
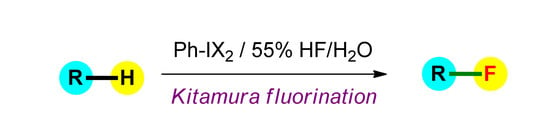
Kitamura Electrophilic Fluorination Using HF as a Source of Fluorine

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Fluorination of 1,3-Dicarbonyl Compounds
2.1. Iodosylbenzene-Mediated Fluorination
2.2. Catalytic Iodoarene-Mediated Fluorination
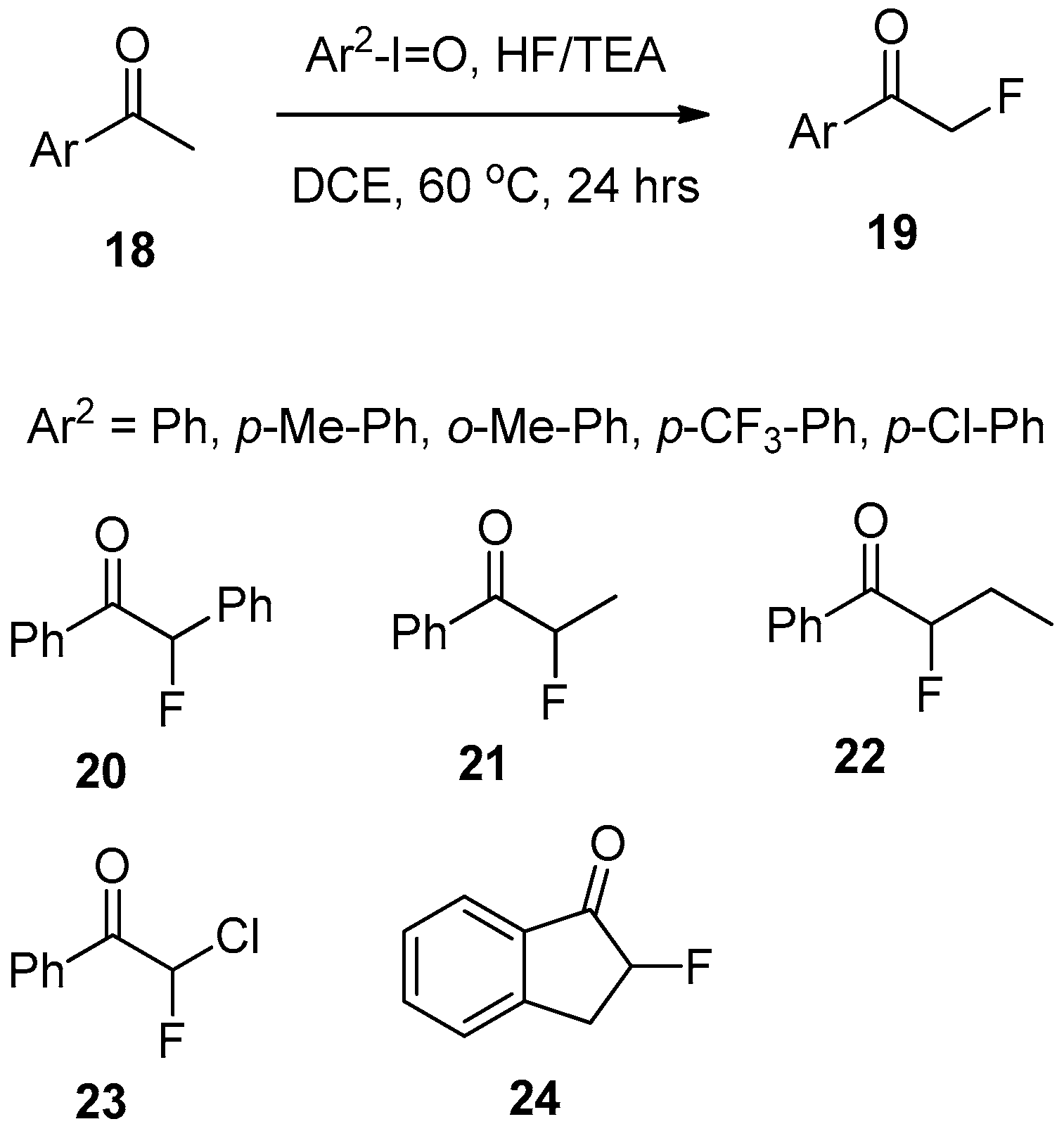
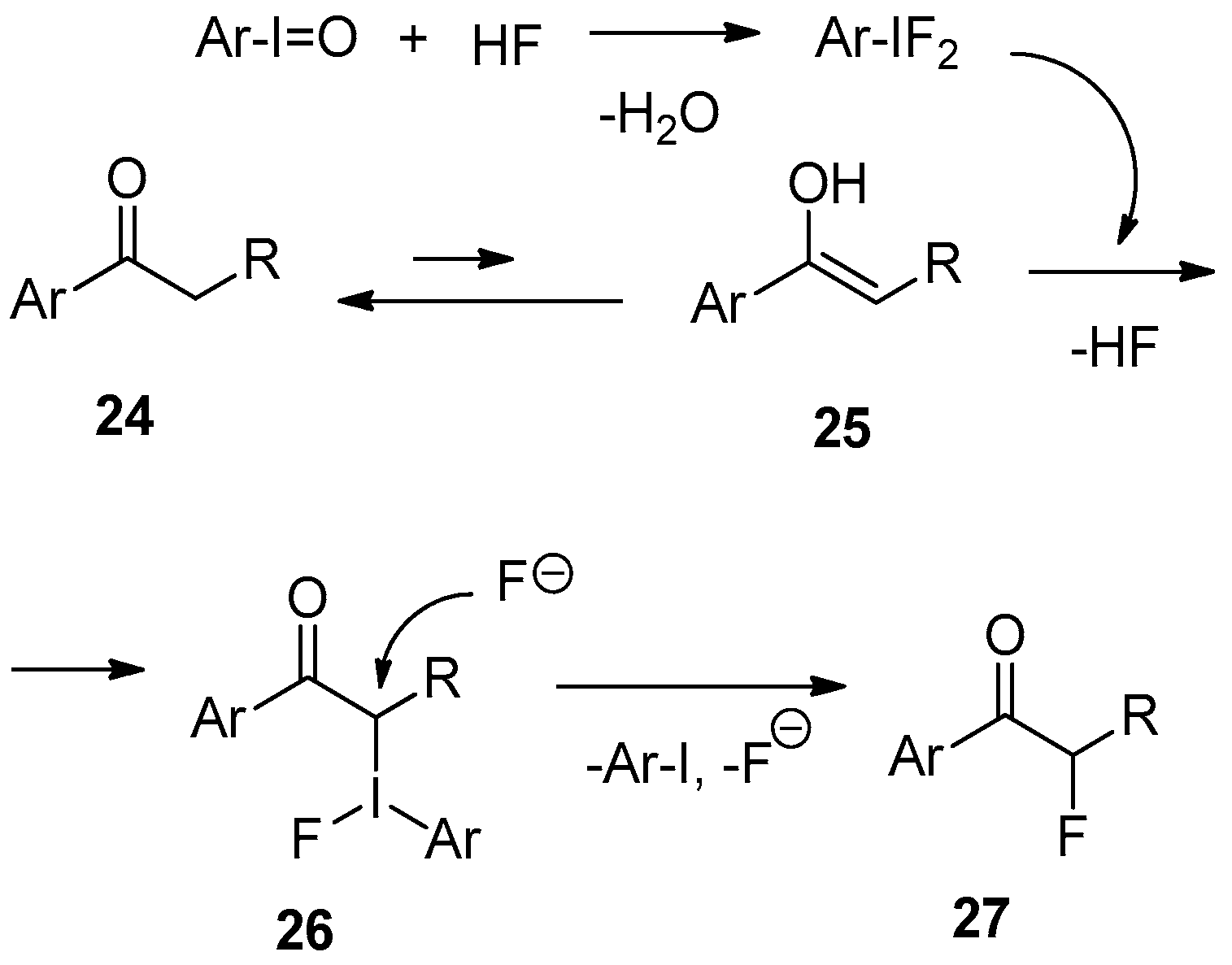
3. Fluorination of Aryl-Alkyl Ketones
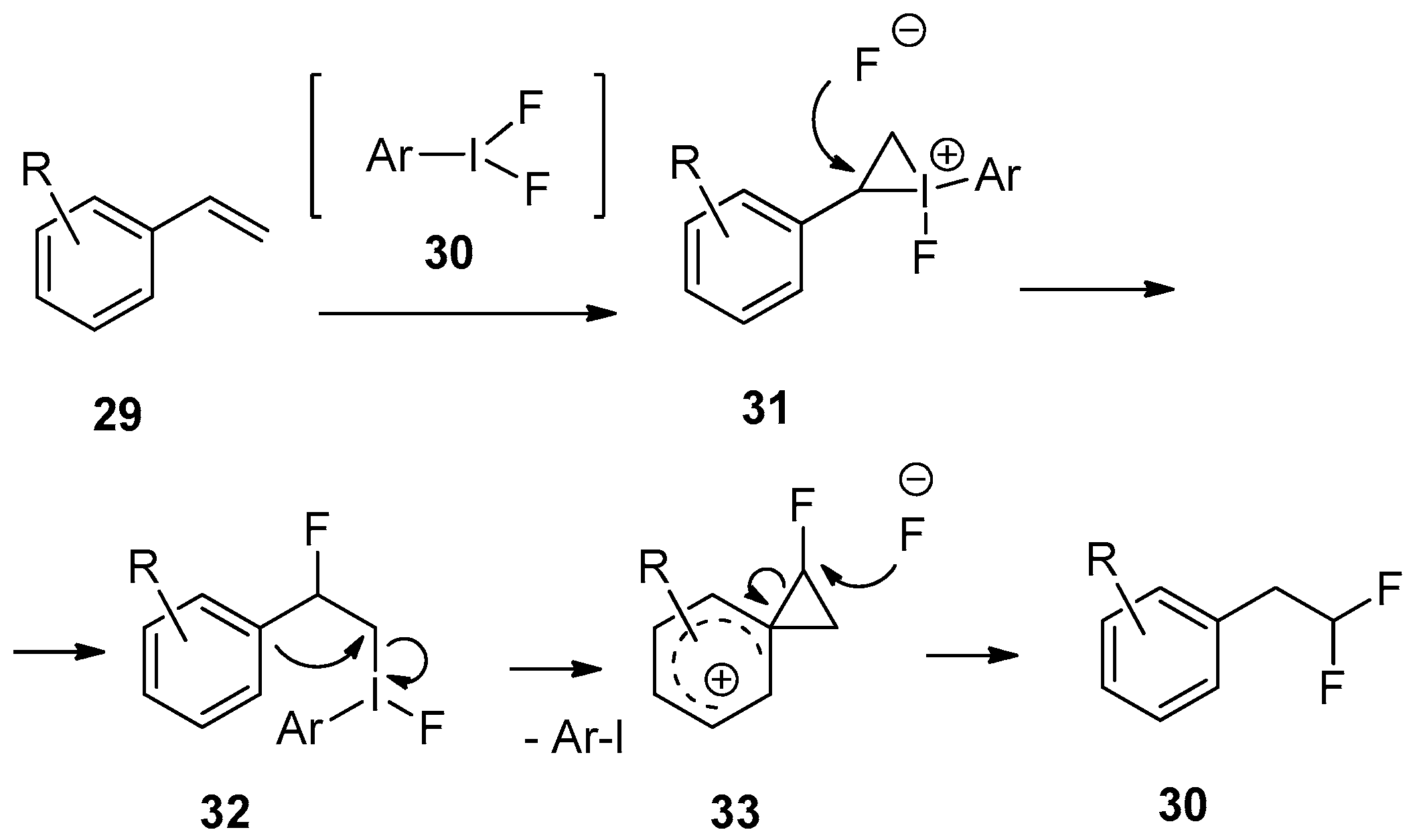
4. Fluorination of Styrene Derivatives
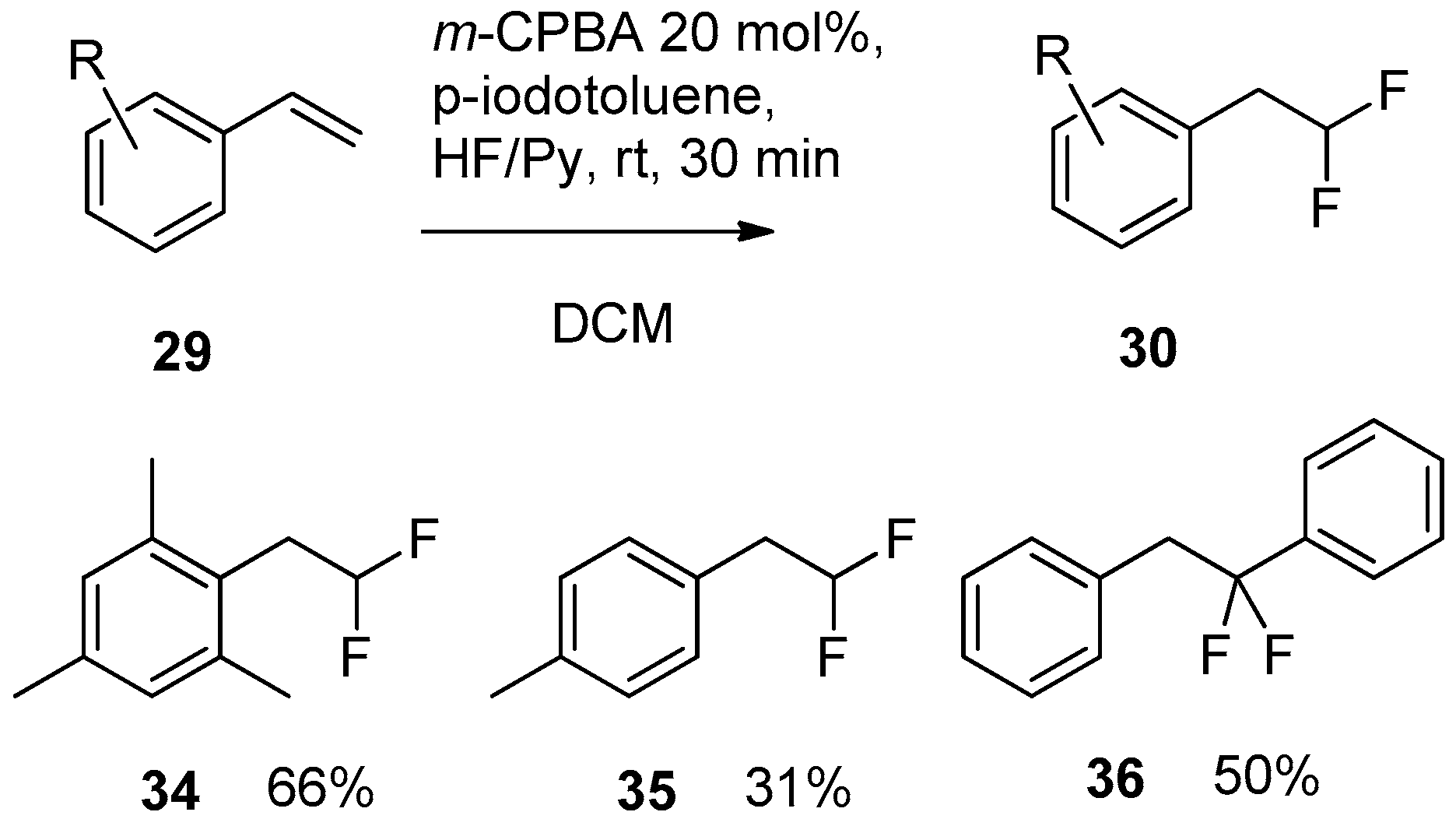
4.1. Hypervalent Iodine-Mediated Fluorination
4.1.1. Stoichiometric Hypervalent Iodine Reagents
4.1.2. Catalytic Hypervalent Iodine Reactions
5. Fluorination of α,β-Unsaturated Ketones and Alcohols
6. Cyclization–Fluorination Cascade
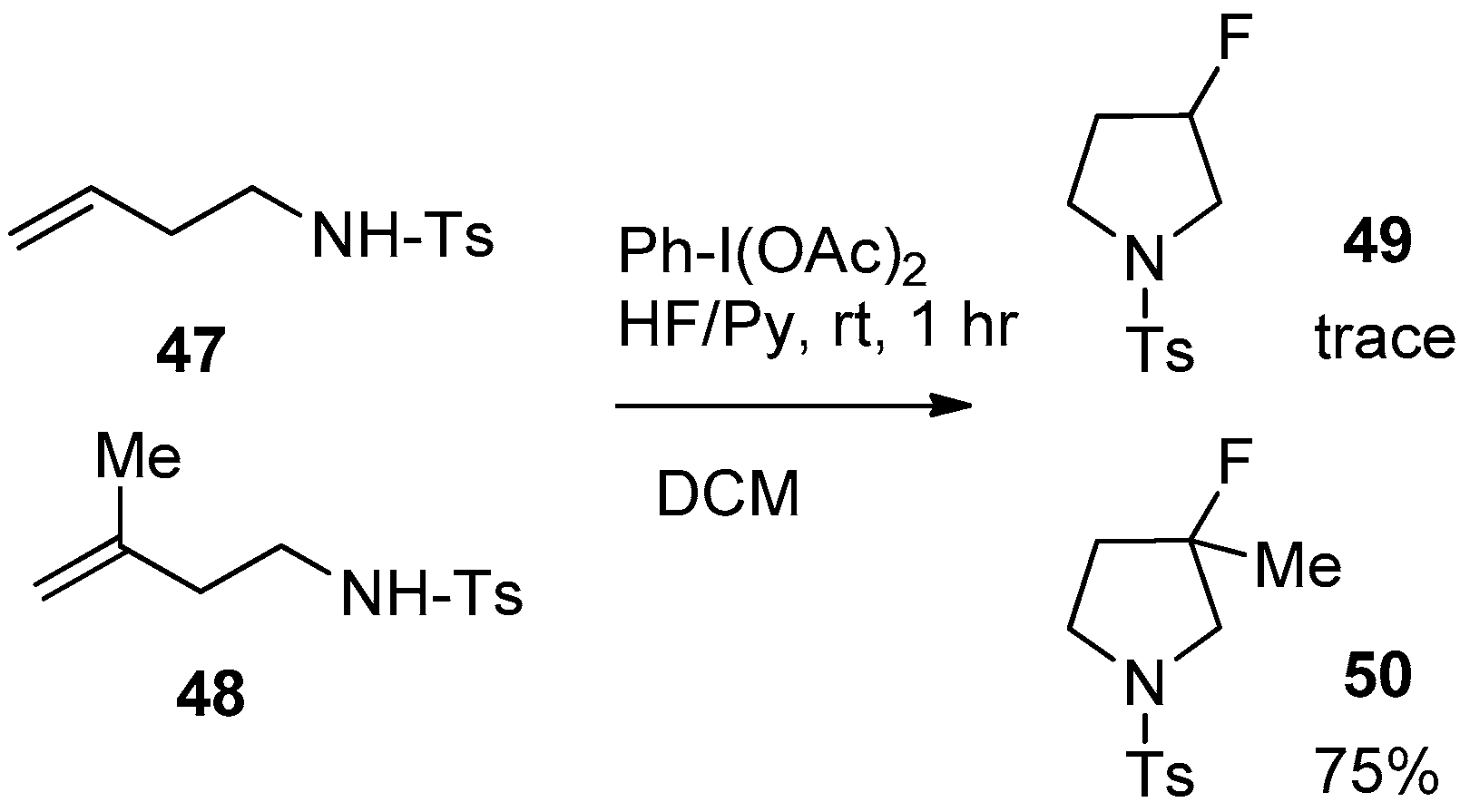
6.1. Homoallyl Amine Derivatives
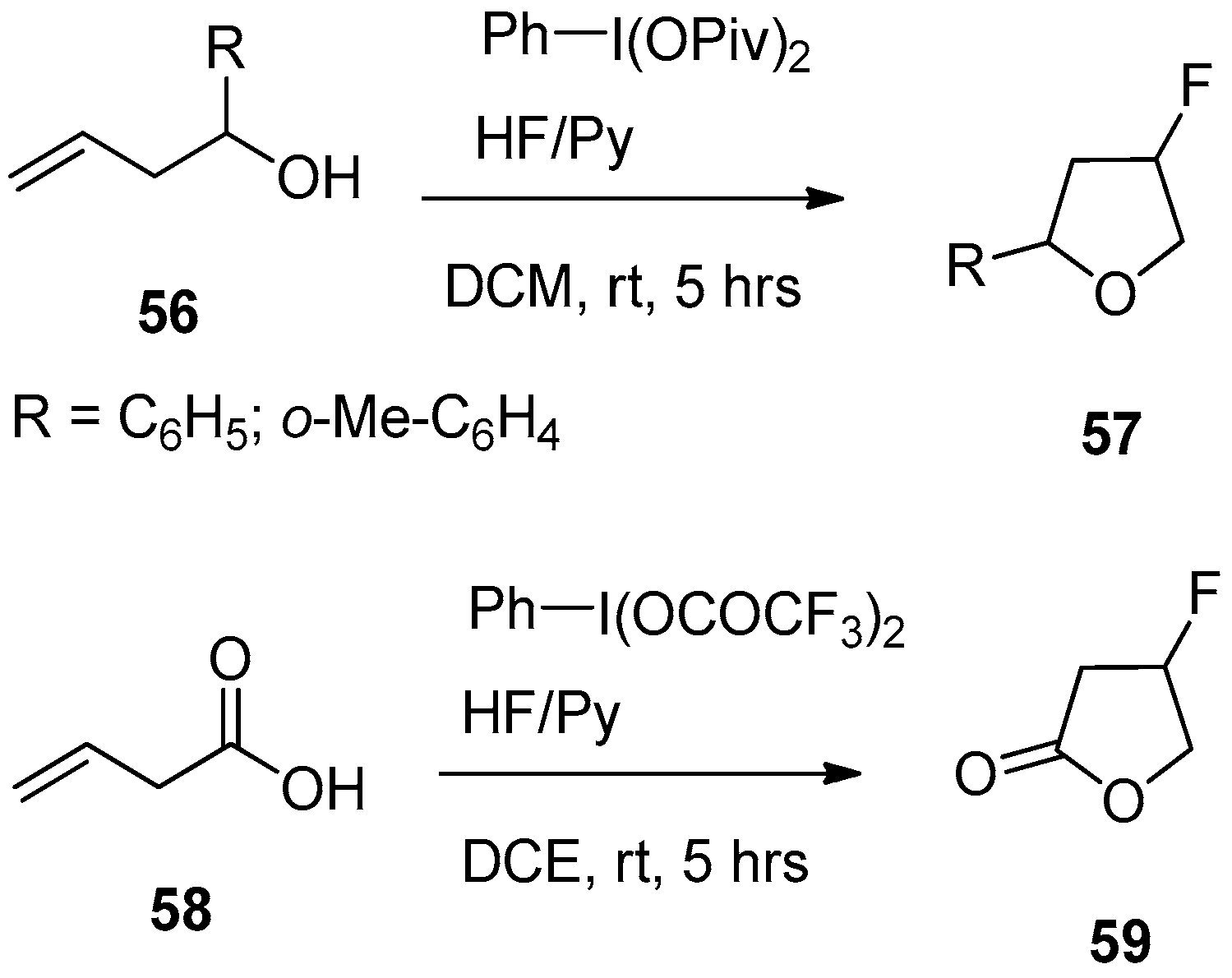
6.2. Homoallyl Alcohol and 3-butenoic Acid Derivatives
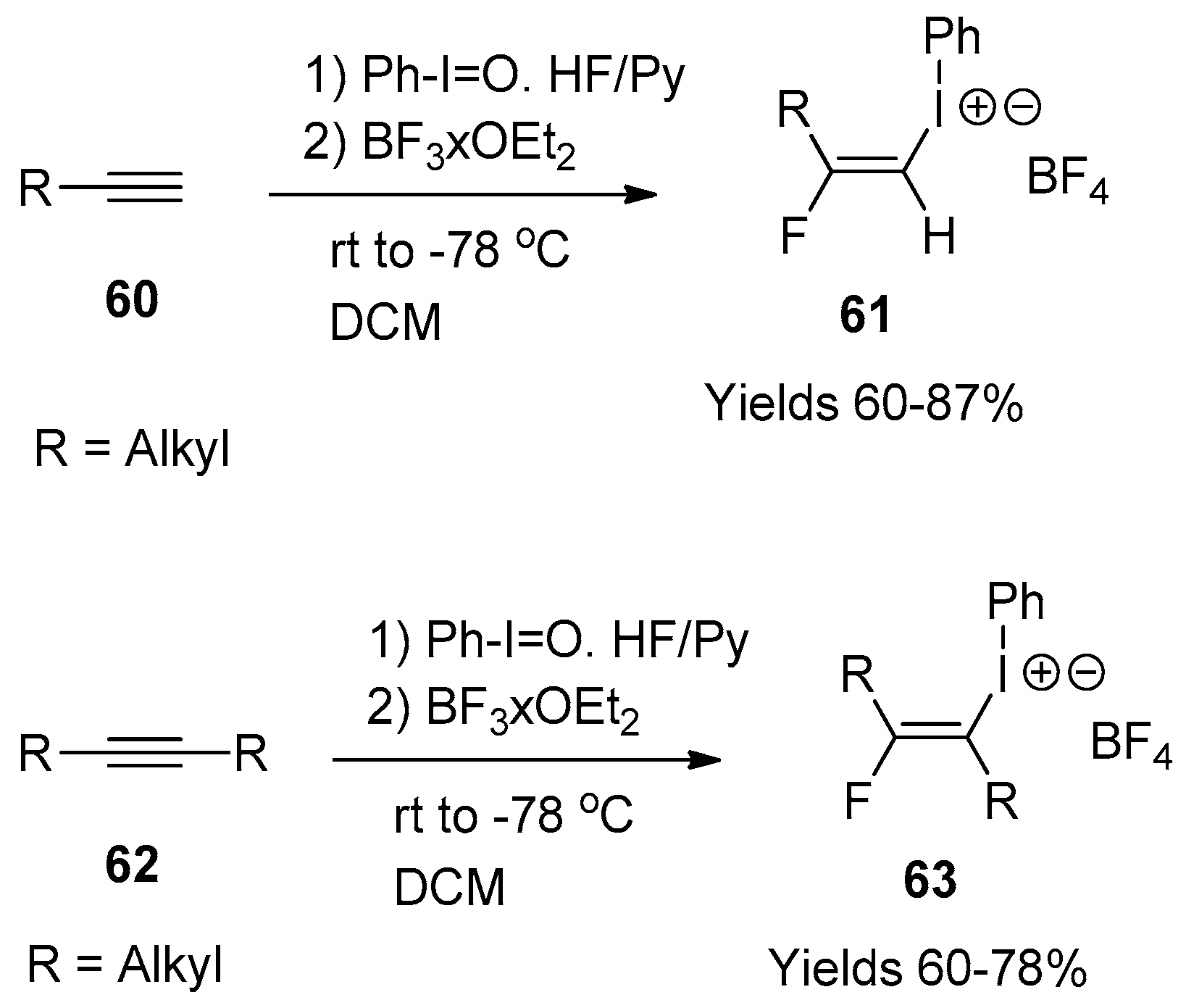
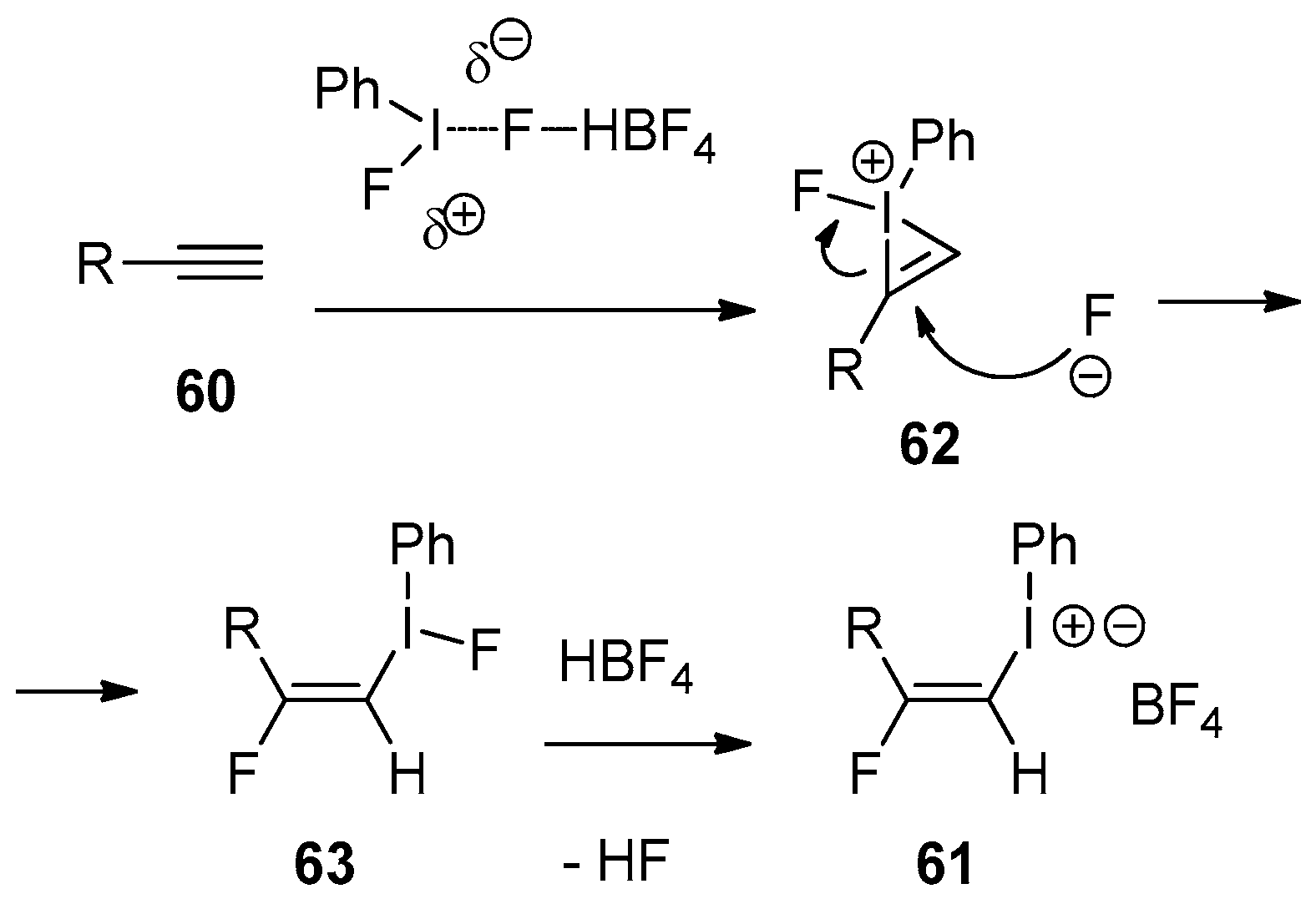
7. Reactions with Alkynes
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Aspern, N.; Röser, S.; Rad, B.R.; Murmann, P.; Streipert, B.; Mönnighoff, X.; Tillmann, S.D.; Shevchuk, M.; Stubbmann-Kazakova, O.; Röschenthaler, G.V.; et al. Phosphorus additives for improving high voltage stability and safety of lithium ion batteries. J. Fluorine Chem. 2017, 198, 24–33. [Google Scholar] [CrossRef]
- Gschwind, F.; Rodriguez-Garcia, G.; Sandbeck, D.J.S.; Gross, A.; Weil, M.; Fichtner, M.; Hörmann, N. Fluoride ion batteries: Theoretical performance, safety, toxicity, and a combinatorial screening of new electrodes. J. Fluorine Chem. 2016, 182, 76–90. [Google Scholar] [CrossRef]
- Jeong, E.; Lee, B.H.; Doh, S.J.; Park, I.J.; Lee, Y.S. Multifunctional surface modification of an aramid fabric via direct fluorination. J. Fluorine Chem. 2012, 141, 69–75. [Google Scholar] [CrossRef]
- Simone, C.D.; Vaccaro, E.; Scola, D.A. The synthesis and characterization of highly fluorinated aromatic polyimides. J. Fluorine Chem. 2019, 224, 100–112. [Google Scholar] [CrossRef]
- Miyajima, H.; Kasuya, M.C.Z.; Hatanaka, K. New fluorous gelators for perfluorodecalin. J. Fluorine Chem. 2019. [Google Scholar] [CrossRef]
- Fersing, C.; Bouhlel, A.; Cantelli, C.; Garrigue, P.; Lisowski, V.; Guillet, B. A Comprehensive Review of Non-Covalent Radiofluorination Approaches Using Aluminum [18F] fluoride: Will [18F] AlF Replace 68Ga for Metal Chelate Labeling? Molecules 2019, 24, 2866. [Google Scholar] [CrossRef] [Green Version]
- Norton, R.S.; Leung, E.W.W.; Chandrashekaran, I.R.; MacRaild, C.A. Applications of 19F-NMR in fragment-based drug discovery. Molecules 2016, 21, 860. [Google Scholar] [CrossRef]
- Bernard-Gauthier, V.; Bailey, J.J.; Berke, S.; Schirrmacher, R. Recent advances in the development and application of radiolabeled kinase inhibitors for PET imaging. Molecules 2015, 20, 22000–22027. [Google Scholar] [CrossRef] [Green Version]
- Pretze, M.; Pietzsch, D.; Mamat, C. Recent trends in bioorthogonal click-radiolabeling reactions using fluorine-18. Molecules 2013, 18, 8618–8665. [Google Scholar] [CrossRef] [Green Version]
- De la Torre, D.G.; Albericio, F. The pharmaceutical industry in 2018. An analysis of FDA drug approvals from the perspective of molecules. Molecules 2019, 24, 809. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; O’Hagan, D. Successful fluorine-containing herbicide agrochemicals. J. Fluorine Chem. 2014, 167, 16–29. [Google Scholar] [CrossRef]
- Jeschke, P. The unique role of halogen substituents in the design of modern agrochemicals. Pest Manag. Sci. 2010, 66, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Theodoridis, G. Chapter 4: Fluorine-Containing Agrochemicals: An Overview of Recent Developments. In Advance in Fluorine Science; Tressaud, A., Ed.; Elsevier: Amsterdam, The Netherlands; Volume 2, pp. 121–175. Available online: https://www.sciencedirect.com/science/article/pii/S1872035806020045 (accessed on 30 April 2020).
- Isanbor, C.; O’Hagan, D. Fluorine in medicinal chemistry: A review of anticancer agents. J. Fluorine Chem. 2006, 127, 303–319. [Google Scholar] [CrossRef]
- O’Hagan, D. Fluorine in health care: Organofluorine containing blockbuster drugs. J. Fluorine Chem. 2010, 131, 1071–1081. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, J.; Wang, S.; Gu, Z.; Aceña, J.L.; Izawa, K.; Liu, H.; Soloshonok, V.A. Recent advances in the trifluoromethylation methodology and new CF3-containing drugs. J. Fluorine Chem. 2014, 167, 37–54. [Google Scholar] [CrossRef]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef]
- Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V.A.; Coelho, J.A.S.; Toste, F.D. Modern Approaches for Asymmetric Construction of Carbon-Fluorine Quaternary Stereogenic Centers: Synthetic Challenges and Pharmaceutical Needs. Chem. Rev. 2018, 118, 3887–3964. [Google Scholar] [CrossRef]
- Mei, H.; Han, J.; Fustero, S.; Medio-Simon, M.; Sedgwick, D.M.; Santi, C.; Ruzziconi, R.; Soloshonok, V.A. Fluorine-Containing Drugs Approved by the FDA in 2018. Chem. Eur. J. 2019, 25, 11797–11819. [Google Scholar] [CrossRef]
