.M.; et al. Approved Drugs Containing Thiols as Inhibitors of Metallo-β-lactamases: Strategy To Combat Multidrug-Resistant Bacteria. J. Med. Chem. 2015, 58, 3626–3630. [Google Scholar] [CrossRef]
- Reading, C.; Cole, M. Clavulanic Acid: A Beta-Lactamase-Inhibiting Beta-Lactam from Streptomyces clavuligerus. Antimicrob. Agents Chemother. 1977, 11, 852–857. [Google Scholar] [CrossRef] [Green Version]
- Drawz, S.M.; Bonomo, R.A. Three Decades of β-Lactamase Inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef] [Green Version]
- Buynak, J.D.; Rao, A.; Doppalapudi, V.R.; Adam, G.; Petersen, P.J.; Nidamarthy, S.D. The synthesis and evaluation of 6-alkylidene-2′β-substituted penam sulfones as β-lactamase inhibitors. Bioorganic Med. Chem. Lett. 1999, 9, 1997–2002. [Google Scholar] [CrossRef]
- Vallejo, J.A.; Guitián, M.M.; Vázquez-Ucha, J.C.; Gonzalez-Bello, C.; Poza, M.; Buynak, J.D.; Bethel, C.R.; Bonomo, R.A.; Bou, G.; Beceiro, A. LN-1-255, a penicillanic acid sulfone able to inhibit the class D carbapenemase OXA-48. J. Antimicrob. Chemother. 2016, 71, 2171–2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangoni, A.A.; Guillou, C.; Vanden Eynde, J.J.; Hulme, C.; Jampilek, J.; Li, W.; Prokai-Tatrai, K.; Rautio, J.; Collina, S.; Tuccinardi, T.; et al. Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes (-)4. Molecules 2018, 2, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, K. Diazabicyclooctanes (DBOs): A potent new class of non-β-lactam β-lactamase inhibitors. Curr. Opin. Microbiol. 2011, 14, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Lagacé-Wiens, P.; Walkty, A.; A Karlowsky, J. Ceftazidime–avibactam: An evidence-based review of its pharmacology and potential use in the treatment of Gram-negative bacterial infections. Core Évid. 2014, 9, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez-Ucha, J.C.; Maneiro, M.; Martínez-Guitián, M.; Buynak, J.; Bethel, C.R.; Bonomo, R.A.; Bou, G.; Poza, M.; Gonzalez-Bello, C.; Beceiro, A. Activity of the β-Lactamase Inhibitor LN-1-255 against Carbapenem-Hydrolyzing Class D β-Lactamases from Acinetobacter baumannii. Antimicrob. Agents Chemother. 2017, 61, e01172-17. [Google Scholar] [CrossRef] [Green Version]
- Leone, S.; Damiani, G.; Pezone, I.; Kelly, M.E.; Cascella, M.; Alfieri, A.; Pace, M.C.; Fiore, M. New antimicrobial options for the management of complicated intra-abdominal infections. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 819–827. [Google Scholar] [CrossRef]
- Wright, H.; Bonomo, R.A.; Paterson, D.L. New agents for the treatment of infections with Gram-negative bacteria: Restoring the miracle or false dawn? Clin. Microbiol. Infect. 2017, 23, 704–712. [Google Scholar] [CrossRef] [Green Version]
- Sader, H.S.; Castanheira, M.; Huband, M.; Jones, R.N.; Flamm, R.K. WCK 5222 (Cefepime-Zidebactam) Antimicrobial Activity against Clinical Isolates of Gram-Negative Bacteria Collected Worldwide in 2015. Antimicrob. Agents Chemother. 2017, 61, e00072-17. [Google Scholar] [CrossRef] [Green Version]
