Isostructural Inorganic–Organic Piperazine-1,4-diium Chlorido- and Bromidoantimonate(III) Monohydrates: Octahedral Distortions and Hydrogen Bonds


Abstract
:
1. Introduction
2. Results and Discussion
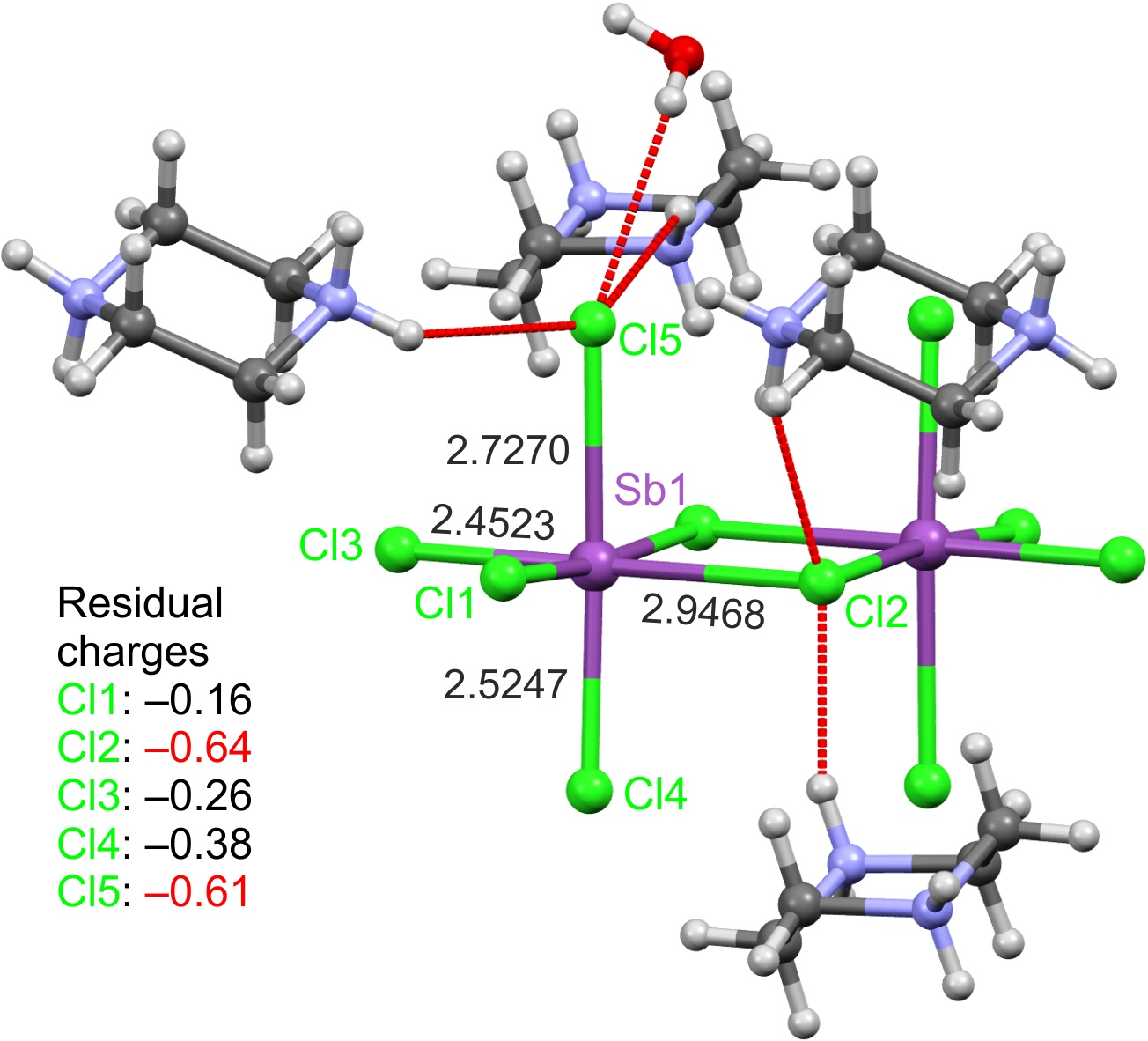
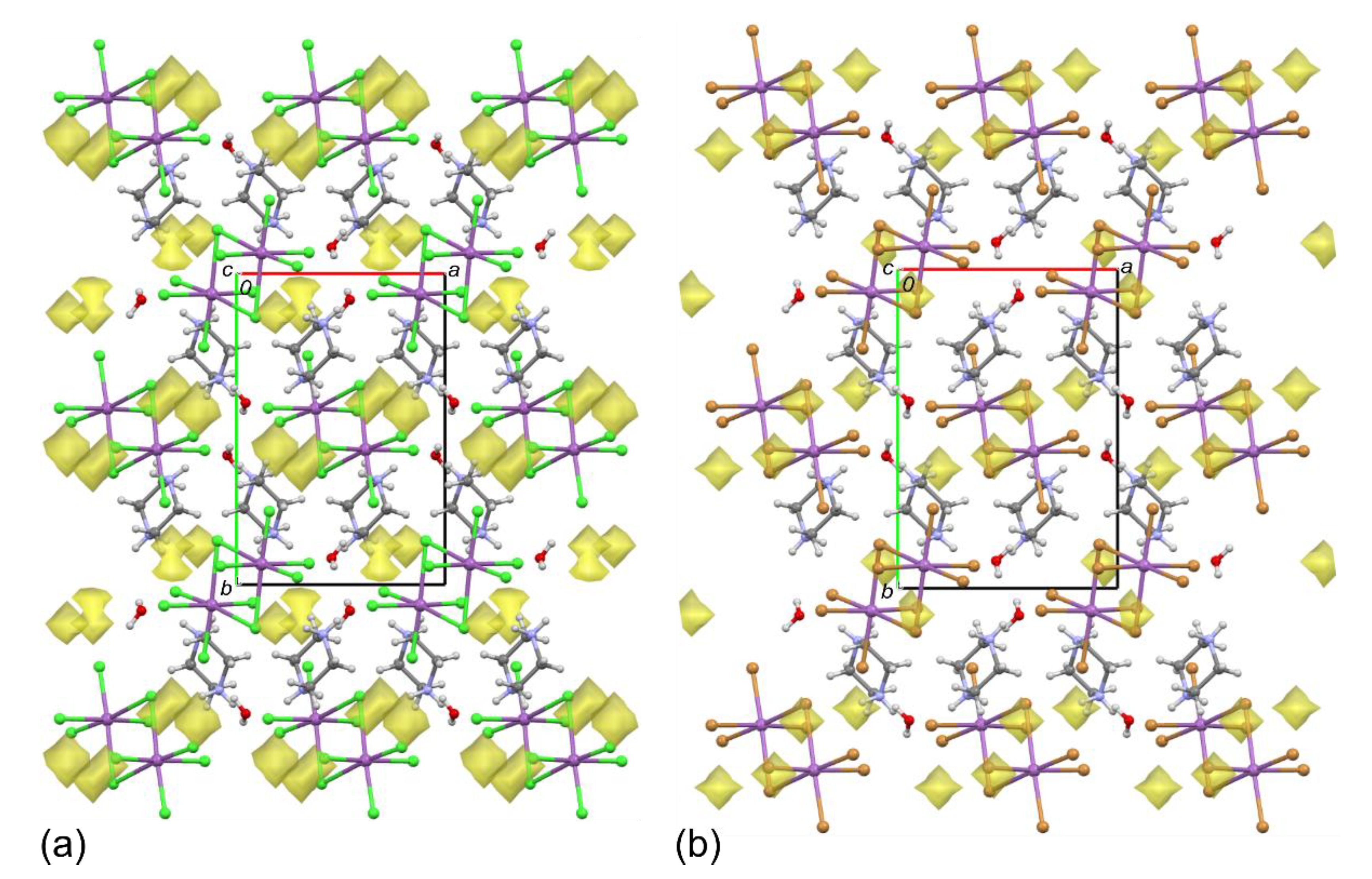
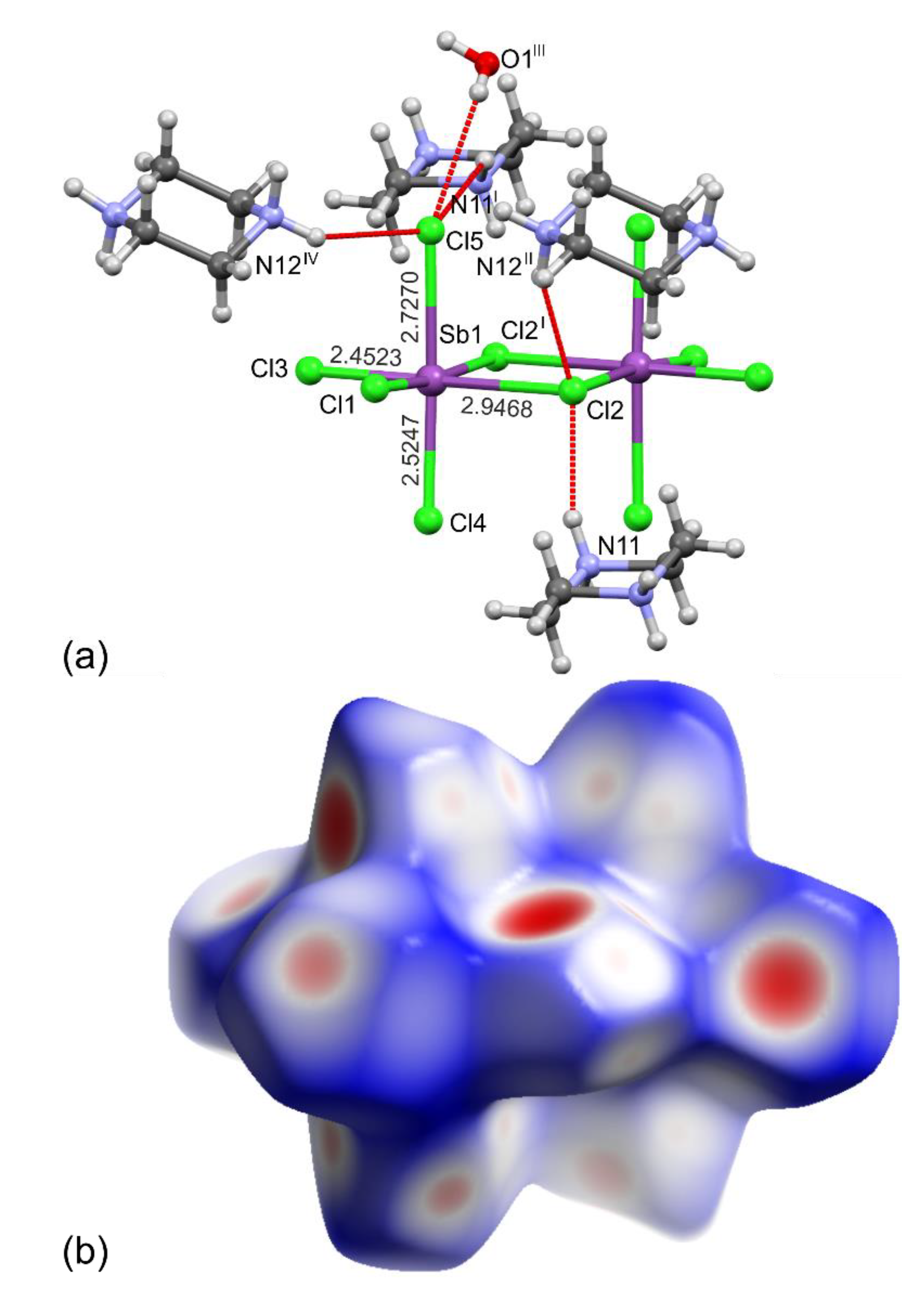
2.1. Structures of 1 and 2
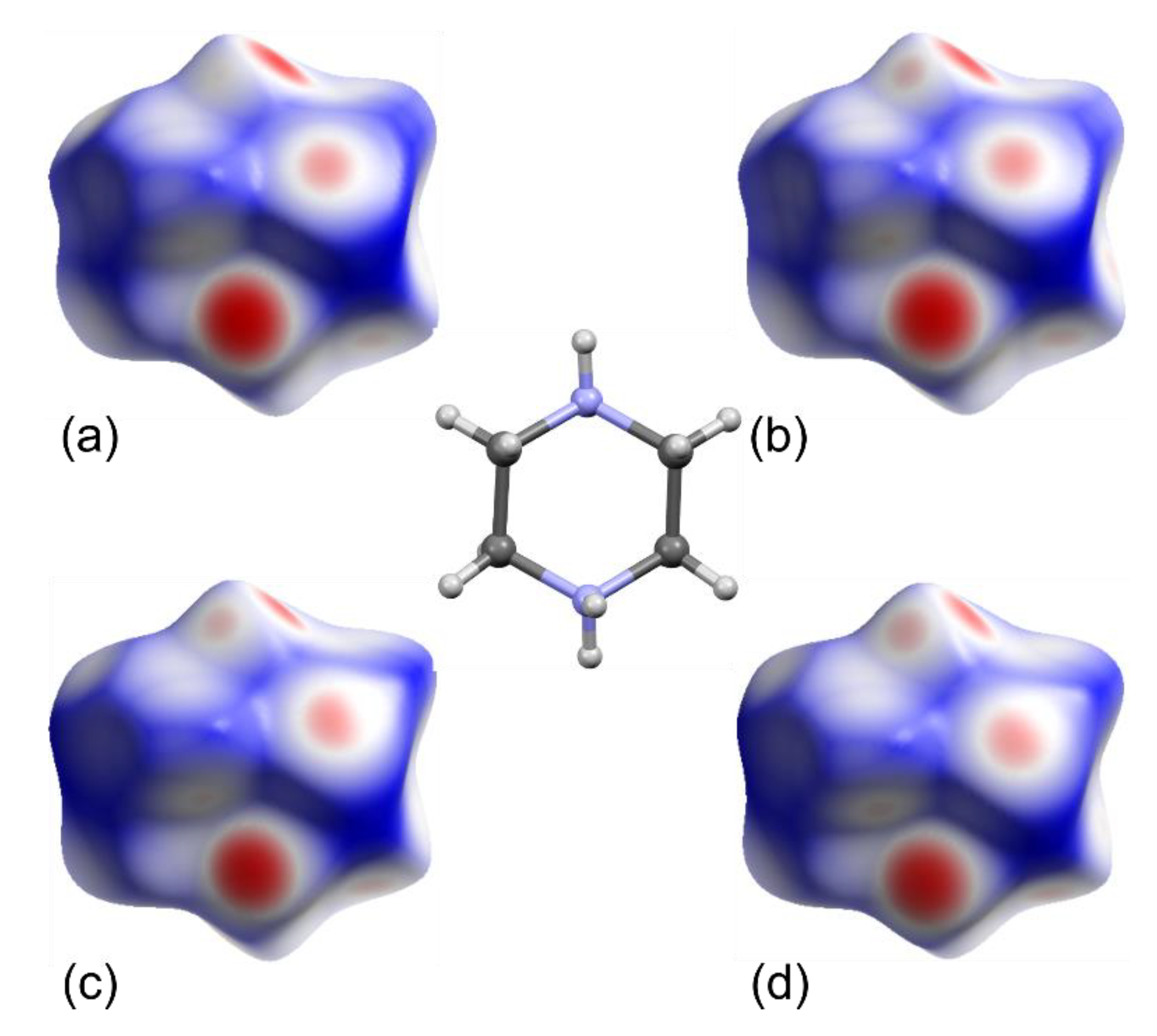
2.2. Octahedral Distortions in 1 and 2
3. Materials and Methods
3.1. Preparation of 1 and 2
3.2. X-ray Structure Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fischer, G.A.; Norman, N.C. The structures of the group 15 element(III) halides and halogenoanions. Adv. Inorg. Chem. 1994, 41, 233–271. [Google Scholar]
- Sobczyk, L.; Jakubas, R.; Zaleski, J. Self-Assembly of Sb(III) and Bi(III) Halo-Coordinated Octahedra in Salts of Organic Cations. Structure, Properties and Phase Transitions. Pol. J. Chem. 1997, 71, 265–330. [Google Scholar]
- Bujak, M.; Zaleski, J. Synthesis of chloroantimonates(III) with selected organic cations. X-ray studies of phase transition in ferroelectric tris(trimethylammonium) nonachlorodiantimonate(III) at 125 K. J. Solid State Chem. 2004, 177, 3202–3211. [Google Scholar] [CrossRef]
- Rhaiem, T.B.; Elleuch, S.; Boughzala, H.; Abid, Y. Synthesis, crystal structure, vibrational and optical properties of chlorometalate hybrids incorporating (DABCOH2)2+ cations. Inorg. Chem. Commun. 2019, 105, 230–239. [Google Scholar] [CrossRef]
- Olivier-Fourcade, J.; Lbanez, A.; Jumas, J.C.; Maurin, M.; Lefebvre, I.; Lippens, P.; Lannoo, M.; Allan, G. Chemical Bonding and Electronic Properties in Antimony Chalcogenides. J. Solid State Chem. 1990, 87, 366–377. [Google Scholar] [CrossRef]
- Wang, X.; Liebau, F. Influence of polyhedron distortions on calculated bond-valence sums for cations with one lone electron pair. Acta Crystallogr. B 2007, 63, 216–228. [Google Scholar] [CrossRef]
- Wheeler, R.A.; Kumar, P.N.V.P. Stereochemically Active or Inactive Lone Pair Electrons in Some Six-Coordinate, Group 15 Halides. J. Am. Chem. Soc. 1992, 114, 4776–4784. [Google Scholar] [CrossRef]
- Gillespie, R.J.; Nyholm, R.S. Inorganic stereochemistry. Q. Rev. Chem. Soc. 1957, 11, 339–380. [Google Scholar] [CrossRef]
