
Cooperative Analysis of Structural Dynamics in RNA-Protein Complexes by Single-Molecule Förster Resonance Energy Transfer Spectroscopy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Spliceosome
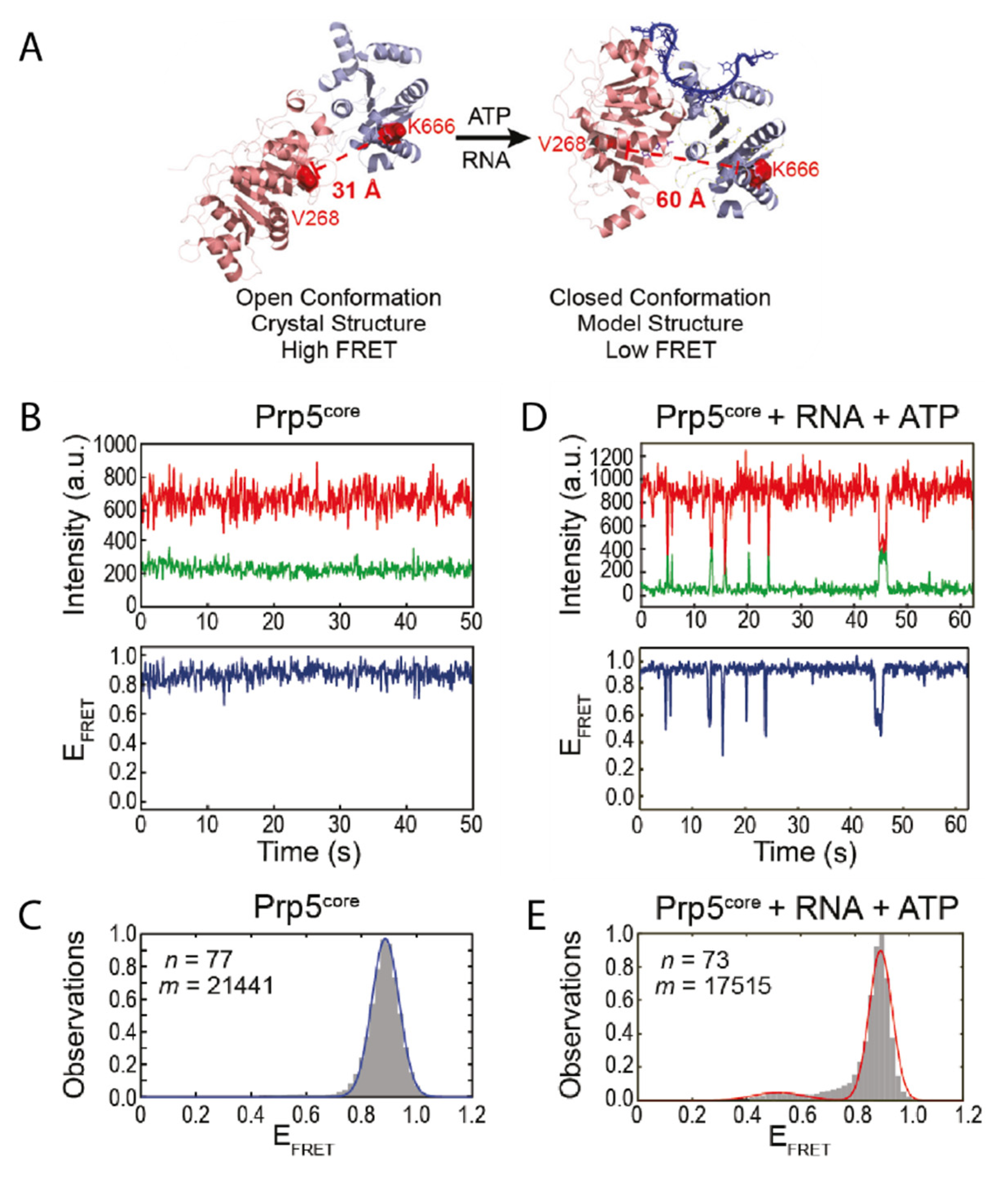
3. The Role of Prp5 on Spliceosome Assembly
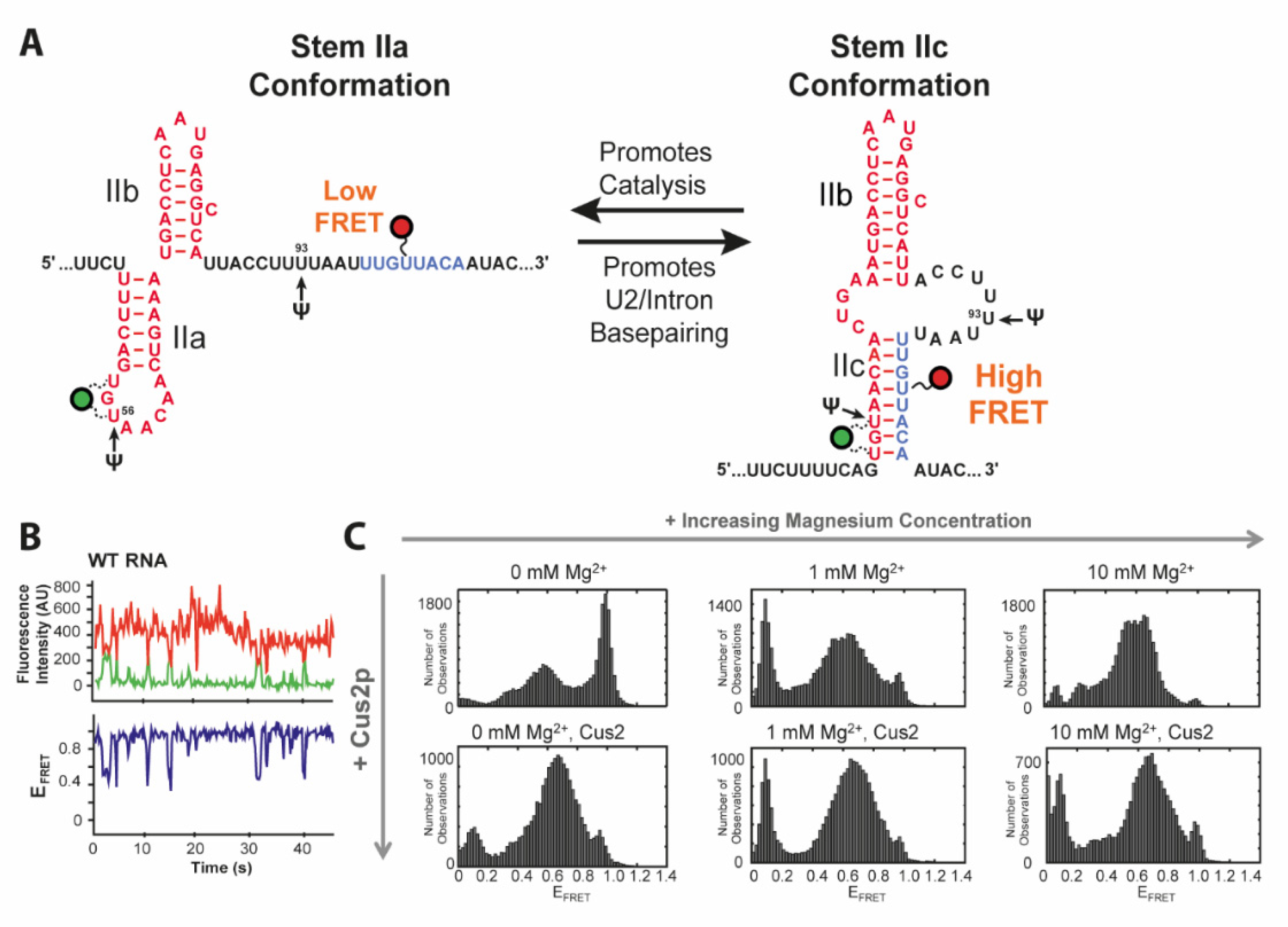
4. U2 Toggling
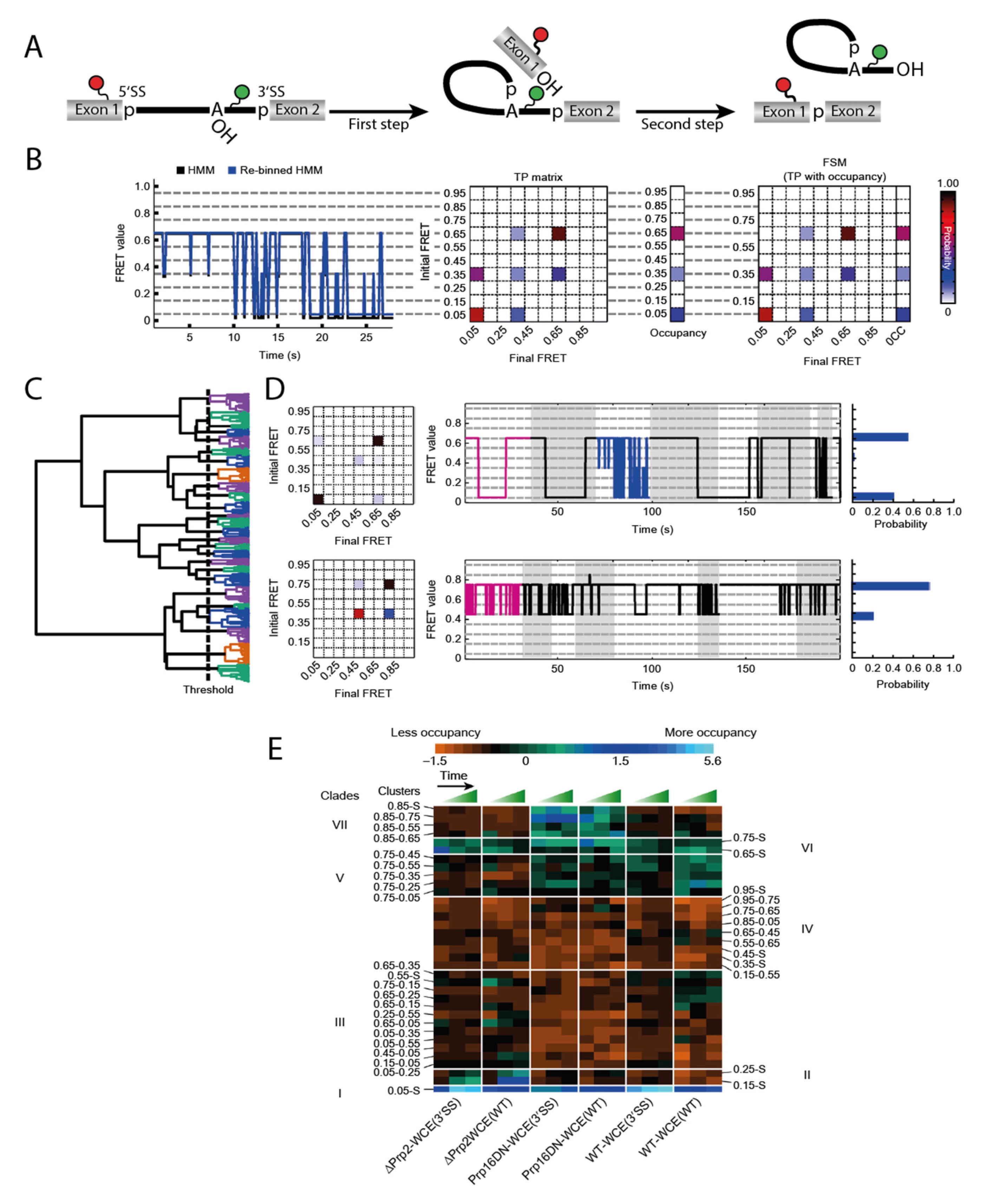
5. Observation of the Full Splicing Cycle
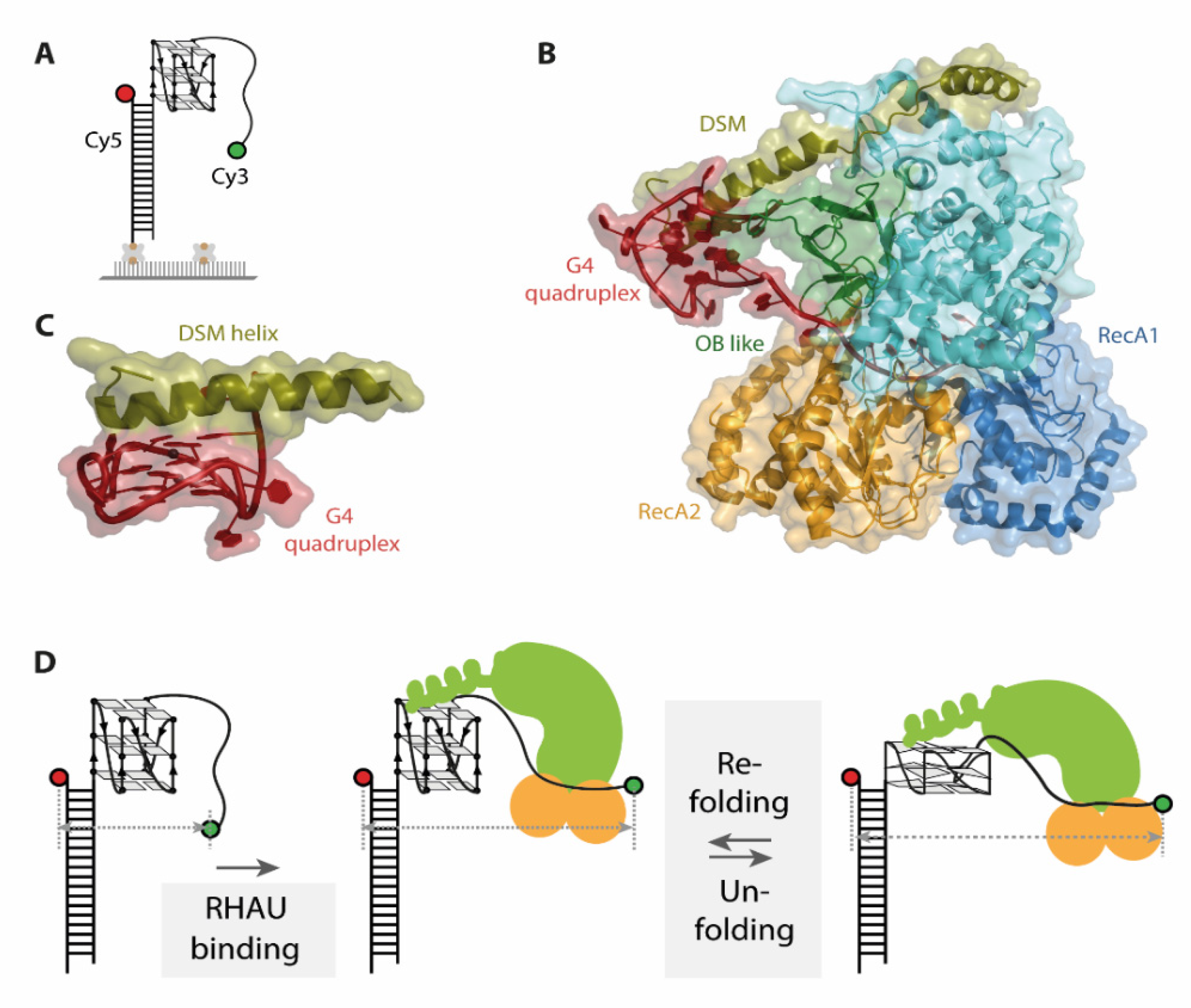
6. G-Quadruplex Helicase
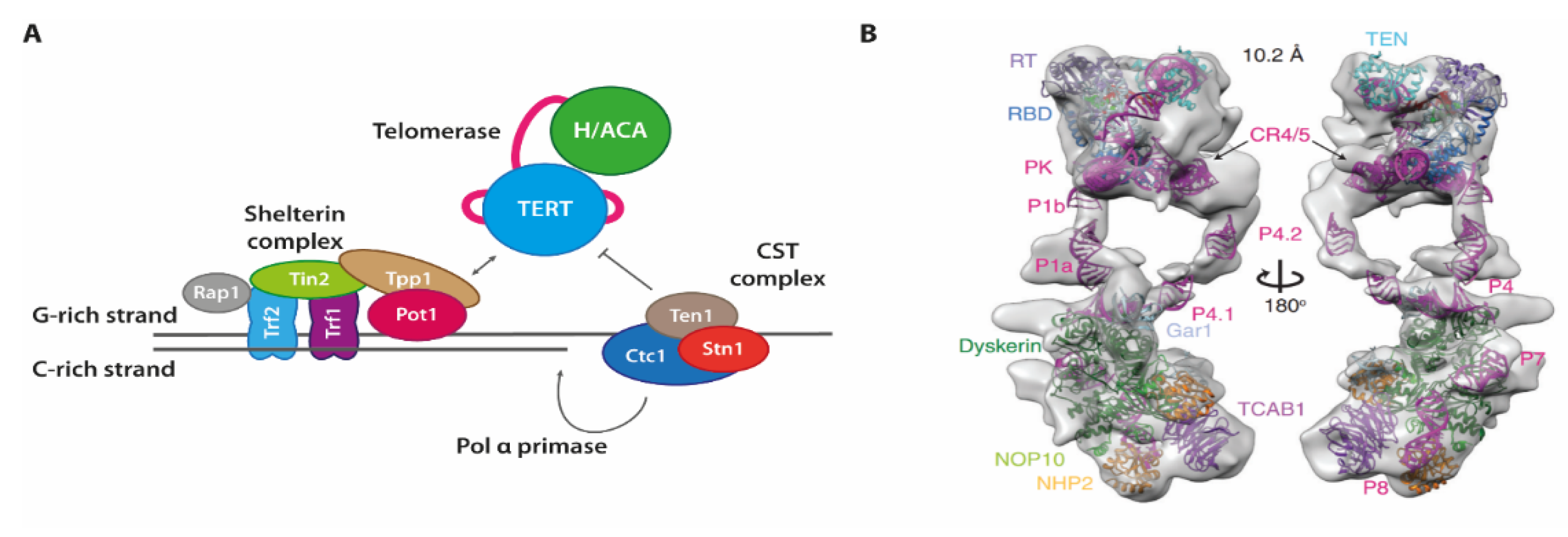
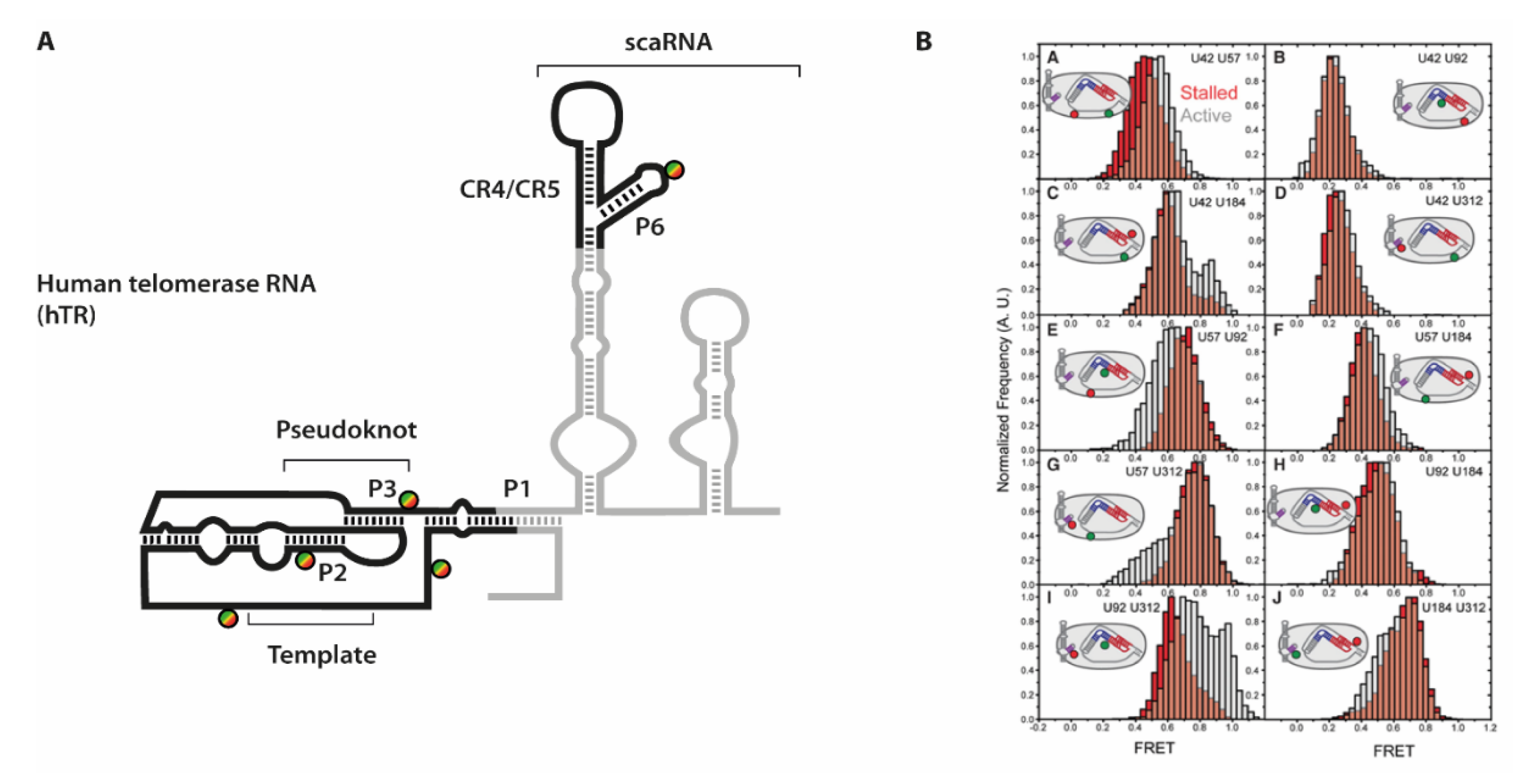
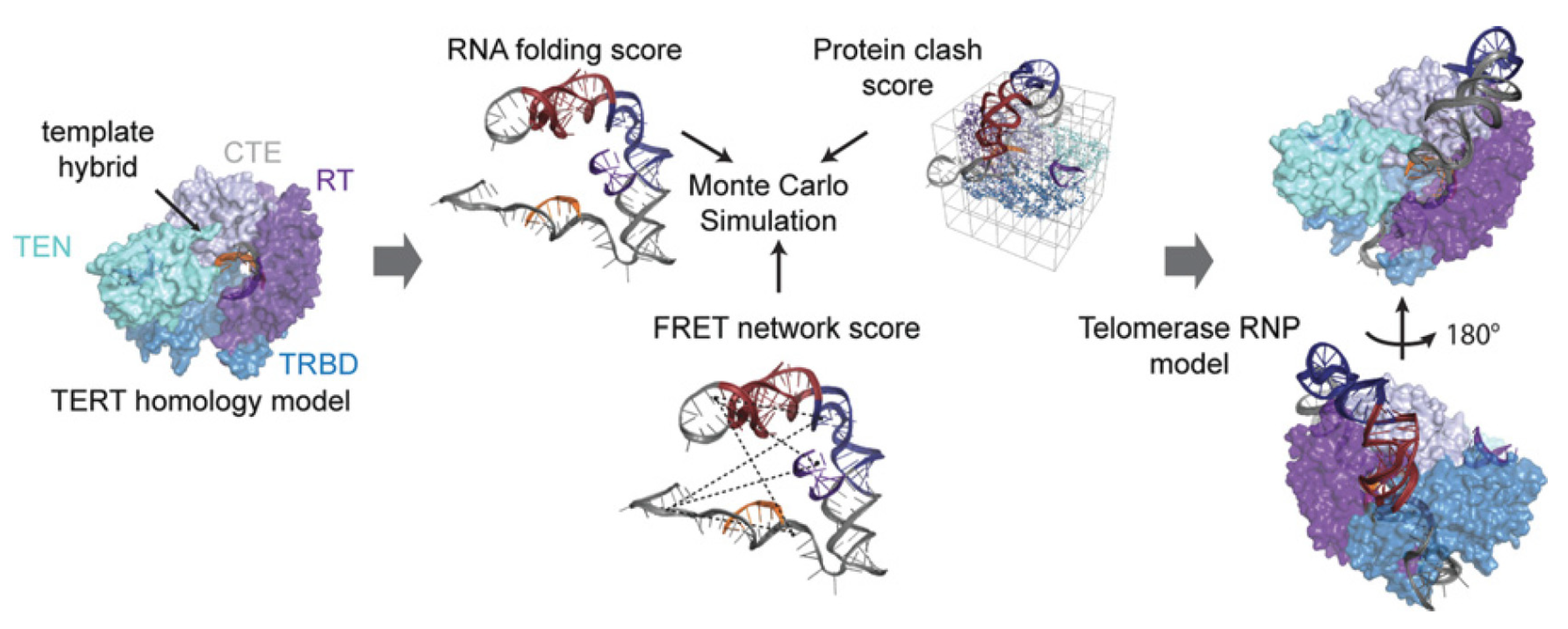
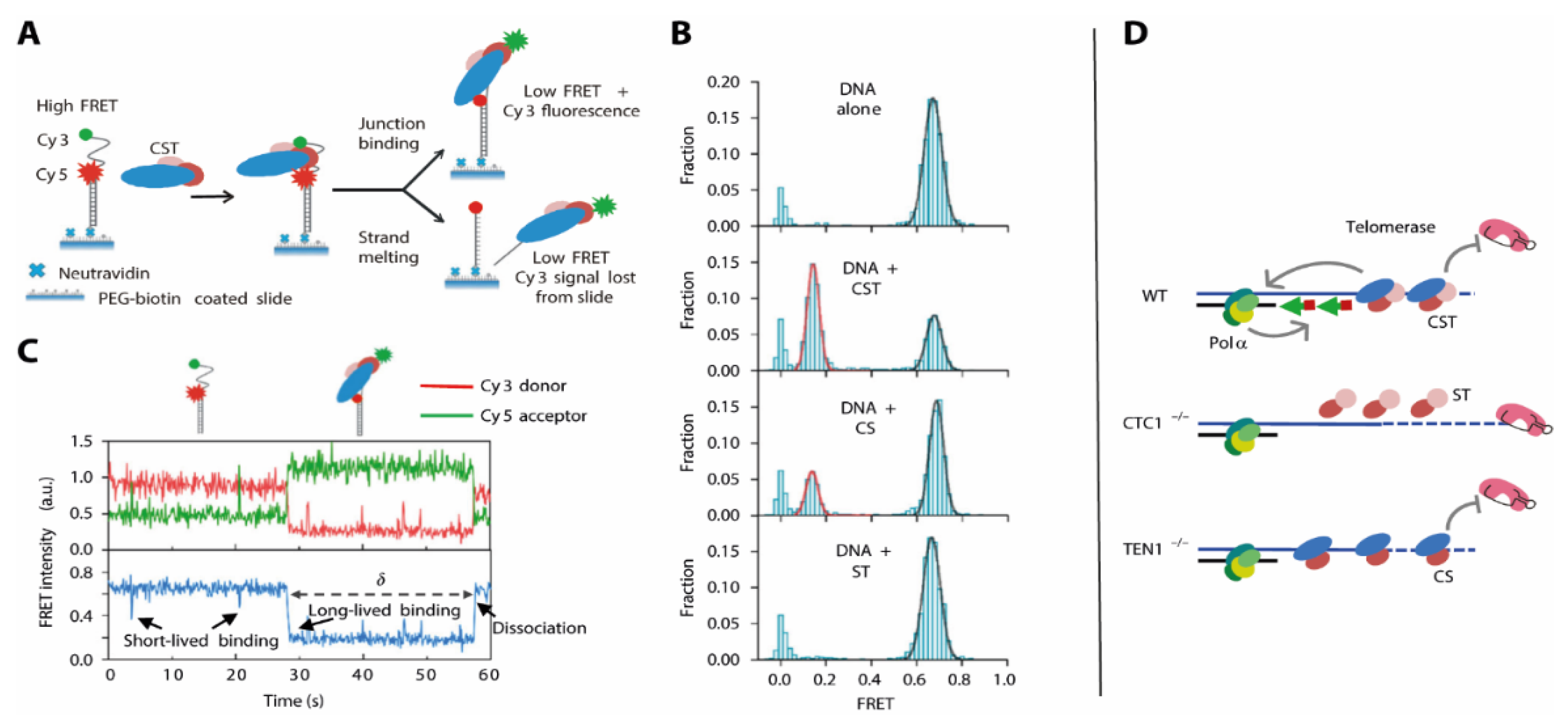
7. Telomere Maintenance
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yusupov, M.M.; Yusupova, G.Z.; Baucom, A.; Lieberman, K.; Earnest, T.N.; Cate, J.H.D.; Noller, H.F. Crystal structure of the ribosome at 5.5 Å resolution. Science 2001, 292, 883–896. [Google Scholar] [CrossRef]
- Jalihal, A.P.; Lund, P.E.; Walter, N.G. Coming together: Rnas and proteins assemble under the single-molecule fluorescence microscope. Cold Spring Harb. Perspect. Biol. 2019, 11, a032441. [Google Scholar] [CrossRef] [Green Version]
- Hanspach, G.; Trucks, S.; Hengesbach, M. Strategic labelling approaches for RNA single-molecule spectroscopy. RNA Biol. 2019, 16, 1119–1132. [Google Scholar] [CrossRef]
- Milles, S.; Tyagi, S.; Banterle, N.; Koehler, C.; Vandelinder, V.; Plass, T.; Neal, A.P.; Lemke, E.A. Click strategies for single-molecule protein fluorescence. J. Am. Chem. Soc. 2012, 134, 5187–5195. [Google Scholar] [CrossRef] [PubMed]
- Jakob, L.; Gust, A.; Grohmann, D. Evaluation and optimisation of unnatural amino acid incorporation and bioorthogonal bioconjugation for site-specific fluorescent labelling of proteins expressed in mammalian cells. Biochem. Biophys. Rep. 2019, 17, 1–9. [Google Scholar] [CrossRef]
- Schuler, B.; Soranno, A.; Hofmann, H.; Nettels, D. Single-Molecule FRET Spectroscopy and the Polymer Physics of Unfolded and Intrinsically Disordered Proteins. Annu. Rev. Biophys. 2016, 45, 207–231. [Google Scholar] [CrossRef] [Green Version]
- Andreou, A.Z.; Klostermeier, D. EIF4B and eIF4G jointly stimulate eIF4A ATPase and unwinding activities by modulation of the eIF4A conformational cycle. J. Mol. Biol. 2014, 426, 51–61. [Google Scholar] [CrossRef]
- Andreou, A.Z.; Harms, U.; Klostermeier, D. Single-stranded regions modulate conformational dynamics and ATPase activity of eIF4A to optimize 5′-UTR unwinding. Nucleic Acids Res. 2019, 47, 5260–5275. [Google Scholar] [CrossRef]