- Smoum, R.; Rubinstein, A.; Dembitsky, V.M.; Srebnik, M. Boron Containing Compounds as Protease Inhibitors. Chem. Rev. 2012, 112, 4156–4220. [Google Scholar] [CrossRef]
- Beesley, T.; Gascoyne, N.; Knott-Hunziker, V.; Petursson, S.; Waley, S.G.; Jaurin, B.; Grundstrom, T. The inhibition of class C β-lactamases by boronic acids. Biochem. J. 1983, 209, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Hecker, S.J.; Reddy, K.R.; Totrov, M.; Hirst, G.C.; Lomovskaya, O.; Griffith, D.C.; King, P.; Tsivkovski, R.; Sun, N.; Sabet, M.; et al. Discovery of a Cyclic Boronic Acid β-Lactamase Inhibitor (RPX7009) with Utility vs Class A Serine Carbapenemases. J. Med. Chem. 2015, 58, 3682–3692. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, E.J.C.; Citron, D.M.; Tyrrell, K.L.; Merriam, C.V. In Vitro Activity of Biapenem plus RPX7009, a Carbapenem Combined with a Serine β-Lactamase Inhibitor, against Anaerobic Bacteria. Antimicrob. Agents Chemother. 2013, 57, 2620–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Herelle, F. Sur un microbe invisible antagoniste des bacilles dysentériques. CR Acad. Sci. Paris 1917, 165, 373–375. [Google Scholar]
- Chanishvili, N.; Aminov, R.I. Bacteriophage therapy: Coping with the growing antibiotic resistance problem. Microbiol. Aust. 2019. [Google Scholar] [CrossRef]
- Kortright, K.E.; Chan, B.K.; Koff, J.L.; Turner, P.E. Phage Therapy: A Renewed Approach to Combat Antibiotic-Resistant Bacteria. Cell Host Microbe 2019, 25, 219–232. [Google Scholar] [CrossRef] [Green Version]
- McCallin, S.; Alam Sarker, S.; Barretto, C.; Sultana, S.; Berger, B.; Huq, S.; Krause, L.; Bibiloni, R.; Schmitt, B.; Reuteler, G.; et al. Safety analysis of a Russian phage cocktail: From MetaGenomic analysis to oral application in healthy human subjects. Virology 2013, 443, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Międzybrodzki, R.; Borysowski, J.; Weber-Dąbrowska, B.; Fortuna, W.; Letkiewicz, S.; Szufnarowski, K.; Pawełczyk, Z.; Rogóż, P.; Kłak, M.; Wojtasik, E.; et al. Clinical Aspects of Phage Therapy. In Advances in Clinical Chemistry; Elsevier: Amsterdam, The Netherlands, 2012; Volume 83, pp. 73–121. [Google Scholar]
- Wright, A.; Hawkins, C.; Änggård, E.; Harper, D. A controlled clinical trial of a therapeutic bacteriophage preparation in chronic otitis due to antibiotic-resistantPseudomonas aeruginosa; a preliminary report of efficacy. Clin. Otolaryngol. 2009, 34, 349–357. [Google Scholar] [CrossRef]
- Alam Sarker, S.; Sultana, S.; Reuteler, G.; Moine, D.; Descombes, P.; Charton, F.; Bourdin, G.; McCallin, S.; Ngom-Bru, C.; Neville, T.; et al. Oral Phage Therapy of Acute Bacterial Diarrhea With Two Coliphage Preparations: A Randomized Trial in Children From Bangladesh. EBioMedicine 2016, 4, 124–137. [Google Scholar] [CrossRef] [Green Version]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.-P.; Que, Y.-A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Le Floch, R.; et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): A randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 2019, 19, 35–45. [Google Scholar] [CrossRef]
