On the Use of Iron in Organic Chemistry


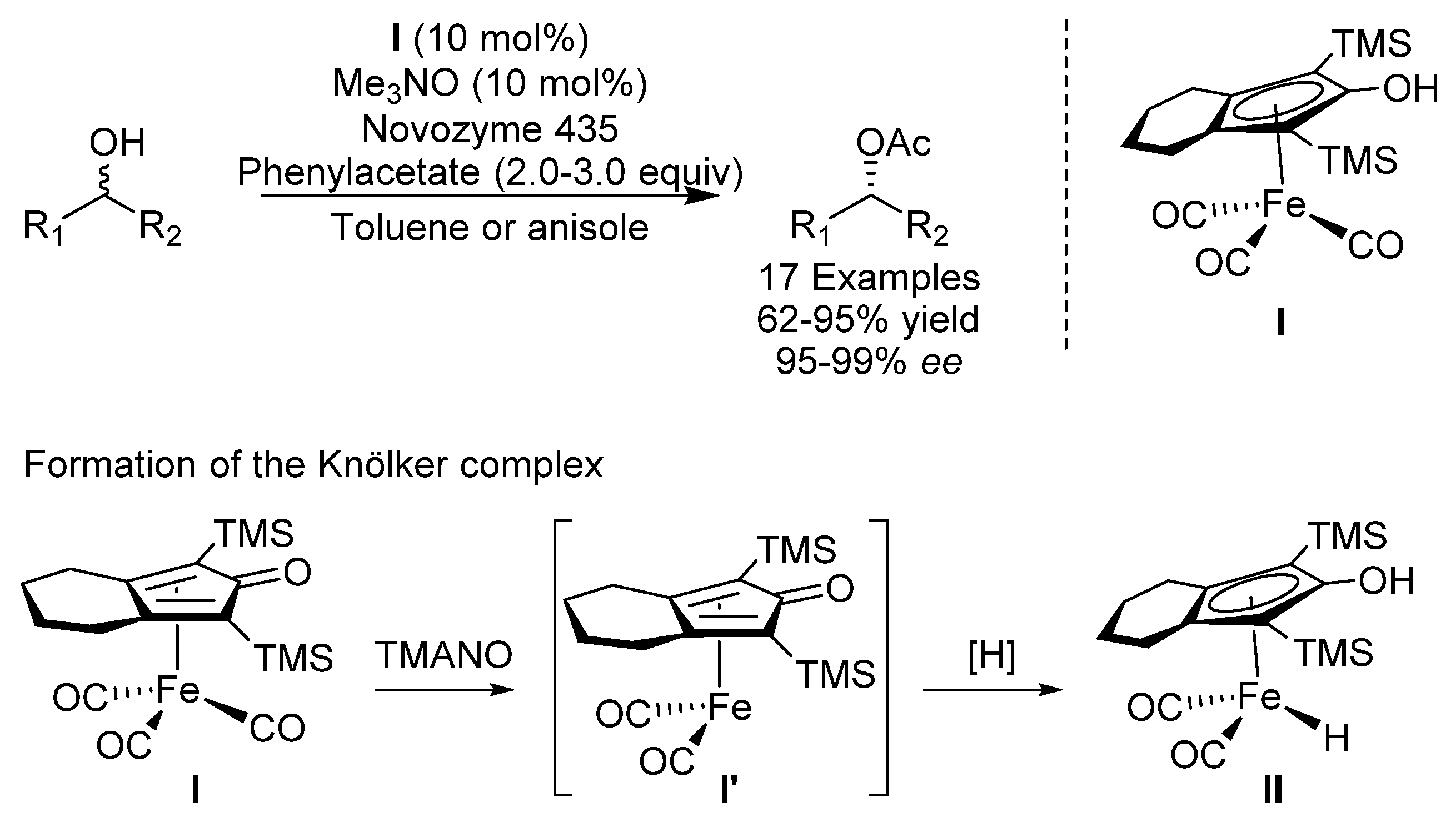{kind=link}
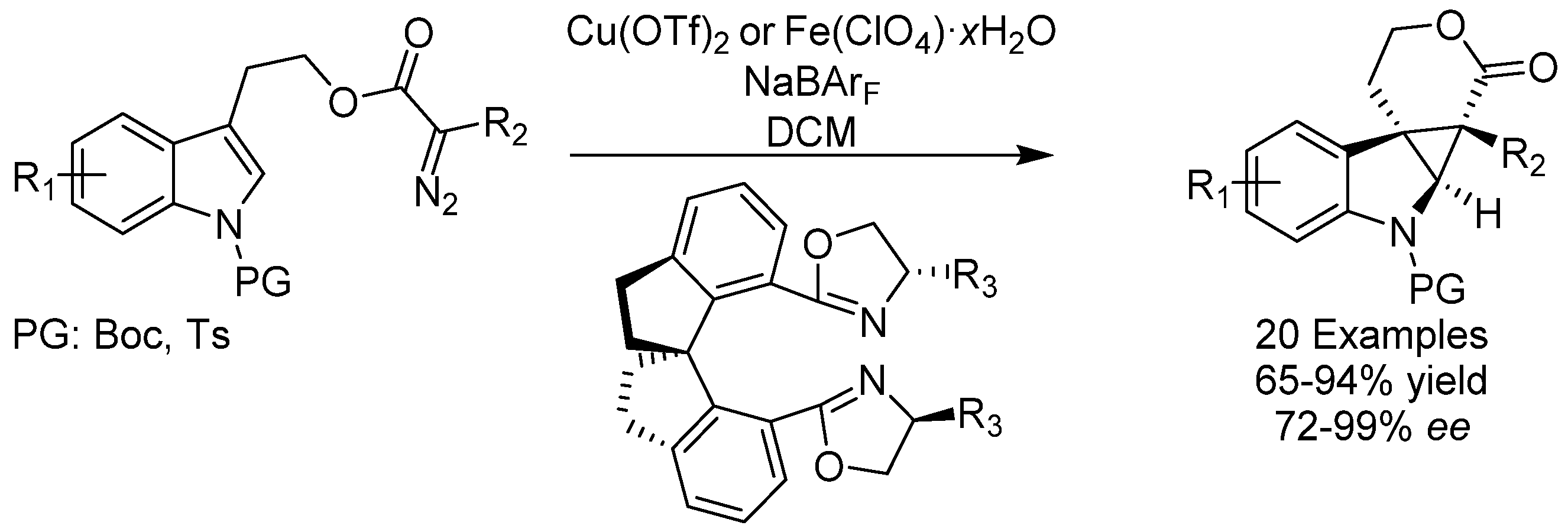{kind=link}
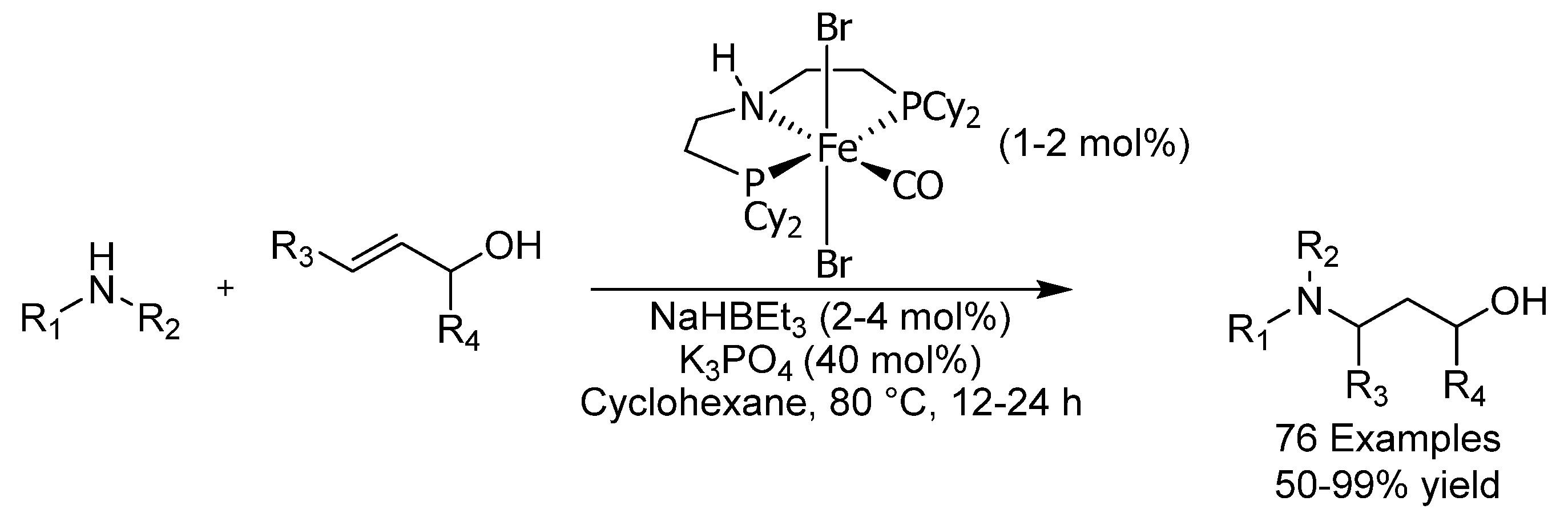{kind=link}
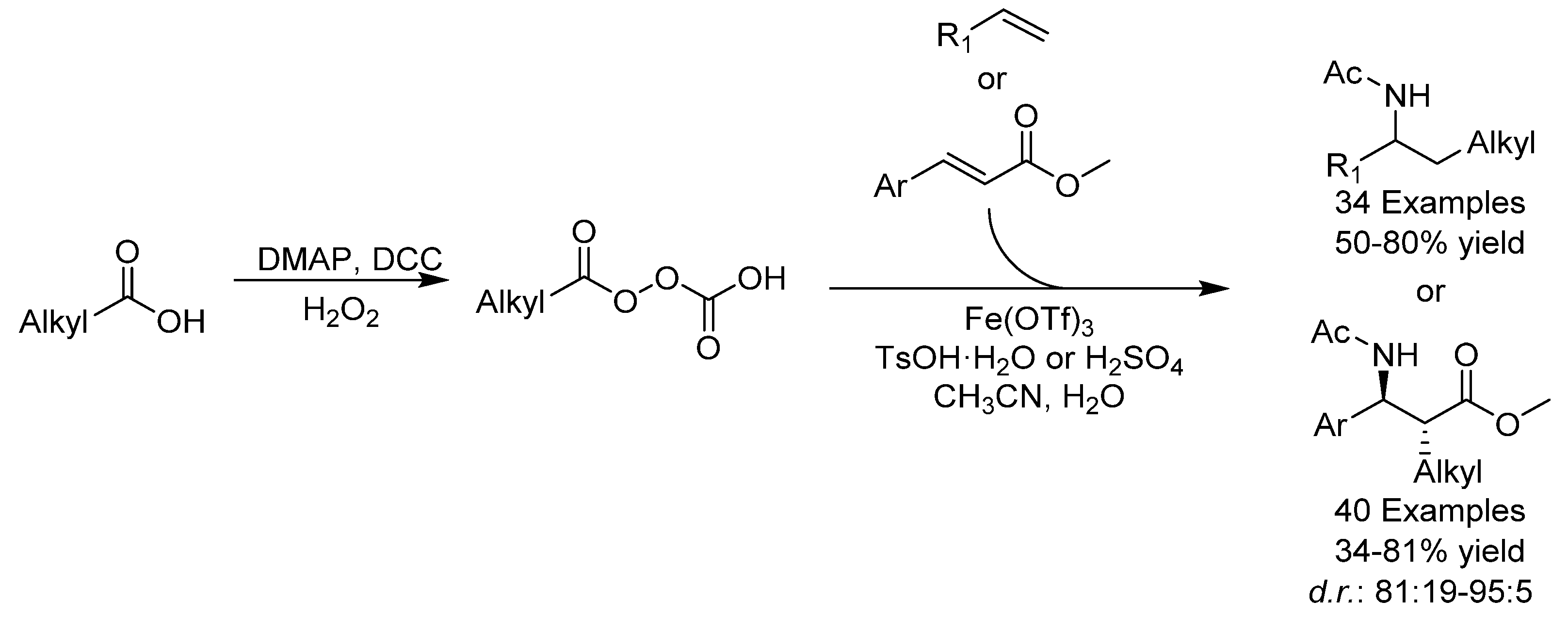{kind=link}
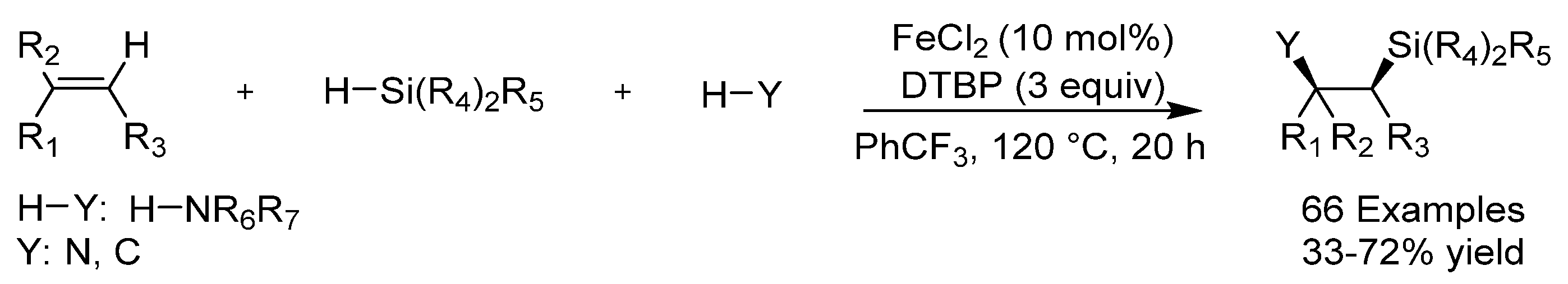{kind=link}
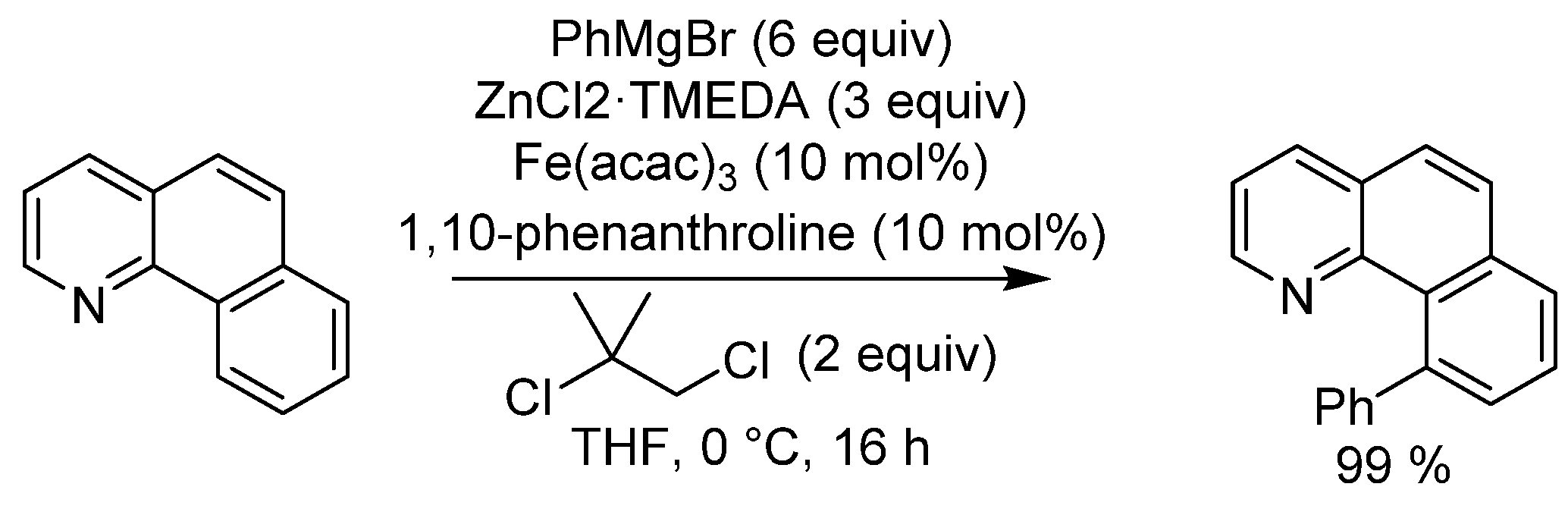{kind=link}
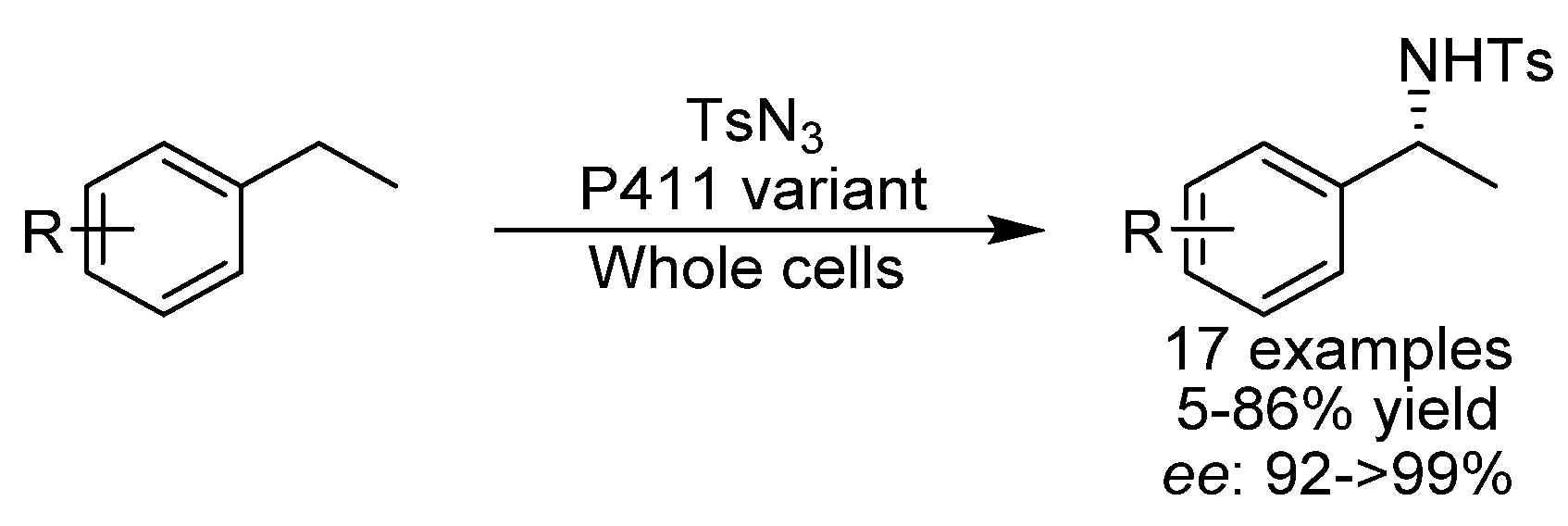{kind=link}
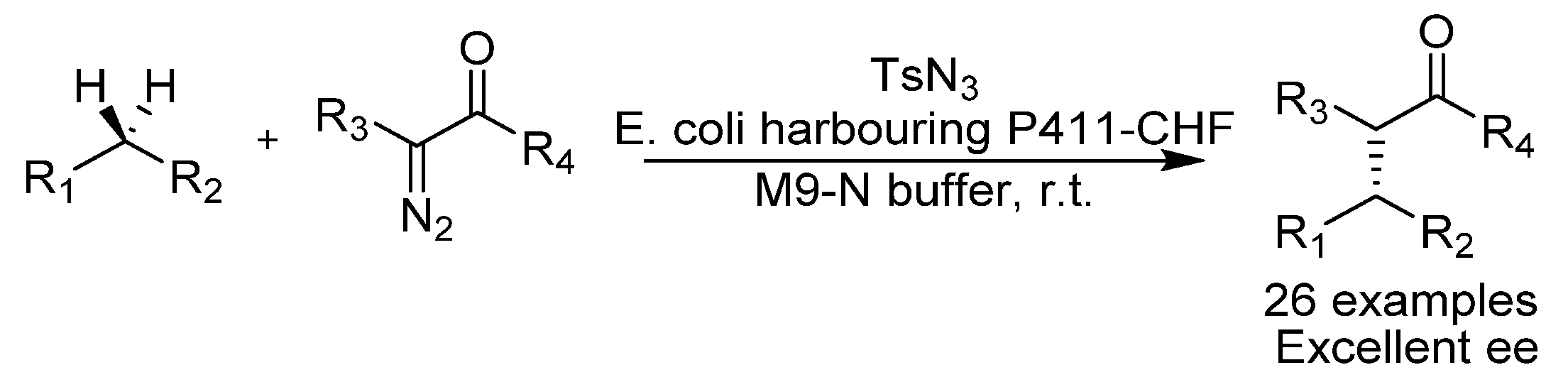{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
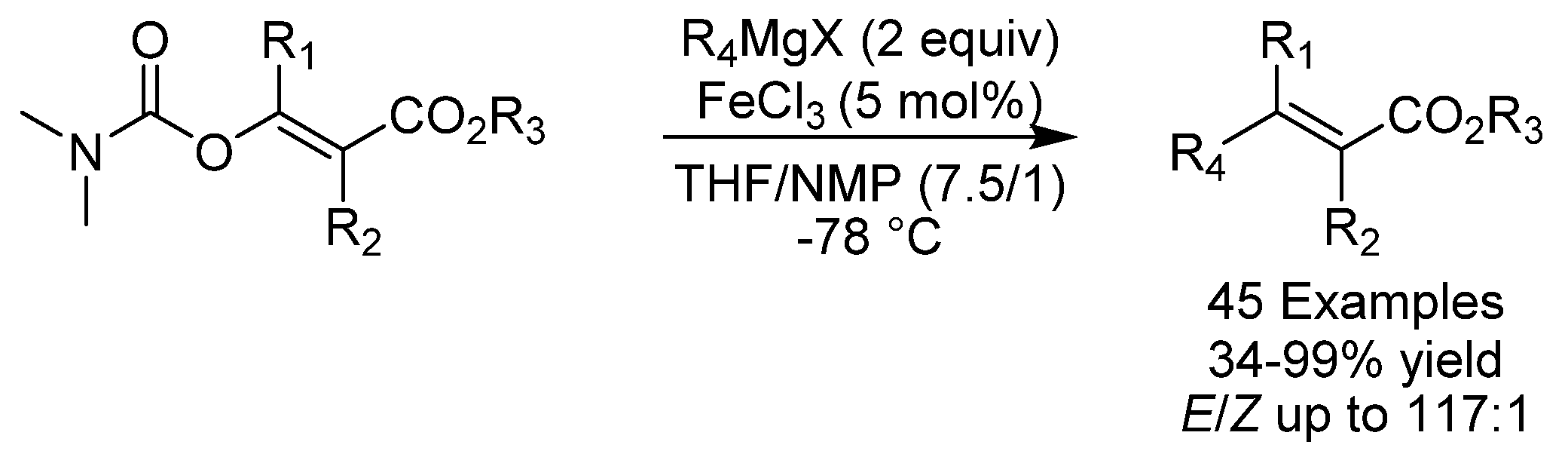{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Iron in Organic Synthesis
2.1. Addition Reactions
2.2. C-H Bond Activation
2.3. Cross-Coupling Reactions
2.4. Cycloadditions
2.5. Isomerizations
2.6. Redox Reactions
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Frey, P.A.; Reed, G.H. The Ubiquity of Iron. ACS Chem. Biol. 2012, 7, 1477–1481. [Google Scholar] [CrossRef] [PubMed]
- Sessions, A.L.; Doughty, D.M.; Welander, P.V.; Summons, R.E.; Newman, D.K. The Continuing Puzzle of the Great Oxidation Event. Curr. Biol. 2009, 19, R567–R574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enthaler, S.; Junge, K.; Beller, M. Sustainable Metal Catalysis with Iron: From Rust to a Rising Star. Angew. Chem. Int. Ed. 2008, 47, 3317–3321. [Google Scholar] [CrossRef]
- Bolm, C. A new iron age. Nat. Chem. 2009, 1, 420. [Google Scholar] [CrossRef] [Green Version]
- Fürstner, A. Iron Catalysis in Organic Synthesis: A Critical Assessment of What It Takes To Make This Base Metal a Multitasking Champion. ACS Cent. Sci. 2016, 2, 778–789. [Google Scholar] [CrossRef]
- Bauer, I.; Knölker, H.-J. Iron Catalysis in Organic Synthesis. Chem. Rev. 2015, 115, 3170–3387. [Google Scholar] [CrossRef]
- Mond, L.; Quinke, F. Note on a Volatile Compound of Iron with Carbonic Oxide. J. Chem. Soc. 1891, 59, 604–607. [Google Scholar] [CrossRef] [Green Version]
- Berthelot, M.C.R. Sur une combinaison volatile de fer et d’oxyde de carbone, le fer carbonyl, et sur le nickel-carbonyle. Compt. Rend. Acad. Sci. 1891, 112, 1343–1348. [Google Scholar]
- Reppe, W.; Vetter, H. Über die Umsetzung von Acetylen mit Kohlenoxyd und Verbindungen mit reationsfähigen Wasserstoffatomen Synthesen α,β-ungesättigter Carbonsäuren und ihrer Derivate. Liebigs. Ann. Chem 1953, 582, 1–37. [Google Scholar] [CrossRef]