- Munro, J.B.; Altman, R.B.; O’Connor, N.; Blanchard, S.C. Identification of Two Distinct Hybrid State Intermediates on the Ribosome. Mol. Cell 2007, 25, 505–517. [Google Scholar] [CrossRef] [Green Version]
- Prokhorova, I.; Altman, R.B.; Djumagulov, M.; Shrestha, J.P.; Urzhumtsev, A.; Ferguson, A.; Chang, C.W.T.; Yusupov, M.; Blanchard, S.C.; Yusupova, G.; et al. Aminoglycoside interactions and impacts on the eukaryotic ribosome. Proc. Natl. Acad. Sci. USA 2017, 114, E10899–E10908. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wasserman, M.R.; Feldman, M.B.; Altman, R.B.; Blanchard, S.C. Mechanistic insights into antibiotic action on the ribosome through single-molecule fluorescence imaging. Ann. N. Y. Acad. Sci. 2011, 1241, E1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, T.J.; Joo, C. Facilitated diffusion of Argonaute-mediated target search. RNA Biol. 2019, 16, 1093–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, T.J.; Klein, M.; Hegge, J.W.; Chandradoss, S.D.; van der Oost, J.; Depken, M.; Joo, C. Argonaute bypasses cellular obstacles without hindrance during target search. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kramm, K.; Endesfelder, U.; Grohmann, D. A Single-Molecule View of Archaeal Transcription. J. Mol. Biol. 2019, 431, 4116–4131. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.R.; Martin, J.S.; Kahlscheuer, M.L.; Krishnan, R.; Abelson, J.; Laederach, A.; Walter, N.G. Single Molecule Cluster Analysis dissects splicing pathway conformational dynamics. Nat. Methods 2015, 12, 1077–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, M.L.; Tretbar, U.S.; Dehaven, A.; Alwan, A.A.; Luo, G.; Mast, H.M.; Hoskins, A.A. Conformational dynamics of stem II of the U2 snRNA. RNA 2016, 22, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Van der Feltz, C.; DeHaven, A.C.; Hoskins, A.A. Stress-induced Pseudouridylation Alters the Structural Equilibrium of Yeast U2 snRNA Stem II. J. Mol. Biol. 2018, 430, 524–536. [Google Scholar] [CrossRef]
- Beier, D.H.; Carrocci, T.J.; van der Feltz, C.; Tretbar, U.S.; Paulson, J.C.; Grabowski, N.; Hoskins, A.A. Dynamics of the DEAD-box ATPase Prp5 RecA-like domains provide a conformational switch during spliceosome assembly. Nucleic Acids Res. 2019, 47, 10842–10851. [Google Scholar] [CrossRef]