- Oechslin, F.; Piccardi, P.; Mancini, S.; Gabard, J.; Moreillon, P.; Entenza, J.M.; Resch, G.; Que, Y.-A. Synergistic Interaction Between Phage Therapy and Antibiotics Clears Pseudomonas Aeruginosa Infection in Endocarditis and Reduces Virulence. J. Infect. Dis. 2017, 215, 703–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huff, W.E.; Huff, G.R.; Rath, N.C.; Balog, J.M.; Donoghue, A.M. Therapeutic efficacy of bacteriophage and Baytril (enrofloxacin) individually and in combination to treat colibacillosis in broilers. Poult. Sci. 2004, 83, 1944–1947. [Google Scholar] [CrossRef] [PubMed]
- Calero-Caceres, W.; Ye, M.; Balcázar, J.L. Bacteriophages as Environmental Reservoirs of Antibiotic Resistance. Trends Microbiol. 2019, 27, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Thanbichler, M.; Shapiro, L. MipZ, a Spatial Regulator Coordinating Chromosome Segregation with Cell Division in Caulobacter. Cell 2006, 126, 147–162. [Google Scholar] [CrossRef] [Green Version]
- Eun, Y.J.; Foss, M.H.; Kiekebusch, D.; Pauw, D.A.; Westler, W.M.; Thanbichler, M.; Weibel, D. DCAP: A Broad-Spectrum Antibiotic That Targets the Cytoplasmic Membrane of Bacteria. J. Am. Chem. Soc. 2012, 134, 11322–11325. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, V.; Hurley, K.; Santos, T.; Weibel, D. DCAP: A broad-spectrum antibiotic that targets the cytoplasmic membrane of bacteria. FASEB J. 2015, 29, 575.6. [Google Scholar]
- Hurley, K.A.; Heinrich, V.; Hershfield, J.R.; Demons, S.T.; Weibel, D. Membrane-Targeting DCAP Analogues with Broad-Spectrum Antibiotic Activity against Pathogenic Bacteria. ACS Med. Chem. Lett. 2015, 6, 466–471. [Google Scholar] [CrossRef] [Green Version]
- Polikanov, Y.S.; A Aleksashin, N.; Beckert, B.; Wilson, D.N. The Mechanisms of Action of Ribosome-Targeting Peptide Antibiotics. Front. Mol. Biosci. 2018, 5, 48. [Google Scholar] [CrossRef] [Green Version]
- Bérdy, J. Bioactive Microbial Metabolites. J. Antibiot. 2005, 58, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Pantel, L.; Florin, T.; Dobosz-Bartoszek, M.; Racine, E.; Sarciaux, M.; Serri, M.; Houard, J.; Campagne, J.-M.; De Figueiredo, R.M.; Midrier, C.; et al. Odilorhabdins, Antibacterial Agents that Cause Miscoding by Binding at a New Ribosomal Site. Mol. Cell 2018, 70, 83–94.e7. [Google Scholar] [CrossRef] [Green Version]
- Bansal, Y.; Silakari, O. The therapeutic journey of benzimidazoles: A review. Bioorganic Med. Chem. 2012, 20, 6208–6236. [Google Scholar] [CrossRef] [PubMed]
- Boiani, M.; Gonzalez, M. Imidazole and benzimidazole derivatives as chemotherapeutic agents. Mini-Reviews Med. Chem. 2005, 5, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Chen, D.; White, R.J.; Patel, D.V.; Yuan, Z. Bacterial Peptide deformylase inhibitors: A new class of antibacterial agents. Curr. Med. Chem. 2005, 12, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Schlichting, I.; Kabsch, W.; Schultz, S.; Wagner, A.F.V. Structure of Peptide Deformylase and Identification of the Substrate Binding Site. J. Boil. Chem. 1998, 273, 11413–11416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, K.T.; Hu, X.; Colton, C.; Chakrabarti, R.; Zhu, M.X.; Pei, D. Characterization of a Human Peptide Deformylase: Implications for Antibacterial Drug Design†. Biochemistry 2003, 42, 9952–9958. [Google Scholar] [CrossRef]
