2. Results and Discussion
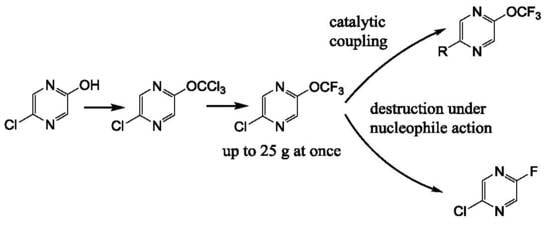
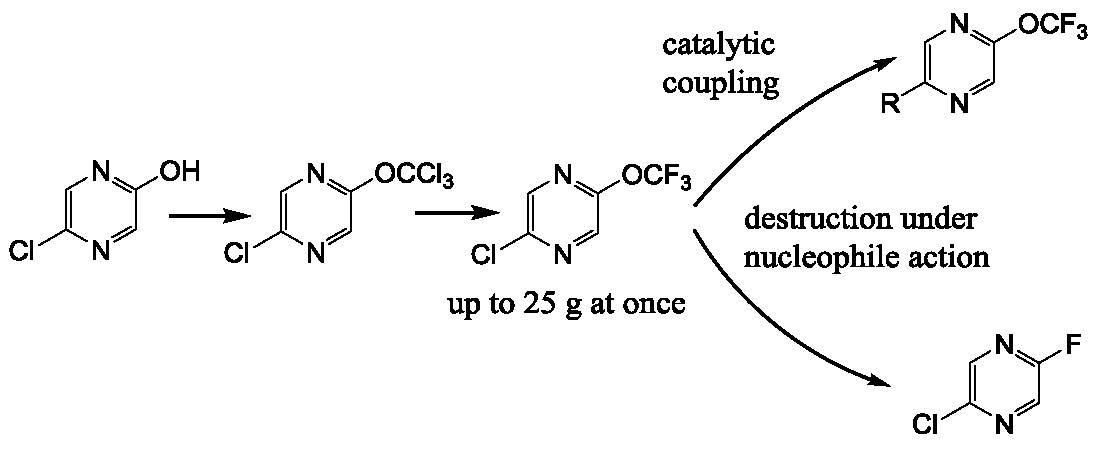
We have prepared 2-chloro-5-trifluoromethoxypyrazine
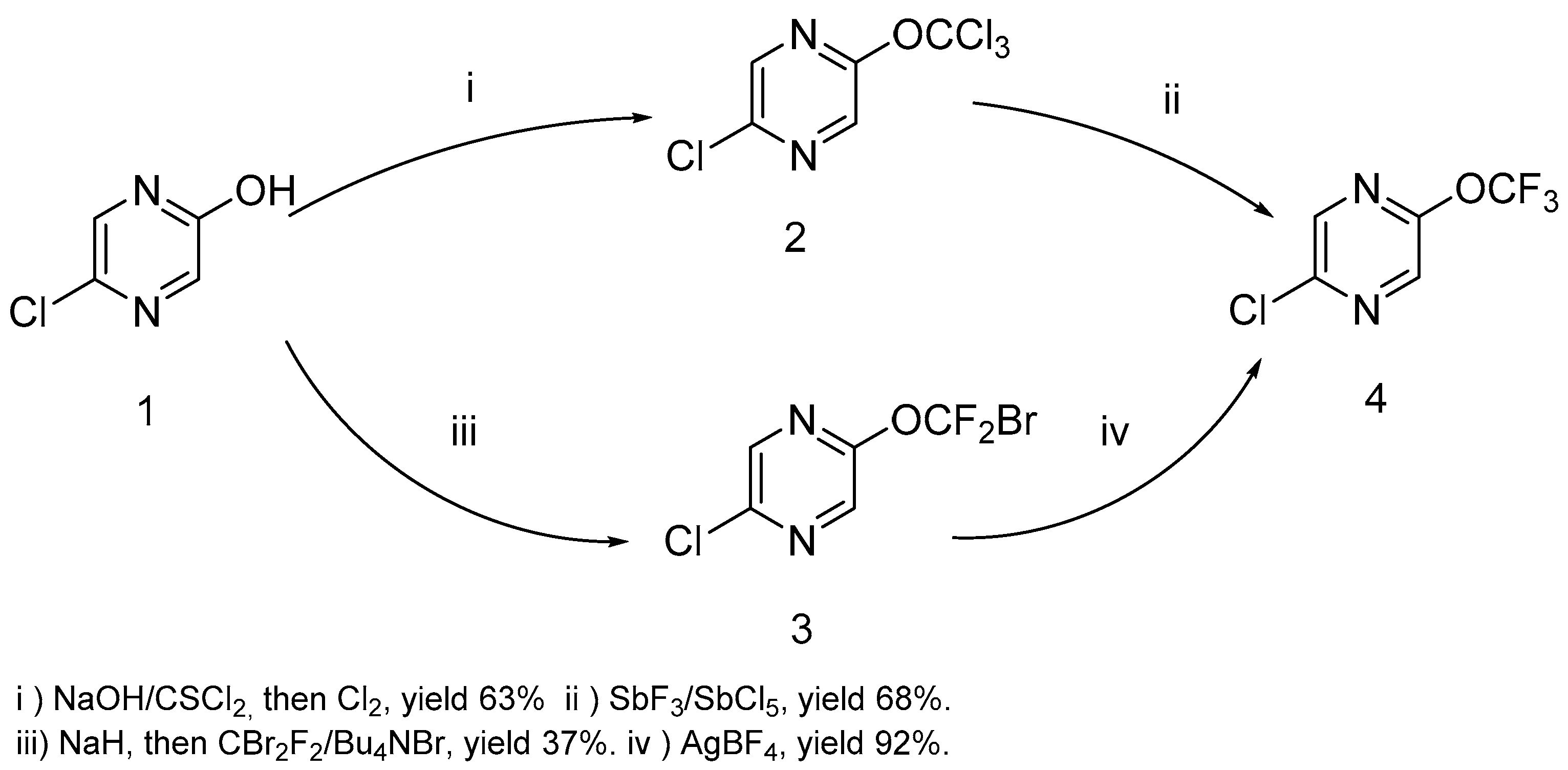
4 by the action of thiophosgene on hydroxypyrazine, with further chlorination and a chlorine/fluorine exchange using antimony trifluoride (
Scheme 1). We used an alternative route based on the halophilic alkylation of pyrazine by dibromodifluoromethane, followed by a bromine/fluorine exchange with silver tetrafluoroborate (
Scheme 1). The last method is less favorable due to the low yield in the first stage and the expensive reagents used. Nonetheless, both methods are suitable for a large scale experiment (pyrazine
4 was prepared in 20–25 g scale) in common laboratory glass equipment.
It must be noted that the presence of chlorine atoms in the pyrazine ring is critical for the success of both methods. We have found that trichloromethoxy and bromodifluoromethoxypyrazine cannot be prepared by these methods starting from 2-hydroxypyrazine. The same peculiarity was shown by Leroux and coworkers for a trifluoromethoxypyridine preparation [
8].