- Tippana, R.; Hwang, H.; Opresko, P.L.; Bohr, V.A.; Myong, S. Single-molecule imaging reveals a common mechanism shared by G-quadruplex–resolving helicases. Proc. Natl. Acad. Sci. USA 2016, 113, 8448–8453. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.C.; Tippana, R.; Demeshkina, N.A.; Murat, P.; Balasubramanian, S.; Myong, S.; Ferré-D’amaré, A.R. Structural basis of G-quadruplex unfolding by the DEAH/RHA helicase DHX36. Nature 2018, 558, 465–483. [Google Scholar] [CrossRef]
- Tippana, R.; Chen, M.C.; Demeshkina, N.A.; Ferré-D’Amaré, A.R.; Myong, S. RNA G-quadruplex is resolved by repetitive and ATP-dependent mechanism of DHX36. Nat. Commun. 2019, 10, 1855. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.F.; Rety, S.; Guo, H.L.; Dai, Y.X.; Wu, W.Q.; Liu, N.N.; Auguin, D.; Liu, Q.W.; Hou, X.M.; Dou, S.X.; et al. Molecular Mechanistic Insights into Drosophila DHX36-Mediated G-Quadruplex Unfolding: A Structure-Based Model. Structure 2018, 26, 403–415.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Hsu, S.J.; Bhattacharjee, A.; Wang, Y.; Diao, J.; Price, C.M. CTC1-STN1 terminates telomerase while STN1-TEN1 enables C-strand synthesis during telomere replication in colon cancer cells. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, J.W.; Kappel, K.; Das, R.; Stone, M.D. Single-molecule FRET-Rosetta reveals RNA structural rearrangements during human telomerase catalysis. Rna 2017, 23, 175–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharjee, A.; Wang, Y.; Diao, J.; Price, C.M. Dynamic DNA binding, junction recognition and G4 melting activity underlie the telomeric and genome-wide roles of human CST. Nucleic Acids Res. 2017, 45, 12311–12324. [Google Scholar] [CrossRef] [Green Version]
- Wan, R.; Yan, C.; Bai, R.; Wang, L.; Huang, M.; Wong, C.C.L.; Shi, Y. The 3.8 Å structure of the U4/U6.U5 tri-snRNP: Insights into spliceosome assembly and catalysis. Science 2016, 351, 466–475. [Google Scholar] [CrossRef]
- Agafonov, D.E.; Kastner, B.; Dybkov, O.; Hofele, R.V.; Liu, W.T.; Urlaub, H.; Lührmann, R.; Stark, H. Molecular architecture of the human U4/U6.U5 tri-snRNP. Science 2016, 351, 1416–1420. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.H.D.; Galej, W.P.; Bai, X.C.; Oubridge, C.; Newman, A.J.; Scheres, S.H.W.; Nagai, K. Cryo-EM structure of the yeast U4/U6.U5 tri-snRNP at 3.7 Å resolution. Nature 2016, 530, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, R.; Blanco, M.R.; Kahlscheuer, M.L.; Abelson, J.; Guthrie, C.; Walter, N.G. Biased Brownian ratcheting leads to pre-mRNA remodeling and capture prior to first-step splicing. Nat. Struct. Mol. Biol. 2013, 20, 1450–1457. [Google Scholar] [CrossRef] [Green Version]