- Kealy, T.J.P.; Pauson, P.L. A New Type of Organo-Iron Compound. Nature 1951, 168, 1039. [Google Scholar] [CrossRef]
- Wilkinson, G.; Rosenblum, M.; Whiting, M.C.; Woodward, R.B. The Structure of Iron Bis-Cyclopentadienyl. J. Am. Chem. Soc. 1952, 74, 2125–2126. [Google Scholar] [CrossRef]
- Fischer, E.O.; Pfab, W. Cyclopentadien-Metallkomplexe, ein neuer Typ metallorganischer Verbindungen. Z. Neturforsch. B 1952, 7, 676. [Google Scholar] [CrossRef] [Green Version]
- Haber, F.; Le Rossignol, R. Über die technische Darstellung von Ammoniak aus den Elementen. Elektrochem. Angew. Phys. Chem. 1913, 19, 53. [Google Scholar]
- Cherkasov, N.; Ibhadon, A.O.; Fitzpatrick, P. A review of the existing and alternative methods for greener nitrogen fixation. Chem. Eng. Process. 2015, 90, 24–33. [Google Scholar] [CrossRef]
- Bornschein, C.; Gustafson, K.; Verho, O.; Beller, M.; Bäckvall, J.-E. Evaluation of Fe and Ru Pincer-Type Complexes as Catalysts for the Racemi¬zation of Secondary Benzylic Alcohols. Chem. Eur. J. 2016, 22, 11583–11586. [Google Scholar] [CrossRef]
- Gustafson, K.P.J.; Guðmundsson, A.; Lewis, K.; Bäckvall, J.-E. Chemoenzymatic Dynamic Kinetic Resolution of Secondary Alochols Using an Air- and Moisture-Stable Iron Racemization Catalyst. Chem. Eur. J. 2017, 23, 1048–1051. [Google Scholar] [CrossRef] [Green Version]
- El-Sepelgy, O.; Alandini, N.; Rueping, M. Merging Iron Catalysis and Biocatalysis-Iron Carbonyl Complexes as Efficient Hydrogen Autotransfer Catalysts in Dynamic Kinetic Resolutions. Angew. Chem. Int. Ed. 2016, 55, 13602–13605. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, N.; Liu, M.; Zhou, S. New air-stable iron catalyst for efficient dynamic kinetic resolution of secondary benzylic and aliphatic alcohols. Tetrahederon. Lett. 2017, 58, 2487–2489. [Google Scholar] [CrossRef]
- Quintard, A.; Rodriguez, J. Iron Cyclopentadienone Complexes: Discovery, Properties, and Catalytic Reactivity. Angew. Chem. Int. Ed. 2014, 53, 4044–4055. [Google Scholar] [CrossRef]
- Xu, H.; Li, Y.-P.; Cai, Y.; Wang, G.-P.; Zhu, S.-F.; Zhou, Q.-L. Highly Enantioselective Copper- and Iron-Catalyzed Intramolecular Cyclopropanation of Indoles. J. Am. Chem. Soc. 2017, 139, 7697–7700. [Google Scholar] [CrossRef]