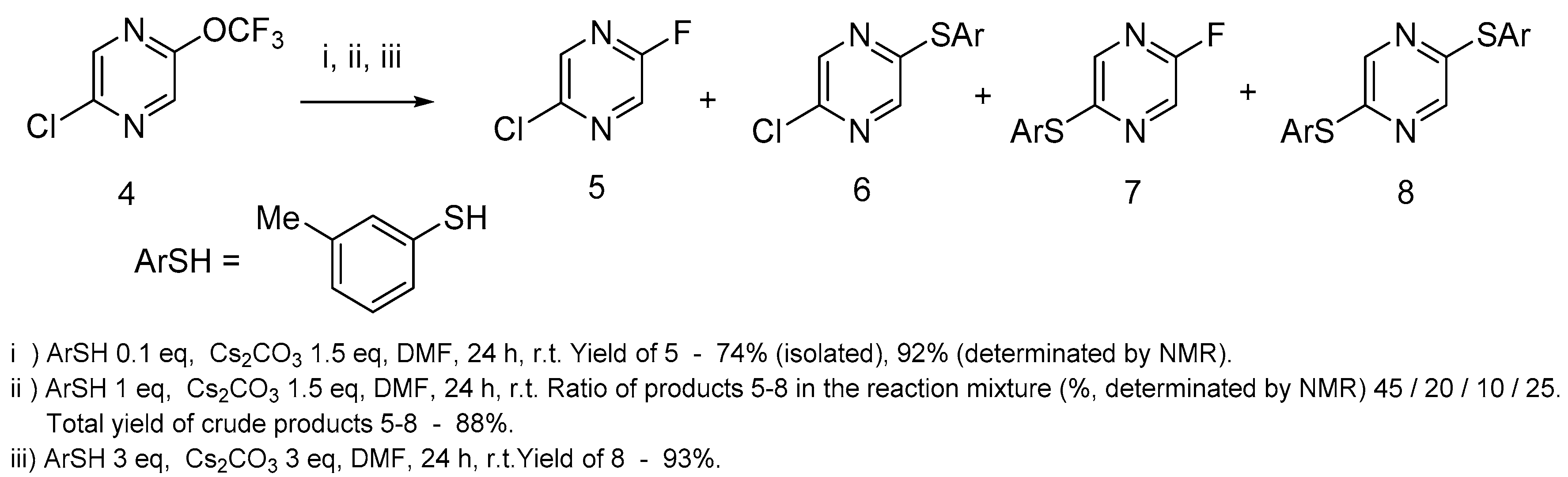
We have studied the 2-chloro-5-trifluoromethoxypyrazine behavior with nucleophiles using 3-methylthiophenol as an example of
S-nucleophile. If one equivalent of 3-methylthiophenol was used in this reaction, 2-chloro-5-fluoropyrazine
5 was the main fluorinated product (
Scheme 2), but a range of products with thiophenol moiety were also formed in these conditions. Surprisingly, 2-chloro-5-fluoropyrazine
5 was the main product when pyrazine
4 reacted with 10 mol% 3-methylthiophenol and an excess of cesium carbonate at room temperature. In the absence of thiophenol, this reaction did not occur even after 1 week of stirring at r.t. Pyrazine
8 was the single product when this reaction was performed with an excess of 3-methylthiophenol.
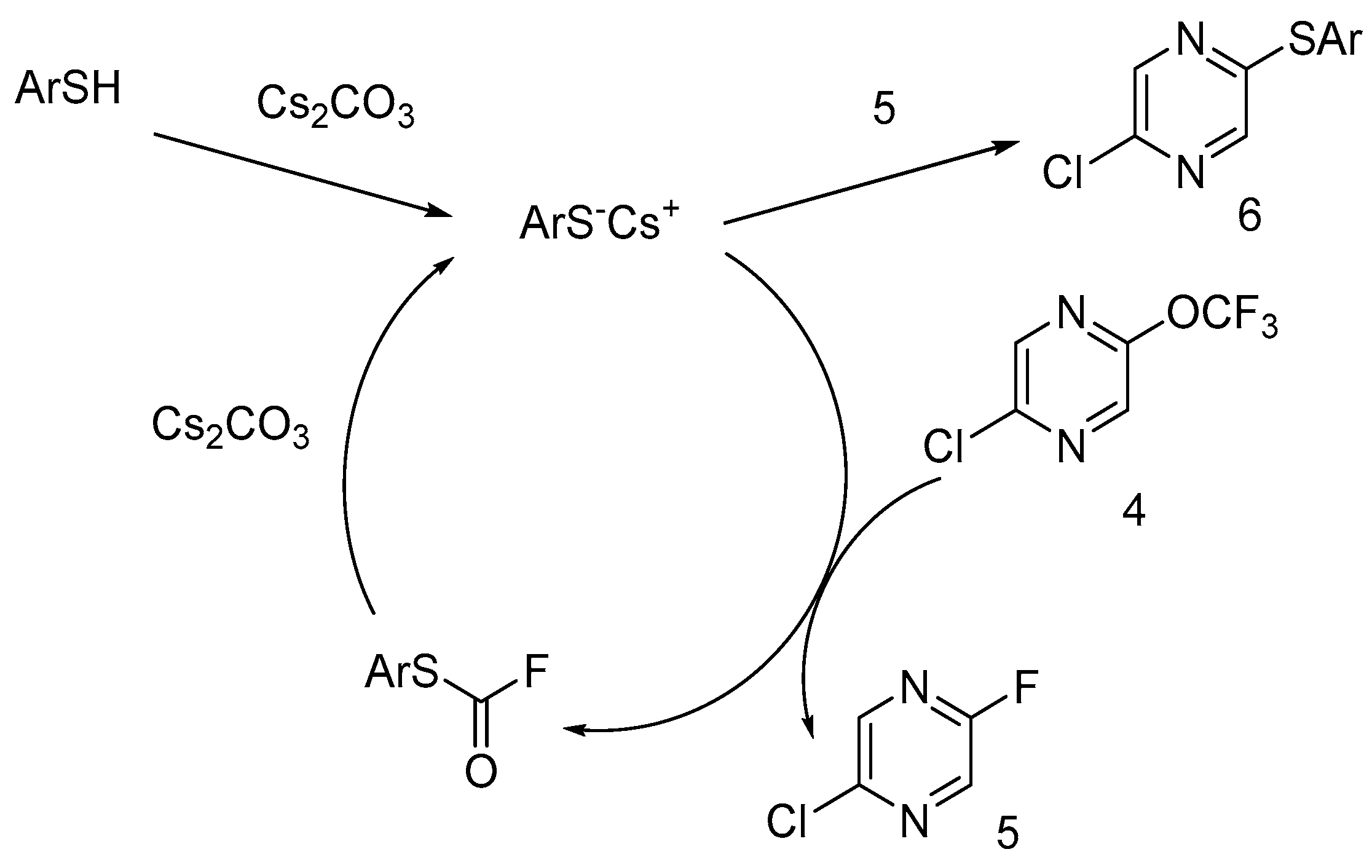
Taking into account the destruction of OCF
3 moiety in the presence of catalytic amounts of thiophenolate anions, we proposed the following scheme for trifluorometoxy group degradation (
Scheme 3): the thiophenolate anion reacts with trifluorometoxy pyrazine
4, with an S-aryl fluorocarbamate formation that can be recovered to thiophenolate by the action of cesium carbonate. The nucleophilic substitution of the fluorine atom of pyrazins
5 and
7 or the chlorine atom of pyrazines
5 and
6 is the route of removing thiophenol from the catalytic cycle (only pyrazine
5 was shown in
Scheme 3 as an example). A high yield of fluoropyrazine
5 is evidence of the high catalytic activity of thiophenol for this reaction.
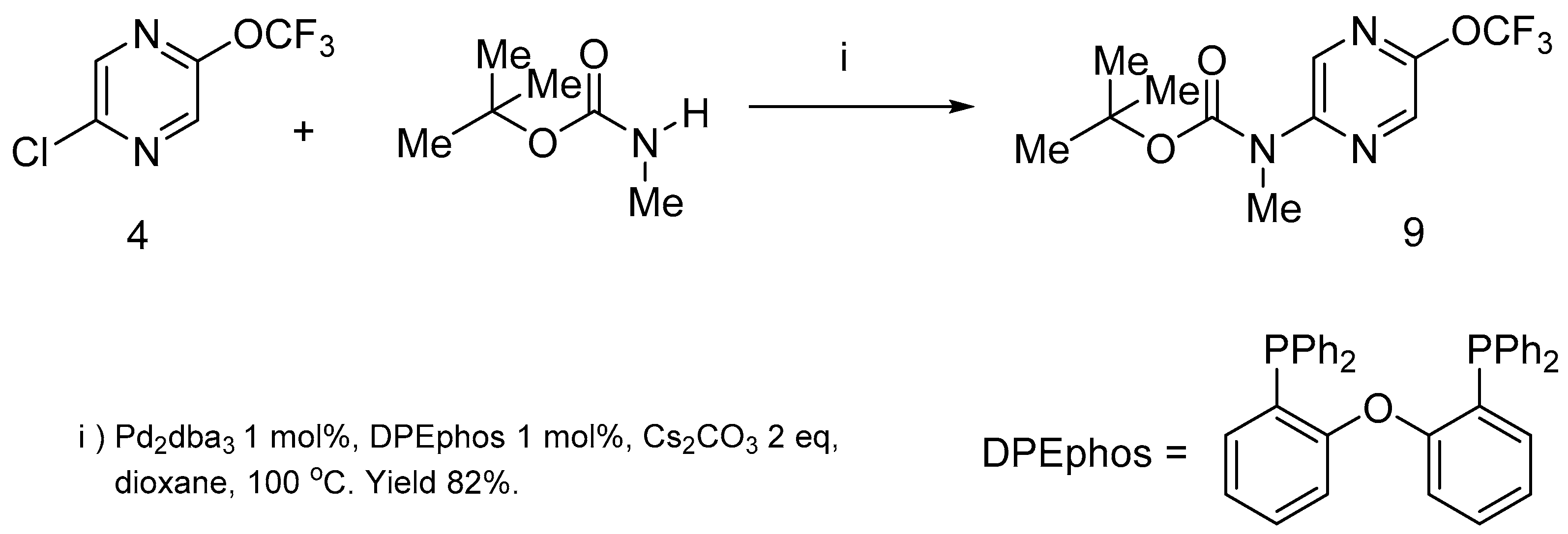
The nucleophilic substitution of chlorine atoms in pyrazine
4 by diethylamine as an example of
N- nucleophiles did not occur at room temperature. The prolonged heating (24 h) of the reaction mixture in various solvents at 100 °C led to the complete destruction of the starting material. Nevertheless, the chlorine atom of this molecule was successfully replaced by nitrogen nucleophile under Buchwald-Hartwig amination reaction conditions [
35,
36,
37]. Palladium dibenzoylacetonate (Pd
2dba
3)/DPEphos catalysis took place and the Boc-protected amine
9 was prepared in high yield (
Scheme 4).
