
DFT Study of Molecular and Electronic Structure of Y, La and Lu Complexes with Porphyrazine and Tetrakis(1,2,5-thiadiazole)porphyrazine


Abstract
:Abstract
Sample Availability
1. Introduction
2. Results and Discussion
2.1. Geometric Structure
2.2. Electron Density Distribution and Chemical Bonding in MClPz and MClTTDPz
2.3. Electronic Absorption Spectra
2.4. Molecular Orbitals
2.5. Vibrational Spectra
3. Materials and Methods
Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sorokin, A.B. Recent progress on exploring µ-oxo bridged binuclear porphyrinoid complexes in catalysis and material science. Coord. Chem. Rev. 2019, 389, 141–160. [Google Scholar] [CrossRef]
- Yu, Z.; Hagfeldt, A.; Sun, L. The application of transition metal complexes in hole-transporting layers for perovskite solar cells: Recent progress and future perspectives. Coord. Chem. Rev. 2020, 406, 213143. [Google Scholar] [CrossRef]
- Almeida-Marrero, V.; Van De Winckel, E.; Anaya-Plaza, E.; Torres, T.; De La Escosura, A. Porphyrinoid biohybrid materials as an emerging toolbox for biomedical light management. Chem. Soc. Rev. 2018, 47, 7369–7400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wöhrle, D.; Schnurpfeil, G.; Makarov, S.G.; Kazarin, A.; Suvorova, O.N. Practical Applications of Phthalocyanines—From Dyes and Pigments to Materials for Optical, Electronic and Photo-electronic Devices. Macroheterocycles 2012, 5, 191–202. [Google Scholar] [CrossRef]
- Lukyanets, E.A.; Nemykin, V.N. The key role of peripheral substituents in the chemistry of phthalocyanines and their analogs. J. Porphyr. Phthalocyanines 2010, 14, 1–40. [Google Scholar] [CrossRef] [Green Version]
- Longevial, J.; Clément, S.; Wytko, J.A.; Ruppert, R.; Weiss, J.; Richeter, S. Peripherally Metalated Porphyrins with Applications in Catalysis, Molecular Electronics and Biomedicine. Chem. Eur. J. 2018, 24, 15442–15460. [Google Scholar] [CrossRef] [Green Version]
- Bettini, S.; Valli, L.; Giancane, G. Applications of Photoinduced Phenomena in Supramolecularly Arranged Phthalocyanine Derivatives: A Perspective. Molecules 2020, 25, 3742. [Google Scholar] [CrossRef]