- Dalbadie-McFarland, G.; Abelson, J. PRP5: A helicase-like protein required for mRNA splicing in yeast. Proc. Natl. Acad. Sci. USA 1990, 87, 4236–4240. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.M.; Yang, F.; Zhang, J.; Tang, Q.; Li, J.; Gu, J.; Zhou, J.; Xu, Y.Z. Crystal Structure Of Prp5P Reveals Interdomain Interactions That Impact Spliceosome Assembly. Cell Rep. 2013, 5, 1269–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, W.W.; Cheng, S.C. A novel mechanism for Prp5 function in prespliceosome formation and proofreading the branch site sequence. Genes Dev. 2015, 29, 81–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.Z.; Query, C.C. Competition between the ATPase Prp5 and Branch Region-U2 snRNA Pairing Modulates the Fidelity of Spliceosome Assembly. Mol. Cell 2007, 28, 838–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karow, A.R.; Klostermeier, D. A conformational change in the helicase core is necessary but not sufficient for RNA unwinding by the DEAD box helicase YxiN. Nucleic Acids Res. 2009, 37, 4464–4471. [Google Scholar] [CrossRef] [PubMed]
- Perriman, R.; Barta, I.; Voeltz, G.K.; Abelsont, J.; Ares, M. ATP requirement for Prp5p function is determined by Cus2p and the structure of U2 small nuclear RNA. Proc. Natl. Acad. Sci. USA 2003, 100, 13857–13862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talkish, J.; Igel, H.; Hunter, O.; Horner, S.W.; Jeffery, N.N.; Leach, J.R.; Jenkins, J.L.; Kielkopf, C.L.; Ares, M. Cus2 enforces the first ATP-dependent step of splicing by binding to yeast SF3b1 through a UHM–Ulm interaction. Rna 2019, 25, 1020–1037. [Google Scholar] [CrossRef] [Green Version]
- Hilliker, A.K.; Mefford, M.A.; Staley, J.P. U2 toggles iteratively between the stem IIa and stem IIc conformations to promote pre-mRNA splicing. Genes Dev. 2007, 21, 821–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perriman, R.J.; Ares, M. Rearrangement of competing U2 RNA helices within the spliceosome promotes multiple steps in splicing. Genes Dev. 2007, 21, 811–820. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Perriman, R.; Igel, H.; Howe, K.J.; Neville, M.; Ares, M. CUS2, a Yeast Homolog of Human Tat-SF1, Rescues Function of Misfolded U2 through an Unusual RNA Recognition Motif. Mol. Cell. Biol. 1998, 18, 5000–5009. [Google Scholar] [CrossRef] [Green Version]
