
Molecular and Cellular Mechanisms of Cytotoxic Activity of Vanadium Compounds against Cancer Cells


Abstract
:1. Introduction
2. Mechanisms of Cytotoxicity
2.1. DNA: The Classical Target
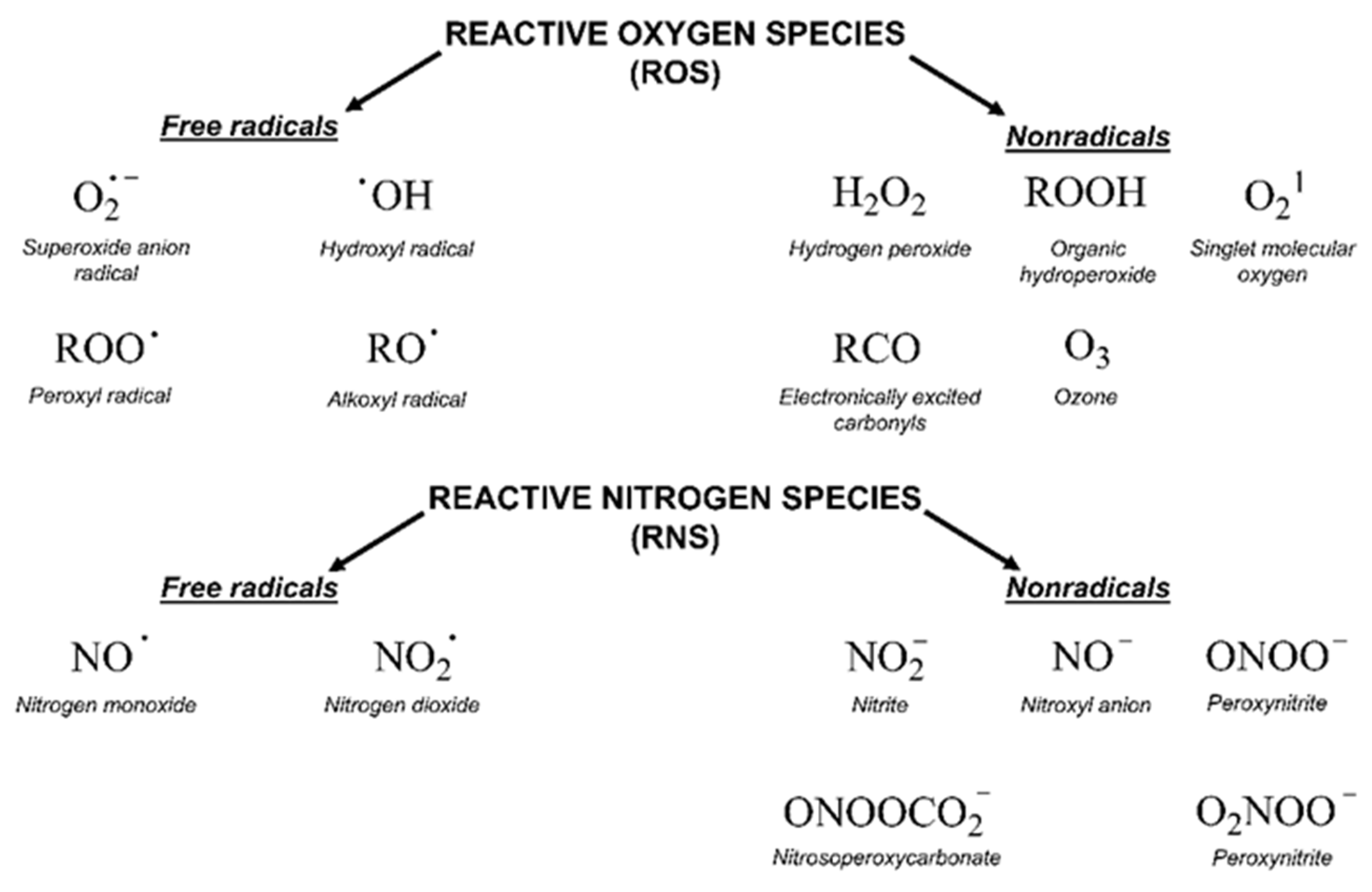
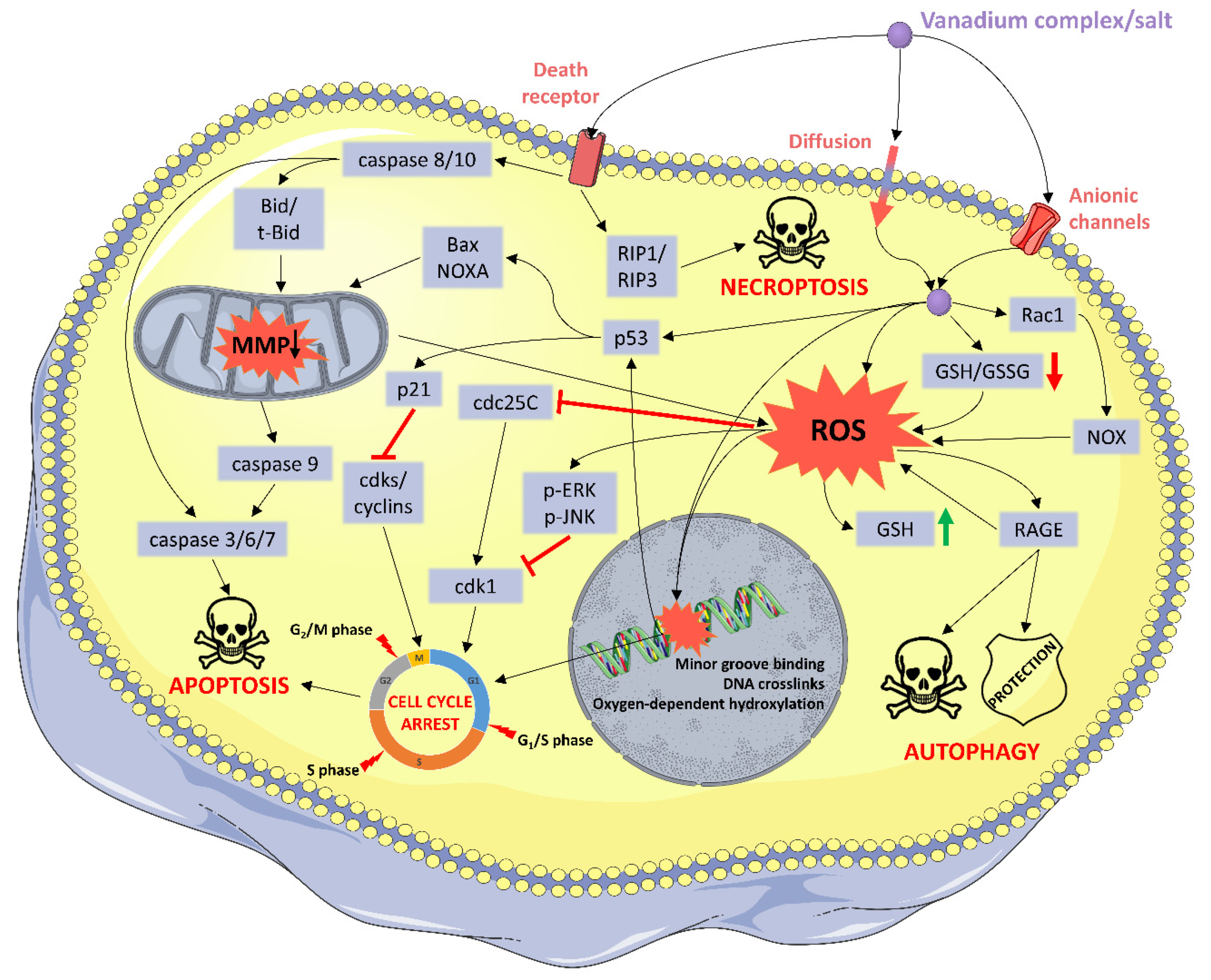
2.2. Oxidative Stress
2.3. Cell Cycle Arrest
2.4. Programed Cell Death
2.5. Other Mechanims
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Glossary
| Bax, Bid | The Bcl-2 family proteins promote apoptosis by governing mitochondrial outer membrane permeabilization (MOMP). |
| Bcl-2, Mcl-1, Bcl-xL | The Bcl-2 family proteins promote apoptosis by governing mitochondrial outer membrane permeabilization (MOMP). |
| Cdc2 | Cell division cycle protein 2 (also known as CDK1, cyclin-dependent kinase 1). It is a key player in cell cycle regulation. The activity of CDK1 oscillates during each cell cycle, which peaks at the G2/M phase and remains low at G1/S phase. |
| Cdc25C | A tyrosine phosphatase protein belonging to the Cdc25 phosphatase family. It directs dephosphorylation of cyclin B/CDK1 and triggers entry into mitosis. |
| Cyclin D1 | Cyclin which forms a complex with CDK4 or CDK6, whose activity is required for cell cycle G1/S transition. |
| DAPK | Death-associated protein kinase. Depletion of DAPK1 results in inhibition of tumor cell count. |
| DIABLO | A mitochondrial protein, inhibitor of apoptosis proteins (IAPs), thus freeing caspases to activate apoptosis. |
| H2AX | A type of histone protein. An phosphorylated form (γH2AX) is formed when double-strand breaks appear. |
| MAPK | A mitogen-activated protein kinase (including ERK, JNK and others). MAPKs regulate cell functions including proliferation, gene expression, differentiation, mitosis, cell survival and apoptosis. |
| mTOR | The mammalian target of rapamycin protein is a kinase that regulates cell growth, cell proliferation, cell motility, cell survival, protein synthesis, autophagy and transcription. mTOR also senses cellular nutrient, oxygen and energy levels. Phosphorylation of ribosomal S6 (S6R) protein is considered a robust readout for mTOR activation. The PI3K/AKT/mTOR pathway is an intracellular signaling pathway important in regulating the cell cycle. |
| Notch 1 | Notch family members play a role in a variety of developmental processes by controlling cell fate decisions. |
| NOXA | Promotes activation of caspases, mitochondrial membrane changes and efflux of apoptogenic proteins from the mitochondria. |
| p21 | Cyclin-dependent kinase inhibitor 1. It represents a major target of p53 activity and thus is associated with linking DNA damage to cell cycle arrest. |
| p53 | Protein plays a role in regulation or progression through the cell cycle, apoptosis, and genomic stability. |
| PARP | Poly (ADP-ribose) polymerase is involved in a number of cellular processes such as DNA repair, genomic stability, and programed cell death. |
| PI3K | Phosphoinositide 3-kinases. a family of enzymes involved in cellular functions such as cell growth, proliferation, differentiation, motility, survival. The PI3K/AKT/mTOR pathway is an intracellular signalling pathway important in regulating the cell cycle. |
| PKB/Akt | Protein kinase B (PKB or Akt) plays a key role in multiple cellular processes such as glucose metabolism, apoptosis, cell proliferation, transcription, and cell migration. Activation of PKB was shown to overcome cell cycle arrest in G1 and G2 phases. |
| Rac-1 | Small signaling G proteins which are pleiotropic regulators of many cellular processes, including the cell cycle, cell–cell adhesion, motility. Rac-1 activates NADPH oxidase inducing ROS generation. |
| RAGE | Receptor for advanced glycation end products. Overexpression of RAGE in pancreatic cancer cells is associated with enhanced autophagy, diminished apoptosis and greater tumor cell viability. |
| Rb | The retinoblastoma protein prevents excessive cell growth by inhibiting cell cycle progression until a cell is ready to divide. Rb is phosphorylated to pRb leading to the inactivation of the activity of Retinoblastoma protein. It allows cells to enter into the cell cycle state. Rb is dysfunctional in many cancers. |
| RET/PTC1 | The most prevalent type of gene rearrangement found in papillary thyroid carcinoma. |
| VEGF | Vascular endothelial growth factor is a signal protein produced by cells that stimulates the formation of blood vessels. |
References
- Bradford, S.S.; Cowan, J.A. From traditional drug design to catalytic metallodrugs: A brief history of the use of metals in medicine. Metallodrugs 2014, 1. [Google Scholar] [CrossRef]
- Elledge, S.J.; Zhou, Z.; Allen, J.B. Ribonucleotide reductase: Regulation, regulation, regulation. Trends Biochem. Sci. 1992, 17, 119–123. [Google Scholar] [CrossRef]
- Parisi, A.F.; Vallee, B.L. Zinc metalloenzymes: Characteristics and significance in biology and medicine. Am. J. Clin. Nutr. 1969, 22, 1222–1239. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, B.; Van Camp, L.; Krigas, T. Inhibition of cell division in escherichia coli by electrolysis products from a platinum electrode. Nature 1965, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Prestayko, A.W.; D’Aoust, J.C.; Issell, B.F.; Crooke, S.T. Cisplatin (Cis-Diamminedichloroplatinum II). Cancer Treat. Rev. 1979, 6, 17–39. [Google Scholar] [CrossRef]
- Cleare, M.J.; Hoeschele, J.D. Studies on the antitumor activity of group VIII transition metal complexes. Part I. Platinum (II) complexes. Bioinorg. Chem. 1973, 2, 187–210. [Google Scholar] [CrossRef]
- Kidani, Y.; Noji, M.; Tashiro, T. Antitumor activity of Platinum(II) complexes of 1,2-Diamino-Cyclohexane isomers. Gann Jpn. J. Cancer Res. 1980, 71, 637–643. [Google Scholar] [CrossRef]