- Donzello, M.P.; Ercolani, C.; Novakova, V.; Zimcik, P.; Stuzhin, P.A. Tetrapyrazinoporphyrazines and their metal derivatives. Part I: Synthesis and basic structural information. Coord. Chem. Rev. 2016, 309, 107–179. [Google Scholar] [CrossRef]
- Novakova, V.; Donzello, M.P.; Ercolani, C.; Zimcik, P.; Stuzhin, P.A. Tetrapyrazinoporphyrazines and their metal derivatives. Part II: Electronic structure, electrochemical, spectral, photophysical and other application related properties. Coord. Chem. Rev. 2018, 361, 1–73. [Google Scholar] [CrossRef]
- Donzello, M.P.; Ercolani, C.; Stuzhin, P.A. Novel families of phthalocyanine-like macrocycles-Porphyrazines with annulated strongly electron-withdrawing 1,2,5-thia/selenodiazole rings. Coord. Chem. Rev. 2006, 250, 1530–1561. [Google Scholar] [CrossRef]
- Suzuki, Y.; Fujimori, M.; Yoshikawa, H.; Awaga, K. Packing motifs and magneto-structural correlations in crystal structures of metallo-tetrakis(1,2,5-thiadiazole)porphyrazine series, MTTDPz (M=H 2, Fe, Co, Ni, Cu, Zn). Chem. Eur. J. 2004, 10, 5158–5164. [Google Scholar] [CrossRef] [PubMed]
- Koifman, O.I.; Ageeva, T.A.; Beletskaya, I.P.; Averin, A.D.; Yakushev, A.A.; Tomilova, L.G.; Dubinina, T.V.; Tsivadze, A.Y.; Gorbunova, Y.G.; Stuzhin, P.A.; et al. Macroheterocyclic Compounds—A Key Building Block in New Functional Materials and Molecular Devices. Macroheterocycles 2020. [Google Scholar] [CrossRef]
- Weiss, R.; Fischer, J. Lanthanide Phthalocyanine Complexes. In The Porphyrin Handbook: Phthalocyanines: Spectroscopic and Electrochemical Characterization; Elsevier Inc.: Cambridge, MA, USA, 2003; Volume 16, pp. 171–246. ISBN 9780123932266. [Google Scholar]
- Bouvet, M.; Gaudillat, P.; Suisse, J.-M. Lanthanide macrocyclic complexes: From molecules to materials and from materials to devices. J. Porphyr. Phthalocyanines 2013, 17, 628–635. [Google Scholar] [CrossRef]
- Stuzhin, P.A.; Ivanova, S.S.; Hamdoush, M.; Kirakosyan, G.A.; Kiselev, A.; Popov, A.; Sliznev, V.; Ercolani, C. Tetrakis(1,2,5-thiadiazolo)porphyrazines. 9. Synthesis and spectral and theoretical studies of the lithium(i) complex and its unusual behaviour in aprotic solvents in the presence of acids. Dalton Trans. 2019, 48, 14049–14061. [Google Scholar] [CrossRef]