- Newby, M.I.; Greenbaum, N.L. A conserved pseudouridine modification in eukaryotic U2 snRNA induces a change in branch-site architecture. Rna 2001, 7, 833–845. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Adachi, H.; Ge, J.; Stephenson, D.; Query, C.C.; Yu, Y. Pseudouridines in U2 snRNA stimulate the ATPase activity of Prp5 during spliceosome assembly. EMBO J. 2016, 35, 654–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; McPheeters, D.S.; Yu, Y.T. Ψ35 in the branch site recognition region of U2 small nuclear RNA is important for pre-mRNA splicing in Saccharomyces cerevisiae. J. Biol. Chem. 2005, 280, 6655–6662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Radwan, M.K.; Xiao, M.; Adachi, H.; Fan, J.; Yu, Y.T. The TOR signaling pathway regulates starvation-induced pseudouridylation of yeast U2 snRNA. Rna 2016, 22, 1146–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Xiao, M.; Yang, C.; Yu, Y.T. U2 snRNA is inducibly pseudouridylated at novel sites by Pus7p and snR81 RNP. EMBO J. 2011, 30, 79–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronson, J.E.; Fei, J.; Hofman, J.M.; Gonzalez, R.L.; Wiggins, C.H. Learning rates and states from biophysical time series: A Bayesian approach to model selection and single-molecule FRET data. Biophys. J. 2009, 97, 3196–3205. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, D.E.; Feigon, J. Multistranded DNA structures. Curr. Opin. Struct. Biol. 1999, 9, 305–314. [Google Scholar] [CrossRef]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA secondary structures: Stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef] [Green Version]
- Lam, E.Y.N.; Beraldi, D.; Tannahill, D.; Balasubramanian, S. G-quadruplex structures are stable and detectable in human genomic DNA. Nat. Commun. 2013, 4, 1–8. [Google Scholar] [CrossRef]
- Rhodes, D.; Lipps, H.J. Survey and summary G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [Green Version]
- Luke, B.; Lingner, J. TERRA: Telomeric repeat-containing RNA. EMBO J. 2009, 28, 2503–2510. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.U.; Bartel, D.P. RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science 2016, 353, aaf5371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tippana, R.; Xiao, W.; Myong, S. G-quadruplex conformation and dynamics are determined by loop length and sequence. Nucleic Acids Res. 2014, 42, 8106–8114. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, O.; Bourdoncle, A.; Boulé, J.B.; Brosh, R.M.; Mergny, J.L. G-quadruplexes and helicases. Nucleic Acids Res. 2016, 44, 1989–2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millevoi, S.; Moine, H.; Vagner, S. G-quadruplexes in RNA biology. Wiley Interdiscip. Rev. RNA 2012, 3, 495–507. [Google Scholar] [CrossRef]