The replacement of chlorine atom in compound
4 with the nitrile group using potassium cyanide failed, however pirazinenitrile
10 was prepared with zinc cyanide under Pd
2dba
3/diphenylphosphinoferrocene (
dppf) catalysis (
Scheme 5).
Alkylpyrazines, especially bearing methoxy group ones, are compounds of great interest due to their odor properties [
38,
39,
40]. For instance, the odor threshold of 2-methoxy-3-
sec-buthyl-pyrazine in water solution is 1 part per 10
12 parts of water [
39].
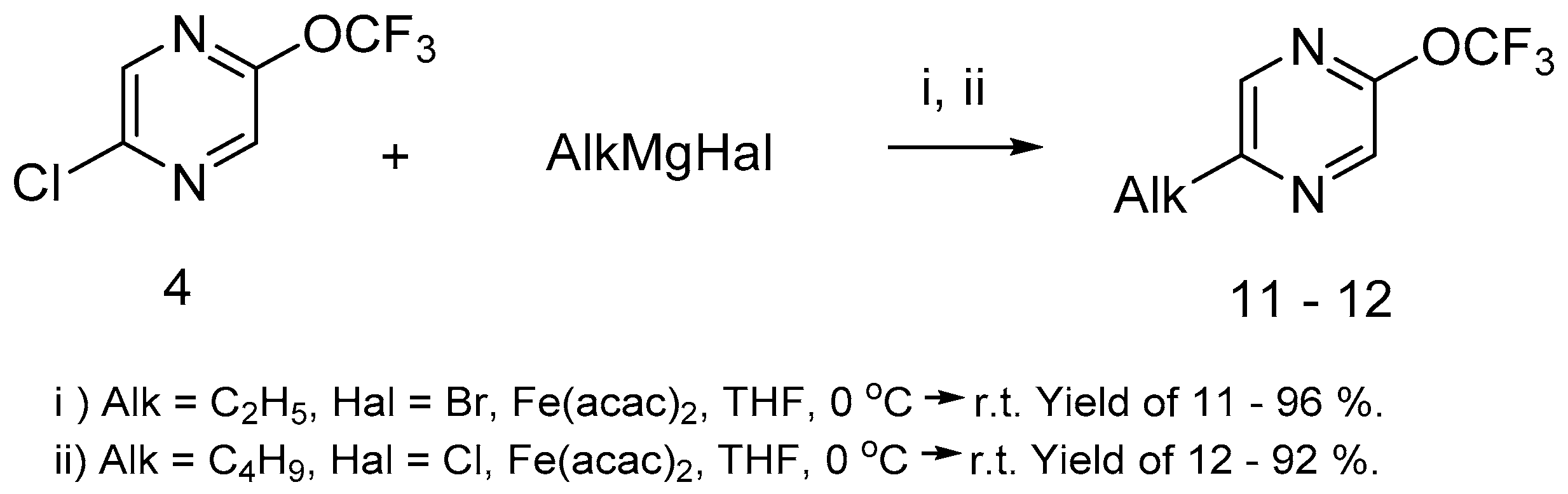
We have shown that the alkyl pirazines-bearing trifluoromethoxy group can be prepared in high yield by Kumada–Corriu coupling [
41,
42,
43] with iron acetylacetonate (Fe(acac)
2) as a catalyst (
Scheme 6). It must be noted that both alkylmagnesium halides (bromide or chloride) are suitable for this reaction.
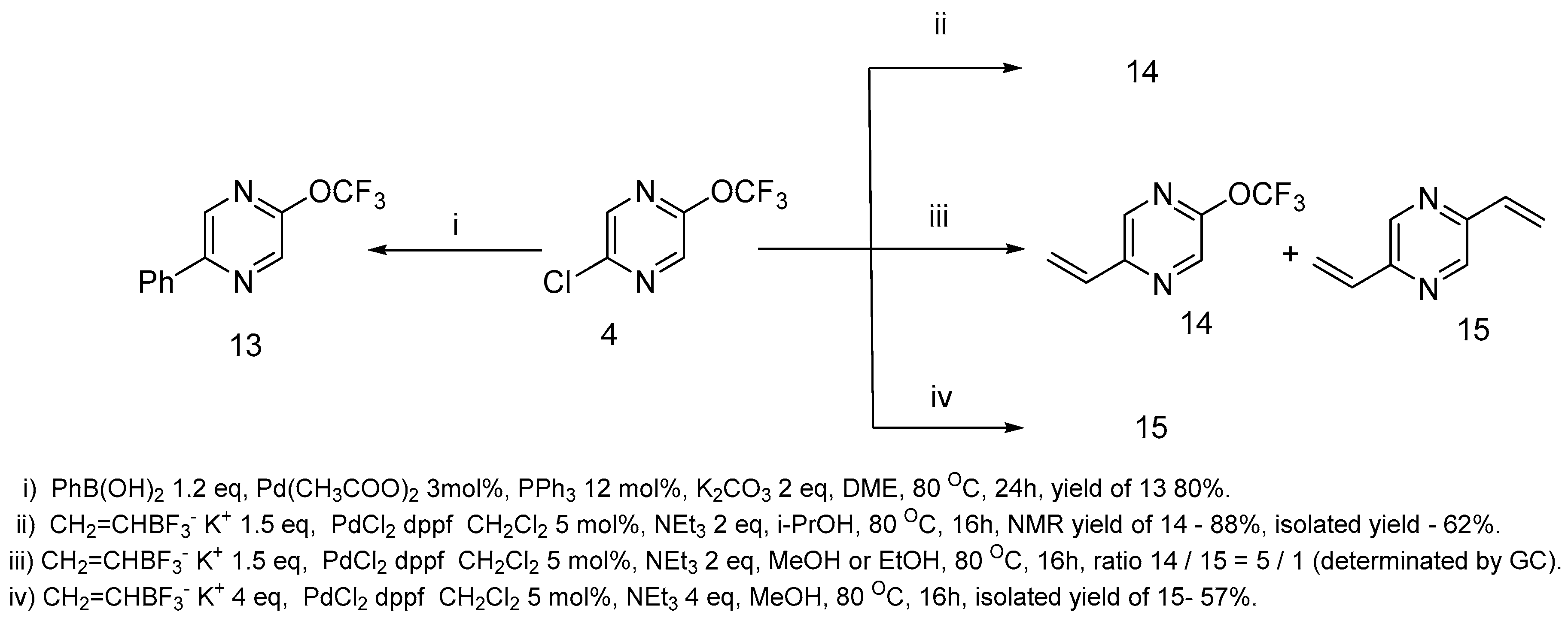
Suzuki coupling [
44] is a powerful method for pyrazine derivatization [
35]. We have found that 2-chloro-5-trifluoromethoxypyrazine
4 is a suitable substrate for coupling with boronic acids as well as with trifluoroborates. The reaction with phenylboronic acid resulted in phenylpyrazine
13 formation in a high yield (
Scheme 7). In the case of potassium ethenyl trifluoroborate, the result of the reaction is dependent on the solvents used (
Scheme 7). We tested various solvents for this reaction (DMF, MeOH, EtOH,
i-PrOH). In DMF, unidentified products were formed. Meanwhile, in the cases of MeOH and EtOH, the reaction resulted in the substitution of the chlorine atom to give product
14, as well as both the chlorine atom and trifluoromethoxy group removal with the pyrazine
15 formation. Bis-ethenyl pyrazine
15 was formed selectively if a large excess of potassium ethenyl trifluoroborate was used. Monoethenyl pyrazine
14 was produced selectively when
i-PrOH was used as the solvent. Unfortunately, it is difficult to isolate the product in a pure state in this case, and pyrazine
14 was obtained only with a 62% yield.