- Bird, M.; Silvestri, A.P.; Dawson, P.E. Expedient on-resin synthesis of peptidic benzimidazoles. Bioorganic Med. Chem. Lett. 2018, 28, 2679–2681. [Google Scholar] [CrossRef]
- Bugday, N.; Kucukbay, F.Z.; Apohan, E.; Küçükbay, H.; Serindag, A.; Yesilada, O. Synthesis and Evaluation of Novel Benzimidazole Conjugates Incorporating Amino Acids and Dipeptide Moieties. Lett. Org. Chem. 2017, 14, 198–206. [Google Scholar] [CrossRef]
- Stotani, S.; Gatta, V.; Medda, F.; Padmanaban, M.; Karawajczyk, A.; Tammela, P.; Giordanetto, F.; Tzalis, D.; Collina, S. A Versatile Strategy for the Synthesis of 4,5-Dihydroxy-2,3-Pentanedione (DPD) and Related Compounds as Potential Modulators of Bacterial Quorum Sensing. Molecules 2018, 23, 2545. [Google Scholar] [CrossRef] [Green Version]
- Stotani, S.; Gatta, V.; Medarametla, P.; Padmanaban, M.; Karawajczyk, A.; Giordanetto, F.; Tammela, P.; Laitinen, T.; Poso, A.; Tzalis, D.; et al. DPD-Inspired Discovery of Novel LsrK Kinase Inhibitors: An Opportunity To Fight Antimicrobial Resistance. J. Med. Chem. 2019, 62, 2720–2737. [Google Scholar] [CrossRef] [Green Version]
- Gasser, G. Metal Complexes and Medicine: A Successful Combination. Chim. Int. J. Chem. 2015, 69, 442–446. [Google Scholar] [CrossRef] [Green Version]
- Medici, S.; Peana, M.F.; Crisponi, G.; Nurchi, V.M.; Lachowicz, J.I.I.; Remelli, M.; Zoroddu, M.A. Silver coordination compounds: A new horizon in medicine. Co-ord. Chem. Rev. 2016, 327, 349–359. [Google Scholar] [CrossRef]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.W.; Ramek, A.; Wang, Y.; Kaya, T.; Wilson, J.A.; Clemons, P.A.; Young, D.W. Route to three-dimensional fragments using diversity-oriented synthesis. Proc. Natl. Acad. Sci. 2011, 108, 6799–6804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Collins, J.G.; Keene, F.R. Ruthenium complexes as antimicrobial agents. Chem. Soc. Rev. 2015, 44, 2529–2542. [Google Scholar] [CrossRef] [Green Version]
- Smitten, K.L.; Southam, H.M.; De La Serna, J.B.; Gill, M.R.; Jarman, P.; Smythe, C.; Poole, R.K.; Thomas, J.A. Using Nanoscopy To Probe the Biological Activity of Antimicrobial Leads That Display Potent Activity against Pathogenic, Multidrug Resistant, Gram-Negative Bacteria. ACS Nano 2019, 13, 5133–5146. [Google Scholar] [CrossRef] [Green Version]
- Abebe, A.; Hailemariam, T. Synthesis and Assessment of Antibacterial Activities of Ruthenium(III) Mixed Ligand Complexes Containing 1,10-Phenanthroline and Guanide. Bioinorg. Chem. Appl. 2016, 2016, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Bonchi, C.; Imperi, F.; Minandri, F.; Visca, P.; Frangipani, E. Repurposing of gallium-based drugs for antibacterial therapy. BioFactors 2014, 40, 303–312. [Google Scholar] [CrossRef]
- A Phase 2 IV Gallium Study for Patients With Cystic Fibrosis (IGNITE Study). Available online: https://clinicaltrials.gov/ct2/show/NCT02354859 (accessed on 1 February 2020).
