Two experimental examples of application are reported. The first employs a precursor ion scan (the reporter molecular fragment is an ion, identified by the m/z of Q3), the second a constant neutral loss scan (the reporter molecular fragment is a neutral species, identified as the fixed m/z difference between Q1 and Q3).
2.2.1. Detection of Dabsyl-Amino Acids by iso-Energetic CID and Precursor Ion Scan
The first proof-of-principle experiment of the use of
iso-energetic collision-induced dissociation for the analysis of small organic molecules by Precursor Ion scan is obtained by derivatizing alpha-amino acids with a very popular chromogenic tag, dimethylamino-azobenzene (dabsyl, DABS-), linked to the amino-group as the sulphonyl amide (
Scheme 3).
Some amino-acid derivatives were prepared individually (list in
Table S1) and the fragment spectra of their deprotonated molecules were measured in the negative ion mode over a range of collision energy up to 70 eV
lab.
Figure 3a,c shows as examples the integrated spectra recorded for the derivatives of the lowest- and highest-mass natural amino acids, glycine and tryptophan, respectively. The most intense fragment in the spectra, at
m/
z 240, is generated from the appended chromophore, as indicated in the general structure of the examined derivatives, and due to its common occurrence and high intensity qualifies as reporter ion for analytical purposes.
The generation efficiency curves of
m/
z 240 are displayed in the corresponding right-hand panels
Figure 3b,d, and show the position of the maxima calculated as described in the Materials and Methods
Section 4.5. In the laboratory frame, the values of the maxima are 29.50 eV
lab (center-of-mass frame, 2.12 eV) for DABS-Gly, and 35.66 eV
lab (center-of-mass frame, 1.93 eV) for DABS-Trp. Results of the measurements in a few other DABS-AA are collected in
Table 1.
Least-squares calculation of center-of-mass collision energy vs. precursor ion
m/
z affords a best-estimate value of 2.15 eV for the representative value of CE
max for the characteristic [M–H]
− to
m/
z 240 transition (
Figures S2 and S3). The obtained value corresponds to a scan-line that starts at 29.9 eV
lab for
m/
z 361 of deprotonated DABS-Gly and ends at 39.8 eV
lab for
m/
z 490 of deprotonated DABS-Trp (see parameters used in the example of
Figure 1).
One interesting observation stems from the comparison of the scatter around the best-fit scan line of actual maxima measured in the individual production efficiency curves of
m/
z 240 (
Figure S4). While differences as large as 3.9 eV
lab are found (the outlying Trp being that with the largest difference), this value should be compared to the actual span of the maxima of the round-topped curves recorded for these compounds. As can be appreciated from the right-column curves of
Figure 4, the width of the 95%–100%–95% round-topped curve maxima is of 5 to 9 eV
lab. The uncertainty of appreciation for the apex (the intersection of the two least-squares ascending and descending lines) results in the order of fractions of eV
lab (0.2–0.4 eV
lab), less than the employed potential step in the accurate ramp-CE experiment (0.5 eV
lab). Therefore, the method employed to obtain the representative value of center-of-mass CE
max yields a reliable value for method setup.
To test whether this experiment is amenable, and useful, to real applications in bio-analysis, a DABS-derivatized urine was analyzed with an un-optimized fast gradient elution linked to a 1-s
m/
z 240 precursor ion scan from 350 to 500 and to a synchronized scan of the collision voltage (CElab, q2-Q1) from −28.4 to −39.6 eV
lab. Main results are displayed in
Figure 4a–d.
Under the un-optimized “shotgun” chromatographic conditions of this experiment, several DABS-derivatized alpha-amino acids of urine elute as partially separated chromatographic peaks between 3.5 and 5.5 min (
Figure 4a) and the integrated spectrum (merged spectra) of the corresponding chromatographic time-frame contains the molecular signals of the main physiologically expected amino acids (
Figure 4b).
This un-optimized, very fast chromatographic condition does not separate the isomers of Leucine, nor isobaric hydroxy-proline, at
m/
z 417. The mono-DABS derivatized histidine signal is observed (
m/
z 441), and the signal at
m/
z 455 may correspond to the unseparated, isomeric methyl-histidines. Methionine can be identified from the co-occurrence of the molecular species at m/z 435 (32S) and −437 (
34S). By analogy, a weaker signal at
m/
z 421 can be attributed to the trace amino acid S-Methyl-cysteine (SMC) [
21], based on the appearance of a corresponding signal at
m/
z 423 of the corresponding
34S-species. This observation, in particular, opens the way to a non-target screening of cysteine thioethers in adductomics applications. No signals are observed, nor were likely expected, for those that yield a doubly dabsylated derivative, Cysteine and Lysine. Only one chromatographic peak is observed in the XIC of
m/
z 432 (Glutamine), and a very weak signal at
m/
z 460 is observed for mono-DABS-Arginine. Precursor Ion spectra extracted corresponding to the XIC for the main identified amino acids feature the expected [M-H]
- molecular signal expected for the individual eluting species (
Figure 4c,d).