- Ma, W.; Zhang, X.; Fan, J.; Liu, Y.; Tang, W.; Xue, D.; Li, C.; Xiao, J.; Wang, C. Iron-Catalyzed Anti-Markovnikov Hydroamination and Hydroamidation of Allylic Alcohols. J. Am. Chem. Soc. 2019, 141, 13506–13515. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.; Chen, S.; Wang, T.; Zhang, X.; Bao, H. Iron-Catalyzed Carboamination of Olefins: Synthesis of Amines and Disubstituted β-Amino Acids. J. Am. Chem. Soc. 2017, 139, 13076–13082. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Song, R.-J.; Ouyang, X.-H.; Wang, C.-Y.; Li, J.-H.; Luo, S. Iron-Catalyzed Intermolecular 1,2-Difunctionalization of Styrenes and Conjugated Alkenes with Silanes and Nucleophiles. Angew. Chem. Int. Ed. 2017, 56, 7916–7919. [Google Scholar] [CrossRef] [PubMed]
- Dimroth, O. Ueber die Mercurirung aromatischer Verbindungen. Ber. Dtsch. Chem. Ges. 1902, 35, 2853–2873. [Google Scholar] [CrossRef] [Green Version]
- Murahashi, S. Synthesis of Phthalimidines from Schiff Bases and Carbon Monoxide. J. Am. Chem. Soc. 1955, 77, 6403–6404. [Google Scholar] [CrossRef]
- Khrushch, A.P.; Tokina, L.A.; Shilov, A.E. Kinetika i Kataliz; Pleiades Publishing: Warrensburg, MO, USA, 1966; Volume 7, p. 901. [Google Scholar]
- Goldman, A.S.; Goldberg, K.I. Organometallic C-H Bond Activation: An Introduction. In Activation and Functionalization of C-H Bonds; Goldberg, K.I., Goldman, A.S., Eds.; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2004; pp. 4–8. [Google Scholar]
- Norinder, J.; Matsumoto, A.; Yoshikai, N.; Nakamura, E. Iron-Catalyzed Direct Arylation through Directed C-H Bond Activation. J. Am. Chem. Soc. 2008, 130, 5858–5859. [Google Scholar] [CrossRef]
- Shang, R.; Ilies, L.; Nakamura, E. Iron-Catalyzed C-H Bond Activation. Chem. Rev. 2017, 117, 9086–9139. [Google Scholar] [CrossRef]
- Jia, F.; Li, Z. Iron-catalyzed/mediated oxidative transformation of C-H bonds. Org. Chem. Front. 2014, 1, 194–214. [Google Scholar] [CrossRef]
- Landwehr, M.; Hochrein, L.; Otey, C.R.; Kasrayan, A.; Bäckvall, J.-E.; Arnold, F.H. Enantioselective α-Hydroxylation of 2-Arylacetic Acid Derivatives and Buspirone Catalyzed by Engineered Cytochrome P450 BM-3. J. Am. Chem. Soc. 2006, 128, 6058–6059. [Google Scholar] [CrossRef] [Green Version]