- Sharma, H.; Thatcher, N.; Baer, J.; Zaki, A.; Smith, A.; McAucliffe, C.A.; Crowther, D.; Owens, S.; Fox, B.W. Blood clearance of radioactively labelled Cis-Diammine 1,1-Cyclobutane Dicarboxylate Platinum(II) (CBDCA) in cancer patients. Cancer Chemother. Pharmacol. 1983, 11, 5–7. [Google Scholar] [CrossRef]
- Tashiro, T.; Kawada, Y.; Sakurai, Y.; Kidani, Y. Antitumor activity of a new platinum complex, oxalato (Trans-l-1,2-Diaminocyclohexane)Platinum (II): New experimental data. Biomed. Pharmacother. 1989, 43, 251–260. [Google Scholar] [CrossRef]
- Hayes, D.M.; Cvitkovic, E.; Golbey, R.B.; Scheiner, E.; Helson, L.; Krakoff, I.H. High dose cis-platinum diammine dichloride: Amelioration of renal toxicity by mannitol diuresis. Cancer 1977, 39, 1372–1381. [Google Scholar] [CrossRef]
- Safirstein, R.; Winston, J.; Goldstein, M.; Moel, D.; Dikman, S.; Guttenplan, J. Cisplatin nephrotoxicity. Am. J. Kidney Dis. 1986, 8, 356–367. [Google Scholar] [CrossRef]
- Komeda, S.; Casini, A. Next-generation anticancer Metallodrugs. Curr. Top. Med. Chem. 2012, 12, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Bruijnincx, P.C.; Sadler, P.J. New trends for metal complexes with anticancer activity. Curr. Opin. Chem. Biol. 2008, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, E.A.; Wickline, S.A. Contrast agents for MRI. Basic Res. Cardiol. 2008, 114–121. [Google Scholar] [CrossRef]
- Price, T.W.; Greenman, J.; Stasiuk, G.J. Current advances in ligand design for inorganic positron emission tomography tracers 68 Ga, 64 Cu, 89 Zr and 44 Sc. Dalton Trans. 2016, 45, 15702–15724. [Google Scholar] [CrossRef]
- Feng, L.; Geisselbrecht, Y.; Blanck, S.; Wilbuer, A.; Atilla-Gokcumen, G.E.; Filippakopoulos, P.; Kräling, K.; Celik, M.A.; Harms, K.; Maksimoska, J.; et al. Structurally sophisticated octahedral metal complexes as highly selective protein kinase inhibitors. J. Am. Chem. Soc. 2011, 133, 5976–5986. [Google Scholar] [CrossRef] [Green Version]
- Rehder, D. The role of vanadium in biology. Metallomics 2015, 7, 730–742. [Google Scholar] [CrossRef] [Green Version]
- Anke, M. Spurenelement Symposium: New Trace Elements; Anke, M., Baumann, W., Braunlich, H., Bruckner, C., Groppel, B., Eds.; FriedrichSchiller-Universitat: Jena, Germany, 1986; pp. 1266–1275. [Google Scholar]
- Pessoa, J.C.; Etcheverry, S.; Gambino, D. Vanadium compounds in medicine. Coord. Chem. Rev. 2015, 301–302, 24–48. [Google Scholar] [CrossRef]
- Kioseoglou, E.; Petanidis, S.; Gabriel, C.; Salifoglou, A. The chemistry and biology of vanadium compounds in cancer therapeutics. Coord. Chem. Rev. 2015, 301, 87–105. [Google Scholar] [CrossRef]
- Leon, E.I.; Cadavid-Vargas, F.J.; Di Virgilio, L.A.; Etcheverry, B.S. Vanadium, ruthenium and copper compounds: A new class of nonplatinum metallodrugs with anticancer activity. Curr. Med. Chem. 2017, 24, 112–148. [Google Scholar] [CrossRef]
- Rehder, D. Implications of vanadium in technical applications and pharmaceutical issues. Inorg. Chim. Acta 2017, 455, 378–389. [Google Scholar] [CrossRef]
- Fuertes, M.; Castilla, J.; Alonso, C.; Pérez, J. Cisplatin biochemical mechanism of action: From cytotoxicity to induction of cell death through interconnections between apoptotic and necrotic pathways. Curr. Med. Chem. 2012, 10, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Anastassopoulou, J. Metal-DNA interactions. J. Mol. Struct. 2003, 651–653, 19–26. [Google Scholar] [CrossRef]
- Mohamadi, M.; Yousef Ebrahimipour, S.; Torkzadeh-Mahani, M.; Foro, S.; Akbari, A. A mononuclear Diketone-based Oxido-Vanadium(IV) complex: Structure, DNA and BSA binding, molecular docking and anticancer ActIVities against MCF-7, HPG-2, and HT-29 cell lines. RSC Adv. 2015, 5, 101063–101075. [Google Scholar] [CrossRef]
- Inamdar, P.R.; Sheela, A. Exploration of DNA binding mode, chemical nuclease, cytotoxic and apoptotic potentials of diketone based Oxovanadium(IV) complexes. Int. J. Biol. Macromol. 2015, 76, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Dankhoff, K.; Ahmad, A.; Weber, B.; Biersack, B.; Schobert, R. Anticancer properties of a new Non-Oxido Vanadium(IV) complex with a catechol-modified 3,3′-Diindolylmethane ligand. J. Inorg. Biochem. 2019, 194, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Fik, M.A.; Gorczyński, A.; Kubicki, M.; Hnatejko, Z.; Wadas, A.; Kulesza, P.J.; Lewińska, A.; Giel-Pietraszuk, M.; Wyszko, E.; Patroniak, V. New vanadium complexes with 6,6″-Dimethyl-2,2′:6′,2″-Terpyridine in terms of structure and biological properties. Polyhedron 2015, 97, 83–93. [Google Scholar] [CrossRef]
- Correia, I.; Roy, S.; Matos, C.P.; Borovic, S.; Butenko, N.; Cavaco, I.; Marques, F.; Lorenzo, J.; Rodríguez, A.; Moreno, V.; et al. Vanadium(IV) and Copper(II) complexes of salicylaldimines and aromatic heterocycles: Cytotoxicity, DNA binding and DNA cleavage properties. J. Inorg. Biochem. 2015, 147, 134–146. [Google Scholar] [CrossRef]
- Rui, W.; Tian, X.; Zeng, P.; Liu, W.; Ying, P.; Chen, H.; Lu, J.; Yang, N.; Chen, H. The anti-tumor activity of novel Oxovanadium complexes derived from Thiosemicarbazones and Fluoro-phenanthroline derivatives. Polyhedron 2016, 117, 803–816. [Google Scholar] [CrossRef]
- Bai, Y.L.; Zhang, Y.W.; Xiao, J.Y.; Guo, H.W.; Liao, X.W.; Li, W.J.; Zhang, Y.C. Oxovanadium Phenanthroimidazole derivatives: Synthesis, DNA binding and antitumor activities. Transit. Met. Chem. 2018, 43, 171–183. [Google Scholar] [CrossRef]
- Mal, S.K.; Chattopadhyay, T.; Fathima, A.; Purohit, C.S.; Kiran, M.S.; Nair, B.U.; Ghosh, R. Synthesis and structural characterization of a Vanadium(V)-Pyridylbenzimidazole complex: DNA binding and anticancer activity. Polyhedron 2017, 126, 23–27. [Google Scholar] [CrossRef]