- Stuzhin, P.A.; Bauer, E.M.; Ercolani, C. Tetrakis(thiadiazole)porphyrazines. 1. Syntheses and Properties of Tetrakis(thiadiazole)porphyrazine and Its Magnesium and Copper Derivatives. Inorg. Chem. 1998, 37, 1533–1539. [Google Scholar] [CrossRef]
- Otlyotov, A.A.; Ryzhov, I.V.; Kuzmin, I.A.; Zhabanov, Y.A.; Mikhailov, M.S.; Stuzhin, P.A. DFT Study of Molecular and Electronic Structure of Ca(II) and Zn(II) Complexes with Porphyrazine and tetrakis(1,2,5-thiadiazole)porphyrazine. Int. J. Mol. Sci. 2020, 21, 2923. [Google Scholar] [CrossRef]
- Donzello, M.P.; Agostinetto, R.; Ivanova, S.S.; Fujimori, M.; Suzuki, Y.; Yoshikawa, H.; Shen, J.; Awaga, K.; Ercolani, C.; Kadish, K.M.; et al. Tetrakis(thiadiazole)porphyrazines. 4. Direct template synthesis, structure, general physicochemical behavior, and redox properties of Al III, GaIII, and InIII complexes. Inorg. Chem. 2005, 44, 8539–8551. [Google Scholar] [CrossRef]
- Bauer, E.M.; Cardarilli, D.; Ercolani, C.; Stuzhin, P.A.; Russo, U. Tetrakis(thiadiazole)porphyrazines. 2. Metal complexes with Mn(II), Fe(II), Co(II), Ni(II), and Zn(II). Inorg. Chem. 1999, 38, 6114–6120. [Google Scholar] [CrossRef]
- Tarakanova, E.N.; Hamdoush, M.; Eroshin, A.V.; Ryzhov, I.V.; Zhabanov, Y.A.; Stuzhin, P.A. Tetra(1,2,5-thiadiazolo)porphyrazines. 10. Synthesis, spectral characterization and DFT study of complexes with yttrium(III) and lutetium(III). Polyhedron 2021, 193, 114877. [Google Scholar] [CrossRef]
- Khelevina, O.G.; Voinov, A.A. Synthesis and kinetic stability of dysprosium(III), europium(III), and neodymium(III) complexes with tetraazaporphine in acetic acid solutions. Russ. J. Gen. Chem. 2000, 70, 778–783. [Google Scholar]
- Khelevina, O.G.; Voinov, A.A. Kinetic stability of the dysprosium(III) complex with tetraazaporphine in water-acetic acid and methanol-acetic acid mixtures. Russ. J. Coord. Chem. Khimiya 1999, 25, 445–449. [Google Scholar]