- Gillespie, R.J. Fifty years of the VSEPR model. Coord. Chem. Rev. 2008, 252, 1315–1327. [Google Scholar] [CrossRef]
- Brown, I.D. The Chemical Bond in Inorganic Chemistry: The Bond Valence Model; Oxford University Press: New York, NY, USA, 2002; pp. 90–98. [Google Scholar]
- Brown, I.D. Recent Developments in the Methods and Applications of the Bond Valence Model. Chem. Rev. 2009, 109, 6858–6919. [Google Scholar] [CrossRef] [Green Version]
- Bujak, M.; Zaleski, J. Dependence of the Distortion of the Square Pyramids in N,N-Dimethylethylenediammonium Pentachloroantimonate(III) on the Geometry of Hydrogen Bonds. Z. Naturforsch. 2001, 56, 521–525. [Google Scholar] [CrossRef]
- Bujak, M.; Osadczuk, P.; Zaleski, J. Aminoguanidinium(2+) aminoguanidinium(1+) hexachloroantimonate(III) at 295 and 92 K. Acta Crystallogr. C 2001, 57, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Bujak, M. Primary- and secondary-octahedral distortion factors in bis(1,4-H2-1,2,4-triazolium) pentabromidoantimonate(III)–1,4-H2-1,2,4-triazolium bromide. Polyhedron 2015, 85, 499–505. [Google Scholar] [CrossRef]
- Bujak, M. Formation and distortion of iodidoantimonates(III): The first isolated [SbI6]3– octahedron. Acta Crystallogr. B 2017, 73, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Bator, G.; Jakubas, R.; Zaleski, J.; Mróz, J. Crystal structure and dielectric relaxation studies of the [N(CH3)3H]3Sb2(1–x)Bi2xCl9 mixed crystals. J. Appl. Phys. 2000, 88, 1015–1023. [Google Scholar] [CrossRef]
- Wojtaś, M.; Bator, G.; Jakubas, R.; Zaleski, J. Crystal structure, phase transition and ferroelectric properties of the [(CH3)3NH]3[Sb2Cl9(1−x)Br9x] (TMACBA) mixed crystals. J. Phys. Condens. Matter 2003, 15, 5765–5781. [Google Scholar] [CrossRef]
- Bujak, M.; Angel, R.J. Single crystal X-ray diffraction studies on [(CH3)nNH4–n]3[Sb2Cl9] (n = 2, 3) chloroantimonates(III) in their low-temperature ferroelectric phases—structures and phase transitions. J. Solid State Chem. 2005, 178, 2237–2246. [Google Scholar] [CrossRef]