- Harrington, C.; Lan, Y.; Akman, S.A. The Identification and Characterization of a G4-DNA Resolvase Activity. J. Biol. Chem. 1997, 272, 24631–24636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creacy, S.D.; Routh, E.D.; Iwamoto, F.; Nagamine, Y.; Akman, S.A.; Vaughn, J.P. G4 Resolvase 1 Binds Both DNA and RNA Tetramolecular Quadruplex with High Affinity and Is the Major Source of Tetramolecular Quadruplex G4-DNA and G4-RNA Resolving Activity in HeLa Cell Lysates. J. Biol. Chem. 2008, 283, 34626–34634. [Google Scholar] [CrossRef] [Green Version]
- Giri, B.; Smaldino, P.J.; Thys, R.G.; Creacy, S.D.; Routh, E.D.; Hantgan, R.R.; Lattmann, S.; Nagamine, Y.; Akman, S.A.; Vaughn, J.P. G4 Resolvase 1 tightly binds and unwinds unimolecular G4-DNA. Nucleic Acids Res. 2011, 39, 7161–7178. [Google Scholar] [CrossRef] [Green Version]
- Lattmann, S.; Stadler, M.B.; Vaughn, J.P.; Akman, S.A.; Nagamine, Y. The DEAH-box RNA helicase RHAU binds an intramolecular RNA G-quadruplex in TERC and associates with telomerase holoenzyme. Nucleic Acids Res. 2011, 39, 9390–9404. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-L. Template boundary definition in mammalian telomerase. Genes Dev. 2003, 17, 2747–2752. [Google Scholar] [CrossRef] [Green Version]
- Heddi, B.; Cheong, V.V.; Martadinata, H.; Phan, A.T. Insights into G-quadruplex specific recognition by the DEAH-box helicase RHAU: Solution structure of a peptide–quadruplex complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9608–9613. [Google Scholar] [CrossRef] [Green Version]
- De Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.; Wang, Y.; Feigon, J. Progress in Human and Tetrahymena Telomerase Structure Determination. Annu. Rev. Biophys. 2017, 46, 199–225. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Sušac, L.; Feigon, J. Structural biology of telomerase. Cold Spring Harb. Perspect. Biol. 2019, 11, a032383. [Google Scholar] [CrossRef] [PubMed]
- Egan, E.D.; Collins, K. Biogenesis of telomerase ribonucleoproteins. RNA 2012, 18, 1747–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, M.; Gillis, A.; Futahashi, M.; Fujiwara, H.; Skordalakes, E. Structural basis for telomerase catalytic subunit TERT binding to RNA template and telomeric DNA. Nat. Struct. Mol. Biol. 2010, 17, 513–518. [Google Scholar] [CrossRef]
