In Situ Assessment of Intrinsic Strength of X-I⋯OA-Type Halogen Bonds in Molecular Crystals with Periodic Local Vibrational Mode Theory


 , , and
, , and 
Abstract
:
1. Introduction
- The binding energy is a reaction parameter, summarizing all effects leading to bonding in a cumulative way. Even in a simple dimer the binding energy cannot serve as a measure for the intrinsic strength of a bond; it is contaminated with the stabilization energy of the two fragments caused by geometry relaxation and reorganization of the electron density of the fragments upon bond breakage [36]. This applies even more to complex systems with halogen bonding at work (e.g., a halogenated drug in a protein or halogen bonding in crystals);
- We need a bond strength measure that is not based on bond breaking and that follows Levine’s suggestion that chemistry is local [37].
- The I⋯O halogen bonds account for a large portion of all halogen bonds ever discovered;
- The oxygen acceptor atom is more common in molecular crystals than the higher chalcogens (e.g., S, Se and Te);
- Dihalogen/interhalogen compounds X-I consist of only two atoms and therefore are the simplest halogen bond donors;
- Recently, Rosokha and co-workers investigated the I-I⋯O-N-type halogen bonding in crystals with N-oxide acceptors via crystallography and theoretical calculations [96,97]. Their analysis based on molecular dimer models suggests that the charge transfer is a key factor in the I⋯O halogen bonding besides electrostatic attraction. Our work here based on a collection of crystal structures should provide a more detailed and comprehensive understanding of halogen bonding in materials.
- To create a comprehensive set of local stretching force constants for X-I⋯OA halogen bonds in different crystals describing the intrinsic halogen bond strength in these systems;
- To derive a more realistic model description considering the crystal packing effect explicitly and to understand factors that affect the solid state halogen bond strength by an in situ investigation of halogen bonding in a crystalline environment;
2. Computational Details
3. Results and Discussion
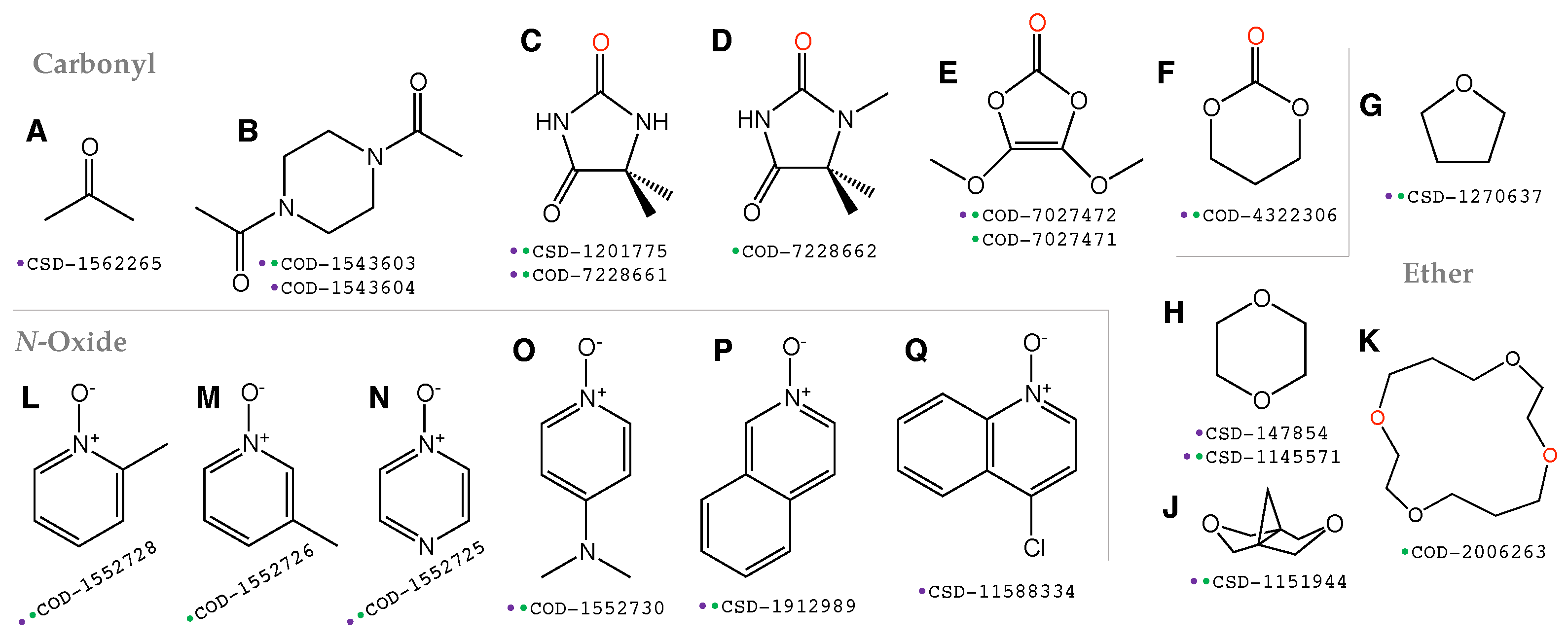
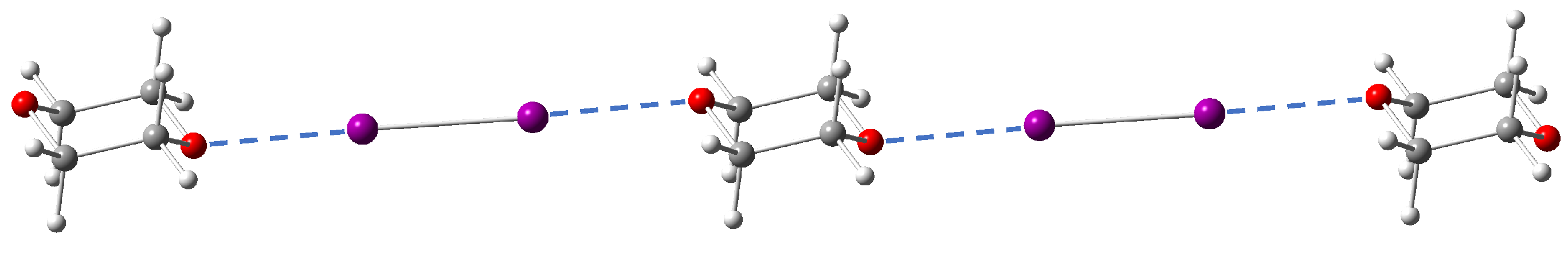
3.1. Selection of Molecular Crystals
- The molecular crystal should contain only the elements C, N, H, O/S/Se and X while excluding metal atoms;
- The total number of atoms in the primitive cell of the crystal should be preferably smaller than 80 to save computational cost;
- The dihalogen/interhalogen X-I should exist as neutral diatomic molecules instead of trihalogen cations.