- Mei, H.; Remete, A.M.; Zou, Y.; Moriwaki, H.; Fustero, S.; Kiss, L.; Soloshonok, V.A.; Han, J.L. Fluorine-containing drugs approved by the FDA in 2019. Chin. Chem. Lett. 2020. [Google Scholar] [CrossRef]
- Liu, J.; Li, Z.; Mei, H.; Soloshonok, V.A.; Han, J.L. Detrifluoroacetylative in Situ Generated Cyclic Fluorinated Enolates for the Preparation of Compounds Featuring a C–F Stereogenic Center. ACS Omega 2019, 4, 19505–19512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noel, M.; Suryanarayanan, V.; Chellammal, S. A review of recent developments in the selective electrochemical fluorination of organic compounds. J. Fluorine Chem. 1997, 83, 31–40. [Google Scholar] [CrossRef]
- Wu, J. Review of recent advances in nucleophilic C–F bond-forming reactions at sp3 centers. Tetrahedron Lett. 2014, 55, 4289–4294. [Google Scholar] [CrossRef] [Green Version]
- Kuehnel, M.F.; Lentz, D.; Braun, T. Synthesis of Fluorinated Building Blocks by Transition-Metal-Mediated Hydrodefluorination Reactions. Angew. Chem. Int. Ed. 2013, 52, 3328–3348. [Google Scholar] [CrossRef]
- Lectard, S.; Hamashima, Y.; Sodeoka, M. Recent Advances in Catalytic Enantioselective Fluorination Reactions. Adv. Synth. Catal. 2010, 352, 2708–2732. [Google Scholar] [CrossRef]
- Yerien, D.E.; Bonesi, S.; Postigo, A. Fluorination methods in drug discovery. Org. Biomol. Chem. 2016, 14, 8398–8427. [Google Scholar] [CrossRef] [Green Version]
- Soloshonok, V.A.; Cai, C.; Hruby, V.J.; Meervelt, L.V. Asymmetric Synthesis of Novel Highly Sterically Constrained (2S,3S)-3-Methyl-3-Trifluoromethyl- and (2S,3S,4R)-3-Trifluoromethyl-4-Methylpyroglutamic Acids. Tetrahedron 1999, 55, 12045–12058. [Google Scholar] [CrossRef]
- Han, J.; Sorochinsky, A.E.; Ono, T.; Soloshonok, V.A. Biomimetic Transamination — A Metal-Free Alternative to the Reductive Amination. Application for Generalized Preparation of Fluorine-Containing Amines and Amino Acids. Curr. Org. Synth. 2011, 8, 281–294. [Google Scholar]
- Aceña, J.L.; Sorochinsky, A.E.; Soloshonok, V.A. Asymmetric synthesis of α-amino acids via homologation of Ni(II) complexes of glycine Schiff bases. Part 3: Michael addition reactions and miscellaneous transformations. Amino Acids 2014, 46, 2047–2073. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Sorochinsky, A.E. Practical Methods for the Synthesis of Symmetrically α,α-Disubstituted-α-Amino Acids. Synthesis 2010, 2319–2344. [Google Scholar] [CrossRef]
- Mikami, K.; Fustero, S.; Sánchez-Roselló, M.; Aceña, J.L.; Soloshonok, V.A.; Sorochinsky, A.E. Synthesis of Fluorine Containing β-Amino Acids. Synthesis 2011, 3045–3079. [Google Scholar]
- Aceña, J.L.; Sorochinsky, A.E.; Moriwaki, H.; Sato, T.; Soloshonok, V.A. Synthesis of fluorine-containing α-amino acids in enantiomerically pure form via homologation of Ni(II) complexes of glycine and alanine Schiff bases. J. Fluorine Chem. 2013, 155, 21–38. [Google Scholar] [CrossRef]
- Turcheniuk, K.V.; Kukhar, V.P.; Roeschenthaler, G.V.; Acena, J.L.; Soloshonok, V.A.; Sorochinsky, A.E. Recent advances in the synthesis of fluorinated aminophosphonates and aminophosphonic acids. RSC Adv. 2013, 3, 6693–6716. [Google Scholar] [CrossRef]
- Kukhar, V.P. Fluorine-containing amino acids. J. Fluorine Chem. 1994, 69, 199–205. [Google Scholar] [CrossRef]
- Furin, G.G. Some new aspects in the application of perfluoroalkyl halides in the synthesis of fluorine-containing organic compounds. Russ. Chem. Rev. 2000, 69, 491–522. [Google Scholar] [CrossRef]
- Kiss, L.; Fülöp, F. Selective Synthesis of Fluorine-Containing Cyclic β-Amino Acid Scaffolds. Chem. Rec. 2018, 18, 266–281. [Google Scholar] [CrossRef]
- Turcheniuk, K.V.; Poliashko, K.O.; Kukhar, V.P.; Rozhenko, A.B.; Soloshonok, V.A.; Sorochinsky, A.E. Efficient asymmetric synthesis of trifluoromethylated β-aminophosphonates and their incorporation into dipeptides. Chem. Commun. 2012, 48, 11519–11521. [Google Scholar] [CrossRef]
- Röschenthaler, G.V.; Kukhar, V.P.; Kulik, I.B.; Belik, M.Y.; Sorochinsky, A.E.; Rusanov, E.B.; Soloshonok, V.A. Asymmetric synthesis of phosphonotrifluoroalanine and its derivatives using N-tert-butanesulfinyl imine derived from fluoral. Tetrahedron Lett. 2012, 53, 539–542. [Google Scholar] [CrossRef]
- Yin, Z.; Moriwaki, H.; Abe, H.; Miwa, T.; Han, J.; Soloshonok, V.A. Large-Scale Asymmetric Synthesis of Fmoc-(S)-2-Amino-6,6,6-Trifluorohexanoic Acid. ChemistryOpen 2019, 8, 701–704. [Google Scholar] [CrossRef] [Green Version]
- Mei, H.; Yin, Z.; Miwa, T.; Moriwaki, H.; Abe, H.; Han, J.; Soloshonok, V.A. Convenient Asymmetric Synthesis of Fmoc-(S)-6,6,6-trifluoro-Norleucine. Symmetry 2019, 11, 578. [Google Scholar] [CrossRef] [Green Version]
- Osipov, S.N.; Kolomiets, A.F.; Bruneau, C.; Picquet, M.; Dixneuf, P.H. Synthesis of fluorine-containing cyclic amino acid derivatives via ring closing olefin metathesis. Chem. Commun. 1998, 2053–2054. [Google Scholar] [CrossRef]
- Osipov, S.N.; Artyushin, O.I.; Kolomiets, A.F.; Bruneau, C.; Picquet, M.; Dixneuf, P.H. Synthesis of Fluorine-Containing Cyclic α-Amino Acid and α-Amino Phosphonate Derivatives by Alkene Metathesis. Eur. J. Org. Chem. 2001, 3891–3897. [Google Scholar] [CrossRef]