- Wang, Y.; Han, B.; Xie, Y.; Wang, H.; Wang, R.; Xia, W.; Li, H.; Sun, H. Combination of gallium(iii) with acetate for combating antibiotic resistant Pseudomonas aeruginosa. Chem. Sci. 2019, 10, 6099–6106. [Google Scholar] [CrossRef] [Green Version]
- Fock, K.M.; Graham, D.Y.; Malfertheiner, P. Helicobacter pylori research: Historical insights and future directions. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 495–500. [Google Scholar] [CrossRef]
- Li, H.; Wang, R.; Sun, H. Systems Approaches for Unveiling the Mechanism of Action of Bismuth Drugs: New Medicinal Applications beyond Helicobacter Pylori Infection. Accounts Chem. Res. 2018, 52, 216–227. [Google Scholar] [CrossRef]
- Wang, R.; Lai, T.-P.; Gao, P.; Zhang, H.; Ho, P.-L.; Woo, P.C.-Y.; Ma, G.; Kao, R.Y.; Li, H.; Sun, H. Bismuth antimicrobial drugs serve as broad-spectrum metallo-β-lactamase inhibitors. Nat. Commun. 2018, 9, 439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luqman, A.; Blair, V.L.; Brammananth, R.; Crellin, P.K.; Coppel, R.L.; Andrews, P.C. Homo- and Heteroleptic Bismuth(III/V) Thiolates from N-Heterocyclic Thiones: Synthesis, Structure and Anti-Microbial Activity. Chem. A Eur. J. 2014, 20, 14362–14377. [Google Scholar] [CrossRef] [PubMed]
- Luqman, A.; Blair, V.L.; Brammananth, R.; Crellin, P.K.; Coppel, R.L.; Andrews, P.C. The Importance of Heterolepticity in Improving the Antibacterial Activity of Bismuth(III) Thiolates. Eur. J. Inorg. Chem. 2016, 2016, 2738–2749. [Google Scholar] [CrossRef]
- Werrett, M.V.; Herdman, M.E.; Brammananth, R.; Garusinghe, U.; Batchelor, W.; Crellin, P.K.; Coppel, R.L.; Andrews, P.C. Bismuth Phosphinates in Bi-Nanocellulose Composites and their Efficacy towards Multi-Drug Resistant Bacteria. Chem. A Eur. J. 2018, 24, 12938–12949. [Google Scholar] [CrossRef] [PubMed]
- Muller, N.; Amiot, A.; Le Thuaut, A.; Bastuji-Garin, S.; Deforges, L.; Delchier, J.C. Rescue therapy with bismuth-containing quadruple therapy in patients infected with metronidazole-resistant Helicobacter pylori strains. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.W. History of the Medical Use of Silver. Surg. Infect. 2009, 10, 289–292. [Google Scholar] [CrossRef] [Green Version]
- Aziz, Z.; Abu, S.; Chong, N.J. A systematic review of silver-containing dressings and topical silver agents (used with dressings) for burn wounds. Burns 2012, 38, 307–318. [Google Scholar] [CrossRef]
- Sierra, M.A.; Casarrubios, L.; La Torre, M.C. Bio-Organometallic Derivatives of Antibacterial Drugs. Chem. A Eur. J. 2019, 25, 7232–7242. [Google Scholar] [CrossRef]
- Yan, J.; AbdelGawad, A.; Rojas, O.J.; El-Naggar, M. Antibacterial activity of silver nanoparticles synthesized In-situ by solution spraying onto cellulose. Carbohydr. Polym. 2016, 147, 500–508. [Google Scholar] [CrossRef]
- Mijnendonckx, K.; Leys, N.; Mahillon, J.; Silver, S.; Van Houdt, R. Antimicrobial silver: Uses, toxicity and potential for resistance. BioMetals 2013, 26, 609–621. [Google Scholar] [CrossRef]
- Frei, A. Metal Complexes, an Untapped Source of Antibiotic Potential? Antibiotics 2020, 9, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Liu, C.; Qu, D.; Chen, Y.; Huang, M.; Liu, Y. Antibacterial evaluation of sliver nanoparticles synthesized by polysaccharides from Astragalus membranaceus roots. Biomed. Pharmacother. 2017, 89, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Al-Hamadani, A.H.; Kareem, A.A. Combination effect of edible mushroom–sliver nanoparticles and antibiotics against selected multidrug biofilm pathogens. Iraq Med. J. 2017, 1, 68–74. [Google Scholar]