- Iodine as the donor atom has relatively large polarizability and will more easily form a -hole than chlorine, bromine or fluorine;
- Although stronger halogen bonding is expected for iodine monofluoride (IF) as the donor molecule, this species is unstable and cannot form co-crystals under ambient conditions [115].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Label | Donor Bond | r | Halogen Bond e | r | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| CSD-1562265 [116] | A | 24 | I-I | 2.7598 | 1.257 | I⋯O=C(C) | 2.8133 | 0.087 | ||
| B1-1 | I-I | 2.7994 | 1.049 | 2.5281 | 0.309 | |||||
| COD-1543603 [117] | B1-2 | 30 | Cl-I | 2.4685 | 2.4142 | 1.252 | 2.3731 | 2.3864 | 0.575 | |
| COD-1543604 [117] | B2 | 56 | I-I | 2.7753 | 2.7057 | 1.199 | I⋯O=C(C,N) | 2.7886 | 2.8195 | 0.143 |
| CSD-1201775 * [118] | C1-1 | 38 | I-I | 2.7715 | 1.134 | 2.5170 | 0.370 | |||
| C1-2 | Cl-I | 2.4661 | 1.225 | 2.3888 | 0.588 | |||||
| C2-1 | I-I | 2.7714 | 1.136 | 2.5195 | 0.368 | |||||
| COD-7228661 * [119] | C2-2 | 38 | Cl-I | 2.4661 | 1.224 | 2.3888 | 0.588 | |||
| COD-7228662 * [119] | D | 88 | Cl-I | 2.4447 | 1.335 | I⋯O=C(N) | 2.4616 | 0.416 | ||
| COD-7027472 * [120] | E1-1 | 36 | I-I | 2.7317 | 1.364 | 2.7340 | 0.149 | |||
| E1-2 | Cl-I | 2.4147 | 1.526 | 2.4958 | 0.412 | |||||
| COD-7027471 * [120] | E2 | 72 | Cl-I | 2.4207 | 1.513 | 2.5118 | 0.374 | |||
| COD-4322306 * [121] | F-1 | 60 | I-I | 2.7667 | 1.146 | 2.6585 | 0.159 | |||
| F-2 | Cl-I | 2.4426 | 1.374 | I⋯O=C(O) | 2.4469 | 0.461 | ||||
| CSD-1270637 * [122] | G-1 | 60 | I-I | 2.7724 | 1.176 | 2.5575 | 0.321 | |||
| G-2 | Cl-I | 2.4553 | 1.326 | 2.4145 | 0.561 | |||||
| CSD-147854 [123] | H1 | 16 | I-I | 2.7685 | 2.6926 | 1.229 | 2.7565 | 2.8078 | 0.203 | |
| CSD-1145571 * [124] | H2-1 | 36 | I-I | 2.7718 | 1.115 | 2.6949 | 0.212 | |||
| H2-2 | Cl-I | 2.4277 | 1.454 | 2.5125 | 0.409 | |||||
| J-1a | 2.7790 | 1.090 | 2.6410 | 0.236 | ||||||
| J-1b | I-I | 2.7745 | 1.123 | 2.6347 | 0.258 | |||||
| J-2a | 2.4493 | 1.330 | 2.4529 | 0.501 | ||||||
| CSD-1151944 * [125] | J-2b | 46 | Cl-I | 2.4263 | 1.477 | 2.4901 | 0.436 | |||
| COD-2006263 * [126] | K | 42 | Cl-I | 2.4319 | 1.415 | I⋯O(C) | 2.5616 | 0.341 | ||
| COD-1552728 [97] | L-1 | 68 | I-I | 2.8168 | 2.7509 | 0.999 | 2.4488 | 2.4803 | 0.413 | |
| L-2 | Cl-I | 2.5022 | 1.120 | 2.3281 | 0.716 | |||||