- Prier, C.K.; Zhang, R.K.; Buller, A.R.; Brinkmann-Chen, S.; Arnold, F.H. Enantioselective, intermolecular benzylic C-H amination catalyzed by an engineered iron-haem enzyme. Nat. Chem. 2017, 9, 629–634. [Google Scholar] [CrossRef]
- Zhou, X.-G.; Yu, X.-Q.; Huang, J.-S.; Che, C.-M. Asymmetric amidation of saturated C-H bonds catalyzed by chiral ruthenium and manganese porphyrins. Chem. Commun. 1999, 2377–2378. [Google Scholar] [CrossRef]
- Zhang, R.K.; Chen, K.; Huang, X.; Wohlschlager, L.; Renata, H.; Arnold, F.H. Enzymatic assembly of carbon-carbon bonds via iron-catalyzed sp3 C-H functionalization. Nature 2019, 565, 67–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, J.R.; Wendell, C.I.; Garwin, J.A.; White, M.C. Catalytic C(sp3)-H Alkylation via an Iron Carbene Intermediate. J. Am. Chem. Soc. 2017, 139, 13624–13627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, J.; Muller, T.; Oliveira, J.C.A.; Ackermann, L. 1,4-Iron Migration for Expedient Allene Annulations through Iron-Catalyzed C-H/N-H/C-O/C-H Functionalizations. Angew. Chem. Int. Ed. 2018, 57, 7719–7723. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, J.; Durham, A.C.; Lindberg, H.; Wang, Y.-M. α-C-H Functionalization of π-Bonds Using Iron Complexes: Catalytic Hydroxyalkylation of Alkynes and Alkenes. J. Am. Chem. Soc. 2019, 141, 19594–19599. [Google Scholar] [CrossRef] [PubMed]
- Cutler, A.; Ehnholt, D.; Lennon, P.; Nicholas, K.; Marten, D.F.; Madhavarao, M.; Raghu, S.; Rosan, A.; Rosenblum, M. Chemistry of dicarbonyl η5-Cyclopentadienyliron Complexes. General Syntheses of Monosubstituted η2-Olefin Complexes and of 1-Substituted η1-Allyl Complexes. Conformational Effects on the Course of Deprotonation of (η2-Olefin) Cations. J. Am. Chem. Soc. 1975, 97, 3149. [Google Scholar] [CrossRef]
- Guan, H.; Sun, S.; Mao, Y.; Chen, L.; Lu, R.; Huang, J.; Liu, L. Iron(II)-Catalyzed Site-Selective Functionalization of Unactivated C(sp3)-H Bonds Guided by Alkoxyl Radicals. Angew. Chem. Int. Ed. 2018, 57, 11413–11417. [Google Scholar] [CrossRef]
- Tamura, M.; Kochi, J. Vinylation of Grignard reagents. Catalysis by iron. J. Am. Chem. Soc. 1971, 93, 1487–1489. [Google Scholar] [CrossRef]