- Saparov, B.; Mitzi, D.B. Organic−Inorganic Perovskites: Structural Versatility for Functional Materials Design. Chem. Rev. 2016, 116, 4558–4596. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, Z.; Zhang, J.; Han, S.; Ji, C.; Li, L.; Hong, M.; Luo, J. Thermochromism to tune the optical bandgap of a lead-free perovskite-type hybrid semiconductor for efficiently enhancing photocurrent generation. J. Mater. Chem. C 2017, 5, 9967–9971. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, Z.; Tao, L.; Shen, N.; Hu, B.; Gong, L.; Li, J.; Chen, X.; Huang, X. Hybrid Chloroantimonates(III): Thermally Induced Triple-Mode Reversible Luminescent Switching and Laser-Printable Rewritable Luminescent Paper. Angew. Chem. Int. Ed. 2019, 58, 9974–9978. [Google Scholar] [CrossRef]
- Jakubas, R. Ferroelectric phase transition in tris(dimethylammonium) nonachlorodiantimonate (III), [NH2(CH3)2]3Sb2Cl9. Solid State Commun. 1986, 60, 389–391. [Google Scholar] [CrossRef]
- Gdaniec, M.; Kostrukiewicz, Z.; Jakubas, R.; Sobczyk, L. Structure and mechanism of ferroelectric phase transition in tris(dimethylammonium) nonachlorodiantimonate (III). Ferroelectrics 1988, 77, 31–37. [Google Scholar] [CrossRef]
- Zaleski, J.; Pietraszko, A. Structure at 200 and 298 K and X-ray Investigations of the Phase Transition at 242 K of [NH2(CH3)2]3Sb2Cl9 (DMACA). Acta Crystallogr. B 1996, 52, 287–295. [Google Scholar] [CrossRef]
- Jakubas, R.; Czapla, Z.; Galewski, Z.; Sobczyk, L. Ferroelectric phase transition in [(CH3)3NH]3Sb2Cl9 (TMACA). Ferroelectr. Lett. 1986, 5, 143–148. [Google Scholar] [CrossRef]
- Kruger, F.-J.; Zettler, F.; Schmidt, A. Tri- und TetraMethylammonium-chloroantimonate(III). Z. Anorg. Allg. Chem. 1979, 449, 135–144. [Google Scholar] [CrossRef]
- Kallel, A.; Bats, J.W. Tris(trimethylammonium) Nonachlorodiantimonate(III), [NH(CH3)3]3[Sb2Cl9]. Acta Crystallogr. C 1985, 41, 1022–1024. [Google Scholar] [CrossRef]
- Tomaszewski, P.E. X-ray study of ferroelectric phase transition in (NH(CH3)3)3Sb2Cl9. Acta Phys. Pol. A 1995, 87, 631–634. [Google Scholar] [CrossRef]
- Bujak, M.; Zaleski, J. High temperature ferro-paraelectric phase transition in tris(trimethylammonium) nonachlorodiantimonate(III) (TMACA) studied by X-ray diffraction method. Cryst. Eng. 2001, 4, 241–252. [Google Scholar] [CrossRef]
- Sghaier, M.O.M.; Holderna-Natkaniec, K.; Czarnecki, P.; Wozniak-Braszak, A.; Chaabouni, S. Structure and internal dynamics of bis(piperazine-1,4-diium) pentachloroantimonate (III) monohydrate. Polyhedron 2014, 79, 37–42. [Google Scholar] [CrossRef]