- Patra, D.; Paul, S.; Sepay, N.; Kundu, R.; Ghosh, T. Structure-activity relationship on DNA binding and anticancer activities of a family of mixed-ligand Oxidovanadium(V) hydrazone complexes. J. Biomol. Struct. Dyn. 2018, 36, 4143–4155. [Google Scholar] [CrossRef] [PubMed]
- Al-Hazmi, G.A.; Abou-Melha, K.S.; El-Metwaly, N.M.; Saleh, K.A. Synthesis of novel VO(II)-Perimidine complexes: Spectral, computational, and antitumor studies. Bioinorg. Chem. Appl. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Dixit, A.; Banerjee, S.; Bhattacharyya, A.; Garai, A.; Karande, A.A.; Chakravarty, A.R. Cellular imaging and mitochondria targeted photo-cytotoxicity in visible light by singlet oxygen using a BODIPY-appended Oxovanadium(IV) DNA crosslinking agent. Medchemcomm 2016, 7, 1398–1404. [Google Scholar] [CrossRef]
- Kumar, A.; Pant, I.; Dixit, A.; Banerjee, S.; Banik, B.; Saha, R.; Kondaiah, P.; Chakravarty, A.R. Terpyridyl Oxovanadium(IV) complexes for DNA crosslinking and Mito-targeted Photocytotoxicity. J. Inorg. Biochem. 2017, 174, 45–54. [Google Scholar] [CrossRef]
- Dash, S.P.; Panda, A.K.; Pasayat, S.; Dinda, R.; Biswas, A.; Tiekink, E.R.T.; Mukhopadhyay, S.; Bhutia, S.K.; Kaminsky, W.; Sinn, E. Oxidovanadium(v) complexes of Aroylhydrazones incorporating heterocycles: Synthesis, characterization and study of DNA binding, photo-induced DNA cleavage and cytotoxic activities. RSC Adv. 2015, 5, 51852–51867. [Google Scholar] [CrossRef]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [Green Version]
- León, I.E.; Cadavid-Vargas, J.F.; Tiscornia, I.; Porro, V.; Castelli, S.; Katkar, P.; Desideri, A.; Bollati-Fogolin, M.; Etcheverry, S.B. Oxidovanadium(IV) complexes with Chrysin and Silibinin: Anticancer activity and mechanisms of action in a human colon adenocarcinoma model. JBIC J. Biol. Inorg. Chem. 2015, 20, 1175–1191. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Sies, H.; Sies, H. Oxidative stress: Introductory remarks. N. Y. Acad. J. 1985, 5, 1–8. [Google Scholar]
- Sies, H.; Jones, D. Oxidative stress. In Encyclopedia of Stress; Elsevier Inc.: Amsterdam, The Netherlands, 2007; pp. 45–48. [Google Scholar] [CrossRef]
- Irshad, M.; Chaudhuri, P.S. Oxidant-Antioxidant System: Role and Significance in Human Body; NISCAIR-CSIR: New Delhi, India, 2002; Volume 40, pp. 1233–1239. [Google Scholar]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; LLeonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Palomo, G.; Rendón-Huerta, E.P.; Montaño, L.F.; Fortoul, T.I. Vanadium compounds and cellular death mechanisms in the A549 cell line: The relevance of the compound valence. J. Appl. Toxicol. 2019, 39, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Anwar, T.; Islam, N.S.; Ramasarma, T.; Ramakrishna, G. Growth arrest of lung carcinoma cells (A549) by Polyacrylate-anchored Peroxovanadate by activating Rac1-NADPH oxidase signalling axis. Mol. Cell. Biochem. 2016, 420, 9–20. [Google Scholar] [CrossRef]
- Sinha, A.; Banerjee, K.; Banerjee, A.; Sarkar, A.; Ahir, M.; Adhikary, A.; Chatterjee, M.; Choudhuri, S.K. Induction of apoptosis in human colorectal cancer cell line, HCT-116 by a vanadium- schiff base complex. Biomed. Pharmacother. 2017, 92, 509–518. [Google Scholar] [CrossRef]
- Wu, J.-X.; Hong, Y.-H.; Yang, X.-G. Bis(Acetylacetonato)-Oxidovanadium(IV) and sodium metavanadate inhibit cell proliferation via ROS-induced sustained MAPK/ERK activation but with elevated AKT activity in human pancreatic cancer AsPC-1 cells. JBIC J. Biol. Inorg. Chem. 2016, 21, 919–929. [Google Scholar] [CrossRef]
- Molinuevo, M.S.; Barrio, D.A.; Cortizo, A.M.; Etcheverry, S.B. Antitumoral properties of two new Vanadyl(IV) complexes in osteoblasts in culture: Role of apoptosis and oxidative stress. Cancer Chemother. Pharmacol. 2004, 53, 163–172. [Google Scholar] [CrossRef]
- León, I.E.; Butenko, N.; Di Virgilio, A.L.; Muglia, C.I.; Baran, E.J.; Cavaco, I.; Etcheverry, S.B. Vanadium and cancer treatment: Antitumoral mechanisms of three Oxidovanadium(IV) complexes on a human osteosarcoma cell line. J. Inorg. Biochem. 2014, 134, 106–117. [Google Scholar] [CrossRef]
- Kowalski, S.; Hać, S.; Wyrzykowski, D.; Zauszkiewicz-Pawlak, A.; Inkielewicz-Stępniak, I. Selective cytotoxicity of vanadium complexes on human pancreatic ductal adenocarcinoma cell line by inducing necroptosis, apoptosis and mitotic catastrophe process. Oncotarget 2017, 8, 60324–60341. [Google Scholar] [CrossRef] [Green Version]
- De Cunha Padua, M.M.; Suter Correia Cadena, S.M.; de Oliveira Petkowicz, C.L.; Martinez, G.R.; Merlin Rocha, M.E.; Mercê, A.L.R.; Noleto, G.R. Toxicity of native and Oxovanadium (IV/V) galactomannan complexes on HepG2 cells is related to impairment of mitochondrial functions. Carbohydr. Polym. 2017, 173, 665–675. [Google Scholar] [CrossRef]
- Li, J.; Jiang, M.; Zhou, H.; Jin, P.; Cheung, K.M.C.; Chu, P.K.; Yeung, K.W.K. Vanadium dioxide nanocoating induces tumor cell death through mitochondrial electron transport chain interruption. Glob. Chall. 2019, 3, 1800058. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, T.T.; Fu, Y.; Wang, K.; Yang, X.G. Vanadium compounds discriminate hepatoma and normal hepatic cells by differential regulation of reactive oxygen species. J. Biol. Inorg. Chem. 2010, 15, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Naso, L.G.; Valcarcel, M.; Roura-Ferrer, M.; Kortazar, D.; Salado, C.; Lezama, L.; Rojo, T.; González-Baró, A.C.; Williams, P.A.M.; Ferrer, E.G. Promising antioxidant and anticancer (human breast cancer) Oxidovanadium(IV) complex of chlorogenic acid. synthesis, characterization and spectroscopic examination on the transport mechanism with bovine serum albumin. J. Inorg. Biochem. 2014, 135, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Torel, J.; Cillard, J.; Cillard, P. Antioxidant activity of flavonoids and reactivity with peroxy radical. Phytochemistry 1986, 25, 383–385. [Google Scholar] [CrossRef]