- Mako, T.L.; Byers, J.A. Recent advances in iron-catalyzed cross coupling reactions and their mechanistic underpinning. Inorg. Chem. Front. 2016, 3, 766–790. [Google Scholar] [CrossRef]
- Piontek, A.; Bisz, E.; Szostak, M. Iron-Catalyzed Cross-Couplings in the Synthesis of Pharmaceuticals: In Pursuit of Sustainability. Angew. Chem. Int. Ed. 2018, 57, 11116–11128. [Google Scholar] [CrossRef]
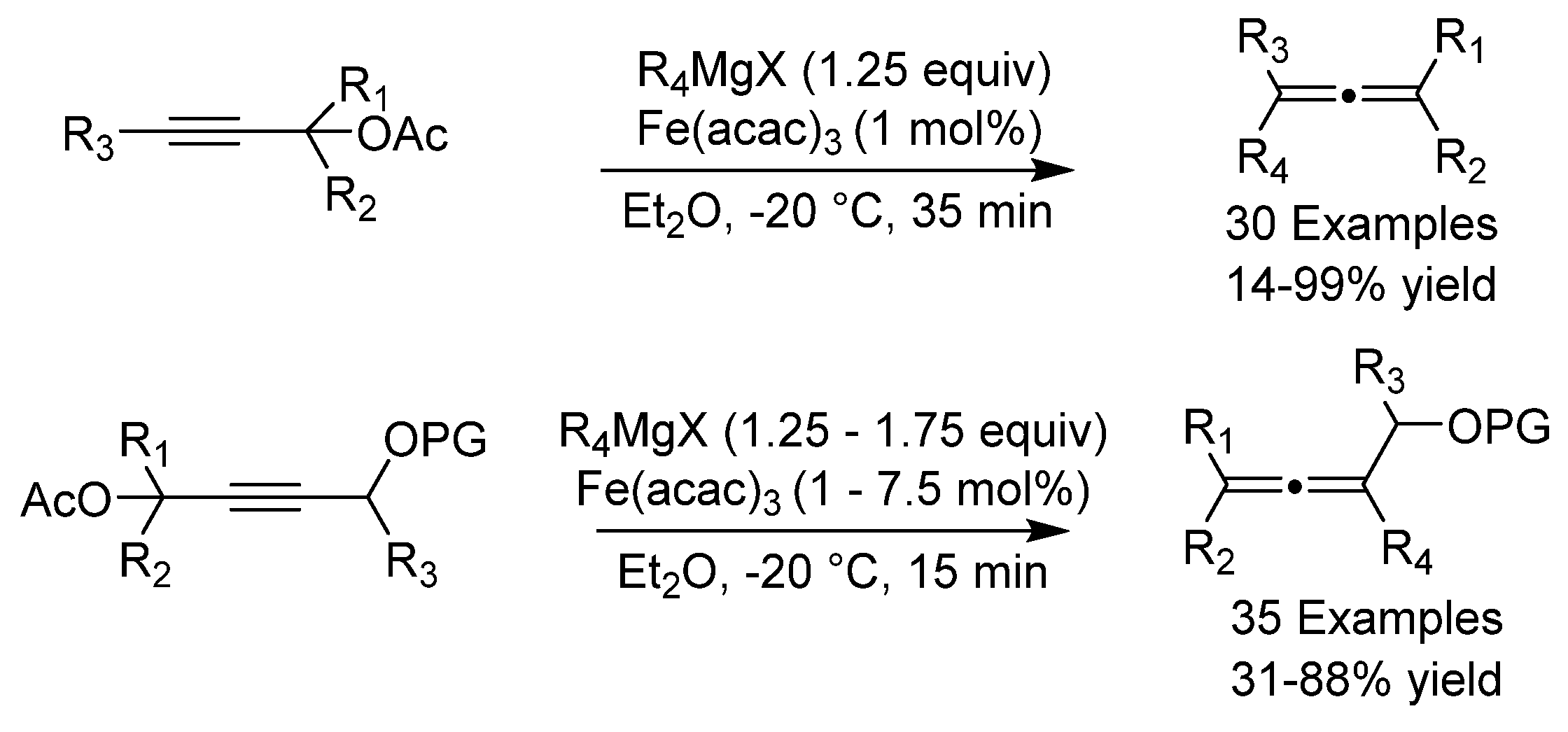
- Kessler, S.N.; Hundemer, F.; Bäckvall, J.-E. A Synthesis of Substituted α -Allenols via Iron-Catalyzed Cross-Coupling of Propargyl Carboxylates with Grignard Reagents. ACS Catal. 2016, 6, 7448–7451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessler, S.N.; Bäckvall, J.-E. Iron-catalyzed Cross-Coupling of Propargyl Carboxylates and Grignard Reagents: Synthesis of Substituted Allenes. Angew. Chem. Int. Ed. 2016, 55, 3734–3738. [Google Scholar] [CrossRef] [PubMed]
- Rivera, A.C.P.; Still, R.; Frantz, D.E. Iron-Catalyzed Stereoselective Cross-Coupling Reactions of Stereodefined Enol Carbamates with Grignard Reagents. Angew. Chem. Int. Ed. 2016, 55, 6689–6693. [Google Scholar] [CrossRef] [PubMed]
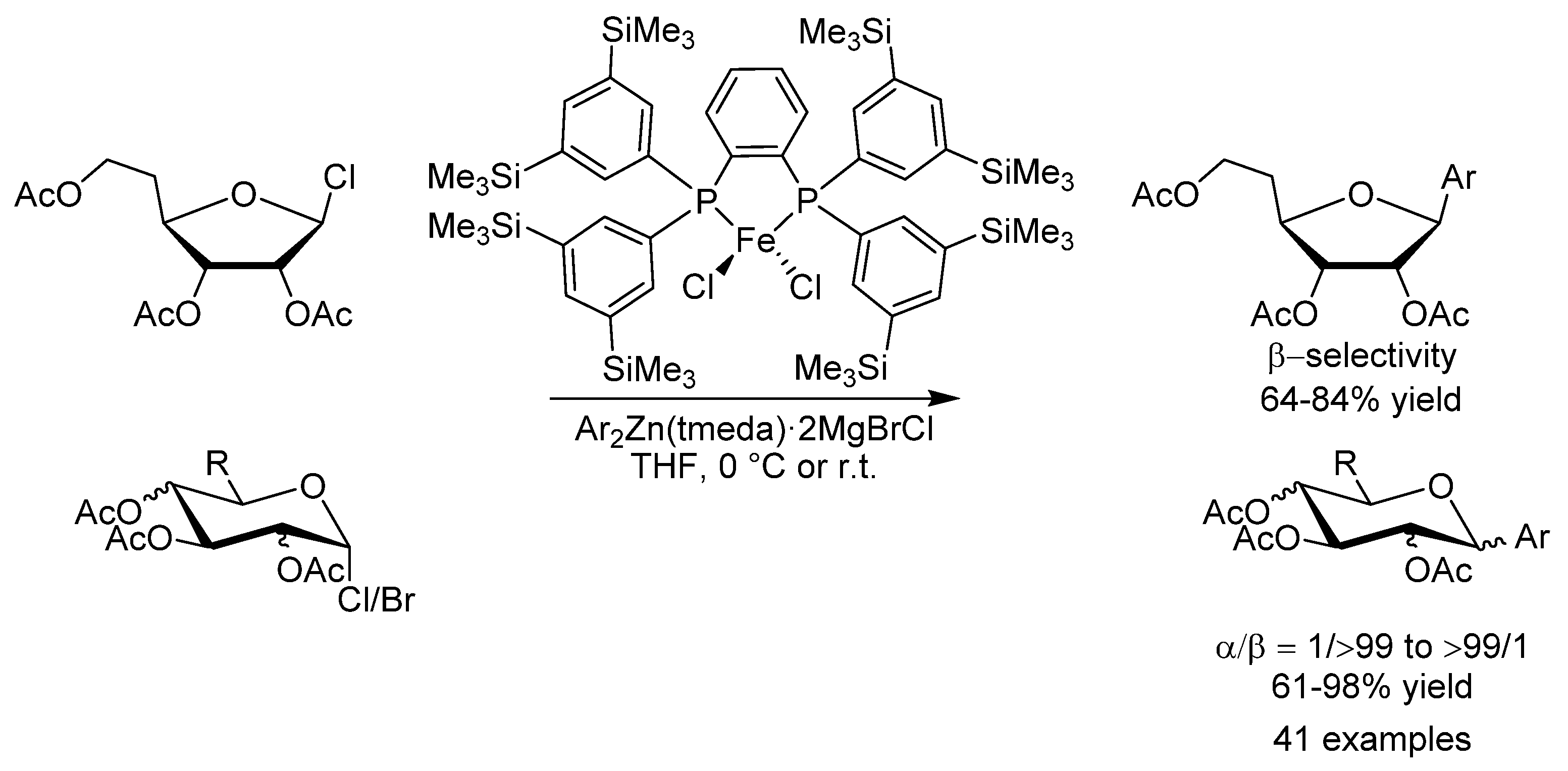
- Adak, L.; Kawamura, S.; Toma, G.; Takenaka, T.; Isozaki, K.; Takaya, H.; Orita, A.; Li, H.C.; Shing, T.K.M.; Nakamura, M. Synthesis of Aryl C-Glycosides via Iron-Catalyzed Cross Coupling of Halosugars: Stereoselective Anomeric Arylation of Glycosyl Radicals. J. Am. Chem. Soc. 2017, 139, 10693–10701. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.; Ball, A.K.; Birkett, J.; Brown, L.; Chappell, B.; Gill, D.M.; Lo, P.K.T.; Patmore, N.J.; Rice, C.R.; Ryan, J.; et al. An iron-catalyzed C-C bond-forming spirocyclization cascade providing sustainable access to new 3D heterocyclic frameworks. Nat. Chem. 2017, 9, 396–401. [Google Scholar] [CrossRef] [PubMed]
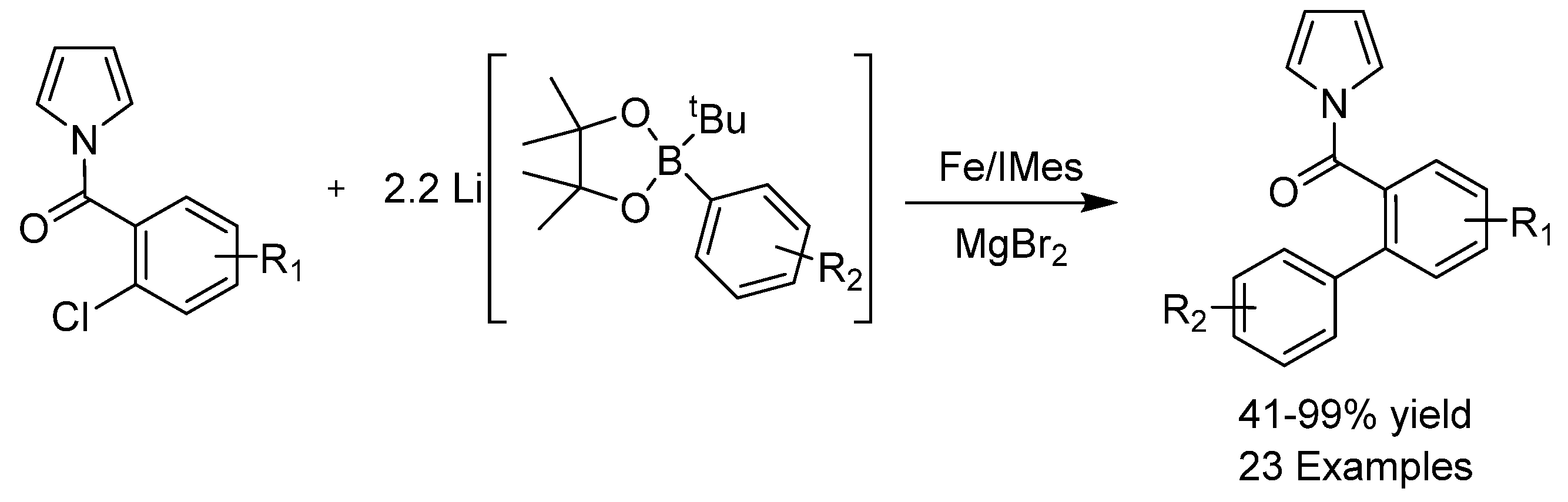
- O’Brien, H.M.; Manzotti, M.; Abrams, R.D.; Elorriaga, D.; Sparkes, H.A.; Davis, S.A.; Bedford, R.B. Iron-Catalyzed substrate-directed Suzuki biaryl cross-coupling. Nat. Catal. 2018, 1, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, C.R.; Zhong, H.; Macaulay, R.L.; Chirik, P.J. Regio- and Diastereoselective Iron-Catalyzed [4+4]-Cycloaddition of 1,3-dienes. J. Am. Chem. Soc. 2019, 141, 8557–8573. [Google Scholar] [CrossRef]