Identification of the amino acids by Data-Dependent Fragment Ion analysis was not performed, since all DABS-derivatives yield the same main fragment ions, all of which deriving from the appended chromophore (see
Figure 4a,c).
2.2.2. Detection of Nucleosides by Iso-Energetic CID and Constant Neutral Fragment Loss Scan
The second proof-of-principle example employs nucleosides, measured in the positive ion mode, to demonstrate that iso-energetic CID is compatible with Constant Neutral fragment Loss Scan (CNL) experiments in the triple quadrupole. In this experiment, all three quadrupoles scan synchronously; in fact, the q2-Q1 voltage difference is “ramped” simultaneously with the scan time of both Q1 and Q3.
Protonated nucleosides (listed in
Table S4) undergo collision-induced fragmentation through a main pathway that involves loss of the N-linked sugar as neutral (loss of 116 Da from the 2-deoxy-ribosides and of 132 Da from the ribosides) and formation of the protonated nucleobase (
Scheme 4). A very minor pathway generates the complementary positively charged sugar species (
m/
z 117 and 133, respectively, for 2-deoxy-ribose and ribose) but their intensity is lower by more than two orders of magnitude with respect to that of the protonated nucleobases, therefore making a Precursor Ion scan utterly unsuitable for analysis.
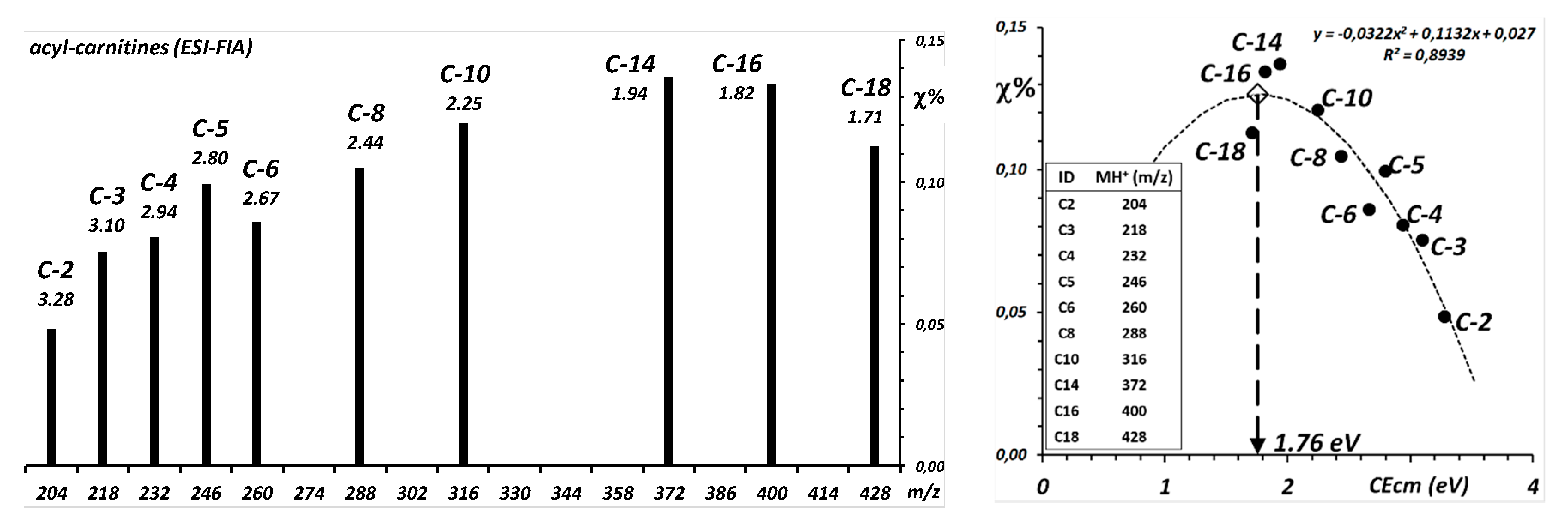
As in the example before, the collision energy corresponding to the maxima of the production efficiency curves of the protonated nucleobases was measured from the fragment ion spectra (measurements in
Table 2 and results of calculation of the representative CE
max in
Figures S6 and S7). The maxima are between approximately 1.5 and 2.0 eV
CM, with no analytically relevant difference between the 2-deoxy-nucleosides (loss of 116 Da; median 1.60 eV) and the nucleosides (loss of 132 Da; median 1.67 eV).