- Huyut, Z.; Beydemir, Ş.; Gülçin, I. Antioxidant and antiradical properties of selected flavonoids and phenolic compounds. Biochem. Res. Int. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Naso, L.G.; Lezama, L.; Rojo, T.; Etcheverry, S.B.; Valcarcel, M.; Roura, M.; Salado, C.; Ferrer, E.G.; Williams, P.A.M. Biological evaluation of morin and its new Oxovanadium(IV) complex as antio-xidant and specific anti-cancer agents. Chem. Biol. Interact. 2013, 206, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Martínez Medina, J.J.; Naso, L.G.; Pérez, A.L.; Rizzi, A.; Okulik, N.B.; Ferrer, E.G.; Williams, P.A.M. Apigenin Oxidovanadium(IV) cation interactions. Synthesis, spectral, bovine serum albumin binding, antioxidant and anticancer studies. J. Photochem. Photobiol. A Chem. 2017, 344, 84–100. [Google Scholar] [CrossRef]
- Leon, I.E.; Di Virgilio, A.L.; Porro, V.; Muglia, C.I.; Naso, L.G.; Williams, P.A.M.; Bollati-Fogolin, M.; Etcheverry, S.B. Antitumor properties of a Vanadyl(Iv) complex with the flavonoid chrysin [VO(Chrysin)2EtOH]2 in a human osteosarcoma model: The role of oxidative stress and apoptosis. Dalton Trans. 2013, 42, 11868. [Google Scholar] [CrossRef]
- Martínez Medina, J.J.; Naso, L.G.; Pérez, A.L.; Rizzi, A.; Ferrer, E.G.; Williams, P.A.M. Antioxidant and anticancer effects and bioavailability studies of the flavonoid baicalin and its Oxidovanadium(IV) complex. J. Inorg. Biochem. 2017, 166, 150–161. [Google Scholar] [CrossRef]
- Basu, A.; Bhattacharjee, A.; Hajra, S.; Samanta, A.; Bhattacharya, S. Ameliorative effect of an Oxovanadium (IV) complex against oxidative stress and nephrotoxicity induced by Cisplatin. Redox Rep. 2017, 22, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Bhattacharjee, A.; Samanta, A.; Bhattacharya, S. Prevention of cyclophosphamide-induced hepatotoxicity and genotoxicity: Effect of an l-Cysteine based Oxovanadium(IV) complex on oxidative stress and DNA damage. Environ. Toxicol. Pharmacol. 2015, 40, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Balaji, B.; Balakrishnan, B.; Perumalla, S.; Karande, A.A.; Chakravarty, A.R. Photocytotoxic Oxovanadium(IV) complexes of Ferrocenyl-Terpyridine and Acetylacetonate derivatives. Eur. J. Med. Chem. 2015, 92, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Jiang, H.; Mao, Y.; Ye, J.; Saffiotti, U. Vanadium(IV)-mediated free radical generation and related 2′-Deoxyguanosine hydroxylation and DNA damage. Toxicology 1996, 106, 27–38. [Google Scholar] [CrossRef]
- Williams, G.H.; Stoeber, K. The cell cycle and cancer. J. Pathol. 2012, 226, 352–364. [Google Scholar] [CrossRef]
- Pietenpol, J.A.; Stewart, Z.A. Cell cycle checkpoint signaling: Cell cycle arrest versus apoptosis. Toxicology 2002, 181–182, 475–481. [Google Scholar] [CrossRef]
- Xiong, Y. Why are there so many CDK inhibitors? Biochim. Biophys. Acta 1996. [Google Scholar] [CrossRef]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for P53 and P21 to sustain G2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef]
- Flatt, P.M.; Tang, L.J.; Scatena, C.D.; Szak, S.T.; Pietenpol, J.A. P53 regulation of G2 checkpoint is retinoblastoma protein dependent. Mol. Cell. Biol. 2000, 20, 4210–4223. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, I.; Hoffmann, I. Cell cycle regulation by the Cdc25 phosphatase family. Prog. Cell Cycle Res. 2000, 107–114. [Google Scholar] [CrossRef]
- Desoize, B. Metals and metal compounds in cancer treatment. Anticancer Res. 2004, 24, 1529–1544. [Google Scholar]
- Liu, T.T.; Liu, Y.J.; Wang, Q.; Yang, X.G.; Wang, K. Reactive-oxygen-species-mediated Cdc25C degradation results in differential antiproliferative activities of vanadate, tungstate, and Molybdate in the PC-3 human prostate cancer cell line. J. Biol. Inorg. Chem. 2012, 17, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Jiang, W.; Li, D.; Gu, M.; Liu, K.; Dong, L.; Wang, C.; Jiang, H.; Dai, W. Sodium Orthovanadate inhibits growth and triggers apoptosis of human anaplastic thyroid carcinoma cells in vitro and in vivo. Oncol. Lett. 2019, 17, 4255–4262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves, A.P.; Videira, A.; Soares, P.; Máximo, V. Orthovanadate-induced cell death in RET/PTC1-harboring cancer cells involves the activation of caspases and altered signaling through PI3K/Akt/MTOR. Life Sci. 2011, 89, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Rozzo, C.; Sanna, D.; Garribba, E.; Serra, M.; Cantara, A.; Palmieri, G.; Pisano, M. Antitumoral effect of vanadium compounds in malignant melanoma cell lines. J. Inorg. Biochem. 2017, 174, 14–24. [Google Scholar] [CrossRef]
- Pisano, M.; Arru, C.; Serra, M.; Galleri, G.; Sanna, D.; Garribba, E.; Palmieri, G.; Rozzo, C. Antiproliferative activity of vanadium compounds: Effects on the major malignant melanoma molecular pathways. Metallomics 2019, 11, 1687–1699. [Google Scholar] [CrossRef]
- Kowalski, S.; Wyrzykowski, D.; Hac, S.; Rychlowski, M.; Radomski, M.W.; Inkielewicz-Stepniak, I.; Kowalski, S.; Wyrzykowski, D.; Hac, S.; Rychlowski, M.; et al. New Oxidovanadium(IV) coordination complex containing 2-Methylnitrilotriacetate ligands induces cell cycle arrest and autophagy in human pancreatic ductal adenocarcinoma cell lines. Int. J. Mol. Sci. 2019, 20, 261. [Google Scholar] [CrossRef] [Green Version]