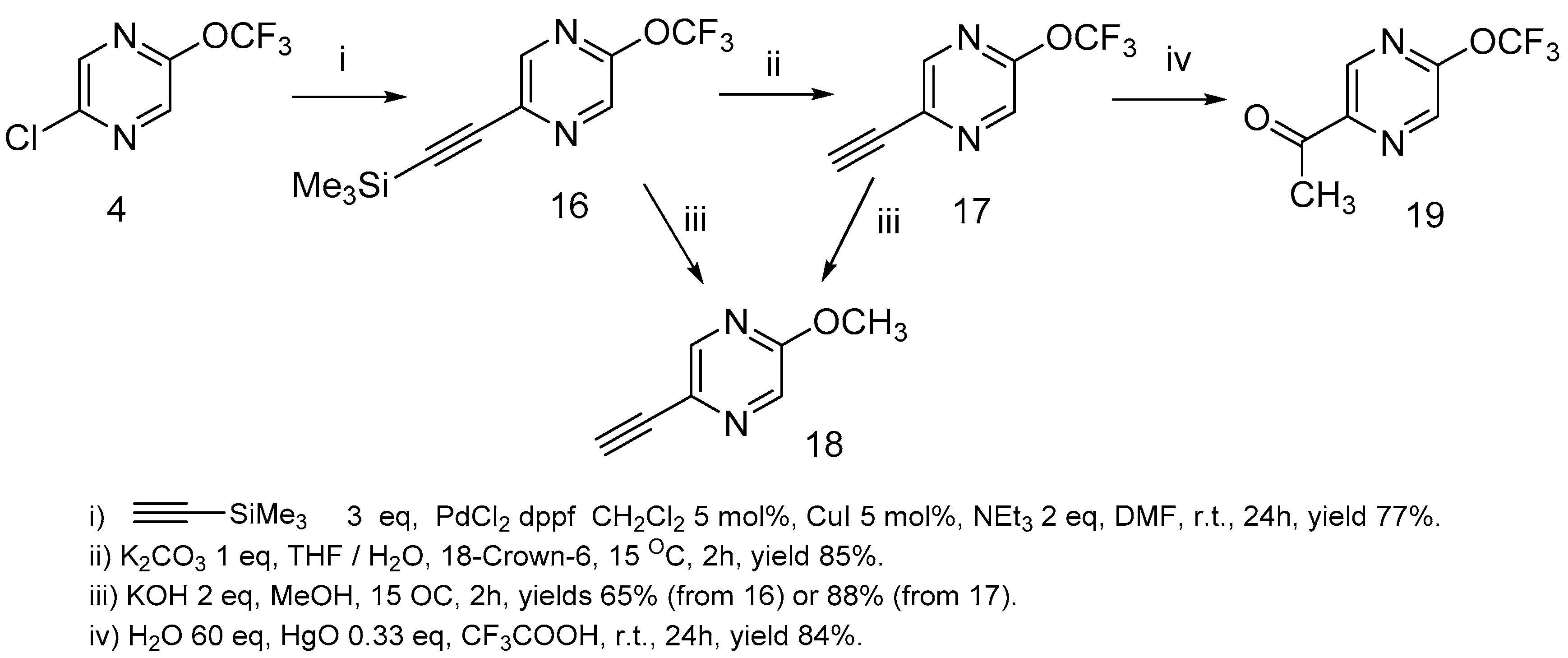
The reaction of 2-chloro-5-trifluoromethoxypyrazine
4 with ethynyltrimethylsilane under Sonogashira [
45] reaction conditions led to 2-ethynyl-5-trifluoromethoxypyrazine
16 formation in a high yield (
Scheme 8).
Ethynylpyrazine
16 can be successfully desililated by the action of potassium carbonate under heterogeneous reaction conditions in the THF/water mixture. When the desililation of pyrazine
16 was performed with KOH in a methanol/water solution, as was described in the literature [
46], a mixture of trifluoromethoxypirazine
17 and methoxypyrazine
18 was obtained. Methoxypyrazine
18 was prepared starting from trifluoromethoxypyrazine
17 or sililated pyrazine
16 by the action of KOH in methanol. The hydration of acetylene
17 via the Kucherov reaction [
47,
48] resulted in 2-acethyl-5-trifluoromethoxy-pyrazine
19 formation in a high yield (
Scheme 8).
3. Materials and Methods
1H-NMR spectra were recorded at 300 MHz with a Varian VXR-300 spectrometer (Varian Inc., Palo Alto, CA, USA), at 500 MHz with a Bruker AVANCE DRX 500 instrument (Bruker, Billerica, MA, USA), or at 400 MHz with a Varian UNITY-Plus 400 spectrometer (Varian Inc., Palo Alto, CA, USA).
13C-NMR-spectra (proton decoupled) were recorded on a Bruker AVANCE DRX 500 instrument at 125 MHz, or at 100 MHz with a Varian UNITY-Plus 400 spectrometer (Varian Inc., Palo Alto, CA, USA).
19F-NMR spectra were recorded at 376 MHz with a Varian UNITY-Plus 400 spectrometer (Varian Inc., Palo Alto, CA, USA). The chemical shifts are given in ppm relative to Me
4Si and CCl
3F, respectively, as internal or external standards. The
1H-,
13C-, and
19F-NMR spectra of the compounds can be found in the
Supplementary Materials. The LC-MS spectra were registered on an “Agilent 1100 Series” instrument with a diode-matrix and mass-selective detector “Agilent 1100 LS/MSD SL” (ionization method: chemical ionization at atmospheric pressure; ionization chamber operation conditions: simultaneous scanning of positive and negative ions in the range 80–1000
m/
z, Agilent Technologies, Santa Clara, CA, USA). The GC-MS spectra were registered on a Hewlett-Packard HP GC/MS 5890/5972 instrument (EI 70 eV) (Philips, Bothell, WA, USA). The melting points were determined in open capillaries using an SMP3 instrument (Stuart Scientific Bibby Sterlin Ltd., Stone, Staffordshire, UK). An elemental analysis was performed in the Analytical Laboratory of the Institute of Organic Chemistry, NAS, Ukraine, Kyiv.
Unless otherwise stated, commercially-available reagents were purchased from Enamine Ltd. (Kyiv, Ukraine) and were used without purification. The solvents were purified according to the standard procedures [
49]. Antimony trifluoride was sublimated immediately prior to use. The 2-Hydroxy-5-chloropyrazine
1 was prepared starting from 2-amino-5-chloropyrazine according to method [
50] in a 37 g scale.
All the reactions were performed in an argon atmosphere. For the column chromatography, Merck Kieselgel 60 silica gel (Merck, Darmstadt, Germany) was used. Thin-layer chromatography (TLC) was carried out on aluminum-backed plates coated with silica gel (Merck Kieselgel 60 F254, Merck, Darmstadt, Germany).
3.1. 2-Chloro-5-(trichloromethoxy)pyrazine 2
5-Chloropyrazin-2-ol 1 (40 g, 0.31 mol) was added in portions to the solution of NaOH (14.1 g, 0.35 mol) in water (250 mL) at 5–10 °C. The mixture was cooled to 0 °C and the solution of thiophosgene (35.3 g, 0.31 mol) in chloroform (250 mL) was added dropwise over 1 h under vigorous stirring. After the addition was complete, the mixture was vigorously stirred for a further 3 h at 0 °C before being extracted with chloroform (5 × 100 mL). The combined organic layers were washed with water and dried with MgSO4. After the dehydration agent had been filtered off, the mixture was saturated with chlorine and stirred for 3 days at 25 °C. The excess of chlorine was removed with nitrogen gas stream, the solvent was distilled off in a vacuum (300 mbar), and the residue was distilled in a vacuum, yielding trichloromethoxypyrazine 2 as a colorless oil or solid. The yield was 47.9 g (63%); b.p. 100–101 °C (1 mbar); m.p. 26–28 °C. 1H-NMR (400 MHz, CDCl3) δ 8.32 (s, 1H, HPy), 8.37 (s, 1H, HPy). 13C-NMR (126 MHz, CDCl3) δ 108.9 (s, OCCl3), 136.0, 140.8, 145.6, 154.0. GC-MS, 70 eV, m/z (rel. int.): 248 (100) [M(335Cl + 37Cl)]+, 246 (80) [M(435Cl)]+, 250 (50) [M(235Cl + 237Cl)]+. Anal. Calcd. for C4H2ClFN2: Cl, 57.20; Found: Cl, 57.26.