- Kareem, A. Combination Effect of Edible Mushroom – Sliver Nanoparticles and Antibioticsagainst selected Multidrug Biofilm Pathogens. Int. J. Res. Pharm. Sci. 2018, 9. [Google Scholar] [CrossRef]
- Rai, M.; Deshmukh, S.; Ingle, A.; Gade, A. Silver nanoparticles: The powerful nanoweapon against multidrug-resistant bacteria. J. Appl. Microbiol. 2012, 112, 841–852. [Google Scholar] [CrossRef]
- Kascatan-Nebioglu, A.; Panzner, M.J.; A Tessier, C.; Cannon, C.L.; Youngs, W.J. N-Heterocyclic carbene–silver complexes: A new class of antibiotics. Co-ord. Chem. Rev. 2007, 251, 884–895. [Google Scholar] [CrossRef]
- Johnson, N.A.; Southerland, M.R.; Youngs, W.J. Recent Developments in the Medicinal Applications of Silver-NHC Complexes and Imidazolium Salts. Molecules 2017, 22, 1263. [Google Scholar] [CrossRef]
- Vincent, M.; Hartemann, P.; Engels-Deutsch, M. Antimicrobial applications of copper. Int. J. Hyg. Environ. Heal. 2016, 219, 585–591. [Google Scholar] [CrossRef]
- Vincent, M.; Duval, R.; Hartemann, P.; Engels-Deutsch, M. Contact killing and antimicrobial properties of copper. J. Appl. Microbiol. 2018, 124, 1032–1046. [Google Scholar] [CrossRef] [Green Version]
- Dalecki, A.G.; Crawford, C.L.; Wolschendorf, F. Copper and Antibiotics. Adv. Microb. Physiol. 2017, 70, 193–260. [Google Scholar]
- Bondarczuk, K.; Piotrowska-Seget, Z. Molecular basis of active copper resistance mechanisms in Gram-negative bacteria. Cell Boil. Toxicol. 2013, 29, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djoko, K.; Goytia, M.M.; Donnelly, P.S.; A Schembri, M.; Shafer, W.M.; McEwan, A. Copper(II)-Bis(Thiosemicarbazonato) Complexes as Antibacterial Agents: Insights into Their Mode of Action and Potential as Therapeutics. Antimicrob. Agents Chemother. 2015, 59, 6444–6453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Živec, P.; Perdih, F.; Turel, I.; Giester, G.; Psomas, G. Different types of copper complexes with the quinolone antimicrobial drugs ofloxacin and norfloxacin: Structure, DNA- and albumin-binding. J. Inorg. Biochem. 2012, 117, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Thummeepak, R.; Pooalai, R.; Harrison, C.; Gannon, L.; Thanwisai, A.; Chantratita, N.; Millard, A.; Sitthisak, S. Essential Gene Clusters Involved in Copper Tolerance Identified in Acinetobacter baumannii Clinical and Environmental Isolates. Pathogens 2020, 9, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-R.; Lee, J.H.; Kang, L.-W.; Jeong, B.C.; Lee, S.H. Educational Effectiveness, Target, and Content for Prudent Antibiotic Use. BioMed Res. Int. 2015, 2015, 1–13. [Google Scholar] [CrossRef] [PubMed]
- De Bont, E.G.P.M.; Alink, M.; Falkenberg, F.C.J.; Dinant, G.-J.; Cals, J. Patient information leaflets to reduce antibiotic use and reconsultation rates in general practice: A systematic review. BMJ Open 2015, 5, 007612. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breijyeh, Z.; Jubeh, B.; Karaman, R. Resistance of Gram-Negative Bacteria to Current Antibacterial Agents and Approaches to Resolve It. Molecules 2020, 25, 1340. https://doi.org/10.3390/molecules25061340
Breijyeh Z, Jubeh B, Karaman R. Resistance of Gram-Negative Bacteria to Current Antibacterial Agents and Approaches to Resolve It. Molecules. 2020; 25(6):1340. https://doi.org/10.3390/molecules25061340
Chicago/Turabian StyleBreijyeh, Zeinab, Buthaina Jubeh, and Rafik Karaman. 2020. "Resistance of Gram-Negative Bacteria to Current Antibacterial Agents and Approaches to Resolve It" Molecules 25, no. 6: 1340. https://doi.org/10.3390/molecules25061340
APA StyleBreijyeh, Z., Jubeh, B., & Karaman, R. (2020). Resistance of Gram-Negative Bacteria to Current Antibacterial Agents and Approaches to Resolve It. Molecules, 25(6), 1340. https://doi.org/10.3390/molecules25061340