- Moskwa, M.; Bator, G.; Piecha-Bisiorek, A.; Jakubas, R.; Medycki, W.; Ciżman, A.; Baran, J. X-ray structure and investigation of molecular motions by dielectric, vibrational and 1H NMR methods for two organic-inorganic hybrid piperazinium compounds: (C4H12N2)2[Sb2Cl10]·2H2O and (C4H12N2)2[Sb2Br10]·2H2O. Mater. Res. Bull. 2018, 104, 202–211. [Google Scholar] [CrossRef]
- Bujak, M. Efficient Diffusion-Controlled Ligand Exchange Crystal Growth of Isostructural Inorganic−Organic Halogenidorhodates(III): The Missing Hexaiodidorhodate(III) Anion. Cryst. Growth Des. 2015, 15, 1295–1302. [Google Scholar] [CrossRef]
- Bajpai, A.; Scott, H.S.; Pham, T.; Chen, K.-J.; Space, B.; Lusi, M.; Perry, M.L.; Zaworotko, M.J. Towards an understanding of the propensity for crystalline hydrate formation by molecular compounds. IUCrJ 2016, 3, 430–439. [Google Scholar] [CrossRef]
- Kálmán, A.; Argay, G.; Scharfenberg-Pfeiffer, D.; Höhne, E.; Ribár, B. ‘Main-Part’ Isostructuralism of Several Cardenolides and Bufadienolides. Structures of Three Cardenolides: (21S)-Methyldigitoxigenin, Uzarigenin and Sarmentogenin Methanol Solvate. Acta Crystallogr. B 1991, 47, 68–77. [Google Scholar] [CrossRef]
- Fábián, L.; Kálmán, A. Volumetric measure of isostructurality. Acta Crystallogr. B 1999, 55, 1099–1108. [Google Scholar] [CrossRef]
- Kubicki, M.; Szafrański, M. Hydrogen bonding in two isomorphous bis-guanidinium salts: Tetrachlorozincate(II) and tetrabromozincate(II). J. Mol. Struct. 1998, 446, 1–9. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallogr. B 2002, 58, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Bujak, M.; Zaleski, J. The Crystal Structure of Bis(ethylammonium) Pentachloroantimonate(III)-ethylammonium Chloride (C2H5NH3)2SbCl5·(C2H5NH3)Cl at 295 and 90 K. On the Deformation of the Octahedral Coordination of SbIII. Pol. J. Chem. 1999, 73, 773–782. [Google Scholar]
- Harris, R.K.; Spragg, R.A. Nuclear Magnetic Resonance Studies of Six-membered Rings. Part I. Ring Inversion in Heterocyclic Compounds. J. Chem. Soc. B 1968, 684–691. [Google Scholar] [CrossRef]
- Krueger, P.J.; Jan, J. Conformational equilibria in some cyclic imines: NH and CH stretching vibrations and the axial lone pair. Can. J. Chem. 1970, 48, 3236–3248. [Google Scholar] [CrossRef]