3.2. 2-(Bromodifluoromethoxy)-5-chloropyrazine 3
To the solution of 5-chloropyrazin-2-ol (40 g, 0.31 mol) and tetrabutylammonium bromide (4.9 g, 15 mmol) in anhydrous DMF (300 mL), NaH (60% in mineral oil) (14.7 g, 0.37 mol) was added in portions at 0 °C and the mixture was stirred for 2 h at r.t. The mixture was cooled to −30 °C and dibromodifluoromethane (257 g, 1.23 mol) was added dropwise. The mixture was carefully warmed and stirred at 30–35 °C for 6 h. Water (900 mL) was added dropwise to the mixture and the excess of CBr2F2 was distilled in a dry ice trap. The residue was extracted with MTBE (tert-butyl-methyl ether) (7 × 100 mL), extract was washed with brine and dried with MgSO4. The solvent was distilled off at atmospheric pressure and the product was distilled in a vacuum. The yield was 29.4 g (37%); colorless liquid or solid; b.p. 80–81 °C (10 mbar); m.p. 14–17 °C. 1H-NMR (400 MHz, CDCl3) δ 8.27 (s, 1H, HPy), 8.35 (s, 1H, HPy). 13C-NMR (126 MHz, CDCl3) δ 111.5 (t, 1JCF = 315.5 Hz, CBrF2), 135.2, 141.0, 145.9, 152.6. 19F-NMR (376.5 MHz, CDCl3) δ −18.4 (s, CBrF2). GC-MS, 70 eV, m/z (rel. int.): 258 (75) [M(79Br + 35Cl)]+, 260 (100) [M(79Br + 37Cl) or (81Br + 35Cl)]+, 262 (25) [M(81Br + 37Cl)]+. Anal. Calcd. for C5H2BrClF2N2O: C, 23.15; H, 0.78; N, 10.80; Found: C, 23.12; H, 0.68; N, 10.72.
3.3. 2-Chloro-5-trifluoromethoxypyrazine 4
Method A. The mixture of freshly sublimated SbF3 (103.7 g, 0.58 mol) and SbCl5 (17.4 g, 0.06 mol) was heated at 125–130 °C for 15 min and 2-chloro-5-(trichloromethoxy)pyrazine 2 (47.9 g, 0.19 mol) was added in portions at 100 °C to this mixture. The reaction mixture was stirred at 145–150 °C for 5 h and then 1 h at 155–160 °C. After cooling to r.t., the mixture was suspended in CH2Cl2 (700 mL), and solutions of K2CO3 (138 g, 1 mol) in 700 mL of water and KF (174 g, 3 mol) in 500 mL of water were carefully added to the mixture. The mixture was filtered through celite, the organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (3 × 100 mL). The combined extracts were washed with water and dried with MgSO4. The solvent was distilled off at atmospheric pressure and the product was distilled in a vacuum.
Method B. Silver tetrafluoroborate (24.3 g, 0.125 mol) was added in portions to the solution of 2-(bromodifluoromethoxy)-5-chloropyrazine 3 (29.4 g, 0.11 mol) in CH2Cl2 (250 mL) at −78 °C. The mixture was warmed to r.t. and stirred for 24 h in darkness. The solution of sodium bicarbonate (11.4 g, 0.136 mol) in 150 mL of water was added to the reaction mixture and the mixture was filtered through celite. The organic layer was separated and the aqueous layer was extracted with CH2Cl2 (3 × 100 mL). The combined extracts were washed with water and dried with MgSO4. The solvent was distilled off at atmospheric pressure and the product was distilled in a vacuum.
The yield (Method A) was 26.0 g (68%); (Method B) 20.7 g (92%); colorless liquid; b.p. 80–81 °C (100 mbar). 1H-NMR (400 MHz, CDCl3) δ 8.23 (s, 1H, HPy), 8.29 (s, 1H, HPy). 13C-NMR (126 MHz, CDCl3) δ 119.5 (q, 1JCF = 262.5 Hz, CF3), 134.6, 140.9, 145.8, 151.8. 19F-NMR (376.5 MHz, CDCl3) δ −57.6 (s, CF3). GC-MS, 70 eV, m/z (rel. int.): 198 (100) [M(35Cl)]+, 20 (30) [37Cl)]+. Anal. Calcd. for C5H2ClF3N2O: C, 30.25; H, 1.02; Cl, 17.86; N, 14.11; Found: C, 30.22; H, 0.98; Cl, 17.93; N, 14.09.
3.4. Reaction of 2-Chloro-5-trifluoromethoxypyrazine 4 with 3-Methylthiophenol and Alternative Route to 2-Chloro-5-fluoropyrazine 5
Method A. The mixture of 2-chloro-5-trifluoromethoxypyrazine 4 (1.0 g 5 mmol), 3-methylthiophenol (0.06 g, 0.5 mmol), and cesium carbonate (2.46 g, 7.5 mmol) in anhydrous DMF (5 mL) was stirred at r.t. under Argon for 24 h. The mixture was poured into water (20 mL) and extracted with ether (5 × 10 mL). The etheral solution was washed with 5% sodium bicarbonate solution and then with brine and dried with MgSO4. The solvent was distilled off at atmospheric pressure and the product was distilled in a vacuum.
Method B. The mixture of 2-chloro-5-trifluoromethoxypyrazine 4 (1.0 g 5 mmol), 3-methylthiophenol (0.62 g, 5 mmol), and cesium carbonate (2.46 g, 7.5 mmol) in anhydrous DMF (5 mL) was stirred at r.t. under Argon for 24 h. The mixture was poured into water (20 mL) and extracted with ether (5 × 10 mL). The etheral solution was washed with 5% sodium bicarbonate solution and then with brine and dried with MgSO4. The solvent was distilled off at atmospheric pressure and the volatile products were distilled into a liquid nitrogen trap in a high vacuum (0.2 mbar) at 35–40 °C (bath temperature). After redistillation at 100 mbar, 2-chloro-5-fluoropyrazine was obtained. The residue after the high vacuum distillation was separated by TLS chromatography using pentane/ethylacetate (5:1 by vol.) as an eluent, yielding a mixture of pirazines 6 and 7 (with ratio 1:2 by weight) and disubstituted pyrazine 8.
Method C. The mixture of 2-chloro-5-trifluoromethoxypyrazine 4 (0.5 g 2.5 mmol), 3-methylthiophenol (0.93 g, 7.5 mmol), and cesium carbonate (2.46 g, 7.5 mmol) in anhydrous DMF (5 mL) was stirred at r.t. under Argon for 24 h. The mixture was poured into water (20 mL) and extracted with ether (5 × 10 mL). The etheral solution was washed with 5% sodium bicarbonate solution and then with brine and dried with MgSO4. The solvent was distilled off in a vacuum and the residue was purified by column chromatography using a mixture of hexane/ethylacetate (5:1 by vol.) as an eluent, yielding disubstituted pyrazine 8.
An alternative route to 2-chloro-5-fluoropyrazine5. To the solution of 2-amino-5-chloropyrazine (5 g, 38.6 mmol) in 50% aqueous HBF4 (20 mL), sodium nitrite (5.3 g, 77.2 mmol) was added in portions at 0–5 °C. The reaction mixture was gently heated to 18–20 °C and stirred for 4 h. The mixture was extracted with ether (5 × 20 mL), and the etheral solution was washed with brine and dried with MgSO4. The solvent was distilled off at atmospheric pressure and the product was distilled in a vacuum.
2-Chloro-5-fluoropyrazine5. The yield was (Method A) 0.49 (74%), (method B) 0.12 g (18%), (alternative method) 4.77 g (93%); colorless liquid, b.p. 80–81 °C (100 mbar). 1H-NMR (500 MHz, CDCl3) δ 8.22 (s, 1H, H3′), 8.24 (d, 1H, 3JHF = 8.5 Hz, H6′). 13C-NMR (126 MHz, CDCl3) δ 132.4 (d, 2JCF = 40.2 Hz, C6′), 141.2 (d, 3JCF = 10.0 Hz, C3′), 145.3 (CCl), 159.3 (d, 1JCF = 252.7 Hz, CF). 19F-NMR (376.5 MHz, CDCl3) δ −85 (s, CF). GC-MS, 70 eV, m/z (rel. int.): 132 (100) [M(35Cl)]+, 134 (32) [M(37Cl)]+. Anal. Calcd. for C4H2ClFN2: C, 36.25; H, 1.52; Cl, 26.75; N, 21.14; Found: C, 36.22; H, 1.53; Cl, 26.73; N, 21.09.