From the perspective of fast method setup, it is appealing to observe that the values of CE
max measured from the fragment intensity curves of the Fragment Ion spectra and those measured from the “reverse” experiment (CNL, in the case of nucleotides) are sufficiently close for the analytical application. As an example,
Figure S7 shows the curves for Guanosine (
m/
z 284 > 152, Fragment Ion spectrum) and (CNL132,
m/
z 284), both recorded from a mixture to ensure that calculation results derive from measurements of the same (noisy) quality. Estimated CE
max is 17.86 and 17.89 eV
lab, respectively.
That such close results can be obtained is advantageous when a few standard compounds are available. In this case, one single “reverse” experiment, rather than several recordings of individual Fragment Ion spectra, may still yield sufficiently reliable data to calculate a starting representative eV
max value for method optimization with the spreadsheet module of
Appendix A.
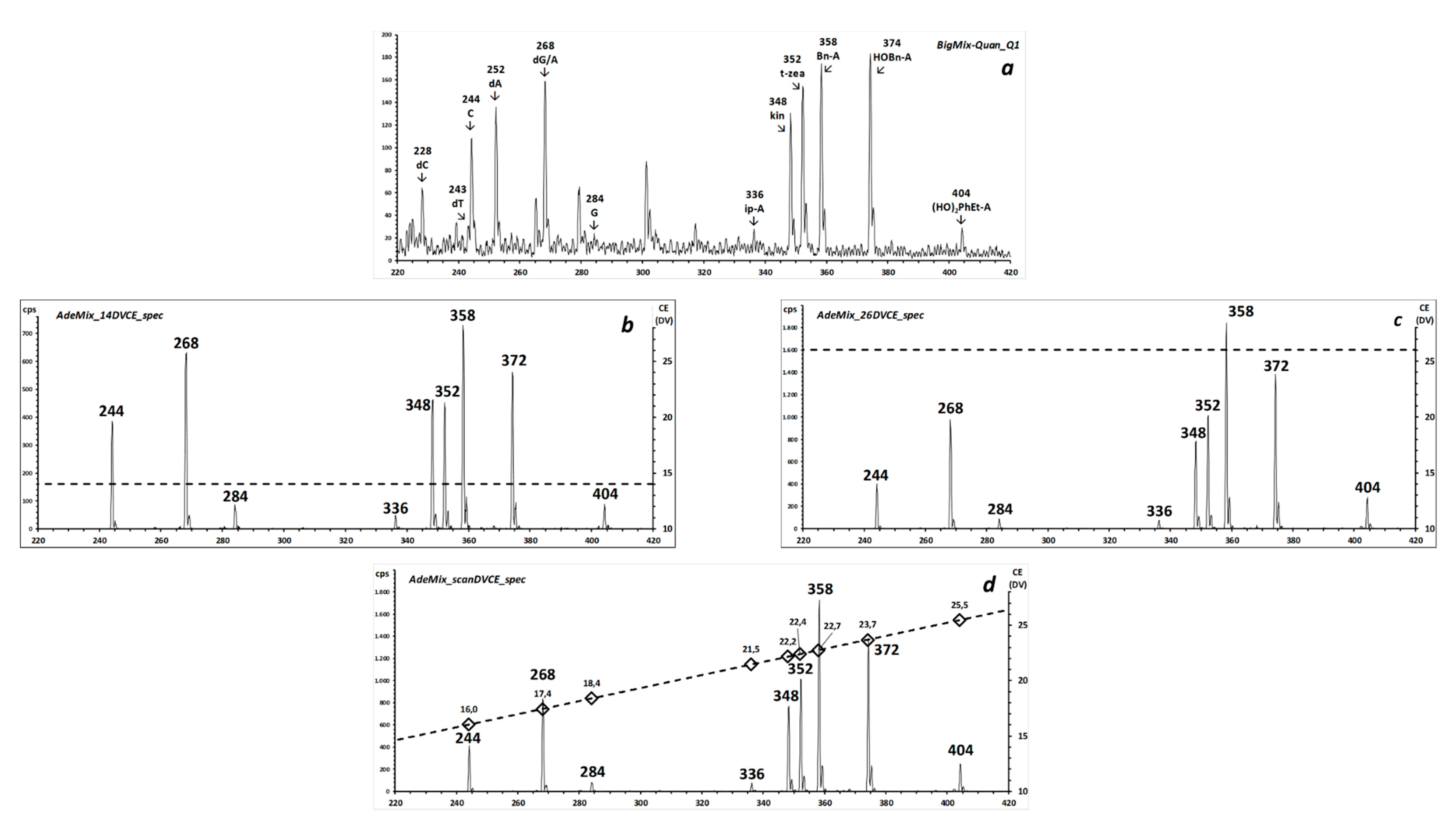
To demonstrate the selectivity of the two CNL scans,
Figure 5 shows the source spectrum of a mixture that contains the four DNA nucleosides (dA, dC, dG, dT) and two RNA nucleosides (only C and G; A was deliberately omitted, since it is isobaric to dG). The two inserts display the results of consecutive alternate 1-s CNL-scans of 116 Da (selective for deoxy-ribonucleosides) and 132 Da (selective for ribonucleosides) at 17 eVlab (1.6 eVCM for precursor
m/
z 270).