- Parkin, A.; Oswald, I.D.H.; Parsons, S. Structures of piperazine, piperidine and morpholine. Acta Crystallogr. B 2004, 60, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Rérat, C. Structure Cristalline du Bichlorhydrate de Pipérazine Monohydraté. Acta Crystallogr. 1960, 13, 459–468. [Google Scholar] [CrossRef]
- Dehghanpour, S.; Mahmoudkhani, A.H.; Langer, V. Redetermination of piperazine dihydrochloride monohydrate. Acta Crystallogr. E 2001, 57, o610–o611. [Google Scholar] [CrossRef]
- Baisch, U.; Rubini, K.; Braga, D. Remarkable structural similarities between organic co-crystals and a metal–organic coordination network—insights into hydrogen bonded aliphatic ammonium chlorides. CrystEngComm 2008, 10, 1939–1947. [Google Scholar] [CrossRef]
- Reiss, G.J.; Bajorat, S. CSD Communication (Private Communication, CCDC 769156, Refcode: UJONEN); The Cambridge Crystallographic Data Centre: Cambridge, UK, 2010. [Google Scholar]
- Flatken, M.A.; Huber, M.J.; Reiss, G.J. The layered crystal structure of piperazine-1,4-diium diiodide–diiodine (1:1), C4H12I4N2. Z. Kristallogr. NCS 2014, 229, 423–424. [Google Scholar] [CrossRef]
- Landrum, G.A.; Hoffmann, R. Secondary Bonding between Chalcogens or Pnicogens and Halogens. Angew. Chem. Int. Ed. 1998, 37, 1887–1890. [Google Scholar] [CrossRef]
- Anderson, K.M.; Orpen, A.G. On the relative magnitudes of cis and trans influences in metal complexes. Chem. Commun. 2001, 2682–2683. [Google Scholar] [CrossRef]
- Bujak, M.; Zaleski, J. Crystal and Molecular Structure of 1,2,4-Triazolium Chloride and its Salt with Antimony Trichloride-Bis(1,2,4-triazolium) pentachloroantimonate(III)-1,2,4-triazolium Chloride. Z. Naturforsch. 2002, 57, 157–164. [Google Scholar] [CrossRef]
- Sheu, H.-L.; Laane, J. Trans. Effect in Halobismuthates and Haloantimonates Revisited. Molecular Structures and Vibrations from Theoretical Calculations. Inorg. Chem. 2013, 52, 4244–4249. [Google Scholar] [CrossRef]
- Podesta, T.J.; Orpen, A.G. Tris(Pyridinium)Triazine in Crystal Synthesis of 3-Fold Symmetric Structures. Cryst. Growth Des. 2005, 5, 681–693. [Google Scholar] [CrossRef]