| COD-1552726 [97] | M | 34 | Cl-I | 2.4930 | 1.114 | 2.3373 | 0.664 | |||
| COD-1552725 [97] | N-1 | 26 | I-I | 2.8073 | 2.7340 | 1.036 | 3.1606 | 3.0345 | 0.164 | |
| N-2 | Cl-I | 2.4462 | 1.361 | 2.4151 | 0.489 | |||||
| O-1 | I-I | 2.8629 | 2.7952 | 0.841 | 2.3857 | 2.3587 | 0.565 | |||
| COD-1552730 [97] | O-2 | 44 | Cl-I | 2.5446 | 0.940 | 2.3030 | 0.783 | |||
| CSD-1912989 [97] | P-1 | 40 | I-I | 2.8188 | 2.7512 | 0.966 | 2.4470 | 2.4637 | 0.435 | |
| P-2 | Cl-I | 2.5096 | 1.059 | 2.3327 | 0.681 | |||||
| CSD-1588334 [96] | Q | 40 | I-I | 2.8118 | 2.7328 | 1.003 | I⋯O-N(C) | 2.4850 | 2.4990 | 0.198 |
| Diiodine | I | 2 | I-I | 2.6919 | 2.6660 | 1.667 | ||||
| Iodine monochloride | ICl | 2 | Cl-I | 2.3413 | 2.3207 | 2.233 |
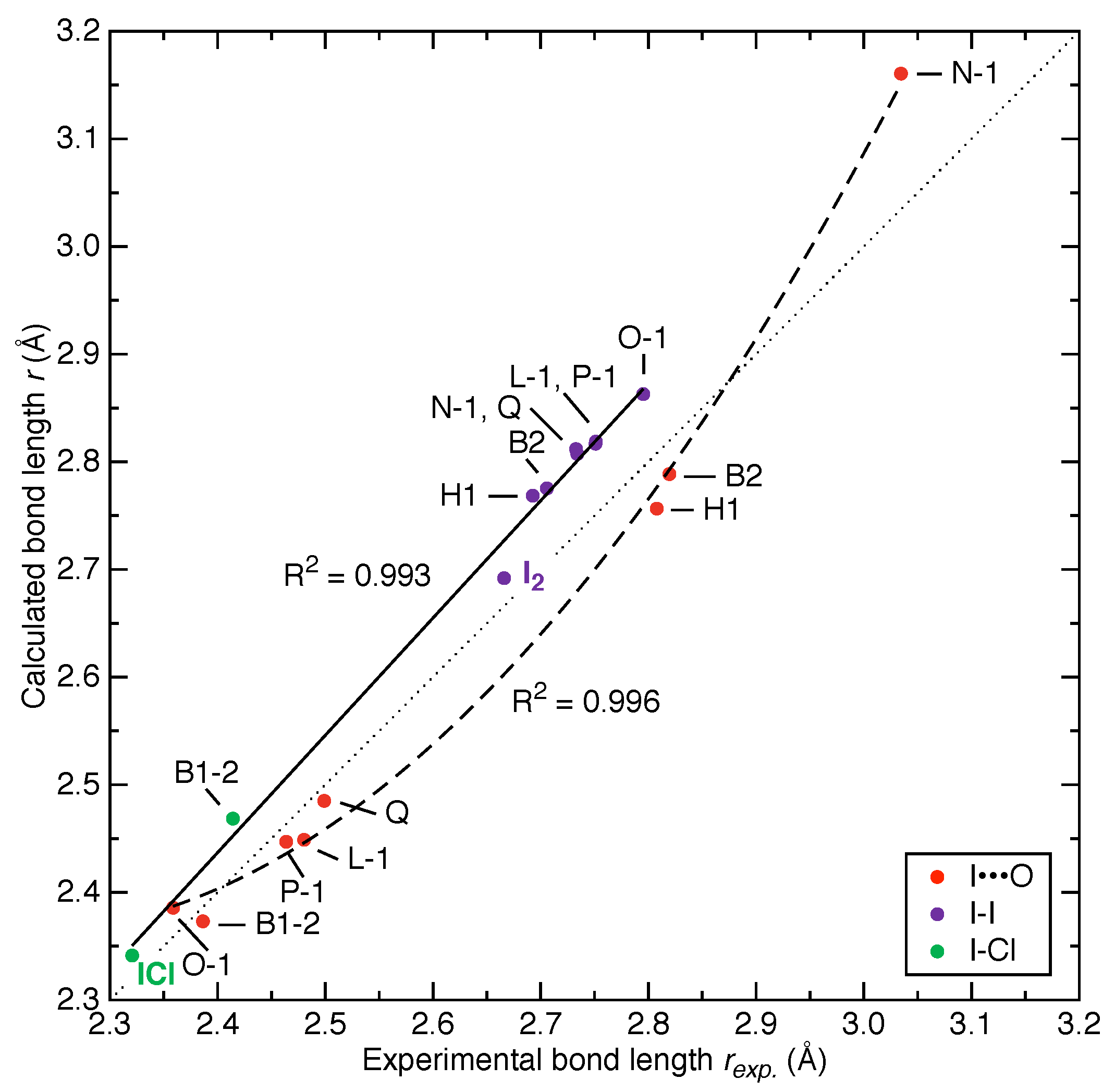
3.2. Comparison of Experimental and Calculated Structures
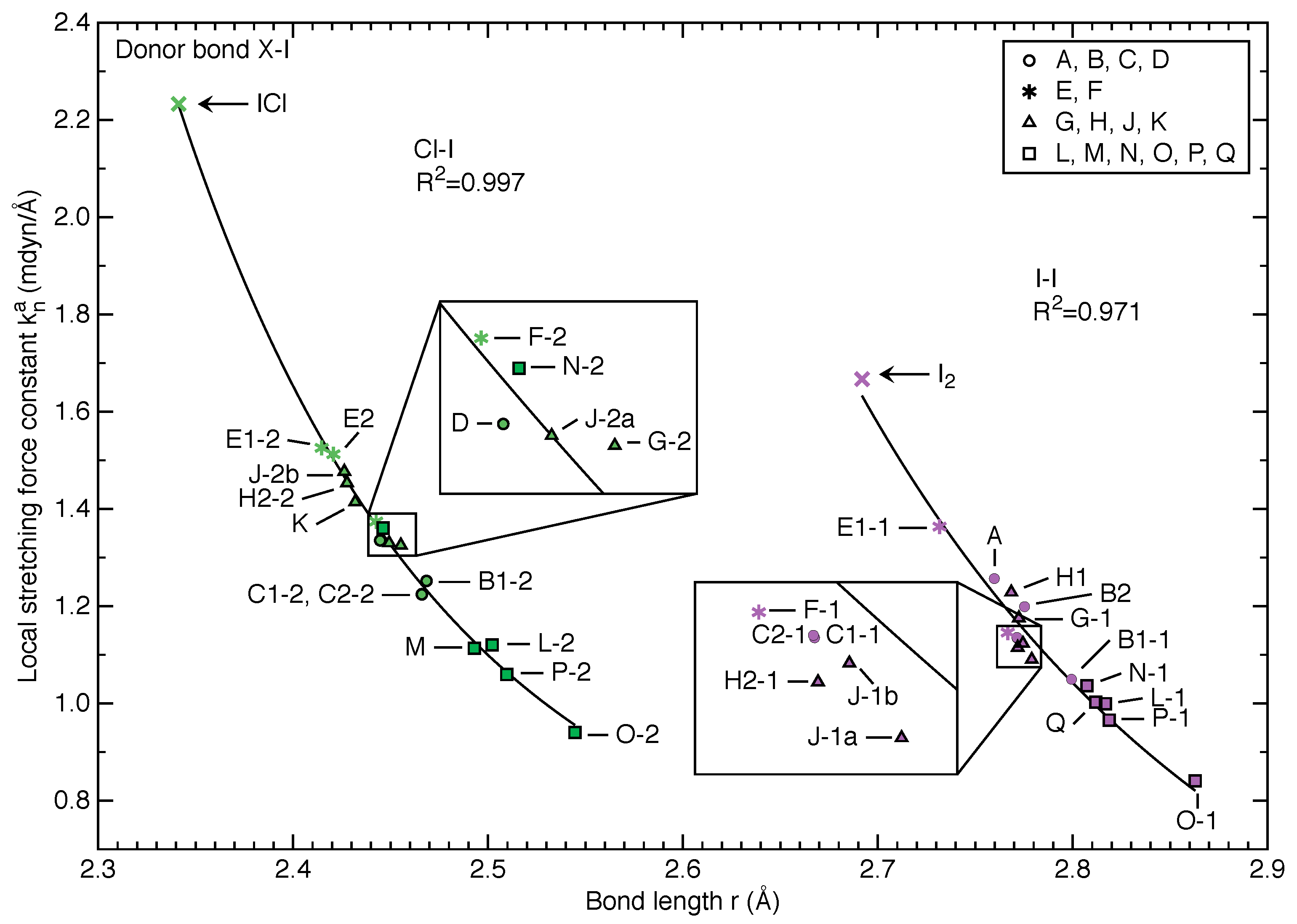
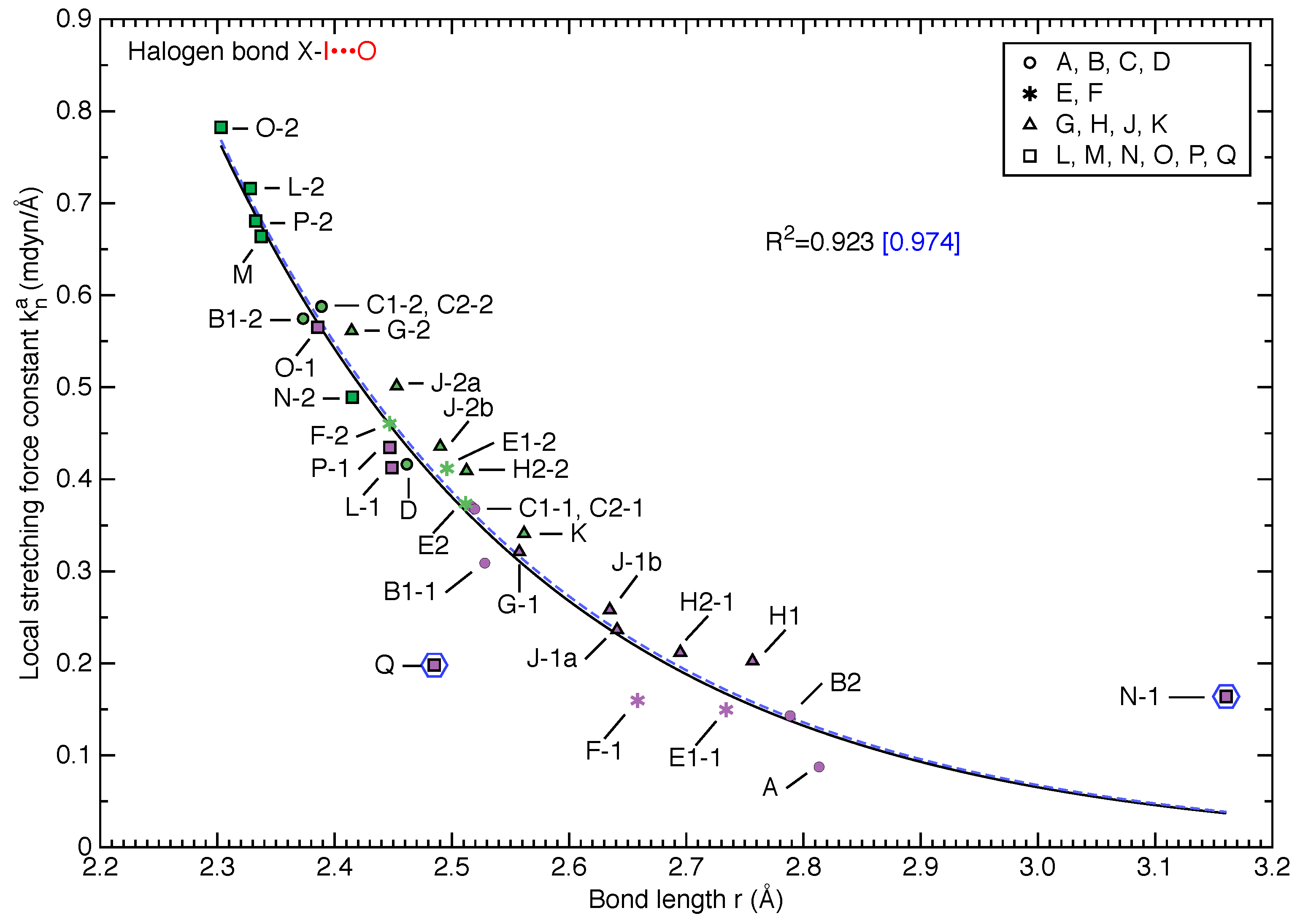
3.3. Intrinsic Strength of Donor Bonds and Halogen Bonds in X-I⋯OA Halogen Bonding
3.3.1. General Trends
3.3.2. Acceptor A–F
3.3.3. Acceptor G–K
3.3.4. Acceptor L–Q
3.3.5. Outliers
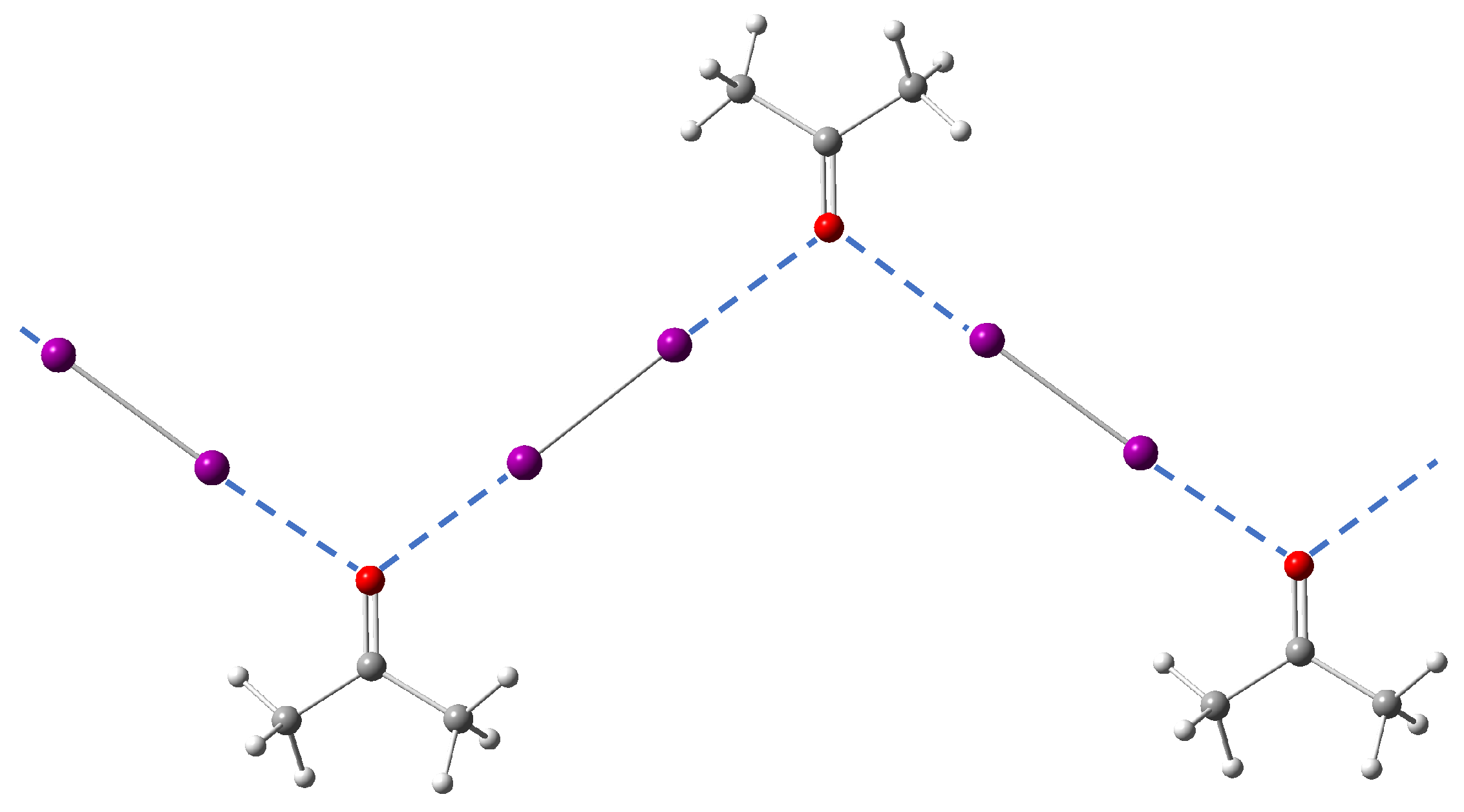
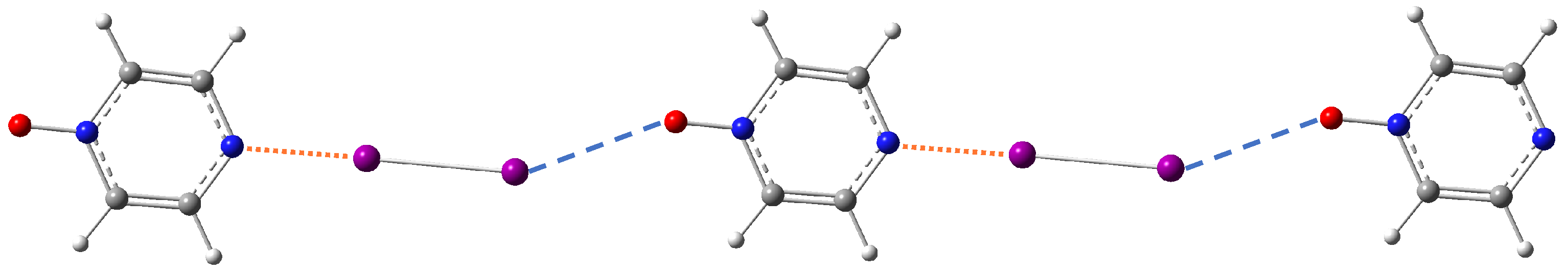
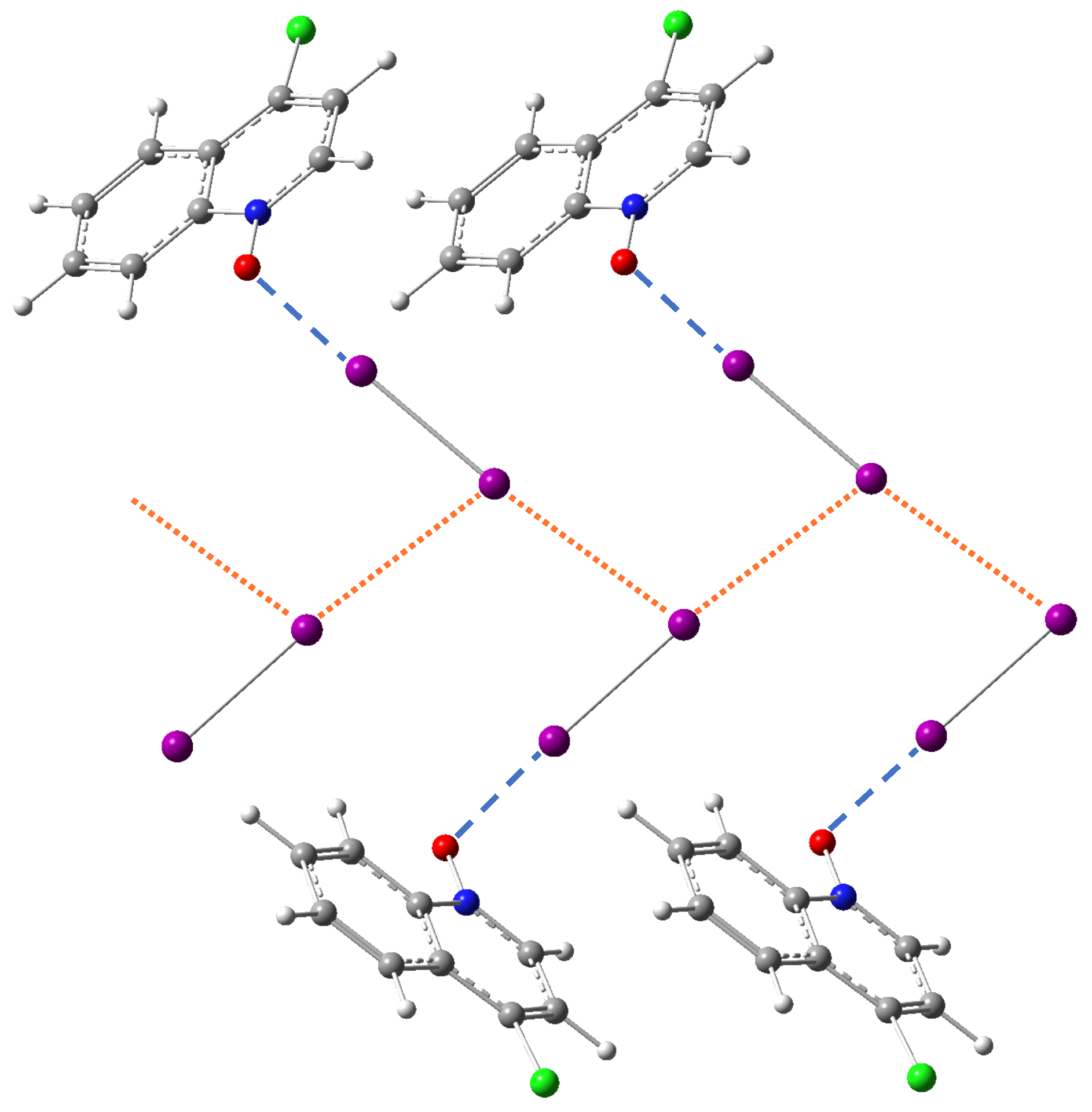
3.3.6. Crystal Packing Effect
- In the case of halogen bonding Q, reference DFT calculations on dimer models by Rosokha and co-workers have demonstrated that the acceptor molecule Q and diiodine could form a halogen bond as strong as the halogen bond between acceptor molecule P and diiodine [97]. However, in the crystal structure, the II interaction network dominates the crystal packing and it directly leads to weaker I⋯O halogen bonds than expected;
- In the case of halogen bonding N-1, the I⋯O halogen bond is weak because the molecule N is a poor acceptor. In this case, the crystal packing must enforce the structure to form a second halogen bonding for compensation to stabilize the whole system.
4. Conclusions
- We observed strong correlations between bond length and local stretching force constant for both X-I donor bonds (i.e., I-I and Cl-I) and I⋯O halogen bonds, which suggests that the generalized Badger’s rule (based on local stretching force constants [61]) originally derived from molecules is also valid for both covalent bonds and non-covalent interactions in crystals;