- Zhabanov, Y.A.; Sliznev, V.V.; Ryzhov, I.V.; Stuzhin, P.A. Peculiarities of electronic structure and chemical bonding in iron and cobalt metal complexes of porphyrazine and tetra(1,2,5-thiadiazole)porphyrazine. J. Porphyr. Phthalocyanines 2020, 24, 1146–1154. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Slee, T.S.; Cremer, D.; Kraka, E. Description of conjugation and hyperconjugation in terms of electron distributions. J. Am. Chem. Soc. 1983, 105, 5061–5068. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Zhabanov, Y.A.; Giricheva, N.I.; Zakharov, A.V.; Islyaikin, M.K. Distinctive features of the structure of hemihexaphyrazine complexes with Y, La, and Lu according to quantum chemical data. J. Mol. Struct. 2017, 1132. [Google Scholar] [CrossRef]
- Giricheva, N.I.; Zakharov, A.V.; Shlykov, S.A.; Girichev, G.V. Molecular structure of GdCl3. Nuclear dynamics of the trichlorides of Gd, Tm, and Lu. J. Chem. Soc. Dalton Trans. 2000, 19, 3401–3403. [Google Scholar] [CrossRef]
- Zakharov, A.V.; Vogt, N.; Shlykov, S.A.; Giricheva, N.I.; Galanin, I.E.; Girichev, G.V.; Vogt, J. The molecular structures of LaCl3 and LaBr3 reinvestigated by gas-phase electron diffraction. J. Mol. Struct. 2004, 707, 147–152. [Google Scholar] [CrossRef]
- Shlykov, S.A.; Giricheva, N.I.; Lapykina, E.A.; Girichev, G.V.; Oberhammer, H. The molecular structure of TbI3, DyI3, HoI3 and ErI3 as determined by synchronous gas-phase electron diffraction and mass spectrometric experiment assisted by quantum chemical calculations. J. Mol. Struct. 2010, 978, 170–177. [Google Scholar] [CrossRef]
- Giricheva, N.I.; Shlykov, S.A.; Lapykina, E.A.; Oberhammer, H.; Girichev, G.V. The molecular structure of PrI3 and GdI3 as determined by synchronous gas-phase electron diffraction and mass spectrometric experiment assisted by quantum chemical calculations. Struct. Chem. 2011, 22, 385–392. [Google Scholar] [CrossRef]
- Ischenko, A.A.; Girichev, G.V.; Tarasov, Y.I. Difraktciya Elektronov: Struktura i Dinamika Svobodnykh Molekul i Kondensirovannogo Sostoyaniya Veshestva; Fizmatlit: Moscow, Russia, 2012. [Google Scholar]
- Giricheva, N.I.; Belova, N.V.; Shlykov, S.A.; Girichev, G.V.; Vogt, N.; Tverdova, N.V.; Vogt, J. Molecular structure of tris(dipivaloylmethanato)lanthanum(III) studied by gas electron diffraction. J. Mol. Struct. 2002, 605, 171–176. [Google Scholar] [CrossRef]