- Tsushima, T.; Kawada, K.; Tsuji, T.; Tawara, K. Fluorine-Containing Amino Acids and Their Derivatives. 4.1 Synthesis and Antibacterial Activity of Threo and Erythro1-Fluorodehydroxylated Chloramphenicol Analogues. J. Med. Chem. 1985, 28, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Osipov, S.N.; Sewald, N.; Kolomiets, A.F.; Fokin, A.V.; Burger, K. Synthesis of α-trifluoromethyl substituted α-amino acid derivatives from methyl 3,3,3-trifluoro-2-diazopropionate. Tetrahedron Lett. 1996, 37, 615–618. [Google Scholar] [CrossRef]
- Konno, T.; Kanda, M.; Ishihara, T.; Yamanaka, H. A novel synthesis of fluorine-containing quaternary amino acid derivatives via palladium-catalyzed allylation reaction. J. Fluorine Chem. 2005, 126, 1517–1523. [Google Scholar] [CrossRef]
- Ayoup, M.S.; Cordes, D.B.; Slawin, A.M.Z.; O’Hagan, D. Fluorine containing amino acids: Synthesis and peptide coupling of amino acids containing the all-cis tetrafluorocyclohexyl motif. Org. Biomol. Chem. 2015, 13, 5621–5624. [Google Scholar] [CrossRef] [Green Version]
- Mei, H.; Han, J.; Klika, K.D.; Izawa, K.; Sato, T.; Meanwell, N.A.; Soloshonok, V.A. Applications of Fluorine-Containing Amino Acids for Drug Design. Eur. J. Med. Chem. 2020, 186, 111826. [Google Scholar] [CrossRef]
- Chen, H.; Viel, S.; Ziarelli, F.; Peng, L. 19F NMR: A valuable tool for studying biological events. Chem. Soc. Rev. 2013, 42, 7971–7982. [Google Scholar] [CrossRef]
- Marsh, E.N.G.; Suzuki, Y. Using 19F NMR to probe biological interactions of proteins and peptides. ACS Chem. Biol. 2014, 9, 1242–1250. [Google Scholar] [CrossRef]
- Cai, L.; Lu, S.; Pike, V.W. Chemistry with [18F]fluoride ion. Eur. J. Org. Chem. 2008, 2008, 2853–2873. [Google Scholar] [CrossRef]
- Brooks, A.F.; Topczewski, J.J.; Ichiishi, N.; Sanford, M.S.; Scott, P.J.H. Late-Stage [18F]Fluorination: New Solutions to Old Problems. Chem. Sci. 2014, 5, 4545–4553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaeckel, C.; Seufert, W.; Thust, S.; Koksch, B. Evaluation of the molecular interactions of fluorinated amino acids with native polypeptides. ChemBioChem 2004, 5, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Salwiczek, M.; Koksch, B. Effects of fluorination on the folding kinetics of a heterodimeric coiled coil. ChemBioChem 2009, 10, 2867–2870. [Google Scholar] [CrossRef] [PubMed]
- Vagt, T.; Nyakatura, E.; Salwiczek, M.; Jaeckel, C.; Koksch, B. Towards identifying preferred interaction partners of fluorinated amino acids within the hydrophobic environment of a dimeric coiled coil peptide. Org. Biomol. Chem. 2010, 8, 1382–1386. [Google Scholar] [CrossRef] [PubMed]
- Nyakatura, E.K.; Reimann, O.; Vagt, T.; Salwiczek, M.; Koksch, B. Accommodating fluorinated amino acids in a helical peptide environment. RSC Adv. 2013, 3, 6319–6322. [Google Scholar] [CrossRef]
- Buer, B.C.; Chugh, J.; Al-Hashimi, H.M.; Marsh, E.N.G. Using fluorine nuclear magnetic resonance to probe the interaction of membrane-active peptides with the lipid bilayer. Biochemistry 2010, 49, 5760–5765. [Google Scholar] [CrossRef]
- De Kort, D.W.; Rombouts, W.H.; Hoeben, F.J.M.; Janssen, H.M.; As, H.V.; van Duynhoven, J.P.M. Scaling behavior of dendritic nanoparticle mobility in semidilute polymer solutions. Macromolecules 2015, 48, 7585–7591. [Google Scholar] [CrossRef]
- Kubyshkin, V.; Afonin, S.; Kara, S.; Budisa, N.; Mykhailiuk, P.K.; Ulrich, A.S. (S)-Trifluoromethyl proline: Evaluation as a structural substitute of proline for solid state 19F-NMR peptide studies. Org. Biomol. Chem. 2015, 13, 3171–3181. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.D.; Kotoris, C.C.; Hum, G. Recent advances in electrophilic fluorination. Tetrahedron 1999, 55, 12431–12477. [Google Scholar] [CrossRef]
- Szpera, R.; Moseley, D.F.J.; Smith, L.B.; Sterling, A.J.; Gouverneur, V. The Fluorination of C−H Bonds: Developments and Perspectives. Angew. Chem. Int. Ed. 2019, 58, 14824–14848. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Fu, L.; Chen, P.; Liu, G. Recent Advances and Perspectives of Transition Metal-Catalyzed Asymmetric Fluorination Reactions. Chin. J. Chem. 2017, 35, 1781–1788. [Google Scholar] [CrossRef]
- Kohlhepp, S.V.; Gulder, T. Hypervalent iodine(III) fluorinations of alkenes and diazo compounds: New opportunities in fluorination chemistry. Chem. Soc. Rev. 2016, 45, 6270–6288. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Dong, T.; Revankar, H.M.; Zhang, C.P. Recent progress on fluorination in aqueous media. Green Chem. 2017, 19, 3951–3992. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Beier, P. Construction of Asymmetric Fluorinated Carbon Centers. Angew. Chem. Int. Ed. 2006, 45, 2172–2174. [Google Scholar] [CrossRef]
- Tredwell, M.; Gouverneur, V. Electrophilic fluorination of organosilanes. Org. Biomol. Chem. 2006, 4, 26–32. [Google Scholar] [CrossRef]
- Davis, F.A.; Han, W.; Murphy, C.K. Selective, electrophilic fluorinations using N-fluoro-o-benzenedisulfonimide. J. Org. Chem. 1995, 60, 4730–4737. [Google Scholar] [CrossRef]