- Nair, R.S.; Kuriakose, M.; Somasundaram, V.; Shenoi, V.; Kurup, M.R.P.; Srinivas, P. The molecular response of vanadium complexes of Nicotinoyl Hydrazone in cervical cancers—A possible interference with HPV oncogenic markers. Life Sci. 2014, 116, 90–97. [Google Scholar] [CrossRef]
- Ni, L.; Zhao, H.; Tao, L.; Li, X.; Zhou, Z.; Sun, Y.; Chen, C.; Wei, D.; Liu, Y.; Diao, G. Synthesis, in vitro cytotoxicity, and structure-ActIVity relationships (SAR) of Multidentate Oxidovanadium(IV) complexes as anticancer agents. Dalton Trans. 2018, 47, 10035–10045. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Wang, X.S.; Fang, W.; Cai, X.Y.; Li, H.Z.; Mao, J.W.; Jin, X.B.; Bai, Y.L.; Lu, J.Z. In vitro study of the Cytotoxicities of two mixed-ligand Oxovanadium complexes on human Hepatoma cells. Pharmazie 2013, 68, 827–834. [Google Scholar] [CrossRef]
- Ying, P.; Zeng, P.; Lu, J.; Chen, H.; Liao, X.; Yang, N. New Oxidovanadium complexes incorporating Thiosemicarbazones and 1, 10-Phenanthroline derivatives as DNA cleavage, potential anticancer agents, and hydroxyl radical scavenger. Chem. Biol. Drug Des. 2015, 86, 926–937. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Z.; Jiang, S.; Zhang, M.; Lu, J.; Huang, L.; Zhang, T.; Gong, K.; Yan, S.; Yang, Z.; et al. Vanadate-induced antiproliferative and apoptotic response in esophageal squamous carcinoma cell line EC109. J. Toxicol. Environ. Health 2016, 79, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Reytman, L.; Hochman, J.; Tshuva, E.Y. Anticancer Diaminotris (Phenolato) Vanadium(V) complexes: Ligand-metal interplay. J. Coord. Chem. 2018, 71, 2003–2011. [Google Scholar] [CrossRef]
- Zimmermann, K.C.; Green, D.R. How cells die: Apoptosis pathways. J. Allergy Clin. Immunol. 2001, 108, S99–S103. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Watari, H.; Abualmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and molecular targeting therapy in cancer. Biomed. Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, A.A.; Jameson, M.J. Sodium orthovanadate inhibits proliferation and triggers apoptosis in oral squamous cell carcinoma in vitro. Biochemistry 2017, 82, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.P.; Suo, F.Z.; Feng, Y.L.; Song, L.L.; Li, Y.; Li, Y.J.; Wang, K.T. Synthesis and biological evaluation of vanadium complexes as novel anti-tumor agents. Eur. J. Med. Chem. 2019, 176, 1–10. [Google Scholar] [CrossRef]
- Roy, S.; Banerjee, S.; Chakraborty, T. Vanadium quercetin complex attenuates mammary cancer by regulating the P53, Akt/MTOR pathway and downregulates cellular proliferation correlated with increased apoptotic events. BioMetals 2018, 31, 647–671. [Google Scholar] [CrossRef]
- Šebestová, L.; Havelek, R.; Řezáčová, M.; Honzíček, J.; Kročová, Z.; Vinklárek, J. Study of antitumor effect of selected vanadium and molybdenum organometallic complexes in human leukemic T-cells. Chem. Biol. Interact. 2015, 242, 61–70. [Google Scholar] [CrossRef]
- Bakhshi Aliabad, H.; Khanamani Falahati-Pour, S.; Ahmadirad, H.; Mohamadi, M.; Hajizadeh, M.R.; Bakhshi, G.; Mahmoodi, M. Vanadium complex induced apoptosis in HepG2 cells by the up-regulation of p53, p21, and caspase-8. WCRJ 2019, 6, e1293. [Google Scholar]
- León, I.E.; Díez, P.; Baran, E.J.; Etcheverry, S.B.; Fuentes, M. Decoding the anticancer activity of VO-clioquinol compound: The mechanism of action and cell death pathways in human osteosarcoma cells. Metallomics 2017, 9, 891–901. [Google Scholar] [CrossRef]
- Banerjee, S.; Dixit, A.; Shridharan, R.N.; Karande, A.A.; Chakravarty, A.R. Endoplasmic reticulum targeted chemotherapeutics: The remarkable photo-cytotoxicity of an Oxovanadium(IV) vitamin-B6 complex in visible light. Chem. Commun. 2014, 50, 5590–5592. [Google Scholar] [CrossRef] [PubMed]
- Chandra, J.; Samali, A.; Orrenius, S. Triggering and modulation of apoptosis by oxidative stress. Free Radic. Biol. Med. 2000, 29, 323–333. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreuzaler, P.; Watson, C.J. Killing a cancer: What are the alternatives? Nat. Rev. Cancer 2012, 411–424. [Google Scholar] [CrossRef]
- Christofferson, D.E.; Yuan, J. Necroptosis as an alternative form of programmed cell death. Curr. Opin. Cell Biol. 2010, 263–268. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S.; Yoshida, T.; Tsujioka, M.; Arakawa, S. Autophagic cell death and cancer. Int. J. Mol. Sci. 2014, 15, 3145–3153. [Google Scholar] [CrossRef] [Green Version]
- Rana, S.; McCaffrey, V.; Journal, B.R.-T.F. Vanadium complex induced cancer cell death via RIPK3 activated necroptosis. FASEB J. 2017, 31, 876.6. [Google Scholar]
- El-Shafey, E.S.; Elsherbiny, E.S. Possible selective cytotoxicity of vanadium complex on breast cancer cells involving pathophysiological pathways. Anti Cancer Agents Med. Chem. 2019, 19, 2130–2139. [Google Scholar] [CrossRef]
- Meyenberg Cunha-de Padua, M.; Noleto, G.R.; de Oliveira Petkowicz, C.L.; Cadena, S.M.S.C.; Bost, F.; Pouysségur, J.; Mazure, N.M. Hypoxia protects against the cell death triggered by Oxovanadium–Galactomannan complexes in HepG2 cells. Cell. Mol. Biol. Lett. 2019, 24, 18. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Chen, D.; Zhang, F.; Willsky, G.R.; Crans, D.C.; Ding, W. Effects of vanadium (III, IV, V)-chlorodipicolinate on glycolysis and antioxidant status in the liver of STZ-induced diabetic rats. J. Inorg. Biochem. 2014, 136, 47–56. [Google Scholar] [CrossRef]
- Kacew, S.; Parulekar, M.R.; Merali, Z. Effects of parenteral vanadium administration on pulmonary metabolism of rats. Toxicol. Lett. 1982, 11, 119–124. [Google Scholar] [CrossRef]