- Belova, N.V.; Girichev, G.V.; Hinchley, S.L.; Kuzmina, N.P.; Rankin, D.W.H.; Zaitzeva, I.G. Molecular structure of tris(dipivaloylmethanato)lutetium(iii) studied by gas electron diffraction and ab initio and DFT calculations. Dalton Trans. 2004, 1715–1718. [Google Scholar] [CrossRef] [PubMed]
- Gouterman, M. Spectra of porphyrins. J. Mol. Spectrosc. 1961, 6, 138–163. [Google Scholar] [CrossRef]
- Gouterman, M.; Wagnière, G.H.; Snyder, L.C. Spectra of porphyrins. Part II. Four orbital model. J. Mol. Spectrosc. 1963, 11, 108–127. [Google Scholar] [CrossRef]
- Weiss, C.; Kobayashi, H.; Gouterman, M. Spectra of porphyrins. Part III. Self-consistent molecular orbital calculations of porphyrin and related ring systems. J. Mol. Spectrosc. 1965, 16, 415–450. [Google Scholar] [CrossRef]
- Zhabanov, Y.A.; Tverdova, N.V.; Giricheva, N.I.; Girichev, G.V.; Stuzhin, P.A. DFT Study of molecular and electronic structure of magnesium (II) tetra(1,2,5-chalcogenadiazolo) porphyrazines, [TXDPzMg] (X = O, S, Se, Te). J. Porphyr. Phthalocyanines 2017, 21, 439–452. [Google Scholar] [CrossRef]
- Jensen, F. Unifying general and segmented contracted basis sets. Segmented polarization consistent basis sets. J. Chem. Theory Comput. 2014, 10, 1074–1085. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis set exchange: A community database for computational sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Peterson, K.A.; Figgen, D.; Dolg, M.; Stoll, H. Energy-consistent relativistic pseudopotentials and correlation consistent basis sets for the 4d elements Y-Pd. J. Chem. Phys. 2007, 126, 124101. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. Des. Assess. Accuracy 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigand, A.; Cao, X.; Yang, J.; Dolg, M. Quasirelativistic f-in-core pseudopotentials and core-polarization potentials for trivalent actinides and lanthanides: Molecular test for trifluorides. Theor. Chem. Acc. 2009, 126, 117–127. [Google Scholar] [CrossRef]
- Dolg, M.; Stoll, H.; Savin, A.; Preuss, H. Energy-adjusted pseudopotentials for the rare earth elements. Theor. Chim. Acta 1989, 75, 173–194. [Google Scholar] [CrossRef]
- Granovsky, A.A. Firefly Version 8. Available online: http://classic.chem.msu.su/gran/firefly/index.html (accessed on 28 December 2020).