- Wang, Q.; Qi, X.; Lu, L.-Q.; Li, T.-R.; Yuan, Z.-G.; Zhang, K.; Li, B.-J.; Lan, Y.; Xiao, W.-J. Iron-Catalyzed Decarboxylative (4+1) Cycloadditions: Exploiting the Reactivity of Ambident Iron-Stabilized Intermediates. Angew. Chem. Int. Ed. 2016, 55, 2840–2844. [Google Scholar] [CrossRef]
- He, R.; Huang, Z.-T.; Zheng, Q.-Y.; Wang, C. Isoquinoline skeleton synthesis via chelation-assisted C-H activation. Tetrahedron Lett. 2014, 55, 5705–5713. [Google Scholar] [CrossRef] [Green Version]
- Jia, T.; Zhao, C.; He, R.; Chen, H.; Wang, C. Iron-Carbonyl-Catalyzed Redox-Neutral [4+2] Annulation of N-H Imines and Internal Alkynes by C-H Bond Activation. Angew. Chem. Int. Ed. 2016, 55, 5268–5271. [Google Scholar] [CrossRef]
- Brenna, D.; Villa, M.; Gieshoff, T.N.; Fischer, F.; Hapke, M.; Jacobi von Wangelin, A. Iron-Catalyzed Cyclotrimerization of Terminal Alkynes by Dual Catalyst Activation in the Absence of Reductants. Angew. Chem. Int. Ed. 2017, 56, 8451–8454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emerson, G.F.; Pettit, R. π-Allyl-Iron Tricarbonyl Cations. J. Am. Chem. Soc. 1962, 84, 4591. [Google Scholar] [CrossRef]
- Guðmundsson, A.; Gustafson, K.P.J.; Mai, B.K.; Yang, B.; Himo, F.; Bäckvall, J.-E. Efficient Formation of 2,3-dihydrofurans via Iron-Catalyzed Cycloisomerization of α-Allenols. ACS Catal. 2018, 8, 12–16. [Google Scholar] [CrossRef]
- Guðmundsson, A.; Gustafson, K.P.J.; Mai, B.K.; Hobiger, V.; Himo, F.; Bäckvall, J.-E. Diastereoselective Synthesis of N-Protected 2,3-Dihydropyrroles via Iron-Catalyzed Cycloisomerization of α-Allenic Sulfonamides. ACS Catal. 2019, 9, 1733–1737. [Google Scholar] [CrossRef]
- El-Sepelgy, O.; Brzozowska, A.; Azofra, L.M.; Jang, Y.K.; Cavallo, L.; Rueping, M. Experimental and Computational Study of an Unexpected Iron-Catalyzed Carboetherification by Cooperative Metal and Ligand Substrate Interaction and Proton Shuttling. Angew. Chem. Int. Ed. 2017, 56, 14863–14867. [Google Scholar] [CrossRef] [PubMed]
- El-Sepelgy, O.; Brzozowska, A.; Sklyaruk, J.; Kyung Jang, Y.; Zubar, V.; Rueping, M. Cooperative Metal-Ligand Catalyzed Intramolecular Hydroamination and Hydroalkoxylation of Allenes Using a Stable Iron Catalyst. Org. Lett. 2018, 20, 696–699. [Google Scholar] [CrossRef]