- Irving, E.; Stoker, A.W. Vanadium compounds as PTP inhibitors. Molecules 2017, 22, 2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, R.J.; Yu, Z.H.; Zhang, R.Y.; Zhang, Z.Y. Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmacol. Sin. 2014, 35, 1227–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Englinger, B.; Pirker, C.; Heffeter, P.; Terenzi, A.; Kowol, C.R.; Keppler, B.K.; Berger, W. Metal drugs and the anticancer immune response. Chem. Rev. 2018, 119, 1519–1624. [Google Scholar] [CrossRef]
- Cohen, M.D.; Parsons, E.; Schlesinger, R.B.; Zelikoff, J.T. Immunotoxicity of in vitro vanadium exposures: Effects on interleukin-1, tumor necrosis factor-α, and prostaglandin E2 production by WEHI-3 macrophages. Int. J. Immunopharmacol. 1993, 15, 437–446. [Google Scholar] [CrossRef]
- Crans, D.C.; Henry, L.; Cardiff, G.; Posner, B.I. Developing vanadium as an antidiabetic or anticancer drug: A clinical and historical perspective. Met. Ions Life Sci. 2019, 203–230. [Google Scholar] [CrossRef]
- Friedrich, A.; Hefele, H.; Mickler, W.; Mönner, A.; Uhlemann, E.; Scholz, F. Voltammetric and potentiometric studies on the stability of vanadium (IV) complexes. A comparison of solution phase voltammetry with the voltammetry of microcrystalline solid compounds. Electroanal. Int. J. Devoted Fundam. Pract. Asp. Electroanal. 1998, 10, 244–248. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Structure | Activity | References |
|---|---|---|
 | Groove binding to salmon sperm DNA accompanied with a partial insertion between the base stacks of the DNA (Kb = 2.3 × 103 M−1) Cytotoxicity (24 h): breast cancer cells MCF-7 (IC50 7.8 µM) liver cancer cells HepG2 (IC50 13.5 µM) colon cancer cells HT-29 (IC50 16.1 µM) | [25] |
 | Oxidative cleavage of DNA through the generation of a hydroxyl radical Minor groove binding to DNA (2: Kb = 1.95 ± 0.16 × 103 M−1 3: Kb = 1.064 ± 0.17 × 103 M−1) Cytotoxicity (48 h): cervical cancer cells HeLa (2: IC50 256.9 µM 3: IC50 480.5 µM) | [26] |
 | Similarities to cisplatin concerning DNA interaction ROS generation, mitochondrial damage, G2/M cell cycle arrest Cytotoxicity (72 h): panel of melanoma, colon, cervical, breast and pancreatic cancer cells IC50 < 10 µM for all cell lines | [27] |
 | Intercalation as the way of DNA binding G2/M cell cycle arrest Cytotoxicity (48 h): cervical cancer cells HeLa (5: IC50 42.9 ± 1.5 µM 6: IC50 33.2 ± 0.9 µM) breast cancer cells T-47D (5: IC50 38.0 ± 1.6 µM 6: IC50 42.3 ± 1.8 µM) Lung cancer cells A549 (5: IC50 87.6 ± 2.4 µM 6: IC50 > 100 µM) | [28] |
 | Phen-containing VIVO compounds display stronger DNA interaction ability than the corresponding bipy analogues Cytotoxicity (72 h): ovarian cancer cells A2780 (7: IC50 20.8 ± 0.5 µM 8: IC50 4.9 ± 1.3 µM 9: IC50 17.1 ± 3.9 µM 10: IC50 4.7 ± 1.8 µM) breast cancer cells MCF-7 (7: IC50 53 ± 2.0 µM 8: IC50 77 ± 1.3 µM 9: IC50 95 ± 3.7 µM 10: IC50 68 ± 1.4 µM) | [29] |
 | Interaction with CT-DNA through a non-classical intercalative mode cleavage plasmid pBR322 DNA upon exposure to ultraviolet light Cytotoxicity (48 h): panel of cervical, breast and esophageal cancer cells IC50 range: 0.31–6.15 μM | [30] |
 | Binding with CT-DNA by an intercalation Kb = 14: 1.53 × 105 M−1 15: 1.41 × 105 M−1 16: 1.05 × 105 M−1 17: 0.95 × 105 M−1 cleave supercoiled plasmid DNA in the presence of H2O2 G0/G1 cell cycle arrest (14) Induction apoptosis in Hela cells (14) Cytotoxicity (24 h): cervical cancer cells HeLa (14: IC50 1.09 ± 0.16 µM 15: IC50 10.36 ± 1.23 µM) bladder cancer cell BIU-87 (14: IC50 4.51 ± 0.68 µM 15: IC50 8.69 ± 1.05 µM) lung cancer cells SPC-A-1 (14: IC50 7.61 ± 0.55 µM 15: IC50 21.43 ± 3.24 µM) | [31] |
 | Interaction with DNA in a intercalative fashion (Kb = 2.76 × 105 M−1) Cytotoxicity (24 h): lung cancer cell A549 breast cancer cells MCF-7 keratinocyte cancer cell A431 IC50 for all cancer cell lines 75 μM normal human keratinocyte cells HaCaT IC50 150 µM | [32] |
 | The intercalative mode of binding to DNA (19: Kb = 6.13 × 105 M−1 20: Kb = 8.69 × 105 M−1) Cytotoxicity (24 h): cervical cancer cell SiHa (19: IC50 33 µM 20: IC50 29 µM) | [33] |
 | Binding to CT-DNA Kb = 21: 6.10 × 104 M−1 22: 7.99 × 104 M−1 23: 6.75 × 104 M−1 24: 6.07 × 104 M−1 25: 8.80 × 104 M−1 Cytotoxicity (48 h): breast cancer cells MCF-7 (25: IC50 11.44 µM 23: IC50 15.50 µM) liver cancer cells HepG2 (25: IC50 9.91 µM 23: IC50 11.01 µM) colon cancer cells HCT 116 (24: IC50 13.27 µM 23: IC50 15.53 µM) | [34] |
  | Light-activated VO2+-DNA crosslink formation (27) singlet oxygen (1O2) induced mitochondria-targeted PDT (27) Cytotoxicity (24 h): breast cancer cells MCF-7 (27: IC50 3.4±0.4 µM in visible light IC50 > 50 µM in the dark) cervical cancer cells HeLa (27: IC50 1.8±0.6 µM in visible light IC50 > 50 µM in the dark) 26: any significant cytotoxicity in light | [36] |
 | Light-activated DNA crosslink formation (in the dark they are partial DNA intercalators) ROS generation in visible light Cytotoxicity (24 h): breast cancer cells MCF-7 (28: IC50 10.4 ± 1.6 µM in visible light IC50 > 50 µM in the dark) (29: IC50 2.3 ± 0.3 µM in visible light IC50 27.6 ± 1.4 µM in the dark) cervical cancer cells HeLa (28: IC50 8.2 ± 0.3 µM in visible light IC50 > 50 µM in the dark) (29: IC50 1.8 ± 0.5 µM in visible light IC50 20.3 ± 1.0 µM in the dark) | [37] |
  | Photo-induced cleavage of pUC19 supercoiled plasmid DNA Interaction with CT-DNA through minor groove binding mode Kb = 30: 8.56 × 104 M−1 31: 1.13 × 105 M−1 32: 4.95 × 104 M−1 33: 5.03 × 103 M−1 Cytotoxicity (72 h): cervical cancer cells HeLa 30: IC50 20 ± 4.52 µM 31: IC50 18 ± 3.38 µM 32: IC50 19.5 ± 3.54 µM 33: IC50 9.9 ± 3.18 µM | [38] |