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Vishnevskiy, Y.V.; Zhabanov, Y.A. New implementation of the first-order perturbation theory for calculation of interatomic vibrational amplitudes and corrections in gas electron diffraction. J. Phys. Conf. Ser. 2015, 633, 12076. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK; Clarendon Press: Oxford, UK, 1990; ISBN 9780198558651. [Google Scholar]
- Some References Related to AIMAll. Available online: http://aim.tkgristmill.com/references.html (accessed on 13 March 2020).
- Zhurko, G.A.; Zhurko, D.A. ChemCraft Version 1.6 (Build 312); Version 1.6 (Build 312) Ed. Available online: http://www.chemcraftprog.com/index.html (accessed on 28 December 2020).







{kind=link}
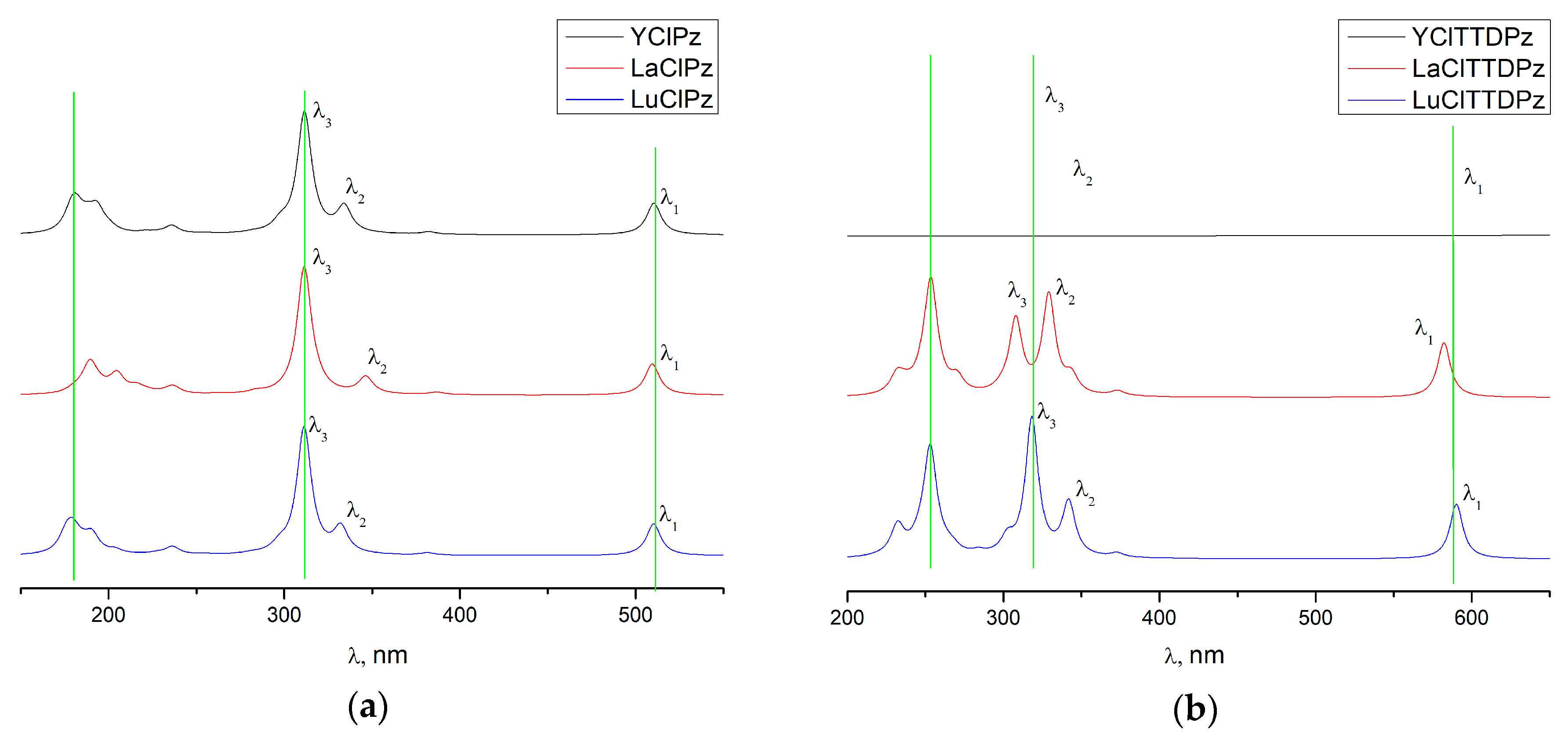{kind=link}
{kind=link}
{kind=link}
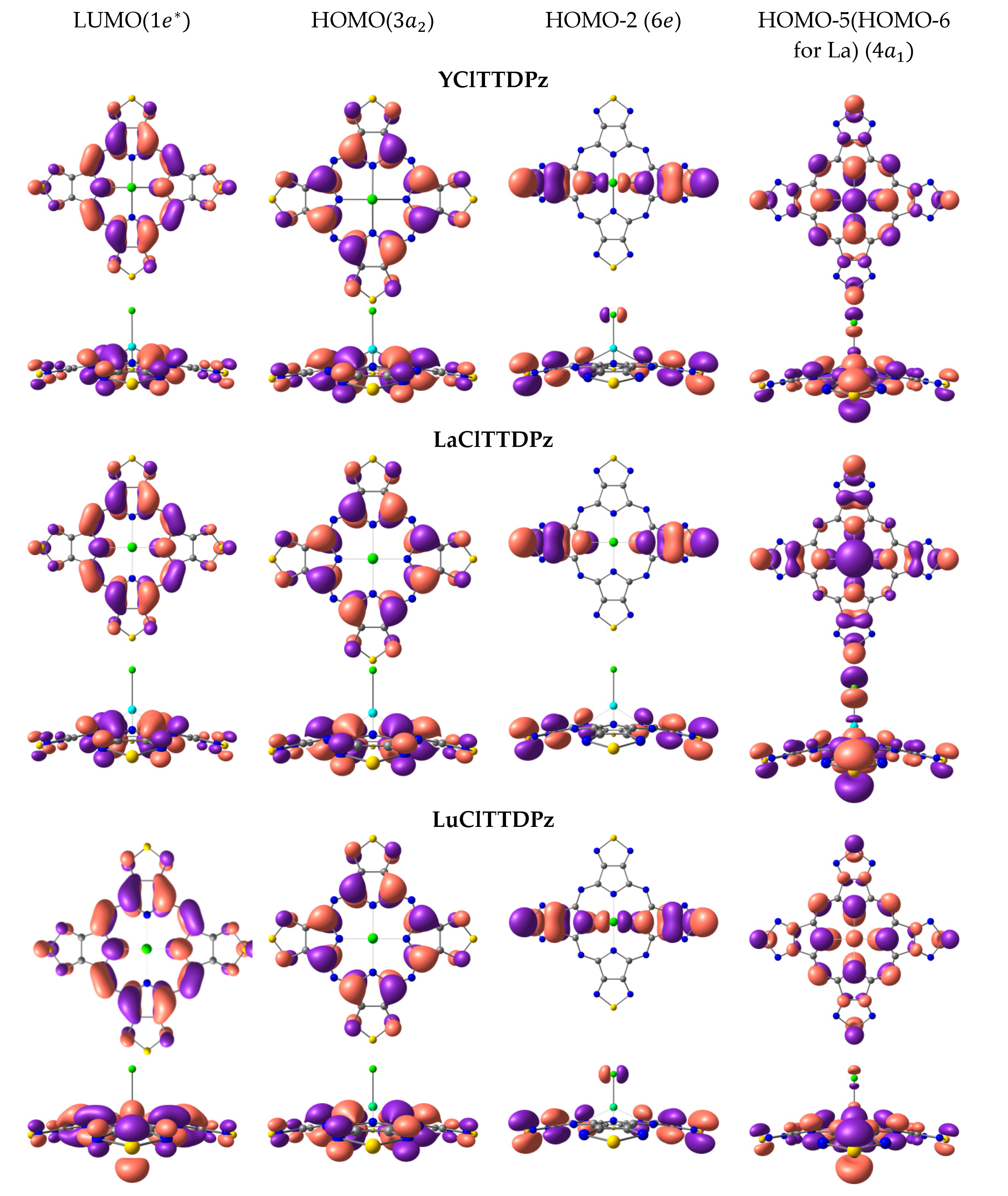{kind=link}
{kind=link}
| LaClPz | LaClTTDPz | LuClPz | LuClTTDPz | YClPz | YClTTDPz | |
|---|---|---|---|---|---|---|
| M-Cl | 2.711 | 2.680 | 2.493 | 2.468 | 2.523 | 2.497 |
| M-Np | 2.462 | 2.486 | 2.260 | 2.284 | 2.291 | 2.313 |
| Np-Cα | 1.369 | 1.377 | 1.370 | 1.379 | 1.370 | 1.379 |
| Cα-Nm | 1.330 | 1.321 | 1.329 | 1.319 | 1.329 | 1.319 |
| Cα-Cβ | 1.456 | 1.460 | 1.454 | 1.458 | 1.454 | 1.458 |
| Cβ-Cβ | 1.355 | 1.423 | 1.355 | 1.422 | 1.355 | 1.422 |
| Cβ-H | 1.077 | 1.077 | 1.077 | |||
| Cβ-Nt | 1.316 | 1.317 | 1.317 | |||
| Nt-S | 1.644 | 1.643 | 1.643 | |||
| (Np…Np)opp | 4.023 | 4.139 | 3.979 | 4.096 | 3.987 | 4.101 |
| (Np…Np)adj | 2.844 | 2.927 | 2.813 | 2.897 | 2.819 | 2.900 |
| ∠ (NpMCl) | 125.2 | 123.6 | 118.3 | 116.3 | 119.5 | 117.6 |
| ∠ (MNpCα) | 124.7 | 122.4 | 125.1 | 123.1 | 125.2 | 122.9 |
| ∠ (NpCαNm) | 127.6 | 128.1 | 127.3 | 127.9 | 127.4 | 128.0 |
| ∠ (CαNmCα) | 125.0 | 127.1 | 124.5 | 126.6 | 124.6 | 126.7 |
| ∠ (CαNpCα) | 107.5 | 111.6 | 107.6 | 111.5 | 107.6 | 111.6 |
| ∠ (NtSNt) | 100.3 | 100.4 | 100.4 | |||
| M-X1 1 | 1.419 | 1.377 | 1.072 | 1.011 | 1.129 | 1.071 |
| X1-X2 2 | 1.028 | 0.775 | 0.767 |
| YClPz | LaClPz | LuClPz | YClTTDPz | LaClTTDPz | LuClTTDPz | |
|---|---|---|---|---|---|---|
| ∇2ρ (M-Np), a.u. | 0.205 | 0.160 | 0.235 | 0.193 | 0.152 | 0.221 |
| q(M|Np) | 0.348 | 0.356 | 0.340 | 0.354 | 0.362 | 0.347 |