The mixture of 2-chloro-5-(m-tolylthio)pyrazine6and 2-fluoro-5(m-tolylthio)pyrazine7. The yield of the mixture was 0.33 g: chloropyrazine 6–11%, fluoropyrazine 7–22% (the yield was determined by NMR and LC analyses of the mixture of pyrazines 6 and 7). Rf = 0.8 pentane/ethylacetate (5:1). Chloropyrazine 6: 1H-NMR (500 MHz, CDCl3) δ 2.36 (s, 3H, CH3), 7.25–7.27 (m, 1H, CHAr), 7.34–7.37 (m, 1H, CHAr), 7.40–7.42 (m, 2H, 2CHAr), 7.96 (s, 1H, H6′Py), 8.37 (s, 1H, H6′Py); LC/MS (CI): m/z = 237 [M(35Cl) + H]+, 239 [M(37Cl) + H]+. Fluoropyrazine 7 1H-NMR (500 MHz, CDCl3) δ 2.36 (s, 3H, CH3), 7.25–7.27 (m, 1H, CHAr), 7.34–7.37 (m, 1H, CHAr), 7.40–7.42 (m, 2H, 2CHAr), 7.84 (s, 1H, H6′Py), 7.26 (d, 1H, 3JHF = 8.5 Hz, H3′Py). 19F-NMR (376.5 MHz, CDCl3) δ −87.9 (s, CF). LC/MS (CI): m/z = 221 [M + H]+.
2,5-Bis(m-tolylthio)pyrazine8. Yield (method B) 0.4 g (25%), (method C) 0.75 g (93%); yellowish powder, m.p. 102–103 °C. Rf = 0.6 pentane/ethylacetate (5:1). 1H-NMR (400 MHz, CDCl3) δ 2.34 (s, 6H, 2CH3), 7.18–7.21 (m, 2H, 2CHAr), 7.27 (t, 2H, 3JHH = 7.6 Hz, 2m-CHAr), 7.33–7.37 (m, 4H, 4CHAr), 8.03 (s, 2H, CHPy). 13C-NMR (126 MHz, CDCl3) δ 21.3, 129.4, 129.6, 130.3, 131.7, 135.2, 139.7, 142.5, 153.6. LC/MS (CI): m/z = 325 [M + H]+. Calcd. for C18H16N2S2: C, 66.63; H, 4.97; N, 8.63; S, 19.76; Found: C, 66.68; H, 5.02; N, 8.59; S, 19.70.
3.5. tert-Butyl Methyl(5-(trifluoromethoxy)pyrazin-2-yl)carbamate 9
To the carefully degassed mixture of 2-chloro-5-trifluoromethoxypyrazine 4 (2 g, 10 mmol), N-Boc-methylamine (2 g, 15 mmol) and Cs2CO3 (6.5 g, 20 mmol) in anhydrous dioxane (30 mL) Pd2dba3 (0.09 g, 0.1 mmol) and bis[(2-diphenylphosphino)phenyl] ether (DPEphos) (0.05 g, 0.1 mmol) were added and the mixture was stirred at 100 °C for 12 h. After cooling to r.t., the mixture was diluted with water (20 mL), filtered through celite, and extracted with ethylacetate (5 × 10 mL). The organic extract was washed with brine, dried with MgSO4, and concentrated in a vacuum. After column chromatography with the mixture of hexane/ethylacetate (5:1 by vol.) as the eluent, pyrazine 9 was obtained. The yield was 2.4 g (82%); colorless powder, m.p. 78–79 °C. 1H-NMR (400 MHz, CDCl3) δ 1.54 (s, 9H, C(CH3)3), 3.39 (s, 3H, CH3), 8.18 (s, 1H, HPy), 8.82 (s, 1H, HPy). 13C-NMR (100 MHz, CDCl3) δ 28.2 (C(CH3)3), 33.8 (NCH3), 82.5 (C(CH3)3), 120.2 (q, 1JCF = 262.5 Hz, CF3), 132.1, 137.6, 148.5, 150.1, 153.6. 19F-NMR (376.5 MHz, CDCl3) δ −57.5 (s, CF3). LC/MS (CI): m/z = 294 [M + H]+. Calcd. for C11H14F3N3O3: C, 45.05; H, 4.81; N, 14.33; Found: C, 45.08; H, 4.88; N, 14.29.
3.6. 5-(Trifluoromethoxy)pyrazine-2-carbonitrile 10
To the carefully degassed mixture of 2-chloro-5-trifluoromethoxypyrazine 4 (1.00 g, 5 mmol), Zn(CN)2 (0.39 g, 3.3 mmol) and Zn dust (0.6 g, 0.9 mmol) in anhydrous dimethylacetamide (10 mL) Pd2dba3 (0.14 g, 0.15 mmol) and dppf (0.17 g, 0.3 mmol) were added, and the mixture was stirred at 100 °C for 8 h. After cooling to r.t., the mixture was diluted with water (30 mL), filtered through celite, and extracted with ether (7 × 10 mL). The etheral solution was washed with brine (3 × 10 mL), dried with MgSO4, and the solvent was distilled off under atmospheric pressure. The residue was purified by TLC chromatography, yielding 0.34 g (36%) cyanopyrazine 10 as a colorless liquid with an intense almond scent, b.p. 95–96 °C (100 mbar). Rf = 0.8 pentane/ethylacetate (10:1). 1H-NMR (400 MHz, CDCl3) δ 8.51 (s, 1H, HPy), 8.62 (s, 1H, HPy). 13C-NMR (125.6 MHz, CDCl3) δ 114.5 (CN), 119.8 (q, 1JCF = 266.5 Hz, CF3), 127.5, 136.9, 145.7, 154.3. 19F-NMR (376.5 MHz, CDCl3) δ −57.5 (s, CF3). GC-MS, 70 eV, m/z (rel. int.): 189 (100) [M]+. Calcd. for C6H2F3N3O: C, 38.11; H, 1.07; N, 22.22; Found: C, 38.18; H, 1.16; N, 22.19.
3.7. Synthesis of Alkylpyrazines-Bearing Trifluoromethoxy Group by Kumada-Corriu Coupling
The solution of ethylmagnesium bromide (18 mL, 18 mmol, 1 mol/L solution in THF) or butylmagnesium chloride (9 mL, 18 mmol, 2 mol/L solution in THF) was added dropwise at 0 °C in an Argon atmosphere to the mixture of 2-chloro-5-trifluoromethoxypyrazine 4 (3.0 g, 15 mmol) and Fe(acac)2 (0.19 g, 0.75 mmol) in 30 mL of anhydrous THF. The mixture was warmed to r.t. and stirred for 3 h (in the case of ethylmagnesium bromide) or 4 h (in the case of butylmagnesium chloride). The reaction mixture was cooled to 0 °C and neutralized with 3% aqueous HCl. The product was extracted with ether (5 × 10 mL) and the etheral solution was washed with brine (5 × 10 mL) and dried with MgSO4. The solvent was distilled off at atmospheric pressure and the product was distilled in a vacuum.
2-Ethyl-5-(trifluoromethoxy)pyrazine11. Yield 2.77 g (96%); colorless liquid with intense fruit scent, b.p. 83–85 °C (100 mbar). 1H-NMR (400 MHz, CDCl3) δ 1.31 (t, 3H, 3JHH = 7.6 Hz, CH3), 2.84 (q, 2H, 3JHH = 7.6 Hz, CH2) 8.13 (s, 1H, HPy), 8.35 (s, 1H, HPy). 13C-NMR (126 MHz, CDCl3) δ 13.3 (s, CH3), 22.8 (s, CH2), 120.1 (q, 1JCF = 262.7 Hz, CF3), 135.0, 140.1, 151.8, 156.7. 19F-NMR (376.5 MHz, CDCl3) δ −57.3 (s, CF3). GC-MS, 70 eV, m/z (rel. int.): 192 (100) [M]+. Anal. Calcd. for C7H7F3N2O: C, 43.76; H, 3.67; N, 14.58; Found: C, 43.62; H, 3.53; N, 14.49.