- The local stretching force constants for I⋯O halogen bonds (Figure 4) span a wide range of 0.1–0.8 mdyn/Å, demonstrating the impressive tunability in bond strength even within a specific type of halogen bonding;
- Our results for some I-I⋯O halogen-bonded crystals previously investigated experimentally and computationally by Rosokha and co-workers [96,97] clearly show the potential of the periodic local mode analysis leading to new deeper insights:
- (a)
- Rosokha’s statement that “besides electrostatic, molecular orbital interactions play a substantial role in XB between diiodine and N-oxides” can be expanded to the I⋯O halogen bonding between dihalogen X-I and any acceptor oxygen atom with lone pair electrons. This generalization is based on the strong correlations between bond length and force constant for both donor bonds and halogen bonds in Figure 3 and Figure 4;
- (b)
- In comparison to the dimer model systems used for DFT calculations by Rosokha and co-workers, our first-principle calculations on crystal models could include the crystal packing effects. On one hand, the overall lattice structure (including donor/halogen bond lengths) of molecular crystals is a direct result of crystal packing. On the other hand, the impact from crystal packing on X-I⋯OA halogen bonding varies in different ways:
- In those cases where the X atom of the X-I halogen donor molecule has no close contact to neighboring atoms in the crystal or is only stabilized by an interaction with the cloud of an adjacent aromatic ring, the I⋯O halogen bonds behave like covalent bonds and rigorously follow a local stretching force constant-bond length relationship;
- When both sides of X-I donor are involved in halogen bonding (only observed for I not for Cl-I), the I⋯O halogen bond is weak. If the acceptor oxygen atom has to accept two halogen bonds simultaneously (e.g., halogen bonding A), the halogen bond strength is even lower. If the X atom forms a non-trivial halogen bond which obscures the target I⋯O halogen bond (e.g., N-1 and Q), the target halogen bond largely deviates from the ideal force constant-bond length relationship.
- However, independent of the engagement of the X atom in additional halogen bonding associated with crystal packing, the donor bonds rigorously follow an ideal force constant-bond length relationship (Figure 3) due to their covalent bond nature.