- Yang, B.; Zhu, C.; Qiu, Y.; Bäckvall, J.-E. Enzyme- and Ruthenium-Catalyzed Enantioselective Transformation of α-Allenic Alcohols into 2,3-Dihydrofurans. Angew. Chem. Int. Ed. 2016, 55, 5568–5572. [Google Scholar] [CrossRef] [Green Version]
- Woodward, R.R.; Hoffmann, R. The Conservation of Orbital Symmetry. Angew. Chem. Int. Ed. Engl. 1969, 8, 781. [Google Scholar] [CrossRef]
- Kramm, F.; Teske, J.; Ullwer, F.; Frey, W.; Plietker, B. Annelated Cyclobutanes by Fe-Catalyzed Cycloisomerization of Enyne Acetates. Angew. Chem. Int. Ed. 2018, 57, 13335–13338. [Google Scholar] [CrossRef]
- Cherkaoui, H.; Soufiaoui, M.; Grée, R. From allylic alcohols to saturated carbonyls using Fe(CO)5 as catalyst: Scope and limitation studies and preparation of two perfume components. Tetrahedron 2001, 57, 2379–2383. [Google Scholar] [CrossRef]
- Xia, T.; Wei, Z.; Spiegelberg, B.; Jiao, H.; Hinze, S.; De Vries, J.G. Isomerization of Allylic Alcohols to Ketones Catalyzed by Well-Defined Iron PNP Pincer Catalysts. Chem. Eur. J. 2018, 24, 4043–4049. [Google Scholar] [CrossRef] [PubMed]
- Escheverria, P.-G.; Fürstner, A. An Iron-Catalyzed Bond-Making/Bond-Breaking Cascade Merges Cycloisomerization and Cross-Coupling Chemistry. Angew. Chem. Int. Ed. 2016, 55, 11188–11192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed-Berendt, B.G.; Polidano, K.; Morrill, L.C. Recent advances in homogeneous borrowing hydrogen catalysis using earth-abundant first row transition metals. Org. Biomol. 2019, 17, 1595–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, D.; Darcel, C. Iron Catalysis in Reduction and Hydrometalation Reactions. Chem. Rev. 2019, 119, 2550–2610. [Google Scholar] [CrossRef]
- Okamura, M.; Kondo, M.; Kuga, R.; Kurashige, Y.; Yanai, T.; Hayami, S.; Praneeth, V.K.K.; Yoshida, M.; Yoneda, K.; Kawata, S.; et al. A pentanuclear iron catalyst designed for water oxidation. Nature 2016, 530, 465–468. [Google Scholar] [CrossRef]
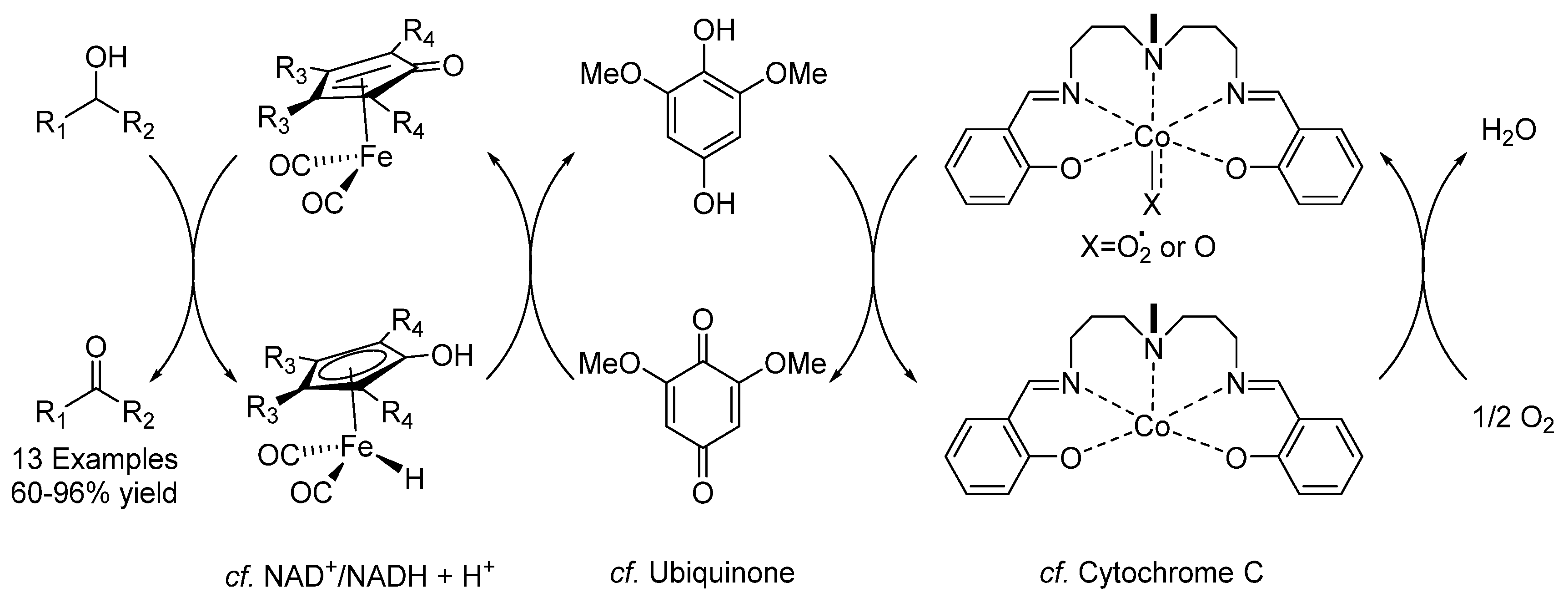
- Guðmundsson, A.; Schlipköter, K.E.; Bäckvall, J.-E. Iron(II)-Catalyzed Biomimetic Aerobic Oxidation of Alcohols. Angew. Chem. Int. Ed. 2020, 59. [Google Scholar] [CrossRef] [Green Version]