2-Butyl-5-(trifluoromethoxy)pyrazine12. 3.03 g (92%); colorless liquid with intense fruit scent, b.p. 83–85 °C (20 mbar). 1H-NMR (400 MHz, CDCl3) δ 0.92 (t, 3H, 3JHH = 7.6 Hz, CH3), 1.37 (sext, 2H, 3JHH = 7.6 Hz, CH2CH3), 1.67 (quint, 2H, 3JHH = 7.6 Hz, CH2CH2CH2), 2.82 (t, 2H, 3JHH = 7.6 Hz, CH2) 8.11 (s, 1H, HPy), 8.34 (s, 1H, HPy). 13C-NMR (126 MHz, CDCl3) δ 13.8 (s, CH3), 22.2 (s, CH2), 31.5 (s, CH2), 34.2 (s, CH2), 120.1 (q, 1JCF = 262.3 Hz, CF3), 135.0, 140.5, 151.8, 155.8. 19F-NMR (376.5 MHz, CDCl3) δ −57.3 (s, CF3). GC-MS, 70 eV, m/z (rel. int.): 220 (100) [M]+. Anal. Calcd. for C9H11F3N2O: C, 49.09; H, 5.04; N, 12.72; Found: C, 49.02; H, 5.03; N, 12.79.
3.8. 2-Phenyl-5-(trifluoromethoxy)pyrazine 13
Palladium acetate (34 mg, 0.15 mmol) and triphenylphosphine (158 mg, 0.6 mmol) were mixed together in carefully degassed dimethoxiethane (25 mL) and stirred for 15 min at r.t. under an Ar atmosphere. Chloropyrazine 4 (1.0 g, 5 mmol), phenylboronic acid (0.7 g, 6 mmol), and K2CO3 (1.4 g, 10 mmol) were added to the reaction mixture and the mixture was stirred at 75 °C for 20 h. After cooling to r.t., the mixture was diluted with water, filtered through celite, and extracted with tret-buthylmethyl ether (3 × 25 mL). The organic solution was washed with brine, dried with MgSO4, and evaporated in a vacuum. The residue was purified by column chromatography with hexane/ethylacetate (10:1 by vol.) as the eluent (Rf = 0.6). The yield was 0.95 g (80%); colorless powder, m.p. 80 °C. 1H-NMR (400 MHz, CDCl3) δ 7.48–7.55 (m, 3H, 3CHPh), 7.92–8.00 (m, 2H, 2CHPh), 8.50 (s, 1H, HPy), 8.70 (s, 1H, HPy). 13C-NMR (126 MHz, CDCl3) δ 120.2 (q, 1JCF = 262.7 Hz, CF3), 126.8, 129.1, 130.0, 135.1, 135.2, 138.4, 150.9, 152.4. 19F-NMR (376.5 MHz, CDCl3) δ −57.2 (s, CF3). GC-MS, 70 eV, m/z (rel. int.): 240 (100) [M]+. Anal. Calcd. for C11H7F3N2O: C, 55.01; H, 2.94; N, 11.66; Found: C, 49.9; H, 3.00; N, 11.69.
3.9. Reaction of 2-Chloro-5-trifluoromethoxypyrazine 4 with Potassium Ethenyl Trifluoroborate
3.9.1. 2-(Trifluoromethoxy)-5-vinylpyrazine 14
To the carefully degassed mixture of 2-chloro-5-trifluoromethoxypyrazine 4 (2.0 g, 10 mmol), potassium ethenyl trifluoroborate (2.0 g, 15 mmol) and NEt3 (2 g, 20 mmol) in anhydrous iso-propanole (10 mL) PdCl2dppf·CH2Cl2 (0.4 g, 0.5 mmol) were added and the mixture was stirred for 20 h at 75 °C. After cooling to r.t., the mixture was diluted with water (20 mL), filtered through celite, and extracted with CH2Cl2 (5 × 10 mL). The organic extract was washed with 1% aqueous HCl, then with water, and dried with CaCl2. After the removal of the solvent at atmospheric pressure, the residue was distilled with deflegmator in a vacuum. The yield was 1.17 g (62%); colorless liquid with a pungent odor, b.p. 90–91 °C (100 mbar). 1H-NMR (500 MHz, CDCl3) δ 5.63 (d, 1H, 3JHH = 11.0 Hz, =CH2), 6.33 (d, 1H, 3JHH = 17.5 Hz, =CH2), 6.83 (dd, 1H, 3JHH-trans = 17.5 Hz, 3JHH-cis = 11.0 Hz, =CH2), 8.27 (s, 1H, HPy), 8.41 (s, 1H, HPy). 13C-NMR (100 MHz, CDCl3) δ 119.9 (q, 1JCF = 262.7 Hz, CF3), 120.6, 131.7, 135.1, 139.1, 148.9, 152.1. 19F-NMR (376.5 MHz, CDCl3) δ −57.3 (s, CF3). GC-MS, 70 eV, m/z (rel. int.): 190 (100) [M]+. Anal. Calcd. for C7H5F3N2O: C, 44.22; H, 2.65; N, 14.73; Found: C, 44.29; H, 2.60; N, 14.69.
3.9.2. 5-Divinylpyrazine 15
The reaction was performed in the glass high pressure tube 50 mL vol. To the carefully degassed mixture of 2-chloro-5-trifluoromethoxypyrazine 4 (2.0 g, 10 mmol), potassium ethenyl trifluoroborate (5.4 g, 40 mmol) and NEt3 (4.1 g, 40 mmol) in anhydrous methanol (10 mL) PdCl2dppf·CH2Cl2 (0.8 g, 15 mmol) were added and the mixture was stirred for 20 h at 75 °C. After cooling to r.t., the mixture was diluted with water (20 mL), filtered through celite, and extracted with CH2Cl2 (5 × 10 mL). The organic extract was washed with 1% aqueous HCl, then with water, and dried with CaCl2. After the removal of the solvent at atmospheric pressure, the residue was purified by flash column chromatography with pentane/ethylacetate (5:1 by vol.) as the eluent (Rf = 0.8). The yield was 0.75 g (57%); colorless liquid, b.p. 45–47 °C (10 mbar). Compound 15 was partly polymerized under distillation and completely polymerized after 1-month storage in a refrigerator. 1H-NMR (500 MHz, CDCl3) δ 5.60 (d, 2H, 3JHH = 11.0 Hz, 2 =CH2), 6.34 (d, 2H, 3JHH = 17.5 Hz, =CH2), 6.83 (dd, 2H, 3JHH-trans = 17.5 Hz, 3JHH-cis =11.0 Hz, =CH2), 8.54 (s, 2H, HPy). 13C-NMR (100 MHz, CDCl3) δ 120.3, 133.3, 142.4, 149.5. GC-MS, 70 eV, m/z (rel. int.): 132 (100) [M]+. Anal. Calcd. for C8H8N2: C, 72.70; H, 6.10; N, 21.21; Found: C, 72.72; H, 6.10; N, 21.19.
3.10. 2-(Trifluoromethoxy)-5-((trimethylsilyl)ethynyl)pyrazine 16
To the carefully degassed mixture of 2-chloro-5-trifluoromethoxypyrazine 4 (3.0 g, 15 mmol), ethynyltrimethylsilane (4.4 g, 45 mmol) and NEt3 (9.2 g, 90 mmol) in anhydrous DMF (25 mL) PdCl2dppf·CH2Cl2 (0.62 g, 0.75 mmol) and CuI (0.14 g, 0.75 mmol) were added and the mixture was stirred for 20 h at r.t. The mixture was diluted with water (75 mL), filtered through celite, and extracted with MTBE (5 × 50 mL). The organic extract was washed with 1% aqueous HCl, then with brine, and dried with MgSO4. After the removal of the solvent, the residue was distilled in a vacuum. An effective deflegmator was necessary, due to difficulties in the separation of 1,4-bis(trimethylsilyl)buta-1,3-diyne that was formed as by-product. The yield was 3.0 g (77%); colorless oil, b.p. 118–120 °C (100 mbar). 1H-NMR (400 MHz, CDCl3) δ 0.26 (s, 9H, SiMe3), 8.35 (s, 2H, 2 HPy). 13C-NMR (100 MHz, CDCl3) δ-0.48 (s, SiMe3), 99.4 (s, C≡C), 100.2 (s, C≡C), 120.0 (q, 1JCF = 263.6 Hz, CF3), 135.4 (s, CPy), 137.2 (s, CPy), 144.5 (s, CPy), 151.8 (s, CPy). 19F-NMR (376.5 MHz, CDCl3) δ −57.3 (s, CF3). GC-MS, 70 eV, m/z (rel. int.): 260 (100) [M]+. Anal. Calcd. for C10H11F3N2OSi: C, 46.14; H, 4.26; N, 10.76; Found: C, 46.19; H, 4.20; N, 10.69.