- Jansson, L.I.; Akiyama, B.M.; Ooms, A.; Lu, C.; Rubin, S.M.; Stone, M.D. Structural basis of template-boundary definition in Tetrahymena telomerase. Nat. Struct. Mol. Biol. 2015, 22, 883–888. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Wang, Z.; Koo, B.K.; Patel, A.; Cascio, D.; Collins, K.; Feigon, J. Structural Basis for Telomerase RNA Recognition and RNP Assembly by the Holoenzyme La Family Protein p65. Mol. Cell 2012, 47, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Kim, N.K.; Peterson, R.D.; Wang, Z.; Feigon, J. Structurally conserved five nucleotide bulge determines the overall topology of the core domain of human telomerase RNA. Proc. Natl. Acad. Sci. USA 2010, 107, 18761–18768. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.-K.; Theimer, C.A.; Mitchell, J.R.; Collins, K.; Feigon, J. Effect of pseudouridylation on the structure and activity of the catalytically essential P6.1 hairpin in human telomerase RNA. Nucleic Acids Res. 2010, 38, 6746–6756. [Google Scholar] [CrossRef] [Green Version]
- Sauerwald, A.; Sandin, S.; Cristofari, G.; Scheres, S.H.W.; Lingner, J.; Rhodes, D. Structure of active dimeric human telomerase. Nat. Struct. Mol. Biol. 2013, 20, 454–460. [Google Scholar] [CrossRef]
- Jiang, J.; Chan, H.; Cash, D.D.; Miracco, E.J.; Loo, R.R.O.; Upton, H.E.; Cascio, D.; Johnson, R.O.B.; Collins, K.; Loo, J.A.; et al. Structure of Tetrahymena telomerase reveals previously unknown subunits, functions, and interactions. Science 2015, 350, aab4070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Wang, Y.; Sušac, L.; Chan, H.; Basu, R.; Zhou, Z.H.; Feigon, J. Structure of Telomerase with Telomeric DNA. Cell 2018, 173, 1179–1190.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.H.D.; Tam, J.; Wu, R.A.; Greber, B.J.; Toso, D.; Nogales, E.; Collins, K. Cryo-EM structure of substrate-bound human telomerase holoenzyme. Nature 2018, 557, 190–195. [Google Scholar] [CrossRef]
- Mihalusova, M.; Wu, J.Y.; Zhuang, X. Functional importance of telomerase pseudoknot revealed by single-molecule analysis. Proc. Natl. Acad. Sci. USA 2011, 108, 20339–20344. [Google Scholar] [CrossRef] [Green Version]
- Stone, M.D.; Mihalusova, M.; O’Connor, C.M.; Prathapam, R.; Collins, K.; Zhuang, X. Stepwise protein-mediated RNA folding directs assembly of telomerase ribonucleoprotein. Nature 2007, 446, 458–461. [Google Scholar] [CrossRef]
- Chen, G.; Wen, J.D.; Tinoco, I. Single-molecule mechanical unfolding and folding of a pseudoknot in human telomerase RNA. RNA 2007, 13, 2175–2188. [Google Scholar] [CrossRef] [Green Version]