- (c)
- Via delicate analysis in terms of substituent effect and other electronic structure factors in acceptor molecules, we are able to explain the majority of the variations in the intrinsic strength of both donor bonds and halogen bonds in X-I⋯OA bonding. Furthermore, the local stretching force constants of the adjacent O-A bonds in acceptor molecules could complement our findings;
- (d)
- In determining the strength of I⋯O halogen bonds, halogen atom X within X-I donor plays a decisive role as the weakest Cl-I⋯O halogen bonds are comparable to the strongest I-I⋯O halogen bonds. The acceptor molecules with different capabilities for (X-I) charge transfer are the second important factor for determining the I⋯O bond strength. Last but not least, the existence of the second halogen bonding via the X atom of the donor X-I bond can substantially weaken the target I⋯O halogen bond in crystals.
- We discovered for the first time a linear correlation for X-I donor bonds along with a quadratic correlation for I⋯O halogen bonds between experimental and calculated bond lengths. One application based on this is to estimate the local stretching force constant of either the X-I donor bond or I⋯O halogen bond of X-I⋯OA halogen bonding directly for a newly resolved crystal structure via the correlations in Figure 2, Figure 3 and Figure 4 given that no second halogen bond exists with atom X of the X-I donor molecule. Such relationship may also hold for other types of non-covalent interactions;
- All calculations in this work were based on projector-augmented wave (PAW) basis. The resulting chemically sound results of local stretching force constants demonstrate that our periodic local mode analysis is not limited to atomic orbital (AO)-based computational results [85], it is generally applicable and independent of how the wavefunction is obtained. Using periodic local vibrational mode theory as a unique tool to investigate the intrinsic strength of other types of halogen bonding (and non-covalent interactions) in crystals/materials will be one of our current and future directions.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| X | Halogen |
| ESP | Electrostatic Potential |
| SAPT | Symmetry-Adapted Perturbation Theory |
| CSD | Cambridge Structural Database |
| COD | Crystallography Open Database |
| VASP | Vienna Ab initio Simulation Package |
| PES | Potential Energy Surface |
| NBO | Natural Bond Orbital |
| DFT | Density Functional Theory |
References
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef]
- Shinada, N.K.; de Brevern, A.G.; Schmidtke, P. Halogens in Protein-Ligand Binding Mechanism: A Structural Perspective. J. Med. Chem. 2019, 62, 9341–9356. [Google Scholar] [CrossRef] [PubMed]
- Heidrich, J.; Sperl, L.E.; Boeckler, F.M. Embracing the Diversity of Halogen Bonding Motifs in Fragment-Based Drug Discovery—Construction of a Diversity-Optimized Halogen-Enriched Fragment Library. Front. Chem. 2019, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Riel, A.M.S.; Rowe, R.K.; Ho, E.N.; Carlsson, A.C.C.; Rappe, A.K.; Berryman, O.B.; Ho, P.S. Hydrogen Bond Enhanced Halogen Bonds: A Synergistic Interaction in Chemistry and Biochemistry. Acc. Chem. Res. 2019, 52, 2870–2880. [Google Scholar] [CrossRef]
- Carlsson, A.C.C.; Scholfield, M.R.; Rowe, R.K.; Ford, M.C.; Alexander, A.T.; Mehl, R.A.; Ho, P.S. Increasing Enzyme Stability and Activity through Hydrogen Bond-Enhanced Halogen Bonds. Biochemistry 2018, 57, 4135–4147. [Google Scholar] [CrossRef] [Green Version]
- Meyer, F.; Dubois, P. Halogen Bonding at Work: Recent Applications in Synthetic Chemistry and Materials Science. CrystEngComm 2013, 15, 3058–3071. [Google Scholar] [CrossRef]
- Berger, G.; Soubhye, J.; Meyer, F. Halogen Bonding in Polymer Science: From Crystal Engineering to Functional Supramolecular Polymers and Materials. Polym. Chem. 2015, 6, 3559–3580. [Google Scholar] [CrossRef]
- Saccone, M.; Catalano, L. Halogen Bonding beyond Crystals in Materials Science. J. Phys. Chem. B 2019, 123, 9281–9290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulfield, D.; Huber, S.M. Halogen Bonding in Organic Synthesis and Organocatalysis. Chem. Eur. J. 2016, 22, 14434–14450. [Google Scholar] [CrossRef] [PubMed]
- Sutar, R.L.; Huber, S.M. Catalysis of Organic Reactions through Halogen Bonding. ACS Catal. 2019, 9, 9622–9639. [Google Scholar] [CrossRef]
- Szell, P.M.; Zablotny, S.; Bryce, D.L. Halogen Bonding as A Supramolecular Dynamics Catalyst. Nat. Commun. 2019, 10, 916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bamberger, J.; Ostler, F.; Mancheño, O.G.M. Frontiers in Halogen and Chalcogen-Bond Donor Organocatalysis. ChemCatChem 2019, 11, 5198–5211. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Tothadi, S.; Desiraju, G.R. Halogen Bonds in Crystal Engineering: Like Hydrogen Bonds yet Different. Acc. Chem. Res. 2014, 47, 2514–2524. [Google Scholar] [CrossRef]
- Lisac, K.; Topić, F.; Arhangelskis, M.; Cepić, S.; Julien, P.A.; Nickels, C.W.; Morris, A.J.; Friščić, T.; Cinčić, D. Halogen-Bonded Cocrystallization with Phosphorus, Arsenic and Antimony Acceptors. Nat. Commun. 2019, 10, 61. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G.; Pilati, T.; Liantonio, R.; Meyer, F. Engineering Functional Materials by Halogen Bonding. J. Polym. Sci., Part A Polym. Chem. 2007, 45, 1–14. [Google Scholar] [CrossRef]
- Wolters, L.P.; Schyman, P.; Pavan, M.J.; Jorgensen, W.L.; Bickelhaupt, F.M.; Kozuch, S. The Many Faces of Halogen Bonding: A Review of Theoretical Models and Methods. WIREs Comput. Mol. Sci. 2014, 4, 523–540. [Google Scholar] [CrossRef]
- Kolář, M.H.; Hobza, P. Computer Modeling of Halogen Bonds and Other σ-Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef] [Green Version]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Li, J.X.; Du, Z.X. A Binuclear Cadmium(II) Cluster Based on π⋯π Stacking and Halogen⋯Halogen Interactions: Synthesis, Crystal Analysis and Fluorescent Properties. J. Clust. Sci. 2019, 31, 507–511. [Google Scholar] [CrossRef]
- Li, J.X.; Du, Z.X.; Wang, J.; Feng, X. Two Mononuclear Zinc(II) Complexes Constructed by Two Types of Phenoxyacetic Acid Ligands: Syntheses, Crystal Structures and Fluorescence Properties. Z. Naturforsch. B J. Chem. Sci. 2019, 74, 839–845. [Google Scholar] [CrossRef]
- Brinck, T.; Murray, J.S.; Politzer, P. Surface Electrostatic Potentials of Halogenated Methanes as Indicators of Directional Intermolecular Interactions. Int. J. Quantum Chem. 1992, 44, 57–64. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen Bonding: The σ-Hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen Bonding: An Electrostatically-Driven Highly Directional Noncovalent Interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Halogen Bonding: An Interim Discussion. ChemPhysChem 2013, 14, 278–294. [Google Scholar] [CrossRef]
- Glendening, E.D.; Streitwieser, A. Natural Energy Decomposition Analysis: An Energy Partitioning Procedure for Molecular Interactions with Application to Weak Hydrogen Bonding, Strong Ionic, and Moderate Donor-Acceptor Interactions. J. Chem. Phys. 1994, 100, 2900–2909. [Google Scholar] [CrossRef]
- Huber, S.M.; Jimenez-Izal, E.; Ugalde, J.M.; Infante, I. Unexpected Trends in Halogen-Bond Based Noncovalent Adducts. Chem. Commun. 2012, 48, 7708. [Google Scholar] [CrossRef]