3.11. 2-Ethynyl-5-(trifluoromethoxy)pyrazine 17
The solution of 2-(trifluoromethoxy)-5-((trimethylsilyl)ethynyl)pyrazine 16 (2 g, 7.7 mmol) and 18-crown-6 (0.05 g, 0.2 mmol) in THF (20 mL) was added dropwise at 5–10 °C to the solution of K2CO3 (1.1 g, 7.7 mmol) in 3 mL of water. The mixture was stirred for 2 h at 10–15 °C, neutralized with 5% aqueous HCl, and extracted with MTBE (5 × 25 mL). The organic extract was washed with brine and dried with MgSO4. After the removal of the solvent at atmospheric pressure, the residue was distilled in a vacuum. The yield was 1.2 g (85%); colorless oil or solid with a grass scent, b.p. 76–77 °C (10 mbar), m.p. 45–46 °C. 1H-NMR (400 MHz, CDCl3) δ 3.33 (s, 1H, ≡CH), 8.39 (s, 2H, 2 HPy). 13C-NMR (100 MHz, CDCl3) δ 78.8 (s, ‒C≡), 81.4 (s, ≡CH), 119.9 (q, 1JCF = 263.6 Hz, CF3), 135.6 (s, CPy), 136.4 (s, CPy), 144.8 (s, CPy), 152.2 (s, CPy). 19F-NMR (376.5 MHz, CDCl3) δ −57.3 (s, CF3). GC-MS, 70 eV, m/z (rel. int.): 188 (100) [M]+. Anal. Calcd. for C7H3F3N2O: C, 44.70; H, 1.61; N, 14.89; Found: C, 44.79; H, 1.60; N, 14.88.
3.12. 2-Ethynyl-5-methoxypyrazine 18
Method A. The solution of 2-(trifluoromethoxy)-5-((trimethylsilyl)ethynyl)pyrazine 16 (1 g, 3.8 mmol) in anhydrous methanol (3 mL) was added to 5 mL of the methanolic solution of KOH (0.43 g, 7.7 mmol) at 0 °C. The mixture was stirred for 2 h at 15 °C and the solvent was removed in a vacuum (without heating). The residue was suspended in ether and neutralized with 3% aqueous HCl. The organic solution was separated and the aqueous layer was extracted with ether (3 × 10 mL). The combined organic solutions were washed with brine and dried with MgSO4. After the removal of the solvent at atmospheric pressure, the residue was distilled in a vacuum.
Method B. The solution of 2-Ethynyl-5-(trifluoromethoxy)pyrazine 17 (1 g, 5.3 mmol) in anhydrous methanol (3 mL) was added to 5 mL of the methanolic solution of KOH (0.43 g, 7.7 mmol) at −10 °C. The mixture was stirred for 2 h at 15 °C and the solvent was removed in a vacuum (without heating). The residue was suspended in ether and neutralized with 3% aqueous HCl. The organic solution was separated and the aqueous layer was extracted with ether (3 × 10 mL). The combined organic solutions were washed with brine and dried with MgSO4. After the removal of the solvent at atmospheric pressure, the residue was distilled in a vacuum.
The yield (Method A) was 0.34 g (65%), (Method B) 0.63 g (88%); colorless solid, m.p. 40–41 °C, b.p. 88–90 °C (10 mbar); 1H-NMR (400 MHz, CDCl3) δ 3.22 (s, 1H, ≡CH), 3.98 (s, 3H, CH3), 8.18 (s, 2H, 2 HPy), 8.25 (s, 2H, 2 HPy). 13C-NMR (100 MHz, CDCl3) δ 53.9 (s, CH3), 78.9 (s, ≡CH), 80.0 (s, ‒C≡), 130.4 (s, CPy), 135.6 (s, CPy), 144.4 (s, CPy), 159.4 (s, CPy). GC-MS, 70 eV, m/z (rel. int.): 134 (100) [M]+. Anal. Calcd. for C7H6N2O: C, 62.68; H, 4.51; N, 20.88; Found: C, 62.62; H, 4.44; N, 20.80.
3.13. 1-(5-(Trifluoromethoxy)pyrazin-2-yl)ethan-1-one 19
Mercury oxide (0.38 g, 1.8 mmol) was added to the mixture of trifluoroacetic acid (60 mL, 800 mmol) and water (6 mL, 332 mmol) and stirred until dissolving (apr 10 min). The mixture was cooled to 10 °C and 2-ethynyl-5-(trifluoromethoxy)pyrazine 17 (1.0 g, 5.3 mmol) was added. The mixture was stirred at 40 °C for 1 h and trifluoroacetic acid was removed in a vacuum (300 mbar). Water (20 mL) was added to the residue and the mixture was neutralized with NaHCO3 to pH 6. The product was extracted with CHCl3 (3 × 10 mL) and the extract was washed with water and dried with MgSO4. After the removal of the solvent at atmospheric pressure, the residue was distilled in a vacuum. The yield was 0.92 g (84%); colorless solid, m.p. 44–45 °C, b.p. 94–95 °C (10 mbar); 1H-NMR (400 MHz, CDCl3) δ 2.69 (s, 3H, CH3), 8.42 (s, 2H, 2 HPy), 8.92 (s, 2H, 2 HPy). 13C-NMR (100 MHz, CDCl3) δ 25.9 (s, CH3), 119.9 (q, 1JCF = 264.6 Hz, CF3), 134.1 (s, CPy), 141.3 (s, CPy), 145.6 (s, CPy), 155.1 (s, CPy), 197.5 (s, CO). 19F-NMR (376.5 MHz, CDCl3) δ −57.3 (s, CF3). GC-MS, 70 eV, m/z (rel. int.): 206 (100) [M]+. Anal. Calcd. for C7H5F3N2O: C, 40.79; H, 2.45; N, 13.59; Found: C, 40.70; H, 2.44; N, 13.60.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
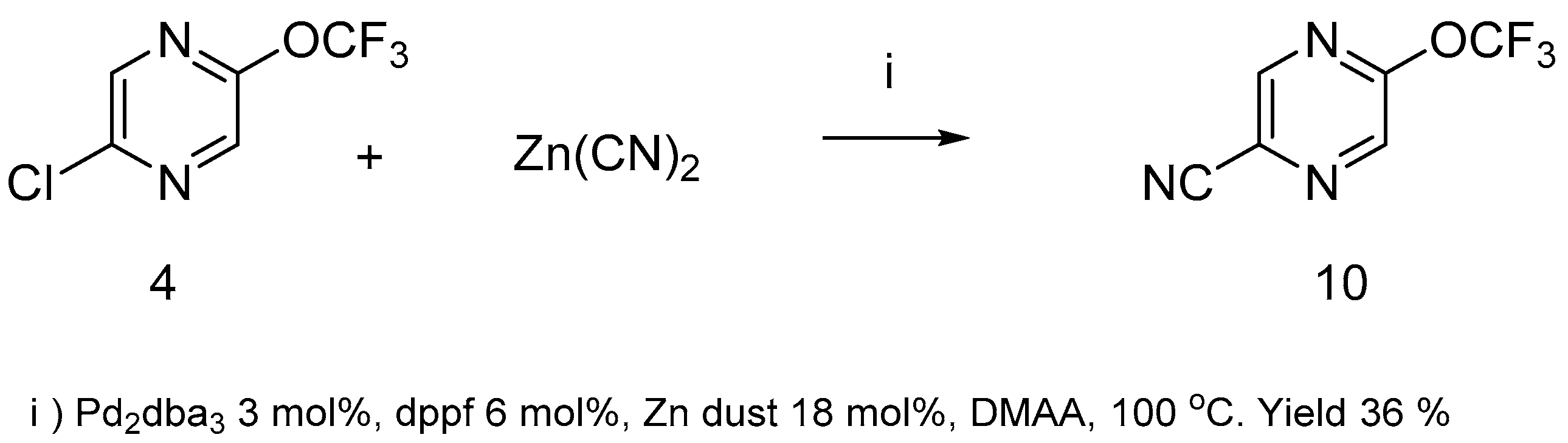{kind=link}
{kind=link}
{kind=link}
{kind=link}