 | The topoisomerase IB inhibition (34) G2/M cell cycle arrest (35) activation caspase 3 and triggering the apoptosis (34) Cytotoxicity (24 h): colon cancer cells HT-29 A concentration-related inhibition from 75 to 100 µM | [40] |
| Structure | Activity | References |
|---|---|---|
 | Lung cancer cells A549 12(36)- and 14(37)-fold increase in ROS generation (100 μM after 48 h) Cytotoxicity: 24 h (36: IC50 >100 µM 37: IC50 >100 µM) 48 h (36: IC50 ~100 µM 37: IC50 >100 µM) | [46] |
[V2O2(O2)4(carboxylate)]-PA | Lung cancer cells A549 Activation of the axis of Rac1-NADPH oxidase leading to oxidative stress Increase in phosphorylation of H2AX (γH2AX), a marker of DNA damage | [47] |
 | Breast cancer cells MCF-7 Glioblastoma cells U-373MG T lymphoblastic leukaemia cells CCRF-CEM and CEM-ADR 5000 Colon cancer cells HCT-116 Depletion of GSH content ROS generation Induction of apoptosis through mitochondrial outer membrane permeabilization but in caspase independent manner | [48] |
 | Pancreatic cancer cells AsPC-1 ROS generation G2/M cell cycle arrest Activation of PI3K/AKT and MAPK/ERK signaling pathways Increased level of phosphorylated Cdc2 at Tyr-15 and the reduced level of Cdc25C | [49] |
GluVO 41 NapVO 42 | Osteosarcoma cells UMR106 Induction of apoptosis ROS generation | [50] |
 | Osteosarcoma cells MG-63 Induction of oxidative stress, apoptosis and DNA cleavage Cytotoxicity (24 h): 43: IC50 >100 µM 44: IC50 >100 µM 45: IC50 58 µM | [51] |
 | Pancreatic cancer cells PANC-1 3-fold increase in ROS generation (45,47) (5 µM 48 h) 8-fold increase in ROS generation (46) (10 µM 48 h) G2/M cell cycle arrest (46) Induction of necroptosis (45, 47) Induction of autophagy (46) Cytotoxicity (48 h): 45: logIC50 0.52 ± 0.28 µM (IC50 3.3 µM) 46: logIC50 1.47 ± 0.07 µM (IC50 29.5 µM) 47: logIC50 1.10 ± 0.11 µM (IC50 12.6 µM) | [52] |
| 48 SAGM:VO SAGM = native galactomannan from S. amazonicum 49 MSAGM:VO MSAGM = modified form native galactomannan from S. amazonicum | Liver cancer cells HepG2 ROS generation Reduction in MMP | [53] |
| VO2-modified quartz surface (releasing ions from surface) | Cholangiocarcinoma cells ROS generation Interruption of the mitochondrial electron transport chain | [54] |
 | Liver cancer cells HepG2 Insignificant ROS generation G1/S cell cycle arrest Immortalized hepatic cells L02 cells ROS generationS and G2/M cell cycle arrest | [55] |
 | Antioxidant activity (inhibitory effects on O2·−, ·OH and ROO· radicals generation) Selective cytotoxicity against breast cancer cells SKBR3 No increase in ROS generation | [56] |
 | Breast cancer cells SKBR3 and T47D Apoptotic cell death process with caspase 3/7 activation Perturbation of the MMP No increase in ROS generation No effect on the normal proliferation of the breast epithelial mammal cells | [59] |
 | Lung cancer cells A549 Cervical cancer cells HeLa ROS generation GSH/GSSH depletion Cytotoxicity: Lung cancer cells A549 24 h (IC50 >100 µM) 48 h (IC50 17.7 ± 3.1 µM) 72 h (IC50 2.2 ± 0.9 µM) Cervical cancer cells HeLa 24 h (IC50 88.1 ± 1.4 µM) 48 h (IC50 115.5 ± 1.6 µM) 72 h (IC50 9.7 ± 1.9 µM) | [60] |
 | Osteosarcoma cells MG-63 ROS generation GSH/GSSH depletion Induction of apoptosis (increased levels of caspase 3) Disruption of the MMP | [61] |
 | Lung cancer cells A549 ROS generation Cytotoxicity: 24 h (IC50 >100 µM) 48 h (IC50 44.7 ± 3.5 µM) 72 h (IC50 21.7 ± 1.3 µM) | [62] |
 | Swiss albino mice Nephroprotective efficacy (against cisplatin) by restoring antioxidant defense mechanism | [63] |
 | Swiss albino mice Prevention of cyclophosphamide-induced hepatotoxicity and genotoxicity Restoration of glutathione level and activities of antioxidant enzymes | [64] |
 | Cervical cancer cells HeLa Breast cancer cells MCF-7 ROS generation Photocleavage of plasmid DNA in green light (568 nm) forming ·OH radicals Low toxicity in normal fibroblast 3T3 cells Cytotoxicity (24 h): cervical cancer cells HeLa (58: IC50 15.5 ± 1.0 µM in visible light IC50 45.1 ± 1.2 µM in the dark) (59: IC50 5.4 ± 0.5 µM in visible light IC50 37.6 ± 1.2 µM in the dark) (60: IC50 19.9 ± 1.1 µM in visible light IC50 40.1 ± 1.1 µM in the dark) breast cancer cells MCF-7 (57: IC50 5.6 ±0.9 µM in visible light IC50 41.2 ± 1.0 µM in the dark) (58: IC50 3.3 ± 0.6 µM in visible light IC50 48.7 ± 1.1 µM in the dark) (59: IC50 2.1 ± 0.3 µM in visible light IC50 38.5 ± 1.1 µM in the dark) | [65] |
| Structure | Activity | References |
|---|---|---|
 | Prostate cancer cells PC-3 G2/M cell cycle arrest ROS-mediated degradation of Cdc25C Increase in the level of phosphorylated Cdc2 at its inactive Tyr-15 site | [74] |
 | Thyroid carcinoma cells 8505C G2/M cell cycle arrest Induction of apoptosis Reduction in MMP Cytotoxicity: 24 h (IC50 3.76 µM) 48 h (IC50 3.55 µM) 72 h (IC50 3.23 µM) 96 h (IC50 1.62 µM) | [75] |
| Papillary thyroid carcinoma-derived cells TPC-1 Decrease in the expression of cyclin D1 Increase in the expression of p21 Undisturbed cell cycle ROS generation Induction of apoptosis (activation of caspase-3) Reduction in MMP Increase in phosphorylation of tyrosine 451 of RET/PTC1 and activation of the mTOR/S6R branch of the PI3K/Akt signaling pathway | [76] | |
 | Malignant melanoma cells A375 and CN-mel Induction of apoptosis G2/M cell cycle arrest (62) G1/S cell cycle arrest (63) Cytotoxicity (72 h): A375 cells 62: IC50 4.7 µM 63: IC50 2.6 µM 64: IC50 4.2 µM 65: IC50 2.4 µM CN-mel cells 62: IC50 6.5 µM 63: IC50 12.4 µM 64: IC50 14.0 µM 65: IC50 10.4 µM | [77] |
| Malignant melanoma cells A375 ROS generation (62, 63) Induction of dephosphorylation of Rb protein (62, 63) Cell cycle arrest by contrasting MAPK pathway activation and strongly inducing p21 expression and Rb hypophosphorylation (62, 63) | [78] | |