| δ(M|Np) | 0.349 | 0.366 | 0.357 | 0.337 | 0.353 | 0.346 |
| q(M|Cl) | 0.779 | 0.793 | 0.763 | 0.757 | 0.770 | 0.740 |
| δ(M|Cl) | 0.536 | 0.564 | 0.552 | 0.574 | 0.606 | 0.588 |
| q(M) | +2.171 | +2.21 | +2.125 | +2.173 | +2.218 | +2.127 |
| q(Np) | −1.191 | −1.179 | −1.187 | −1.164 | −1.153 | −1.16 |
| q(Nm) | −1.121 | −1.123 | −1.121 | −1.124 | −1.125 | −1.123 |
| q(Cα) | 0.928 | 0.920 | 0.929 | 0.963 | 0.958 | 0.963 |
| q(Cβ) | 0.017 | 0.014 | 0.017 | 0.526 | 0.525 | 0.526 |
| q(ligand) 1 | −1.391 | −1.423 | −1.362 | −1.416 | −1.447 | −1.386 |
| State | Composition (%) | Λ (nm) | f | |
|---|---|---|---|---|
| YClPz | ||||
| 11 E | (16) (82) | 510 | 0.15 | λ1 |
| 41 E | (53) (38) (5) | 334 | 0.13 | λ2 |
| 51 E | (41) (42) (10) | 312 | 0.60 | λ3 |
| LaClPz | ||||
| 11 E | (15) (82) | 510 | 0.15 | λ1 |
| 41 E | (47) (44) (4) | 346 | 0.08 | λ2 |
| 61 E | (12) (38) (36) (11) | 311 | 0.62 | λ3 |
| LuClPz | ||||
| 11E | (16) (82) | 510 | 0.16 | λ1 |
| 41E | (56) (35) (5) | 332 | 0.12 | λ2 |
| 51E | (39) (45) (11) | 311 | 0.62 | λ3 |
| YClTTDPz | ||||
| 11 E | (5) (91) | 588 | 0.27 | λ1 |
| 51 E | (43) (45) (10) | 342 | 0.23 | λ2 |
| 61 E | (5) (41) (36) (6) (7) | 319 | 0.67 | λ3 |
| LaClTTDPz | ||||
| 11 E | (6) (91) | 582 | 0.27 | λ1 |
| 51 E | (80) (10) (10) | 344 | 0.08 | |
| 61 E | (21) (7) (59) (5) (6) | 329 | 0.48 | λ2 |
| 71 E | (13) 3 (61) (14) | 308 | 0.36 | λ3 |
| LuClTTDPz | ||||
| 11 E | (5) (91) | 590 | 0.27 | λ1 |
| 51 E | (39) (49) (10) | 342 | 0.26 | λ2 |
| 61 E | (5) (45) (32) (6) (7) | 318 | 0.67 | λ3 |
| Frequency, cm−1 | Irel, % | Symmetry | Assignment 1 | Exp, cm−1 |
|---|---|---|---|---|
| YClTTDPz | ||||
| 1101.6(ω80-ω81) | 59 | E | r(Np-Cα) (24), r(Nm-Cα) (28), r(Cα-Cβ) (15), r(Cβ-Nt) (9), φ(CβCβNt) (6), φ(CβNtS) (7) | 1090 acac [20] |
| 1280.0(ω87-ω88) | 100 | E | r(Np-Cα) (27), r(Cβ-Cβ) (10), r(Cβ-Nt) (10), φ(CαNpCα) (6), φ(NpCαNm) (10), φ(NmCαCβ) (9), φ(CαCβNt) (6) | 1260 acac [20] |
| LaClTTDPz | ||||
| 1100.2(ω80-ω81) | 61 | E | r(Np-Cα) (28), r(Nm-Cα) (27), r(Cα-Cβ) (14), r(Cβ-Nt) (8), φ(CβCβNt) (5), φ(CβNtS) (6) | |
| 1274.8(ω87-ω88) | 100 | E | r(Np-Cα) (29), r(Cβ-Cβ) (8), r(Cβ-Nt) (9), φ(CαNpCα) (6), φ(NpCαNm) (10), φ(CαNmCα) (5), φ(NmCαCβ) (9), φ(CαCβNt) (6) | |