- Differding, E.; Rüegg, G.M. Nucleophilic substitution versus electron transfer: 1. On the mechanism of electrophilic fluorinations. Tetrahedron Lett. 1991, 32, 3815–3818. [Google Scholar] [CrossRef]
- Rozatian, N.; Ashworth, I.W.; Sandford, G.; Hodgson, D.R.W. A quantitative reactivity scale for electrophilic fluorinating reagents. Chem. Sci. 2018, 9, 8692–8702. [Google Scholar] [CrossRef] [Green Version]
- Piana, S.; Devillers, I.; Togni, A.; Rothlisberger, U. The mechanism of catalytic enantioselective fluorination: Computational and experimental studies. Angew. Chem. Int. Ed. 2002, 41, 979–982. [Google Scholar] [CrossRef]
- Banks, R.E.; Mohialdin-Khaffaf, S.N.; Lal, G.S.; Sharif, I.; Syvret, R.G. 1-Alkyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane salts: A novel family of electrophilic fluorinating agents. J. Chem. Soc. Chem. Commun. 1992, 595–596. [Google Scholar] [CrossRef]
- Shibata, N. Development of shelf-stable reagents for fluoro-functionalization reactions. Bull. Chem. Soc. Jpn. 2016, 89, 1307–1320. [Google Scholar] [CrossRef] [Green Version]
- Yasui, H.; Yamamoto, T.; Ishimaru, T.; Fukuzumi, T.; Tokunaga, E.; Akikazu, K.; Shiro, M.; Shibata, N. N-Fluoro-(3,5-di-tert-butyl-4-methoxy)benzenesulfonimide (NFBSI): A sterically demanding electrophilic fluorinating reagent for enantioselective fluorination. J. Fluorine Chem. 2011, 132, 222–225. [Google Scholar] [CrossRef]
- Fukushi, K.; Suzuki, S.; Kamo, T.; Tokunaga, E.; Sumii, Y.; Kagawa, T.; Kawada, K.; Shibata, N. Methyl NFSI: Atom-economical alternative to NFSI shows higher fluorination reactivity under Lewis acid-catalysis and non-catalysis. Green Chem. 2016, 18, 1864–1868. [Google Scholar] [CrossRef]
- Fukushi, K.; Tokunaga, E.; Sumii, Y.; Kagawa, T.; Nagasaki, N.; Shibata, N. Lewis Acid-Catalyzed Selective Mono-fluorination of Malonates Using Me-NFSI. Fluorine Notes 2016, 105, 3–4. [Google Scholar]
- Zhu, C.L.; Maeno, M.; Zhang, F.G.; Shigehiro, T.; Kagawa, T.; Kawada, K.; Shibata, N.; Ma, J.A.; Cahard, D. Chiral N-Fluorodibenzenesulfonimide Analogues for Enantioselective Electrophilic Fluorination and Oxidative Fluorination. Eur. J. Org. Chem. 2013, 29, 6501–6505. [Google Scholar] [CrossRef]
- Shibata, N.; Ishimaru, T.; Nakamura, S.; Toru, T. New approaches to enantioselective fluorination: Cinchona alkaloids combinations and chiral ligands/metal complexes. J. Fluorine Chem. 2007, 128, 469–483. [Google Scholar] [CrossRef]
- Mohar, B.; Baudoux, J.; Plaquevent, J.C.; Cahard, D. Electrophilic Fluorination Mediated by Cinchona Alkaloids: Highly Enantioselective Synthesis of α-Fluoro-α-phenylglycine Derivatives. Angew. Chem. Int. Ed. 2001, 40, 4214–4216. [Google Scholar] [CrossRef]
- Cahard, D.; Audouard, C.; Plaquevent, J.C.; Roques, N. Design, Synthesis, and Evaluation of a Novel Class of Enantioselective Electrophilic Fluorinating Agents: N-Fluoro Ammonium Salts of Cinchona Alkaloids (F-CA-BF4). Org. Lett. 2000, 2, 3699–3701. [Google Scholar] [CrossRef]
- Shibata, N.; Suzuki, E.; Takeuchi, Y. A Fundamentally New Approach to Enantioselective Fluorination Based on Cinchona Alkaloid Derivatives/Selectfluor Combination. J. Am. Chem. Soc. 2000, 122, 10728–10729. [Google Scholar] [CrossRef]
- Umemoto, T.; Nagayoshi, M. N,N′-Difluoro-1,4-diazoniabicyclo[2.2.2]octane Salts, Highly Reactive and Easy-to-Handle Electrophilic Fluorinating Agents. Bull. Chem. Soc. Jpn. 1996, 69, 2287–2295. [Google Scholar] [CrossRef]
- Kitamura, T.; Kuriki, S.; Morshed, M.H.; Hori, Y. A Practical and Convenient Fluorination of 1,3-Dicarbonyl Compounds Using Aqueous HF in the Presence of Iodosylbenzene. Org. Lett. 2011, 13, 2392–2394. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.D.; Greenhall, M.P.; Hutchinson, J. Direct fluorination of 1, 3-dicarbonyl compounds. Tetrahedron 1996, 52, 1–8. [Google Scholar] [CrossRef]
- Zajc, B.; Zupan, M. Room temperature fluorination of 1, 3-diketones and enol acetates with xenon difluoride. J. Chem. Soc. Chem. Commun. 1980, 759–760. [Google Scholar] [CrossRef]
- Yemul, S.S.; Kagan, H.B.; Setton, R. Selective fluorination by C19XeF6. Tetrahedron Lett. 1980, 21, 277–280. [Google Scholar] [CrossRef]
- Zajc, B.; Zupan, M. Fluorination with xenon difluoride. 27. The effect of catalyst on fluorination of 1,3-diketones and enol acetates. J. Org. Chem. 1982, 47, 573–575. [Google Scholar] [CrossRef]
- Inman, C.E.; Oesterling, R.E.; Tyczkowski, E.A. Reactions of Perchloryl Fluoride with Organic Compounds. II. Fluorination of Certain Active Methylene Compounds1. J. Am. Chem. Soc. 1958, 80, 6533–6535. [Google Scholar] [CrossRef]
- Machleidt, H.; Hartmann, V.I. Organische Fluorverbindungen, VI. Reaktionen cyclischer Fluor-carbonyl-Verbindungen. Liebigs Ann. Chem. 1964, 679, 9–19. [Google Scholar] [CrossRef]
- Lerman, O.; Rozen, S. Acetyl hypofluorite, a new moderating carrier of elemental fluorine and its use in fluorination of 1,3-dicarbonyl derivatives. J. Org. Chem. 1983, 48, 724–727. [Google Scholar] [CrossRef]