- Mirochnik, A.G.; Udovenko, A.A.; Storozhuk, T.V.; Karasev, V.E.; Bukvetskii, B.V. Synthesis and Luminescence of Arsenic(III) and Antimony(III) Halide Complexes with N,N’-Diphenylguanidine. Crystal Structures of Tris(N,N’-diphenylguanidinium) Hexachloro- and Hexabromoarsenates(III) and -Antimonates(III). Russ. J. Inorg. Chem. 2003, 48, 961–971. [Google Scholar]
- Pauling, L. The Nature of the Chemical Bond, 3rd ed.; Cornell University Press: Ithaca, NY, USA, 1960; pp. 255–256. [Google Scholar]
- Hall, M.; Nunn, M.; Beglev, M.J.; Sowerby, D.B. Nonahalogenodiantimon(III)ates; Their Preparation and the Crystal Structures of [Hpy]3[Sb2Cl9], [NMe4]3[Sb2Br9], and [NMe4]3[Sb2Br3Cl6]. J. Chem. Soc. Dalton Trans. 1986, 1231–1238. [Google Scholar] [CrossRef]
- Robinson, K.; Gibbs, G.V.; Ribbe, P.H. Quadratic Elongation: A Quantitative Measure of Distortion in Coordination Polyhedra. Science 1971, 172, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Brown, I.D.; Shannon, R.D. Empirical Bond-Strength-Bond-Length Curves for Oxides. Acta Crystallogr. A 1973, 29, 266–282. [Google Scholar] [CrossRef]
- Vezzosi, I.M.; Battaglia, L.P.; Corradi, A.B. The Crystal and Molecular Structure of Diethylenetriammonium Hexachloroantimonate(III). Inorg. Chim. Acta 1984, 89, 151–155. [Google Scholar] [CrossRef]
- Knödler, F.; Ensinger, U.; Schwarz, W.; Schmidt, A. Dimethylammonium-chloroantimonate. Struktur und Schwingungsspektren. Z. Anorg. Allg. Chem. 1988, 557, 208–218. [Google Scholar] [CrossRef]
- Piecha, A.; Gągor, A.; Pietraszko, A.; Jakubas, R. Unprecedented solid-state chemical reaction—from (C3N2H5)3SbBr6·H2O to (C3N2H5)5Sb2Br11. From centrosymmetric to non-centrosymmetric crystal structure. J. Solid State Chem. 2010, 183, 3058–3066. [Google Scholar] [CrossRef]
- Wolff, S.K.; Grimwood, D.J.; McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer 2.0.; University of Western Australia: Perth, Australia, 2007. [Google Scholar]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr. B 2004, 60, 627–668. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J.; Jayatilaka, D. Electrostatic potentials mapped on Hirshfeld surfaces provide direct insight into intermolecular interactions in crystals. CrystEngComm 2008, 10, 377–388. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- CrysAlis CCD, Version 171.33.57; Oxford Diffraction Ltd.: Yarnton, UK, 2010.