- Mulliken, R.S. Structures of Complexes Formed by Halogen Molecules with Aromatic and with Oxygenated Solvents. J. Am. Chem. Soc. 1950, 72, 600–608. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from A Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Rosokha, S.V.; Kochi, J.K. Fresh Look at Electron-Transfer Mechanisms via the Donor/Acceptor Bindings in the Critical Encounter Complex. Acc. Chem. Res. 2008, 41, 641–653. [Google Scholar] [CrossRef]
- Rosokha, S.V.; Stern, C.L.; Ritzert, J.T. Experimental and Computational Probes of the Nature of Halogen Bonding: Complexes of Bromine-Containing Molecules with Bromide Anions. Chem. Eur. J. 2013, 19, 8774–8788. [Google Scholar] [CrossRef] [PubMed]
- Rosokha, S.V.; Stern, C.L.; Swartz, A.; Stewart, R. Halogen Bonding of Electrophilic Bromocarbons With Pseudohalide Anions. Phys. Chem. Chem. Phys. 2014, 16, 12968–12979. [Google Scholar] [CrossRef] [PubMed]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Heßelmann, A.; Jansen, G. Intermolecular Dispersion Energies from Time-Dependent Density Functional Theory. Chem. Phys. Lett. 2003, 367, 778–784. [Google Scholar] [CrossRef]
- Stone, A.J. Are Halogen Bonded Structures Electrostatically Driven? J. Am. Chem. Soc. 2013, 135, 7005–7009. [Google Scholar] [CrossRef] [PubMed]
- Cremer, D.; Kraka, E. From Molecular Vibrations to Bonding, Chemical Reactions, and Reaction Mechanism. Curr. Org. Chem. 2010, 14, 1524–1560. [Google Scholar] [CrossRef]
- Levine, R. Molecular Reaction Dynamics; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory (International Series of Monographs on Chemistry); Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Lu, Y.X.; Zou, J.W.; Wang, Y.H.; Jiang, Y.J.; Yu, Q.S. Ab Initio Investigation of the Complexes between Bromobenzene and Several Electron Donors: Some Insights into the Magnitude and Nature of Halogen Bonding Interactions. J. Phys. Chem. A 2007, 111, 10781–10788. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. Chemical Bonds without Bonding Electron Density? Does the Difference Electron-Density Analysis Suffice for a Description of the Chemical Bond? Angew. Chem. Int. Ed. 1984, 23, 627–628. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. A Description of the Chemical Bond in Terms of Local Properties of Electron Density and Energy. Croat. Chem. Acta 1984, 57, 1259–1281. [Google Scholar]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-Garcia, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D.; Edgecombe, K.E. A Simple Measure of Electron Localization in Atomic and Molecular Systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- de Silva, P.; Corminboeuf, C. Simultaneous Visualization of Covalent and Noncovalent Interactions Using Regions of Density Overlap. J. Chem. Theory Comput. 2014, 10, 3745–3756. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.C.; Contreras-García, J.; Hénon, E. Accurately Extracting the Signature of Intermolecular Interactions Present in the NCI Plot of the Reduced Density Gradient versus Electron Density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef] [PubMed]
- Pinter, B.; Nagels, N.; Herrebout, W.A.; De Proft, F. Halogen Bonding from a Hard and Soft Acids and Bases Perspective: Investigation by Using Density Functional Theory Reactivity Indices. Chem. Eur. J. 2012, 19, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, P.; Varadwaj, A.; Marques, H. Halogen Bonding: A Halogen-Centered Noncovalent Interaction Yet to Be Understood. Inorganics 2019, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Konkoli, Z.; Cremer, D. A New Way of Analyzing Vibrational Spectra. I. Derivation of Adiabatic Internal Modes. Int. J. Quantum Chem. 1998, 67, 1–9. [Google Scholar] [CrossRef]
- Konkoli, Z.; Larsson, J.A.; Cremer, D. A New Way of Analyzing Vibrational Spectra. II. Comparison of Internal Mode Frequencies. Int. J. Quantum Chem. 1998, 67, 11–27. [Google Scholar] [CrossRef]
- Konkoli, Z.; Cremer, D. A New Way of Analyzing Vibrational Spectra. III. Characterization of Normal Vibrational Modes in terms of Internal Vibrational Modes. Int. J. Quantum Chem. 1998, 67, 29–40. [Google Scholar] [CrossRef]
- Konkoli, Z.; Larsson, J.A.; Cremer, D. A New Way of Analyzing Vibrational Spectra. IV. Application and Testing of Adiabatic Modes within the Concept of the Characterization of Normal Modes. Int. J. Quantum Chem. 1998, 67, 41–55. [Google Scholar] [CrossRef]
- Zou, W.; Kalescky, R.; Kraka, E.; Cremer, D. Relating Normal Vibrational Modes to Local Vibrational Modes with the Help of an Adiabatic Connection Scheme. J. Chem. Phys. 2012, 137, 084114. [Google Scholar] [CrossRef] [Green Version]
- Kraka, E.; Cremer, D. Dieter Cremer’s Contribution to the Field of Theoretical Chemistry. Int. J. Quantum Chem. 2019, 119, e25849. [Google Scholar] [CrossRef] [Green Version]
- Wilson, E.B.; Decius, J.C.; Cross, P.C. Molecular Vibrations; McGraw-Hill: New York, NY, USA, 1955. [Google Scholar]
- Woodward, L.A. Introduction to the Theory of Molecular Vibrations and Vibrational Spectroscopy; Oxford University Press: Oxford, UK, 1972. [Google Scholar]
- Herzberg, G. Molecular Spectra and Molecular Structure. Volume II: Infrared and Raman Spectra of Polyatomic Molecules; Krieger Publishing Co.: New York, NY, USA, 1991. [Google Scholar]
- Herzberg, G.; Huber, K.P. Molecular Spectra and Molecular Structure: IV. Constants of Diatomic Molecules; Van Nostrand, R., Ed.; Springer: New York, NY, USA, 1979. [Google Scholar]
- Zou, W.; Cremer, D. C2 in a Box: Determining its Intrinsic Bond Strength for the X1Σ+g Ground State. Chem. Eur. J. 2016, 22, 4087–4097. [Google Scholar] [CrossRef] [PubMed]
- Cremer, D.; Kraka, E. Generalization of the Tolman Electronic Parameter: The Metal-Ligand Electronic Parameter and the Intrinsic Strength of the Metal-Ligand Bond. Dalton Trans. 2017, 46, 8323–8338. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Tian, C.; Verma, N.; Zou, W.; Wang, C.; Cremer, D.; Kraka, E. Recovering Intrinsic Fragmental Vibrations Using the Generalized Subsystem Vibrational Analysis. J. Chem. Theory Comput. 2018, 14, 2558–2569. [Google Scholar] [CrossRef]
- Kraka, E.; Larsson, J.A.; Cremer, D. Generalization of the Badger Rule Based on the Use of Adiabatic Vibrational Modes. In Computational Spectroscopy; Grunenberg, J., Ed.; Wiley: New York, NY, USA, 2010; pp. 105–149. [Google Scholar]
- Kalescky, R.; Kraka, E.; Cremer, D. Identification of the Strongest Bonds in Chemistry. J. Phys. Chem. A 2013, 117, 8981–8995. [Google Scholar] [CrossRef]
- Kraka, E.; Cremer, D. Characterization of CF Bonds with Multiple-Bond Character: Bond Lengths, Stretching Force Constants, and Bond Dissociation Energies. ChemPhysChem 2009, 10, 686–698. [Google Scholar] [CrossRef]
- Kraka, E.; Setiawan, D.; Cremer, D. Re-Evaluation of the Bond Length-Bond Strength Rule: The Stronger Bond Is not Always the Shorter Bond. J. Comput. Chem. 2015, 37, 130–142. [Google Scholar] [CrossRef]
- Setiawan, D.; Sethio, D.; Cremer, D.; Kraka, E. From Strong to Weak NF Bonds: On the Design of a New Class of Fluorinating Agents. Phys. Chem. Chem. Phys. 2018, 20, 23913–23927. [Google Scholar] [CrossRef]
- Sethio, D.; Lawson Daku, L.M.; Hagemann, H.; Kraka, E. Quantitative Assessment of B−B−B, B−Hb−B, and B−Ht Bonds: From BH3 to B12H122-. ChemPhysChem 2019, 20, 1967–1977. [Google Scholar] [CrossRef]
- Freindorf, M.; Kraka, E.; Cremer, D. A Comprehensive Analysis of Hydrogen Bond Interactions Based on Local Vibrational Modes. Int. J. Quantum Chem. 2012, 112, 3174–3187. [Google Scholar] [CrossRef]
- Kalescky, R.; Zou, W.; Kraka, E.; Cremer, D. Local Vibrational Modes of the Water Dimer—Comparison of Theory and Experiment. Chem. Phys. Lett. 2012, 554, 243–247. [Google Scholar] [CrossRef]