- Rozen, S.; Brand, M. Direct fluorination of lithium enolates with acetyl hypofluorite. Synthesis 1985, 665–667. [Google Scholar] [CrossRef]
- Rozen, S.; Hebel, D. Acyl hypofluorites. A new class of organic compounds. J. Org. Chem. 1990, 55, 2621–2623. [Google Scholar] [CrossRef]
- Rozen, S.; Menahem, Y. A novel fluorinating method for the synthesis of α-fluoroketones. Tetrahedron Lett. 1979, 20, 725–728. [Google Scholar] [CrossRef]
- Stavber, S.; Zupan, M. Room-temperature fluorination of alkenes with caesium fluoroxysulphate. J. Chem. Soc. Chem. Commun. 1981, 795–796. [Google Scholar] [CrossRef]
- Banks, R.E.; Murtagh, V.; Tsiliopoulos, E. N-halogeno compounds. Part 12 [1]. Site-specific fluorination of carbanions with perfluoro-N-fluoropiperidine [2]. J. Fluorine Chem. 1991, 52, 389–401. [Google Scholar] [CrossRef]
- Xu, Z.Q.; DesMarteau, D.D.; Gotoh, Y. N-Fluorobis [(perfluoroalkyl) sulfonyl] imides. Efficient reagents for the fluorination of 1, 3-dicarbonyl derivatives. J. Fluorine Chem. 1992, 58, 71–79. [Google Scholar] [CrossRef]
- Banks, R.E.; Lawrence, N.J.; Popplewell, A.L. Efficient electrophilic fluorination of β-dicarbonyl compounds with the selectfluor reagent F-TEDA-BF4{1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate)}. J. Chem. Soc. Chem. Commun. 1994, 343–344. [Google Scholar] [CrossRef]
- Cabrera, I.; Appel, W.K. New fluorinating reagents from acesulfam sweeteners. Tetrahedron 1995, 51, 10205–10208. [Google Scholar] [CrossRef]
- Umemoto, T.; Nagayoshi, M.; Adachi, K.; Tomizawa, G. Synthesis, Properties, and Reactivity of N,N‘-Difluorobipyridinium and Related Salts and Their Applications as Reactive and Easy-To-Handle Electrophilic Fluorinating Agents with High Effective Fluorine Content. J. Org. Chem. 1998, 63, 3379–3385. [Google Scholar] [CrossRef]
- Frantz, R.; Hintermann, L.; Perseghini, M.; Broggini, D.; Togni, A. Titanium-catalyzed stereoselective geminal heterodihalogenation of β-ketoesters. Org. Lett. 2003, 5, 1709–1712. [Google Scholar] [CrossRef]
- Gupta, O.D.; Shreeve, J.M. Reactions of trifluoroamine oxide: A new method for selective fluorination of 1, 3-diketones and β-ketoesters. Tetrahedron Lett. 2003, 44, 2799–2801. [Google Scholar] [CrossRef]
- Stavber, G.; Zupan, M.; Stavber, S. Solvent-free fluorination of organic compounds using N–F reagents. Tetrahedron Lett. 2007, 48, 2671–2673. [Google Scholar] [CrossRef]
- Hara, S.; Sekiguchi, M.; Ohmori, A.; Fukuhara, T.; Yoneda, N. Selective fluorination of β-ketoesters using iodotoluene difluoride and a HF–amine complex. Chem. Commun. 1996, 1899–1900. [Google Scholar] [CrossRef]
- Yoshida, M.; Fujikawa, K.; Sato, S.; Hara, S. α-Fluorination of β-dicarbonyl compounds using p-iodotoluene difluoride under neutral conditions. ARKIVOC 2003, 6, 36–42. [Google Scholar]
- Gondo, K.; Kitamura, T. Reaction of iodonium ylides of 1, 3-dicarbonyl compounds with HF reagents. Molecules 2012, 17, 6625–6632. [Google Scholar] [CrossRef]
- Goudreau, S.R.; Marcoux, D.; Charette, A.B. General method for the synthesis of phenyliodonium ylides from malonate esters: Easy access to 1,1-cyclopropane diesters. J. Org. Chem. 2009, 74, 470–473. [Google Scholar] [CrossRef]
- Kitamura, T.; Kuriki, S.; Muta, K.; Morshed, M.H.; Muta, K. Facile Synthesis of 2-Fluoro-1, 3-dicarbonyl Compounds with Aqueous Hydrofluoric Acid Mediated by Iodosylarenes. Synthesis 2013, 45, 3125–3130. [Google Scholar] [CrossRef]
- Kitamura, T.; Muta, K.; Oyamada, J. A Convenient Synthesis of 2-Fluoro-and 2-Chloromalonic Esters Mediated by Hypervalent Iodine. Synthesis 2015, 47, 3241–3245. [Google Scholar] [CrossRef]
- Kitamura, T.; Muta, K.; Kuriki, S. Catalytic fluorination of 1, 3-dicarbonyl compounds using iodoarene catalysts. Tetrahedron Lett. 2013, 54, 6118–6120. [Google Scholar] [CrossRef]
- Leroy, J. 1, 2-Difluoroethylenes: Synthesis via fluoro ketones. J. Org. Chem. 1981, 46, 206–209. [Google Scholar] [CrossRef]
- Moughamir, K.; Atmani, A.; Mestdagh, H.; Rolando, C.; Francesch, C. Activation of tetrabutylammonium hydrogen difluoride with pyridine: A mild and efficient procedure for nucleophilic fluorination. Tetrahedron Lett. 1998, 39, 7305–7306. [Google Scholar] [CrossRef]
- Makosza, M.; Bujok, R. Cocatalysis in phase-transfer catalyzed fluorination of alkyl halides and sulfonates. J. Fluorine Chem. 2005, 126, 209–216. [Google Scholar] [CrossRef]
- Fuglseth, E.; Thvedt, T.H.K.; Møll, M.F.; Hoff, B.H. Electrophilic and nucleophilic side chain fluorination of para-substituted acetophenones. Tetrahedron 2008, 64, 7318–7323. [Google Scholar] [CrossRef]
- Chen, Z.; Zhu, W.; Zhen, Z.; Zou, X. One-pot α-nucleophilic fluorination of acetophenones in a deep eutectic solvent. J. Fluorine Chem. 2010, 131, 340–344. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Shang, H.; Chen, X.; Ren, X. Diastereoselective Mannich Reactions Using Fluorinated Ketones: Synthesis of Stereogenic Carbon–Fluorine Units. J. Org. Chem. 2016, 81, 9858–9866. [Google Scholar] [CrossRef]
- Xu, Y.S.; Tang, Y.; Feng, H.J.; Liu, J.T.; Hsung, R.P. A Highly Regio-and Stereoselective Synthesis of α-Fluorinated Imides via Fluorination of Chiral Enamides. Org. Lett. 2015, 17, 572–575. [Google Scholar] [CrossRef] [Green Version]