- CrysAlisPro, Version 1.171.39.15e; Rigaku Oxford Diffraction, Rigaku Corporation: The Woodlands, TX, USA, 2015.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 1 | 2 | 2 |
|---|---|---|---|---|
| T (K) | 295(2) | 85.0(5) | 295(2) | 85.0(5) |
| Formula | C4H14Cl5N2OSb | C4H14Cl5N2OSb | C4H14Br5N2OSb | C4H14Br5N2OSb |
| Mr | 405.18 | 405.18 | 627.43 | 627.43 |
| Crystal system | monoclinic | monoclinic | monoclinic | monoclinic |
| Space group, Z | P21/n, 4 | P21/n, 4 | P21/n, 4 | P21/n, 4 |
| a (Å) | 9.54078(13) | 9.45434(13) | 9.9162(3) | 9.82225(15) |
| b (Å) | 14.14260(19) | 14.05399(19) | 14.4090(4) | 14.34740(19) |
| c (Å) | 10.03963(14) | 9.91376(13) | 10.3898(3) | 10.29729(16) |
| β (°) | 99.1119(13) | 99.2085(13) | 99.432(3) | 99.7664(15) |
| V (Å3) | 1337.57(3) | 1300.28(3) | 1464.45(7) | 1430.10(4) |
| ρcalc (g/cm3) | 2.012 | 2.070 | 2.846 | 2.914 |
| R1, I > 2σ(I) | 0.0212 | 0.0140 | 0.0252 | 0.0183 |
| wR2, all data | 0.0525 | 0.0327 | 0.0500 | 0.0368 |
| Compound | 1 | 1 | 2 | 2 |
|---|---|---|---|---|
| T (K) | 295 | 85 | 295 | 85 |
| Sb1–X1 | 2.3914(5) | 2.3936(4) | 2.5687(5) | 2.5732(3) |
| Sb1–X2 | 2.9705(6) | 2.9468(4) | 3.0884(5) | 3.0674(3) |
| Sb1–X2I | 3.2308(6) | 3.2291(4) | 3.2537(5) | 3.2528(3) |
| Sb1–X3 | 2.4433(6) | 2.4523(4) | 2.6090(5) | 2.6247(3) |
| Sb1–X4 | 2.5246(6) | 2.5247(4) | 2.7049(5) | 2.7080(3) |
| Sb1–X5 | 2.7350(6) | 2.7270(4) | 2.8731(5) | 2.8697(3) |
| Sb1···Sb1I | 4.0832(2) | 4.0099(2) | 4.3962(5) | 4.3428(3) |
| X1–Sb1–X2 | 85.66(2) | 85.073(12) | 87.479(14) | 87.133(9) |
| X1–Sb1–X2I | 175.28(2) | 174.247(12) | 176.077(16) | 175.030(11) |
| X1–Sb1–X3 | 91.13(3) | 91.066(14) | 92.652(17) | 92.467(11) |
| X1–Sb1–X4 | 90.50(2) | 90.290(13) | 91.154(15) | 91.122(10) |
| X1–Sb1–X5 | 85.66(2) | 84.948(12) | 86.920(14) | 86.227(10) |
| X2–Sb1–X2I | 97.753(14) | 99.165(9) | 92.277(11) | 93.245(8) |
| X2–Sb1–X3 | 176.70(2) | 176.075(12) | 177.960(15) | 177.964(11) |
| X2–Sb1–X4 | 89.783(15) | 89.840(11) | 90.061(12) | 90.183(9) |
| X2–Sb1–X5 | 89.695(15) | 89.388(10) | 87.614(12) | 87.392(8) |
| X2I–Sb1–X3 | 85.42(2) | 84.640(12) | 87.453(14) | 86.981(9) |
| X2I–Sb1–X4 | 92.771(17) | 93.596(11) | 92.762(12) | 93.832(9) |
| X2I–Sb1–X5 | 91.083(15) | 91.180(10) | 89.157(12) | 88.839(8) |
| X3–Sb1–X4 | 90.981(18) | 90.886(12) | 91.971(14) | 91.820(10) |
| X3–Sb1–X5 | 89.329(18) | 89.570(12) | 90.360(14) | 90.591(10) |
| X4–Sb1–X5 | 176.147(17) | 175.224(12) | 177.043(13) | 176.492(10) |
| Sb1–X2–Sb1I | 82.247(14) | 80.835(9) | 87.723(11) | 86.755(8) |
| Atoms | D–H | H∙∙∙A | D∙∙∙A | D–H∙∙∙A |
|---|---|---|---|---|
| 1, 295 K | ||||
| O1–H11···Cl1I | 0.85(1) | 2.95(3) | 3.449(2) | 119(3) |
| O1–H12···Cl3 | 0.85(1) | 2.71(3) | 3.414(3) | 140(4) |
| O1–H11···Cl5II | 0.85(1) | 2.70(2) | 3.399(3) | 141(3) |
| N11–H112···Cl2 | 0.89(1) | 2.35(2) | 3.201(2) | 163(2) |
| N11–H111···Cl5III | 0.89(1) | 2.76(2) | 3.3444(19) | 125(2) |
| N12–H122···Cl2IV | 0.90(1) | 2.38(2) | 3.256(2) | 165(2) |
| N12–H121···Cl5V | 0.89(1) | 2.51(2) | 3.2606(19) | 143(2) |
| C13–H132···Cl4 | 0.96(1) | 2.90(2) | 3.555(2) | 126(2) |
| C14–H141···Cl3V | 0.97(1) | 2.79(2) | 3.659(2) | 150(2) |
| C14–H142···Cl3VI | 0.96(1) | 2.84(2) | 3.675(3) | 146(2) |
| N11–H111···O1VII | 0.89(1) | 2.19(2) | 2.943(3) | 143(2) |
| 1, 85 K | ||||
| O1–H11···Cl1I | 0.85(1) | 2.80(2) | 3.4198(14) | 131(2) |
| O1–H12···Cl3 | 0.85(1) | 2.57(1) | 3.3512(15) | 155(2) |
| O1–H11···Cl5II | 0.85(1) | 2.67(2) | 3.3493(14) | 138(2) |