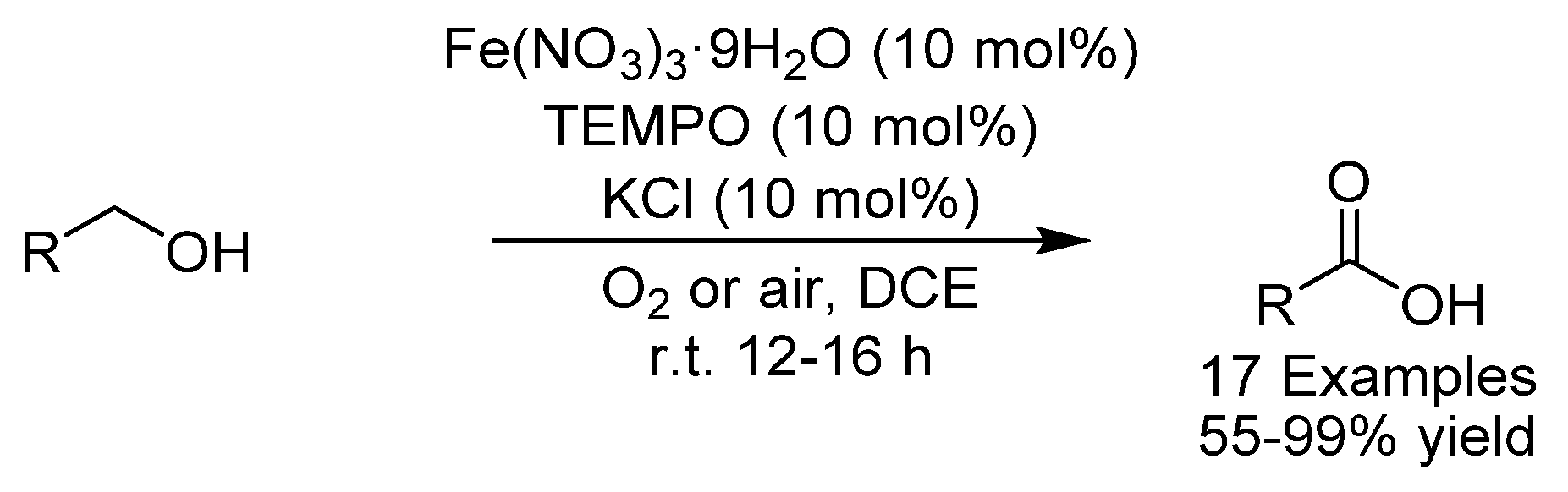
- Jiang, X.; Zhang, J.; Ma, S. Iron Catalysis for Room-Temperature Aerobic Oxidation of Alcohols to Carboxylic Acids. J. Am. Chem. Soc. 2016, 138, 8344–8347. [Google Scholar] [CrossRef]
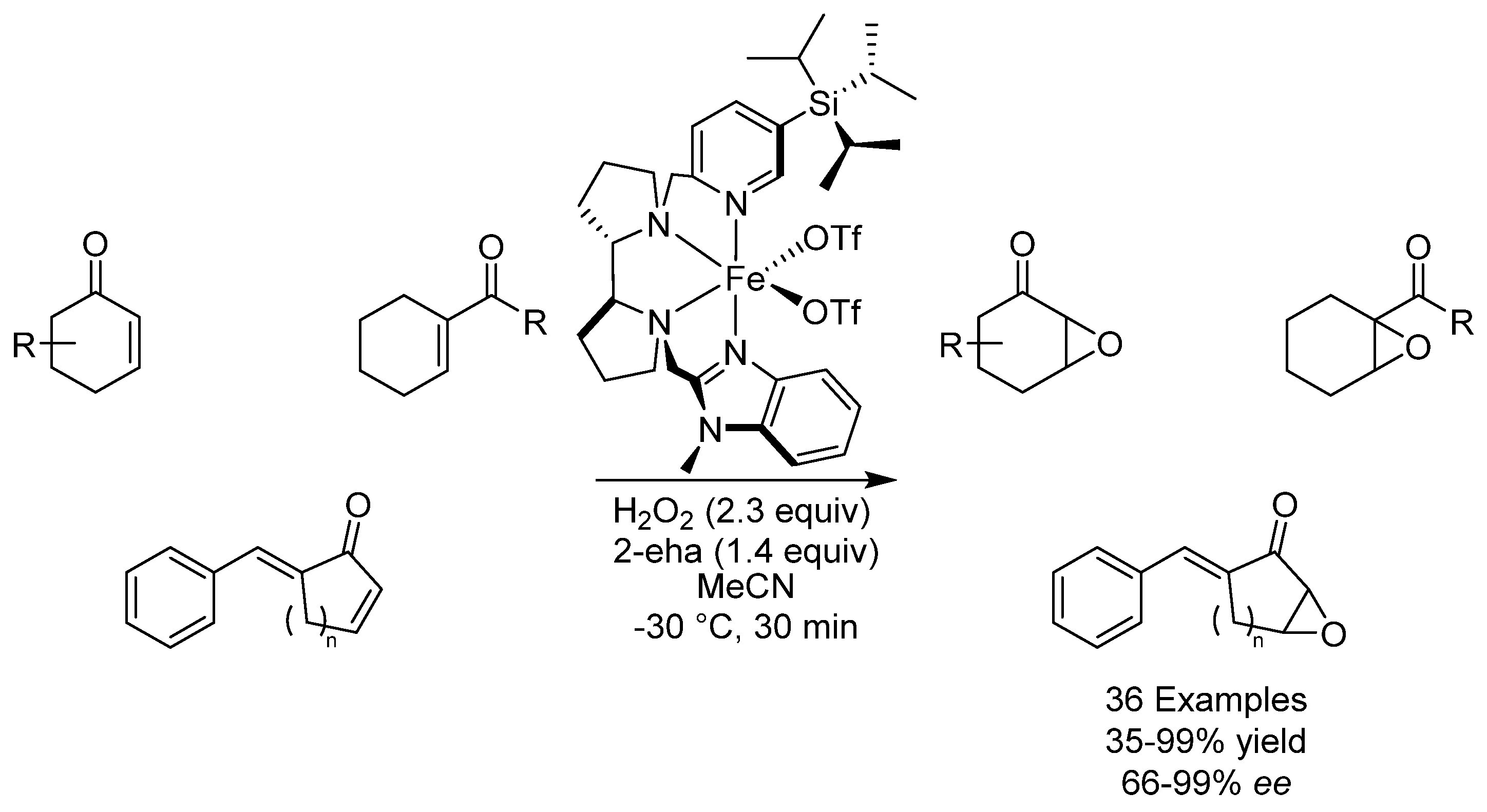
- Cussó, O.; Cianfanelli, M.; Ribas, X.; Gebbink, R.J.M.K.; Costas, M. Iron Catalyzed Highly Enantioselective Epoxidation of Cyclic Aliphatic Enones with Aqueous H2O2. J. Am. Chem. Soc. 2016, 138, 2732–2738. [Google Scholar] [CrossRef] [Green Version]
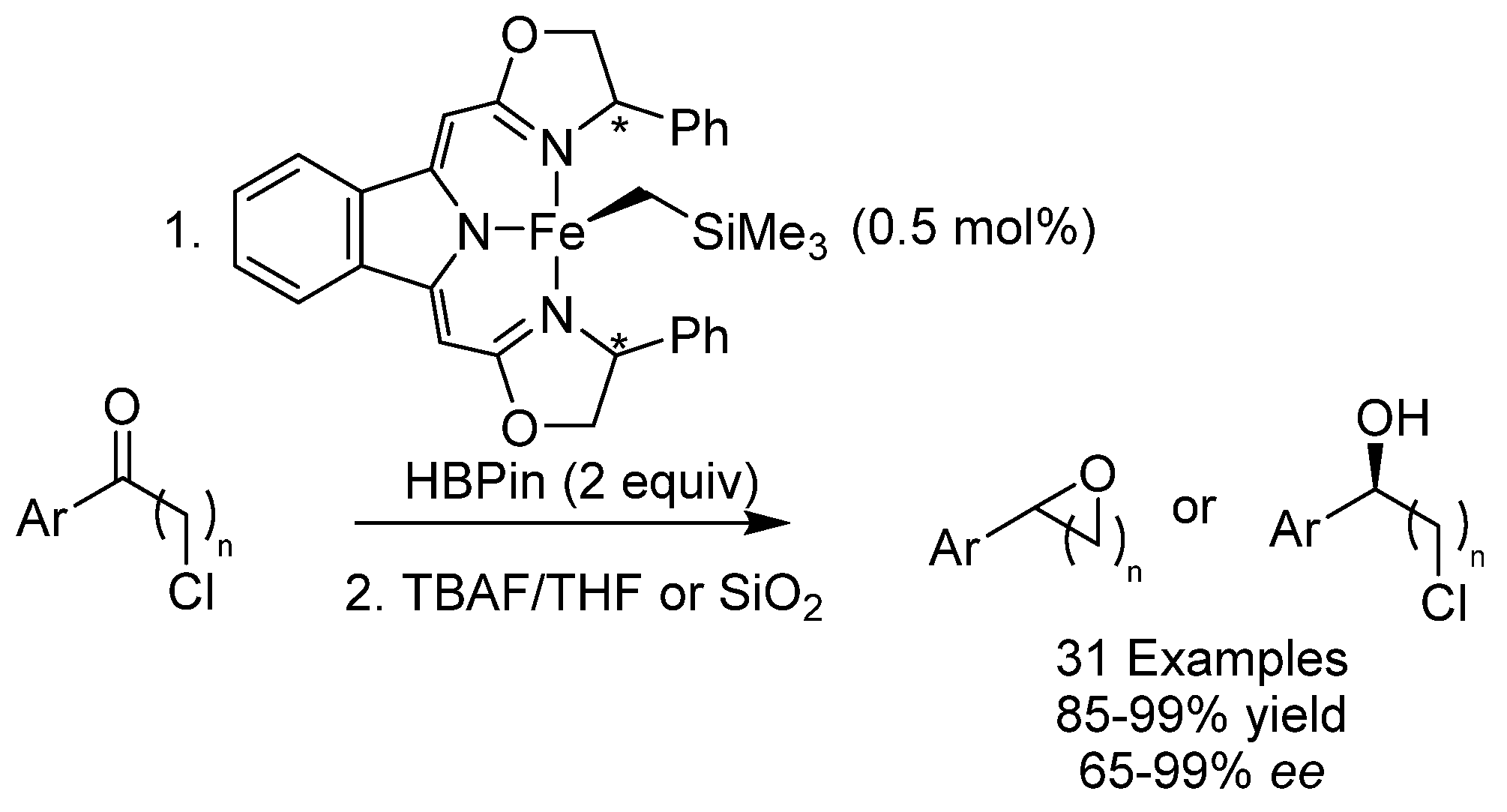
- Blasius, C.K.; Vasilenko, V.; Gade, L.H. Ultrafast Iron-Catalyzed Reduction of Functionalized Ketones: Highly Enantioselective Synthesis of Halohydrines, Oxaheterocycles and Aminoalcohols. Angew. Chem. Int. Ed. 2018, 57, 10231–10235. [Google Scholar] [CrossRef]
- Farrar-Tobar, R.A.; Wozniak, B.; Savini, A.; Hinze, S.; Tin, S.; de Vries, J.G. Base-Free Iron Catalyzed Transfer Hydrogenation of Esters Using EtOH as Hydrogen Source. Angew. Chem. Int. Ed. 2019, 58, 1129–1133. [Google Scholar] [CrossRef]
- Lator, A.; Gaillard, S.; Poater, A.; Renaud, J.-L. Iron-Catalyzed Chemoselective Reduction of α,β-Unsaturated Ketones. Chem. Eur. J. 2018, 24, 5770–5774. [Google Scholar] [CrossRef] [PubMed]
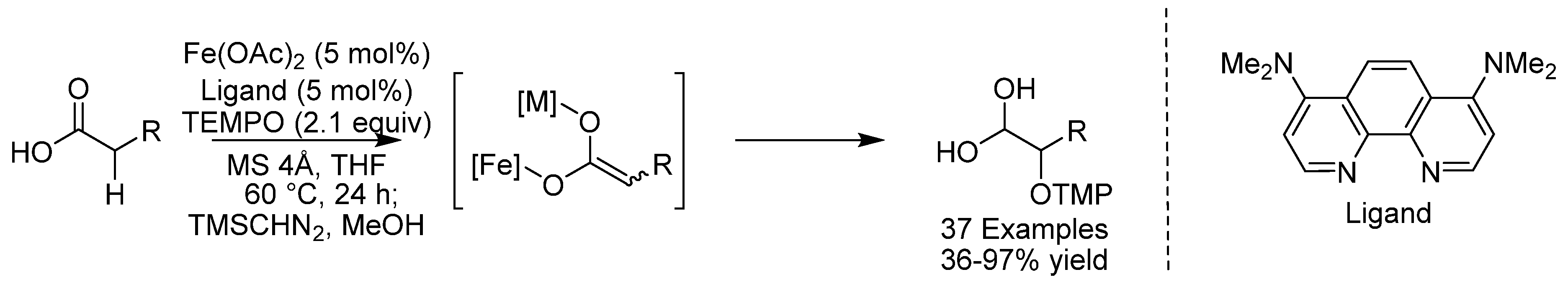
- Tanaka, T.; Yazaki, R.; Ohshima, T. Chemoselective Catalytic α-Oxidation of Carboxylic Acids: Iron/Alkali Metal Cooperative Redox Active Catalysis. J. Am. Chem. Soc. 2020, 142, 4517–4524. [Google Scholar] [CrossRef] [PubMed]
































© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guðmundsson, A.; Bäckvall, J.-E. On the Use of Iron in Organic Chemistry. Molecules 2020, 25, 1349. https://doi.org/10.3390/molecules25061349
Guðmundsson A, Bäckvall J-E. On the Use of Iron in Organic Chemistry. Molecules. 2020; 25(6):1349. https://doi.org/10.3390/molecules25061349
Chicago/Turabian StyleGuðmundsson, Arnar, and Jan-E. Bäckvall. 2020. "On the Use of Iron in Organic Chemistry" Molecules 25, no. 6: 1349. https://doi.org/10.3390/molecules25061349
APA StyleGuðmundsson, A., & Bäckvall, J. -E. (2020). On the Use of Iron in Organic Chemistry. Molecules, 25(6), 1349. https://doi.org/10.3390/molecules25061349