All expected signals that correspond to the protonated molecules of the six compounds are observed in the respective NL spectra. Adenosine was deliberately omitted from this mixture to confirm that the NL116 scan is specific for deoxy-ribonucleosides, since the precursor at m/z 268 from isobaric A is absent from the NL132 spectrum, but that from dG appears in the NL116 scan.
A more complex mixture that contains the four DNA nucleosides (dA, dC, dG, dT), three RNA nucleosides (A, C, G), and six 6N-substituted derivatives of adenine (listed in
Table S4; complete ESI source spectrum in
Figure S8) was further employed to investigate the potentiality of the iso-energetic CID scan for applications in the field of nucleoside analysis. In this mixture, the ratio of the isobaric dG:A is approximately 1:4.
The possibility of screening the sample for modified nucleobases is exemplified by six 6N-substituted derivatives of adenine (8–13) [
22,
23] with masses between 350 and 400 Da, and thus 30% to 50% higher than that of the generating riboside, adenosine. Their fragment spectra (one example is reported in
Figure S9) feature loss of the ribose, and the first-generation protonated adenine further loses the N6-adducted substituent to yield the
m/
z 136 AdeH
+ fragment, and often one from the appended molecular unit.
The experiment reported in
Figure 6a–d is a concept with prospective real applications, such as the identification of natural epigenetic modifications of DNA nucleobases, of xenobiotic DNA adducts, of regulatory t-RNA modified bases, and of synthetic nucleobase tools for research in molecular biology [
24]. Its aim is to evaluate whether the
i-CID approach is feasible in a routine triple quadrupole instrument without compromising mass resolution and at a scan speed compatible with coupling to liquid chromatography. To test whether the use of a CE ramp improves the accuracy in the determination of nucleoside mixtures, the peak intensities of the non-isobaric nucleosides obtained in the fixed-CE and in the ramp-CE Neutral Loss scans were compared to those measured in the ESI source spectra of the same mixture. The cycle of measurement follows the pattern and conditions reported in
Table S10. In the same sample, a consecutive NL116_rampCE experiment, selective for 2-deoxy-ribonucleotides was also performed, with essentially similar results (data not shown).
As is apparent from the ion profiles (insert in
Figure S12), no deterioration of resolution occurs when the q2 voltage is ramped synchronously with the Q1 and Q3 scans to achieve the collision energy scan. The fastest conditions chosen in the employed routine triple quadrupole instrument (infusion mode, CNL scan with synchronized q2 ramping) were a scan speed of 2.5 seconds/1.000 Da (
m/
z 220–420 in 0.5 seconds) and a collision voltage ramp speed of 23.6 eV
lab/s (14.6–26.4 eV
lab in 0.5 seconds). To hint at the analytical potential of this experiment in bio-analysis, the relative signal intensity of the considered ribo-nucleosides was compared between the different analytical conditions, as summarized in
Figures S13 and S14. In particular, the relative signal abundances of the substituted adenines were closer to those measured in the source spectra when the CEscan mode was employed, rather than at any fixed CE value (
Figure S14).
In the employed routine triple quadrupole instrument (infusion mode), scan speed of the Neutral Loss scan with synchronized q2 ramping can be as fast as 0.5 s in a
m/
z range of 200 Da, without any consistent signal instability or loss of resolution (insert in
Figure S12), and allowing for two scan modes to alternate (
i-NL116 and
i-NL132).















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}