- Saidalimu, I.; Fang, X.; Lv, W.; Yang, X.; He, X.; Zhang, J.; Wu, F. Organocatalytic Asymmetric Michael Addition/Carbon-Carbon Bond Cleavage of Trifluoromethyl α-Fluorinated gem-Diols to Nitroolefins. Adv. Synth. Catal. 2013, 355, 857–863. [Google Scholar] [CrossRef]
- Kitahara, K.; Mizutani, H.; Iwasa, S.; Shibatomi, K. Asymmetric Synthesis of α-Chloro-α-halo Ketones by Decarboxylative Chlorination of α-Halo-β-ketocarboxylic Acids. Synthesis 2019, 51, 4385–4392. [Google Scholar] [CrossRef]
- Katada, M.; Kitahara, K.; Iwasa, S.; Shibatomi, K. Catalyst-free decarboxylative fluorination of tertiary β-keto carboxylic acids. Synlett 2018, 29, 2408–2411. [Google Scholar]
- Krespan, C.G.; Smart, B.E. Fluorocarbanion chemistry. A versatile synthesis of functionalized fluoro ketones. J. Org. Chem. 1986, 51, 320–326. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Mikami, K.; Yamazaki, T.; Welch, J.T.; Honek, J. (Eds.) Current Fluoroorganic Chemistry. New Synthetic Directions, Technologies, Materials and Biological Applications; ACS Symposium Series 949; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Xiao, X.; Xie, Y.; Su, C.; Liu, M.; Shi, Y. Organocatalytic Asymmetric Biomimetic Transamination: From α-Keto Esters to Optically Active α-Amino Acid Derivatives. J. Am. Chem. Soc. 2011, 133, 12914–12917. [Google Scholar] [CrossRef]
- Liu, M.; Li, J.; Xie, Y.; Shi, Y. An efficient synthesis of optically active trifluoromethyl aldimines via asymmetric biomimetic transamination. Chem. Commun. 2013, 49, 1404–1406. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Kukhar, V.P. Biomimetic transamination of α-keto perfluorocarboxylic esters. An efficient preparative synthesis of β,β,β-trifluoroalanine. Tetrahedron 1997, 53, 8307–8314. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ohkura, H.; Yasumoto, M. Operationally convenient asymmetric synthesis of (S)- and (R)-3-amino-4,4,4-trifluorobutanoic acid: Part I: Enantioselective biomimetic transamination of isopropyl 4,4,4-trifluoro-3-oxo-butanoate. J. Fluorine Chem. 2006, 127, 924–929. [Google Scholar] [CrossRef]
- Guo, C.; Song, J.; Gong, L.Z. Biomimetic Asymmetric 1,3-Dioplar Cycloaddition: Amino Acid Precursors in Biosynthesis Serve as Latent Azomethine Ylides. Org. Lett. 2013, 15, 2676–2679. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Soloshonok, I.V.; Kukhar, V.P.; Svedas, V.K. Biomimetic Transamination of α-Alkyl β-Keto Carboxylic Esters. Chemoenzymatic Approach to the Stereochemically Defined α-Alkyl β-Fluoroalkyl β-Amino Acids. J. Org. Chem. 1998, 63, 1878–1884. [Google Scholar] [CrossRef]
- Xiao, X.; Liu, M.; Rong, C.; Xue, F.; Li, S.; Xie, Y.; Shi, Y. An Efficient Asymmetric Biomimetic Transamination of α-Keto Esters to Chiral α-Amino Esters. Org. Lett. 2012, 14, 5270–5273. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ono, T.; Soloshonok, I.V. Enantioselective Biomimetic Transamination of β-Keto Carboxylic Acid Derivatives. An Efficient Asymmetric Synthesis of β-Fluoroalkyl-β-Amino Acids. J. Org. Chem. 1997, 62, 7538–7539. [Google Scholar]
- Soloshonok, V.A.; Ono, T. The Effect of Substituents on the Feasibility of Azomethine-Azomethine Isomerization: New Synthetic Opportunities for Biomimetic Transamination. Tetrahedron 1996, 52, 14701–14712. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ohkura, H.; Yasumoto, M. Operationally Convenient Asymmetric Synthesis of (S)- and (R)-3-Amino-4,4,4-trifluorobutanoic Acid. Part II: Enantioselective Biomimetic Transamination of 4,4,4-Trifluoro-3-oxo-N-[(R)-1-phenylethyl)butanamide. J. Fluorine Chem. 2006, 127, 930–935. [Google Scholar] [CrossRef]
- Barnette, W.E. N-Fluoro-N-alkylsulfonamides: Useful reagents for the fluorination of carbanions. J. Am. Chem. Soc. 1984, 106, 452–454. [Google Scholar] [CrossRef]
- Differding, E.; Lang, R.W. New fluorinating reagents-I. The first enantioselective fluorination reaction. Tetrahedron Lett. 1988, 29, 6087–6090. [Google Scholar] [CrossRef]
- Resnati, G.; DesMarteau, D.D. N-fluorobis [(trifluoromethyl) sulfonyl] imide: An efficient reagent for the. alpha.-fluorination of functionalized carbonyl compounds. J. Org. Chem. 1991, 56, 4925–4929. [Google Scholar] [CrossRef]
- Davis, F.A.; Han, W. N-Fluoro-o-benzenedisulfonimide: A useful new fluorinating reagent. Tetrahedron Lett. 1991, 32, 1631–1634. [Google Scholar] [CrossRef]
- Davis, F.A.; Zhou, P.; Murphy, C.K. Asymmetric fluorination of enolates with N-fluoro 2, 10-(3, 3-dichlorocamphorsultam). Tetrahedron Lett. 1993, 34, 3971–3974. [Google Scholar] [CrossRef]
- Middleton, W.J.; Bingham, E.M. alpha-Fluorination of carbonyl compounds with trifluoromethyl hypofluorite. J. Am. Chem. Soc. 1980, 102, 4845–4846. [Google Scholar] [CrossRef]
- Purrington, S.T.; Lazaridis, N.V.; Bumgardner, C.L. Preparation of α-fluoroaldehydes and α-fluoroketones using dilute fluorine. Tetrahedron Lett. 1986, 27, 2715–2716. [Google Scholar] [CrossRef]
- Umemoto, T.; Kawada, K.; Tomita, K. N-fluoropyridinium triflate and its derivatives: Useful fluorinating agents. Tetrahedron Lett. 1986, 27. [Google Scholar] [CrossRef]