| LuClTTDPz | ||||
| 1102.1(ω80-ω81) | 59 | E | r(Np-Cα) (24), r(Nm-Cα) (28), r(Cα-Cβ) (15), r(Cβ-Nt) (9), φ(CβCβNt) (6), φ(CβNtS) (7) | 1090 acac [20] |
| 1281.5(ω87-ω88) | 100 | E | r(Np-Cα) (26), r(Cβ-Cβ) (10), r(Cβ-Nt) (10), φ(CαNpCα) (6), φ(NpCαNm) (10), φ(NmCαCβ) (9), φ(CαCβNt) (8) | 1262 acac [20] |
| YClPz | ||||
| 839.81 (ω48) | 46 | A1 | OPB(Cβ-Np-Nm-Cα) (35), OPB(H-Cα-Cβ-Cβ) (44), θ(Cα-Nm-Cα-Np) (5), θ(Nm-Cα-Cβ-Cβ) (11) | |
| 982.30 (ω53-ω54) | 100 | E | r(Np-Y) (5), r(Np-Cα) (14), r(Cα-Cβ) (53), r(Cα-Cβ) (5) | |
| LaClPz | ||||
| 838.13 (ω48) | 44 | A1 | OPB(Cβ-Np-Nm-Cα) (35), OPB(H-Cα-Cβ-Cβ) (44), θ(Cα-Nm-Cα-Np) (5), θ(Nm-Cα-Cβ-Cβ) (11) | |
| 975.65(ω53-ω54) | 100 | E | r(Np-La) (5), r(Np-Cα) (15), r(Nm-Cα) (5), r(Cα-Cβ) (56) | |
| LuClPz | ||||
| 839.62 (ω48) | 46 | A1 | OPB(Cβ-Np-Nm-Cα) (35), OPB(H-Cα-Cβ-Cβ) (45), θ(Cα-Nm-Cα-Np) (5), θ(Nm-Cα-Cβ-Cβ) (11) | |
| 983.45 (ω53-ω54) | 100 | E | r(Np-Lu) (5), r(Np-Cα) (7), r(Np-Cα) (7), r(Cα-Cβ) (26), r(Cα-Cβ) (27) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhabanov, Y.A.; Ryzhov, I.V.; Kuzmin, I.A.; Eroshin, A.V.; Stuzhin, P.A. DFT Study of Molecular and Electronic Structure of Y, La and Lu Complexes with Porphyrazine and Tetrakis(1,2,5-thiadiazole)porphyrazine. Molecules 2021, 26, 113. https://doi.org/10.3390/molecules26010113
Zhabanov YA, Ryzhov IV, Kuzmin IA, Eroshin AV, Stuzhin PA. DFT Study of Molecular and Electronic Structure of Y, La and Lu Complexes with Porphyrazine and Tetrakis(1,2,5-thiadiazole)porphyrazine. Molecules. 2021; 26(1):113. https://doi.org/10.3390/molecules26010113
Chicago/Turabian StyleZhabanov, Yuriy A., Igor V. Ryzhov, Ilya A. Kuzmin, Alexey V. Eroshin, and Pavel A. Stuzhin. 2021. "DFT Study of Molecular and Electronic Structure of Y, La and Lu Complexes with Porphyrazine and Tetrakis(1,2,5-thiadiazole)porphyrazine" Molecules 26, no. 1: 113. https://doi.org/10.3390/molecules26010113
APA StyleZhabanov, Y. A., Ryzhov, I. V., Kuzmin, I. A., Eroshin, A. V., & Stuzhin, P. A. (2021). DFT Study of Molecular and Electronic Structure of Y, La and Lu Complexes with Porphyrazine and Tetrakis(1,2,5-thiadiazole)porphyrazine. Molecules, 26(1), 113. https://doi.org/10.3390/molecules26010113