- Hengesbach, M.; Kim, N.K.; Feigon, J.; Stone, M.D. Single-molecule FRET reveals the folding dynamics of the human telomerase rna pseudoknot domain. Angew. Chem. Int. Ed. 2012, 51, 5876–5879. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, B.M.; Loper, J.; Najarro, K.; Stone, M.D. The C-terminal domain of Tetrahymena thermophila telomerase holoenzyme protein p65 induces multiple structural changes in telomerase RNA. RNA 2012, 18, 653–660. [Google Scholar] [CrossRef] [Green Version]
- Berman, A.J.; Akiyama, B.M.; Stone, M.D.; Cech, T.R. The RNA accordion model for template positioning by telomerase RNA during telomeric DNA synthesis. Nat. Struct. Mol. Biol. 2011, 18, 1371–1375. [Google Scholar] [CrossRef] [Green Version]
- Wallweber, G.; Gryaznov, S.; Pongracz, K.; Pruzan, R. Interaction of human telomerase with its primer substrate. Biochemistry 2003, 42, 589–600. [Google Scholar] [CrossRef]
- Parks, J.W.; Stone, M.D. Coordinated DNA dynamics during the human telomerase catalytic cycle. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.Y.; Redon, S.; Lingner, J. The human CST complex is a terminator of telomerase activity. Nature 2012, 488, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, B.; Li, T.; Liu, R.; Xiao, Y.; Geng, X.; Li, G.; Liu, Q.; Price, C.M.; Liu, Y.; et al. Mammalian CST averts replication failure by preventing G-quadruplex accumulation. Nucleic Acids Res. 2019, 47, 5243–5259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraud-Panis, M.-J.; Teresa Teixeira, M.; Gé li, V.; Gilson, E. Molecular Cell Review CST Meets Shelterin to Keep Telomeres in Check. Mol. Cell 2010, 39, 665–676. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meiser, N.; Fuks, C.; Hengesbach, M. Cooperative Analysis of Structural Dynamics in RNA-Protein Complexes by Single-Molecule Förster Resonance Energy Transfer Spectroscopy. Molecules 2020, 25, 2057. https://doi.org/10.3390/molecules25092057
Meiser N, Fuks C, Hengesbach M. Cooperative Analysis of Structural Dynamics in RNA-Protein Complexes by Single-Molecule Förster Resonance Energy Transfer Spectroscopy. Molecules. 2020; 25(9):2057. https://doi.org/10.3390/molecules25092057
Chicago/Turabian StyleMeiser, Nathalie, Christin Fuks, and Martin Hengesbach. 2020. "Cooperative Analysis of Structural Dynamics in RNA-Protein Complexes by Single-Molecule Förster Resonance Energy Transfer Spectroscopy" Molecules 25, no. 9: 2057. https://doi.org/10.3390/molecules25092057
APA StyleMeiser, N., Fuks, C., & Hengesbach, M. (2020). Cooperative Analysis of Structural Dynamics in RNA-Protein Complexes by Single-Molecule Förster Resonance Energy Transfer Spectroscopy. Molecules, 25(9), 2057. https://doi.org/10.3390/molecules25092057