- Kitamura, T.; Muta, K.; Muta, K. Hypervalent Iodine-Promoted α-Fluorination of Acetophenone Derivatives with a Triethylamine· HF Complex. J. Org. Chem. 2014, 79, 5842–5846. [Google Scholar] [CrossRef]
- Rueeger, H.; Lueoend, R.; Rogel, O.; Rondeau, J.M.; Möbitz, H.; Machauer, R.; Jacobson, L.; Staufenbiel, M.; Desrayaud, S.; Neumann, U. Discovery of Cyclic Sulfone Hydroxyethylamines as Potent and Selective β-Site APP-Cleaving Enzyme 1 (BACE1) Inhibitors: Structure-Based Design and in Vivo Reduction of Amyloid β-Peptides. J. Med. Chem. 2012, 55, 3364–3386. [Google Scholar] [CrossRef]
- Levin, V.V.; Zemtsov, A.A.; Struchkova, M.I.; Dilman, A.D. Reactions of organozinc reagents with potassium bromodifluoroacetate. J. Fluorine Chem. 2015, 171, 97–101. [Google Scholar] [CrossRef]
- Dolbier, W.R., Jr.; Okamoto, M. Preparation of 1, 1-difluoroalkanes from aldehydes via 1, 1-bistriflates: Advantageous use of HF–Lewis base reagents. J. Fluorine Chem. 2014, 167, 96–100. [Google Scholar] [CrossRef]
- Bujok, R.; Mlkosza, M. Synthesis of 1, 1-Difluoroalkanes via Phase Transfer Catalysed Reaction of 1, 1-bis-Triflates with KF in the Presence of Cocatalyst-Ph3SnF. Synlett 2002, 1285–1286. [Google Scholar]
- Martinez, A.G.; Barcina, J.O.; Rys, A.Z.; Subramanian, L.R. A facile conversion of aliphatic aldehydes to 1, 1-difluoroalkanes. Tetrahedron Lett. 1992, 33, 7787–7788. [Google Scholar] [CrossRef]
- Bloodworth, A.J.; Bowyer, K.J.; Mitchell, J.C. A mild, convenient, halogen-exchange route to gem-difluorides and trifluorides. Tetrahedron Lett. 1987, 28, 5347–5350. [Google Scholar] [CrossRef]
- Patrick, T.B.; Johri, K.K.; White, D.H. Fluoro-decarboxylation with xenon difluoride. J. Org. Chem. 1983, 48, 4158–4159. [Google Scholar] [CrossRef]
- Patrick, T.B.; Johri, K.K.; White, D.H.; Bertrand, W.S.; Mokhtar, R.; Kilbourn, M.R.; Welch, M.J. Replacement of the carboxylic acid function with fluorine. Can. J. Chem. 1986, 64, 138–141. [Google Scholar] [CrossRef]
- Salomon, P.; Zard, S.Z. A Practical Source of Chlorodifluoromethyl Radicals. Convergent Routes to gem-Difluoroalkenes and -dienes and (2,2-Difluoroethyl)-indoles, -azaindoles, and –naphthols. Org. Lett. 2014, 16, 2926–2929. [Google Scholar] [CrossRef]
- Zupan, M.; Pollak, A. Preparation of a polymer-supported aryliodine(III) difluoride and its use to fluorinate olefins to difluorides. J. Chem. Soc. Chem. Commun. 1975, 715–716. [Google Scholar] [CrossRef]
- Carpenter, W.R. Aryliodosodifluorides. J. Org. Chem. 1966, 31, 2688–2689. [Google Scholar] [CrossRef]
- Lermontov, S.A.; Rakov, I.M.; Zefirov, N.S.; Stang, P.J. Hexafluoropropene oxide–a fluorinating reagent for the formation of element–fluorine bonds. J. Fluorine Chem. 1999, 93, 103–106. [Google Scholar] [CrossRef]
- Patrick, T.B.; Qian, S. Rearrangement of Phenylethenes on Reaction with Iodine− Xenon Difluoride. Org. Lett. 2000, 2, 3359–3360. [Google Scholar] [CrossRef]
- Hara, S.; Nakahigashi, J.; Ishi-i, K.; Fukuhara, T.; Yoneda, N. Fluorinative ring-contraction of cyclic alkenes with p-iodotoluene difluoride. Tetrahedron Lett. 1998, 39, 2589–2592. [Google Scholar] [CrossRef]
- Sawaguchi, M.; Hara, S.; Yoneda, N. Fluorination of alkenes by iodoarene difluorides. J. Fluorine Chem. 2000, 105, 313–317. [Google Scholar] [CrossRef]
- Zupan, M.; Pollak, A. Fluorination with xenon difluoride. Preparation of aryliodine/III/difluoride. J. Fluorine. Chem. 1976, 7, 445–447. [Google Scholar] [CrossRef]
- Kitamura, T.; Muta, K.; Oyamada, J. Hypervalent Iodine-Mediated Fluorination of Styrene Derivatives: Stoichiometric and Catalytic Transformation to 2,2-Difluoroethylarenes. J. Org. Chem. 2015, 80, 10431–10436. [Google Scholar] [CrossRef]
- Ilchenko, N.O.; Tasch, B.O.A.; Szabo, K.J. Mild Silver-Mediated Geminal Difluorination of Styrenes Using an Air- and Moisture-Stable Fluoroiodane Reagent. Angew. Chem. Int. Ed. 2014, 53, 12897–12901. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, T.; Yoshida, K.; Mizuno, S.; Miyake, A.; Oyamada, J. Difluorination of functionalized aromatic olefins using hypervalent iodine/HF reagents. J. Org. Chem. 2018, 83, 14853–14860. [Google Scholar] [CrossRef]
- Xu, J.; Wei, L.; Mathvink, R.; He, J.; Park, Y.J.; He, H.; Leiting, B.; Lyons, K.A.; Marsilio, F.; Patel, R.A.; et al. Discovery of potent and selective phenylalanine based dipeptidyl peptidase IV inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 2533–2536. [Google Scholar] [CrossRef]
- Augustyns, K.J.L.; Lambeir, A.M.; Borloo, M.; De Meester, I.; Vedernikova, I.; Vanhoof, G.; Hendriks, D.; Scharpe, S.; Haemers, A. Pyrrolidides: Synthesis and structure-activity relationship as inhibitors of dipeptidyl peptidase IV. Eur. J. Med. Chem. 1997, 32, 301–309. [Google Scholar] [CrossRef]
- Caldwell, C.G.; Chen, P.; He, J.; Parmee, E.R.; Leiting, B.; Marsilio, F.; Patel, R.A.; Wu, J.K.; Eiermann, G.J.; Petrov, A.; et al. Fluoropyrrolidine amides as dipeptidyl peptidase IV inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 1265–1268. [Google Scholar] [CrossRef]