- Kalescky, R.; Kraka, E.; Cremer, D. Local Vibrational Modes of the Formic Acid Dimer—The Strength of the Double H-Bond. Mol. Phys. 2013, 111, 1497–1510. [Google Scholar] [CrossRef]
- Tao, Y.; Zou, W.; Jia, J.; Li, W.; Cremer, D. Different Ways of Hydrogen Bonding in Water—Why Does Warm Water Freeze Faster than Cold Water? J. Chem. Theory Comput. 2017, 13, 55–76. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Zou, W.; Kraka, E. Strengthening of Hydrogen Bonding With the Push-Pull Effect. Chem. Phys. Lett. 2017, 685, 251–258. [Google Scholar] [CrossRef]
- Makoś, M.Z.; Freindorf, M.; Sethio, D.; Kraka, E. New Insights into Fe−H2 and Fe−H- Bonding of a [NiFe] Hydrogenase Mimic—A Local Vibrational Mode Study. Theor. Chem. Acc. 2019, 138, 76. [Google Scholar] [CrossRef]
- Lyu, S.; Beiranvand, N.; Freindorf, M.; Kraka, E. Interplay of Ring Puckering and Hydrogen Bonding in Deoxyribonucleosides. J. Phys. Chem. A 2019, 123, 7087–7103. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, V.; Cremer, D.; Kraka, E. The Many Facets of Chalcogen Bonding: Described by Vibrational Spectroscopy. J. Phys. Chem. A 2017, 121, 6845–6862. [Google Scholar] [CrossRef]
- Oliveira, V.; Kraka, E. Systematic Coupled Cluster Study of Noncovalent Interactions Involving Halogens, Chalcogens, and Pnicogens. J. Phys. Chem. A 2017, 121, 9544–9556. [Google Scholar] [CrossRef]
- Setiawan, D.; Kraka, E.; Cremer, D. Hidden Bond Anomalies: The Peculiar Case of the Fluorinated Amine Chalcogenides. J. Phys. Chem. A 2015, 119, 9541–9556. [Google Scholar] [CrossRef]
- Setiawan, D.; Kraka, E.; Cremer, D. Strength of the Pnicogen Bond in Complexes Involving Group VA Elements N, P, and As. J. Phys. Chem. A 2014, 119, 1642–1656. [Google Scholar] [CrossRef]
- Setiawan, D.; Kraka, E.; Cremer, D. Description of Pnicogen Bonding with the help of Vibrational Spectroscopy-The Missing Link Between Theory and Experiment. Chem. Phys. Lett. 2014, 614, 136–142. [Google Scholar] [CrossRef]
- Setiawan, D.; Cremer, D. Super-Pnicogen Bonding in the Radical Anion of the Fluorophosphine Dimer. Chem. Phys. Lett. 2016, 662, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Sethio, D.; Oliveira, V.; Kraka, E. Quantitative Assessment of Tetrel Bonding Utilizing Vibrational Spectroscopy. Molecules 2018, 23, 2763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, V.; Kraka, E.; Cremer, D. The Intrinsic Strength of the Halogen Bond: Electrostatic and Covalent Contributions Described by Coupled Cluster Theory. Phys. Chem. Chem. Phys. 2016, 18, 33031–33046. [Google Scholar] [CrossRef]
- Oliveira, V.; Kraka, E.; Cremer, D. Quantitative Assessment of Halogen Bonding Utilizing Vibrational Spectroscopy. Inorg. Chem. 2016, 56, 488–502. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, V.; Cremer, D. Transition from Metal-Ligand Bonding to Halogen Bonding Involving a Metal as Halogen Acceptor: A Study of Cu, Ag, Au, Pt, and Hg Complexes. Chem. Phys. Lett. 2017, 681, 56–63. [Google Scholar] [CrossRef]
- Yannacone, S.; Oliveira, V.; Verma, N.; Kraka, E. A Continuum from Halogen Bonds to Covalent Bonds: Where Do λ3 Iodanes Fit? Inorganics 2019, 7, 47. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Zou, W.; Sethio, D.; Verma, N.; Qiu, Y.; Tian, C.; Cremer, D.; Kraka, E. In Situ Measure of Intrinsic Bond Strength in Crystalline Structures: Local Vibrational Mode Theory for Periodic Systems. J. Chem. Theory Comput. 2019, 15, 1761–1776. [Google Scholar] [CrossRef]
- Dronskowski, R.; Bloechl, P.E. Crystal Orbital Hamilton Populations (COHP): Energy-Resolved Visualization of Chemical Bonding in Solids Based on Density-Functional Calculations. J. Phys. Chem. 1993, 97, 8617–8624. [Google Scholar] [CrossRef]
- Hughbanks, T.; Hoffmann, R. Chains of Trans-Edge-Sharing Molybdenum Octahedra: Metal-Metal Bonding in Extended Systems. J. Am. Chem. Soc. 1983, 105, 3528–3537. [Google Scholar] [CrossRef]
- Hoffman, R. Solids and Surfaces: A Chemist’s View of Bonding in Extended Structures; Wiley-VCH: Hoboken, NJ, USA, 1989. [Google Scholar]
- Marzari, N.; Vanderbilt, D. Maximally Localized Generalized Wannier Functions for Composite Energy Bands. Phys. Rev. B 1997, 56, 12847–12865. [Google Scholar] [CrossRef] [Green Version]
- Souza, I.; Marzari, N.; Vanderbilt, D. Maximally Localized Wannier Functions for Entangled Energy Bands. Phys. Rev. B 2001, 65, 035109. [Google Scholar] [CrossRef] [Green Version]
- Leboeuf, M.; Köster, A.M.; Jug, K.; Salahub, D.R. Topological Analysis of the Molecular Electrostatic Potential. J. Chem. Phys. 1999, 111, 4893–4905. [Google Scholar] [CrossRef]
- Andrés, J.; Safont, V.S.; Gracia, L.; Llusar, R.; Longo, E. Bridging Structure and Real-Space Topology: Understanding Complex Molecules and Solid-State Materials. In Recent Advances in Complex Functional Materials; Springer International Publishing: Berlin/Heidelberg, Germany, 2017; pp. 427–454. [Google Scholar]
- Otero-de-la-Roza, A.; Johnson, E.R.; Luaña, V. Critic2: A Program for Real-Space Analysis of Quantum Chemical Interactions in Solids. Comput. Phys. Commun. 2014, 185, 1007–1018. [Google Scholar] [CrossRef]
- Burdett, J.K.; McCormick, T.A. Electron Localization in Molecules and Solids: The Meaning of ELF. J. Phys. Chem. A 1998, 102, 6366–6372. [Google Scholar] [CrossRef]
- Dunnington, B.D.; Schmidt, J.R. Generalization of Natural Bond Orbital Analysis to Periodic Systems: Applications to Solids and Surfaces via Plane-Wave Density Functional Theory. J. Chem. Theory Comput. 2012, 8, 1902–1911. [Google Scholar] [CrossRef]
- Nizhnik, Y.P.; Sons, A.; Zeller, M.; Rosokha, S.V. Effects of Supramolecular Architecture on Halogen Bonding between Diiodine and Heteroaromatic N-Oxides. Cryst. Growth Des. 2018, 18, 1198–1207. [Google Scholar] [CrossRef]
- Borley, W.; Watson, B.; Nizhnik, Y.P.; Zeller, M.; Rosokha, S.V. Complexes of Diiodine with Heteroaromatic N-Oxides: Effects of Halogen-Bond Acceptors in Halogen Bonding. J. Phys. Chem. A 2019, 123, 7113–7123. [Google Scholar] [CrossRef]
- Badger, R.M. A Relation Between Internuclear Distances and Bond Force Constants. J. Chem. Phys. 1934, 2, 128–131. [Google Scholar] [CrossRef] [Green Version]
- Badger, R.M. The Relation Between Internuclear Distances and the Force Constants of Diatomic Molecules. Phys. Rev. 1935, 48, 284–285. [Google Scholar] [CrossRef] [Green Version]
- Badger, R.M. The Relation Between the Internuclear Distances and Force Constants of Molecules and Its Application to Polyatomic Molecules. J. Chem. Phys. 1935, 3, 710–714. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal-Amorphous- Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Hafner, J. Ab-Initio Simulations of Materials using VASP: Density-Functional Theory and Beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef] [PubMed]
- Hamada, I. van der Waals Density Functional Made Accurate. Phys. Rev. B 2014, 89, 121103. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Lee, K.; Murray, É.D.; Kong, L.; Lundqvist, B.I.; Langreth, D.C. Higher-Accuracy van der Waals Density Functional. Phys. Rev. B 2010, 82, 081101. [Google Scholar] [CrossRef] [Green Version]
- Tran, F.; Kalantari, L.; Traoré, B.; Rocquefelte, X.; Blaha, P. Nonlocal van der Waals Functionals for Solids: Choosing an Appropriate One. Phys. Rev. Mater. 2019, 3, 063602. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Kraka, E.; Zou, W.; Filatov, M.; Tao, Y.; Grafenstein, J.; Izotov, D.; Gauss, J.; He, Y.; Wu, A.; Konkoli, Z.; et al. COLOGNE2019. 2019. Available online: http://www.smu.edu/catco (accessed on 14 February 2020).