 | Pancreatic cancer cells PANC-1 and MIA PaCa2 G2/M cell cycle arrest (increase in the expression of cyclinB1 and cdk1) Induction of p53/p21 pathway Induction of autophagy Increase in the expression of RAGE ROS generation Cytotoxicity (48 h): PANC-1 cells: IC50 44.67 µM MIA PaCa2 cells: IC50 72.22 µM | [79] |
 | Cervical cancer cells HeLa and SiHa Induction of p53/p21 pathway Induction of apoptosis | [80] |
 | Liver cancer cells SMMC-7721, HepG2 S and G2/M cell cycle arrest (70) Induction of apoptosis (70) Cytotoxicity (48 h): SMMC-7721 cells 69: IC50 60.19 ± 0.03 µM 70: IC50 5.34 ± 0.03 µM 71: IC50 119.44 ± 0.03 µM 72: IC50 25.55 ± 0.02 µM 47: IC50 42.46 ± 0.03 µM HepG2 cells 69: IC50 52.33 ± 0.02 µM 70: IC50 29.07 ± 0.01 µM 71: IC50 106.13 ± 0.02 µM 72: IC50 39.63 ± 0.03 µM 47: IC50 101.62 ± 0.02 µM | [81] |
 | Liver cancer cells BEL-7402, HUH-7 and HepG2 G1/S cell cycle arrest Reduction in MMP Induction of apoptosis | [82] |
 | Neuroblastoma cells SH-SY5Y and SK-N-SH Breast cancer cells MCF-7 G1/S cell cycle arrest (75) Induction of apoptosis (75) Cleavage plasmid pBR322 DNA Cytotoxicity (48 h): SH-SY5Y cells 75: IC50 1.08 ± 0.26 µM 76: IC50 2.30 ± 0.09 µM 77: IC50 1.69 ± 0.21 µM 78: IC50 3.92 ± 0.43 µM SK-N-SH cells 75: IC50 0.21 ± 0.023 µM 76: IC50 0.48 ± 0.017 µM 77: IC50 0.37 ± 0.025 µM 78: IC50 1.42 ± 0.11 µM MCF-7 cells 75: IC50 6.49 ± 0.28 µM 76: IC50 4.41 ± 1.27 µM 77: IC50 8.99 ± 0.39 µM 78: IC50 8.63 ± 1.31 µM | [83] |
 | Colon cancer cells HT-29 Ovarian cancer cells OVCAR-3, A2780, A2780cis and A2780adr S cell cycle arrest Induction of apoptosis Cytotoxicity (72 h): HT-29 cells 79: IC50 1.4 ± 0.2 µM 80: IC50 2.9 ± 0.7 µM OVCAR-3 cells 79: IC50 0.7 ± 0.3 µM 80: IC50 0.40 ± 0.04 µM A2780 cells 79: IC50 0.17 ± 0.07 µM 80: IC50 0.6 ± 0.1 µM A2780cis cells 79: IC50 0.29 ± 0.15 µM 80: IC50 0.8 ± 0.1 µM | [85] |
| Structure | Mechanism | References |
|---|---|---|
 | Oral squamous cell carcinoma Cal27 Induction of apoptosis: poly(ADPribose)polymerase cleavage Cytotoxicity (72 h): IC50 25 μM | [88] |
 | Gastric cancer cells MGC-803 Induction of apoptosis (upregulation of Bax, PARP and caspase-3/9, downregulation of Bcl-2) Prevention from the colony formation, migration and EMT process Cytotoxicity (72 h): IC50 2.69 μM | [89] |
 | Breast cancer cells MCF-7 Increase in the expression of p53 Decrease in the expression of Akt, mTOR and VEGF Induction of apoptosis (activation of 3 and 9 caspase, DNA fragmentation) In vivo study (Balb/c mice) Increase in apoptotic index Upregulation of Bcl-2 and downregulation of Bax and p53 | [90] |
 | T-leukemic cells (p53 wild-type MOLT-4 and p53-deficient Jurkat) Induction of apoptosis (activation of the caspases 9-intrinsic pathway and 8-extrinsic pathway) Increase in the expression of the tumor-suppressor protein p53 and its form phosphorylated at the serine 15 Cytotoxicity: 24 h: MOLT-4 IC50 3.1 ± 0.4 μM Jurkat IC50 2.9 ± 0.2 μM 48 h: MOLT-4 IC50 2.1 ± 0.2 μM Jurkat IC50 2.8 ± 0.3 μM 72 h: MOLT-4 IC50 2.3 ± 0.2 μM Jurkat IC50 1.7 ± 0.1 ± μM | [91] |
 | Liver cancer cells HepG2 Induction of apoptosis (using the p53/p21 pathway-dependent way) Decrease in the expression of caspase-8 and Bid | [92] |
 | Osteosarcoma cells MG-63 Determination of the relative abundance of 224 proteins Increase in the expression of caspase 3, caspase 6, caspase 7, caspase 10, caspase 11, Bcl-x, DAPK Decrease in the expression of PKB/Akt, DIABLO | [93] |
 | Cervical cancer cells HeLa Breast cancer cells MCF-7 Induction of apoptosis Specific localization to endoplasmic reticulum (ER) Cytotoxicity (48 h): cervical cancer cells HeLa (86: IC50 44.1 ± 1.8 µM in visible light IC50 59.3 ± 1.7 µM in the dark) (87: IC50 0.24 ± 0.02 µM in visible light IC50 > 40 µM in the dark) breast cancer cells MCF-7 (86: IC50 53.3 ± 1.9 µM in visible light IC50 72.5 ± 2.1 µM in the dark) (87: IC50 0.53 ± 0.03 µM in visible light IC50 > 40 µM in the dark) | [94] |
 | Colon cancer cells HT-29 Induction of necroptosis | [100] |
 | Breast cancer cells MDA-MB-231 G2/M cell cycle arrest Induction of autophagy Induction of apoptosis Increase in the expression of Notch 1 | [101] |
| 91 MSAGM:VO MSAGM = Galactomannan from Schizolobium amazonicum | Liver cancer cells HepG2 Induction of apoptosis under normoxic conditions (lost under hypoxic conditions) The expressions of anti-apoptotic Mcl-1 and Bcl-XL increased in hypoxia, whereas the expression of pro-apoptotic Bax decreased Induction of autophagy (elimination of the anti-cancer activity with activation of autophagy under conditions of hypoxia) | [102] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowalski, S.; Wyrzykowski, D.; Inkielewicz-Stępniak, I. Molecular and Cellular Mechanisms of Cytotoxic Activity of Vanadium Compounds against Cancer Cells. Molecules 2020, 25, 1757. https://doi.org/10.3390/molecules25071757
Kowalski S, Wyrzykowski D, Inkielewicz-Stępniak I. Molecular and Cellular Mechanisms of Cytotoxic Activity of Vanadium Compounds against Cancer Cells. Molecules. 2020; 25(7):1757. https://doi.org/10.3390/molecules25071757
Chicago/Turabian StyleKowalski, Szymon, Dariusz Wyrzykowski, and Iwona Inkielewicz-Stępniak. 2020. "Molecular and Cellular Mechanisms of Cytotoxic Activity of Vanadium Compounds against Cancer Cells" Molecules 25, no. 7: 1757. https://doi.org/10.3390/molecules25071757
APA StyleKowalski, S., Wyrzykowski, D., & Inkielewicz-Stępniak, I. (2020). Molecular and Cellular Mechanisms of Cytotoxic Activity of Vanadium Compounds against Cancer Cells. Molecules, 25(7), 1757. https://doi.org/10.3390/molecules25071757