- Fukushima, H.; Hiratate, A.; Takahashi, M.; Mikami, A.; Saito-Hori, M.; Munetomo, E.; Kitano, K.; Chonan, S.; Saito, H.; Suzuki, A.; et al. Synthesis and structure–activity relationships of potent 4-fluoro-2-cyanopyrrolidine dipeptidyl peptidase IV inhibitors. Bioorg. Med. Chem. 2008, 16, 4093–4106. [Google Scholar] [CrossRef]
- Mao, W.; Ning, M.; Liu, Z.; Zhu, Q.; Leng, Y.; Zhang, A. Design, synthesis, and pharmacological evaluation of benzamide derivatives as glucokinase activators. Bioorg. Med. Chem. 2012, 20, 2982–2991. [Google Scholar] [CrossRef] [PubMed]
- Goossens, F.; Vanhoof, G.; Meester, I.; Augustyns, K.; Borloo, M.; Tourwe, D.; Haemers, A.; Scharpe, S. Development and Evaluation of Peptide-Based Prolyl Oligopeptidase Inhibitors- Introduction of N-Benzyloxycarbonyl-Prolyl-3-Fluoropyrrolidine as a Lead in Inhibitor Design. Eur. J. Biochem. 1997, 250, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.M.; Murkin, A.S.; Li, L.; Schramm, V.L.; Gainsford, G.J.; Skelton, B.W. A β-Fluoroamine Inhibitor of Purine Nucleoside Phosphorylase. J. Med. Chem. 2008, 51, 5880–5884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giardina, G.; Dondio, G.; Grugni, M. Facile and Efficient Syntheses of Novel (S)- and (R)-3-Fluoropyrrolidines and 3,3-Difluoropyrrolidine. Synlett 1995, 1995, 55–57. [Google Scholar] [CrossRef]
- Demange, L.; Cluzeau, J.; Ménez, A.; Dugave, C. Synthesis of optically pure N-Boc-protected (2R,3R)- and (2R,3S)-3-fluoroprolines. Tetrahedron Lett. 2001, 42, 651–653. [Google Scholar] [CrossRef]
- Doi, M.; Nishi, Y.; Kiritoshi, N.; Iwata, T.; Nago, M.; Nakano, H.; Uchiyama, S.; Nakazawa, T.; Wakamiya, T.; Kobayashi, Y. Simple and efficient syntheses of Boc- and Fmoc-protected 4(R)- and 4(S)-fluoroproline solely from 4(R)-hydroxyproline. Tetrahedron 2002, 58, 8453–8459. [Google Scholar] [CrossRef]
- Verniest, G.; Piron, K.; Van Hende, E.; Thuring, J.W.; Macdonald, G.; Deroose, F.; De Kimpe, N. Synthesis of aminomethylated 4-fluoropiperidines and 3-fluoropyrrolidines. Org. Biomol. Chem. 2010, 8, 2509–2512. [Google Scholar] [CrossRef]
- Fjelbye, K.; Marigo, M.; Clausen, R.P.; Juhl, K. Diastereodivergent Access to Syn and Anti 3,4-Substituted β-Fluoropyrrolidines: Enhancing or Reversing Substrate Preference. Org. Lett. 2016, 18, 1170–1173. [Google Scholar] [CrossRef]
- Kong, W.; Merino, E.; Nevado, C. Divergent Reaction Mechanisms in the Aminofluorination of Alkenes. Chimia 2014, 68, 430–435. [Google Scholar] [CrossRef]
- Chen, P.; Liu, G. Advancements in Aminofluorination of Alkenes and Alkynes: Convenient Access to β-Fluoroamines. Eur. J. Org. Chem. 2015, 2015, 4295–4309. [Google Scholar] [CrossRef]
- Combettes, L.E.; Lozano, O.; Gouverneur, V. Metal free fluoroamination of allylsilanes: A route to 3-fluoropyrrolidines. J. Fluorine Chem. 2012, 143, 167–176. [Google Scholar] [CrossRef]
- Xu, T.; Qiu, S.; Liu, G. Palladium-Catalyzed Intramolecular Aminofluorination of Styrenes. Chin. J. Chem. 2011, 29, 2785–2790. [Google Scholar] [CrossRef]
- Cui, J.; Jia, Q.; Feng, R.Z.; Liu, S.S.; He, T.; Zhang, C. Boron Trifluoride Etherate Functioning as a Fluorine Source in an Iodosobenzene-Mediated Intramolecular Aminofluorination of Homoallylic Amines. Org. Lett. 2014, 16, 1442–1445. [Google Scholar] [CrossRef]
- Kitamura, T.; Miyake, A.; Muta, K.; Oyamada, J. Hypervalent Iodine/HF Reagents for the Synthesis of 3-Fluoropyrrolidines. J. Org. Chem. 2017, 82, 11721–11726. [Google Scholar] [CrossRef]
- Kitamura, T.; Mizuno, S.; Muta, K.; Oyamada, J. Synthesis of β-Fluorovinyliodonium Salts by the Reaction of Alkynes with Hypervalent Iodine/HF Reagents. J. Org. Chem. 2018, 83, 2773–2778. [Google Scholar] [CrossRef]
- Dolbier, W.R., Jr.; Celewicz, L.; Ohnishi, K. Fluorination reactions with HF/THF medium solvolysis of N-tosyl-O-phenylhydroxylamine. Tetrahedron Lett. 1989, 30, 4929–4930. [Google Scholar] [CrossRef]
- Dolbier, W.R., Jr.; Gray, T.A.; Onnishi, K. Hydrogen Fluoride/Tetrahydrofuran as a Fluorinating Medium. A General Synthesis of 1,1,1,2-Tetrafluoro-2-alkenes. Synthesis 1987, 956–958. [Google Scholar] [CrossRef]






















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.; Butler, G.; Moriwaki, H.; Konno, H.; Soloshonok, V.A.; Kitamura, T. Kitamura Electrophilic Fluorination Using HF as a Source of Fluorine. Molecules 2020, 25, 2116. https://doi.org/10.3390/molecules25092116
Han J, Butler G, Moriwaki H, Konno H, Soloshonok VA, Kitamura T. Kitamura Electrophilic Fluorination Using HF as a Source of Fluorine. Molecules. 2020; 25(9):2116. https://doi.org/10.3390/molecules25092116
Chicago/Turabian StyleHan, Jianlin, Greg Butler, Hiroki Moriwaki, Hiroyuki Konno, Vadim A. Soloshonok, and Tsugio Kitamura. 2020. "Kitamura Electrophilic Fluorination Using HF as a Source of Fluorine" Molecules 25, no. 9: 2116. https://doi.org/10.3390/molecules25092116
APA StyleHan, J., Butler, G., Moriwaki, H., Konno, H., Soloshonok, V. A., & Kitamura, T. (2020). Kitamura Electrophilic Fluorination Using HF as a Source of Fluorine. Molecules, 25(9), 2116. https://doi.org/10.3390/molecules25092116