| N11–H112···Cl2 | 0.89(1) | 2.34(1) | 3.1859(14) | 160(2) |
| N11–H111···Cl5III | 0.90(1) | 2.76(2) | 3.3024(14) | 121(1) |
| N12–H122···Cl2IV | 0.90(1) | 2.38(1) | 3.2316(14) | 158(2) |
| N12–H121···Cl5V | 0.89(1) | 2.49(1) | 3.2287(13) | 140(2) |
| C13–H132···Cl4 | 0.96(1) | 2.86(2) | 3.5277(15) | 128(1) |
| C14–H141···Cl3V | 0.96(1) | 2.77(1) | 3.6256(16) | 148(1) |
| C14–H142···Cl3VI | 0.96(1) | 2.80(1) | 3.6358(16) | 145(1) |
| N11–H111···O1VII | 0.90(1) | 2.09(1) | 2.8926(19) | 148(2) |
| 2, 295 K | ||||
| O1–H11···Br1I | 0.85(1) | 2.92(3) | 3.592(4) | 137(4) |
| O1–H12···Br3 | 0.85(1) | 2.67(2) | 3.505(4) | 169(4) |
| O1–H11···Br5II | 0.85(1) | 3.07(2) | 3.543(4) | 117(2) |
| N11–H112···Br2 | 0.90(1) | 2.56(2) | 3.374(4) | 151(3) |
| N11–H111···Br5III | 0.90(1) | 2.90(3) | 3.480(3) | 124(3) |
| N12–H122···Br2IV | 0.90(1) | 2.61(2) | 3.473(3) | 159(3) |
| N12–H121···Br5V | 0.90(1) | 2.78(3) | 3.441(3) | 132(3) |
| C13–H132···Br4 | 0.97(1) | 2.94(3) | 3.685(4) | 134(3) |
| C14–H141···Br3V | 0.97(1) | 2.87(2) | 3.779(4) | 157(3) |
| C14–H142···Br3VI | 0.97(1) | 2.95(2) | 3.787(4) | 146(3) |
| N11–H111···O1VII | 0.90(1) | 2.17(3) | 2.939(5) | 142(3) |
| 2, 85 K | ||||
| O1–H11···Br1I | 0.85(1) | 2.93(2) | 3.551(2) | 132(3) |
| O1–H12···Br3 | 0.85(1) | 2.61(2) | 3.433(2) | 165(4) |
| O1–H11···Br5II | 0.85(1) | 2.99(2) | 3.502(3) | 121(2) |
| N11–H112···Br2 | 0.90(1) | 2.51(2) | 3.358(2) | 157(3) |
| N11–H111···Br5III | 0.90(1) | 2.96(3) | 3.443(2) | 115(2) |
| N12–H122···Br2IV | 0.91(1) | 2.58(2) | 3.437(2) | 158(3) |
| N12–H121···Br5V | 0.90(1) | 2.75(2) | 3.410(2) | 131(2) |
| C13–H132···Br4 | 0.98(1) | 2.93(2) | 3.657(3) | 133(2) |
| C14–H141···Br3V | 0.97(1) | 2.87(2) | 3.746(3) | 152(2) |
| C14–H142···Br3VI | 0.97(1) | 2.92(2) | 3.743(3) | 143(2) |
| N11–H111···O1VII | 0.90(1) | 2.08(2) | 2.896(3) | 151(3) |
| Compound | 1 | 1 | 2 | 2 |
|---|---|---|---|---|
| Atoms/T (K) | 295 | 85 | 295 | 85 |
| X1 | –0.15 | –0.16 | –0.20 | –0.21 |
| X2 | –0.65 | –0.64 | –0.54 | –0.53 |
| X2I | –0.65 | –0.64 | –0.54 | –0.53 |
| X3 | –0.25 | –0.26 | –0.27 | –0.29 |
| X4 | –0.38 | –0.38 | –0.40 | –0.40 |
| X5 | –0.62 | –0.61 | –0.58 | –0.58 |
| Parameter | Bond Length Distortion, Δ × 103 | Bond Angle Distortion, σ2 | ||
|---|---|---|---|---|
| Compound/T (K) | 295 | 85 | 295 | 85 |
| 1 | 12.35 | 11.93 | 11.88 | 16.31 |
| 2 | 7.77 | 7.34 | 4.90 | 6.91 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bujak, M.; Siodłak, D. Isostructural Inorganic–Organic Piperazine-1,4-diium Chlorido- and Bromidoantimonate(III) Monohydrates: Octahedral Distortions and Hydrogen Bonds. Molecules 2020, 25, 1361. https://doi.org/10.3390/molecules25061361
Bujak M, Siodłak D. Isostructural Inorganic–Organic Piperazine-1,4-diium Chlorido- and Bromidoantimonate(III) Monohydrates: Octahedral Distortions and Hydrogen Bonds. Molecules. 2020; 25(6):1361. https://doi.org/10.3390/molecules25061361
Chicago/Turabian StyleBujak, Maciej, and Dawid Siodłak. 2020. "Isostructural Inorganic–Organic Piperazine-1,4-diium Chlorido- and Bromidoantimonate(III) Monohydrates: Octahedral Distortions and Hydrogen Bonds" Molecules 25, no. 6: 1361. https://doi.org/10.3390/molecules25061361
APA StyleBujak, M., & Siodłak, D. (2020). Isostructural Inorganic–Organic Piperazine-1,4-diium Chlorido- and Bromidoantimonate(III) Monohydrates: Octahedral Distortions and Hydrogen Bonds. Molecules, 25(6), 1361. https://doi.org/10.3390/molecules25061361