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Gražulis, S.; Chateigner, D.; Downs, R.T.; Yokochi, A.F.T.; Quirós, M.; Lutterotti, L.; Manakova, E.; Butkus, J.; Moeck, P.; Le Bail, A. Crystallography Open Database – An Open-Access Collection of Crystal Structures. J. Appl. Crystallogr. 2009, 42, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Haynes, W. CRC Handbook of Chemistry and Physics, 97th ed.; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Marshall, W.; Jones, R.; Knight, K. Negative 2D Thermal Expansion in the Halogen Bonded Acetone Bromine Complex. CrystEngComm 2018, 20, 3246–3250. [Google Scholar] [CrossRef]
- Suponitsky, K.Y.; Burakov, N.; Kanibolotsky, A.L.; Mikhailov, V.A. Multiple Noncovalent Bonding in Halogen Complexes with Oxygen Organics. I. Tertiary Amides. J. Phys. Chem. A 2016, 120, 4179–4190. [Google Scholar] [CrossRef]
- Cristiani, F.; Demartin, F.; Devillanova, F.A.; Isaia, F.; Saba, G.; Verani, G. An X-ray, Spectroscopic and Semiempirical Quantum-Mechanical Study on Complexes of Thiones and Selones with Molecular Diiodine. J. Chem. Soc. Dalton Trans. 1992, 24, 3553–3560. [Google Scholar] [CrossRef]
- Montis, R.; Arca, M.; Aragoni, M.C.; Bauzá, A.; Demartin, F.; Frontera, A.; Isaia, F.; Lippolis, V. Hydrogen- And Halogen-Bond Cooperativity in Determining the Crystal Packing of Dihalogen Charge-Transfer Adducts: A Study Case from Heterocyclic Pentatomic Chalcogenone Donors. CrystEngComm 2017, 19, 4401–4412. [Google Scholar] [CrossRef] [Green Version]
- Bricklebank, N.; Skabara, P.J.; Hibbs, D.E.; Hursthouse, M.B.; Malik, K.A. Reaction of Thiones with Dihalogens; Comparison of the Solid State Structures of 4,5-bis(methylsulfanyl)-1,3-dithiole-2-thione-diiodine, -dibromine and -iodine monobromide. J. Chem. Soc. Dalton Trans. 1999, 17, 3007–3014. [Google Scholar] [CrossRef]
- Bigoli, F.; Deplano, P.; Ienco, A.; Mealli, C.; Mercuri, M.L.; Pellinghelli, M.A.; Pintus, G.; Saba, G.; Trogu, E.F. Structure and Bonding of Diiodine Adducts of the Sulfur-Rich Donors 1,3-dithiacyclohexane-2-thione (ptc) and 4,5-ethylenedithio-1,3-dithiole-2-thione (ttb). Inorg. Chem. 1999, 38, 4626–4636. [Google Scholar] [CrossRef]
- Hope, H.; McCullough, J. The Crystal Structure of the Molecular Complex of Iodine with Tetrahydroselenophene, C4H8Se.I2. Acta Crystallogr. 1964, 17, 712–718. [Google Scholar] [CrossRef]
- Bock, H.; Holl, S. Wechselwirkungen in Molekülkristallen, 167 [1, 2]. Kristallzüchtung und Strukturbestimmung von σ-Donator/Akzeptor-Komplexen zwischen 1, 4-Dioxan und den Polyiod- Molekülen I2, I2C= CI2,(IC) 4S sowie (IC) 4NR (R= H, CH3)/Interaction in Molecular Crystals, 167 [1, 2]. C rystallization and Structure Determination of σ-Donor/Acceptor Complexes between 1, 4-Dioxane and the Polyiodine Molecules I2, I2C= CI2,(IC) 4S and (IC) 4NR (R= H, CH3). Z. Naturforsch. B J. Chem. Sci. 2001, 56, 111–121. [Google Scholar]
- McCullough, J.D.; Knobler, C.; Baker, C.; Hope, H. Crystal and Molecular Structure of the Iodine Monobromide Complex of 1,4-dithiane, C4H8S2.2IBr. Inorg. Chem. 1971, 10, 697–700. [Google Scholar] [CrossRef]
- Herbstein, F.H.; Ashkenazi, P.; Kaftory, M.; Kapon, M.; Reisner, G.M.; Ginsburg, D. Propellanes LXXIX. Comparison of the Geometries of dithia[n.3.3]propellanes (n =1, 2, 3) and dithia(and oxathia)[4.3.3]propellanes. Study of the Influence of Complexation with HgCl2, I2, CdCl2 and PdCl2 and of Formation of Sulfoxides on Some of these Compounds. Demonstration of the ’Klammer’ effect. Structures of Eighteen Crystals. Acta Crystallogr. B 1986, 42, 575–601. [Google Scholar]
- Blake, A.J.; Li, W.S.; Lippolis, V.; Schröder, M. 1,4,8,11-Tetrakis(diiodine)-1,4,8,11-tetrathiacyclotetradecane. Acta Crystallogr. C 1997, 53, 886–888. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-Atom Fragments for Describing Molecular Charge Densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Sirianni, D.A.; Alenaizan, A.; Cheney, D.L.; Sherrill, C.D. Assessment of Density Functional Methods for Geometry Optimization of Bimolecular van der Waals Complexes. J. Chem. Theory Comput. 2018, 14, 3004–3013. [Google Scholar] [CrossRef]
- Boyer, M.A.; Marsalek, O.; Heindel, J.P.; Markland, T.E.; McCoy, A.B.; Xantheas, S.S. Beyond Badger’s Rule: The Origins and Generality of the Structure-Spectra Relationship of Aqueous Hydrogen Bonds. J. Phys. Chem. Lett. 2019, 10, 918–924. [Google Scholar] [CrossRef]
- Brown, A.C.; Gibson, J. XXX.-A Rule for Determining Whether A Given Benzene Mono-derivative Shall Give A Meta-di-derivative or A Mixture of Ortho- and Para-di-derivatives. J. Chem. Soc., Trans. 1892, 61, 367–369. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, L.N. Orientation of Substitution in the Benzene Nucleus. Chem. Rev. 1952, 50, 47–67. [Google Scholar] [CrossRef]
- Solomons, T.W.G.; Fryhle, C.B.; Snyder, S.A. Organic Chemistry, 11th ed.; Wiley: Hoboken, NJ, USA, 2013. [Google Scholar]
- Tao, Y.; Zou, W.; Cremer, D.; Kraka, E. Characterizing Chemical Similarity with Vibrational Spectroscopy: New Insights into the Substituent Effects in Monosubstituted Benzenes. J. Phys. Chem. A 2017, 121, 8086–8096. [Google Scholar] [CrossRef]
- Zhang, X.; da Silva, I.; Godfrey, H.G.W.; Callear, S.K.; Sapchenko, S.A.; Cheng, Y.; Vitórica-Yrezábal, I.; Frogley, M.D.; Cinque, G.; Tang, C.C.; et al. Confinement of Iodine Molecules into Triple-Helical Chains within Robust Metal-Organic Frameworks. J. Am. Chem. Soc. 2017, 139, 16289–16296. [Google Scholar] [CrossRef]








| No. | I-I⋯O=C | (mdyn/Å) | Cl-I⋯O=C | (mdyn/Å) |
|---|---|---|---|---|
| 1 | E1-1 | 10.327 | E1-2 | 9.932 |
| 2 | A | 9.755 | E2 | 9.905 |
| 3 | F-1 | 9.000 | F-2 | 8.807 |
| 4 | C1-1 | 8.945 | C1-2 | 8.579 |
| 5 | C2-1 | 8.937 | C2-2 | 8.575 |
| 6 | B2 | 8.431 | D | 8.561 |
| 7 | B1-1 | 7.917 | B1-2 | 7.651 |
| I-I⋯O−-N+ | (mdyn/Å) | Cl-I⋯O−-N+ | (mdyn/Å) | |
|---|---|---|---|---|
| 1 | O-1 | 4.866 | O-2 | 4.738 |
| 2 | Q | 4.916 | P-2 | 4.785 |
| 3 | P-1 | 4.944 | M | 4.867 |
| 4 | L-1 | 5.061 | L-2 | 4.896 |
| 5 | N-1 | 6.829 | N-2 | 5.297 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tao, Y.; Qiu, Y.; Zou, W.; Nanayakkara, S.; Yannacone, S.; Kraka, E. In Situ Assessment of Intrinsic Strength of X-I⋯OA-Type Halogen Bonds in Molecular Crystals with Periodic Local Vibrational Mode Theory. Molecules 2020, 25, 1589. https://doi.org/10.3390/molecules25071589
Tao Y, Qiu Y, Zou W, Nanayakkara S, Yannacone S, Kraka E. In Situ Assessment of Intrinsic Strength of X-I⋯OA-Type Halogen Bonds in Molecular Crystals with Periodic Local Vibrational Mode Theory. Molecules. 2020; 25(7):1589. https://doi.org/10.3390/molecules25071589
Chicago/Turabian StyleTao, Yunwen, Yue Qiu, Wenli Zou, Sadisha Nanayakkara, Seth Yannacone, and Elfi Kraka. 2020. "In Situ Assessment of Intrinsic Strength of X-I⋯OA-Type Halogen Bonds in Molecular Crystals with Periodic Local Vibrational Mode Theory" Molecules 25, no. 7: 1589. https://doi.org/10.3390/molecules25071589
APA StyleTao, Y., Qiu, Y., Zou, W., Nanayakkara, S., Yannacone, S., & Kraka, E. (2020). In Situ Assessment of Intrinsic Strength of X-I⋯OA-Type Halogen Bonds in Molecular Crystals with Periodic Local Vibrational Mode Theory. Molecules, 25(7), 1589. https://doi.org/10.3390/molecules25071589