
Generation of Mixed Anhydrides via Oxidative Fragmentation of Tertiary Cyclopropanols with Phenyliodine(III) Dicarboxylates



Abstract
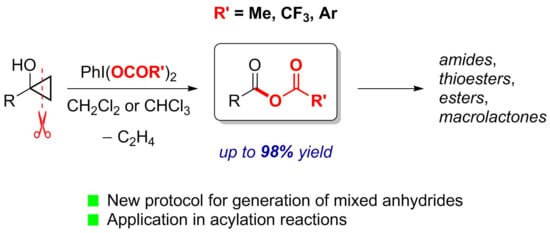
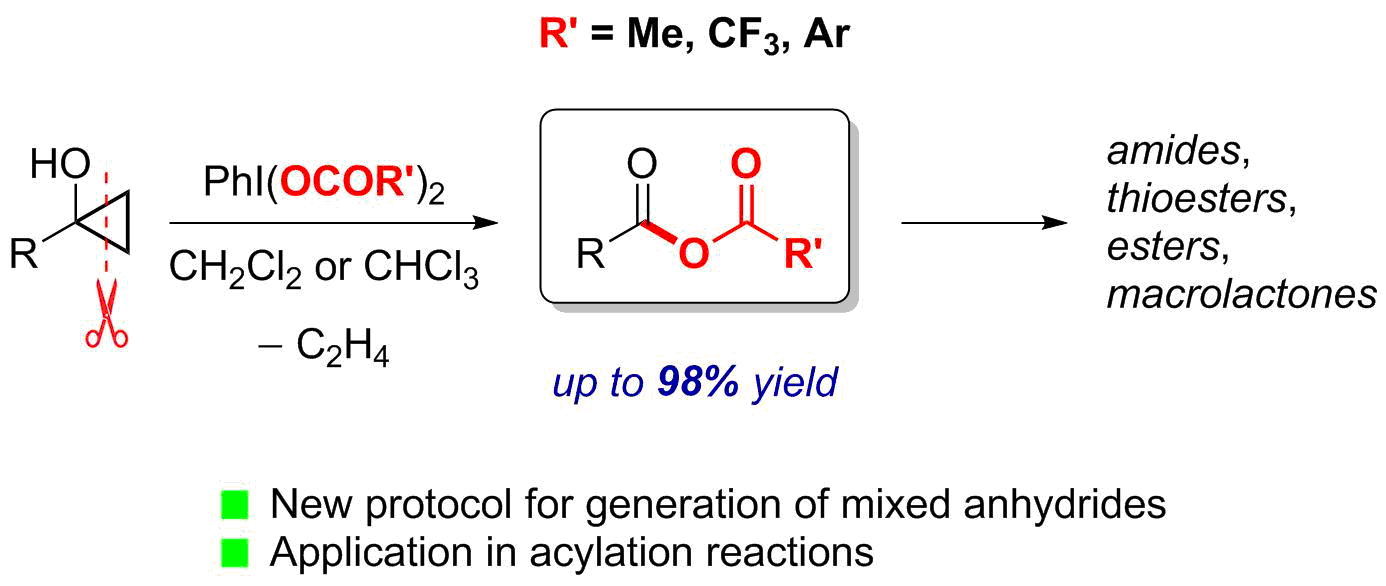
:
1. Introduction
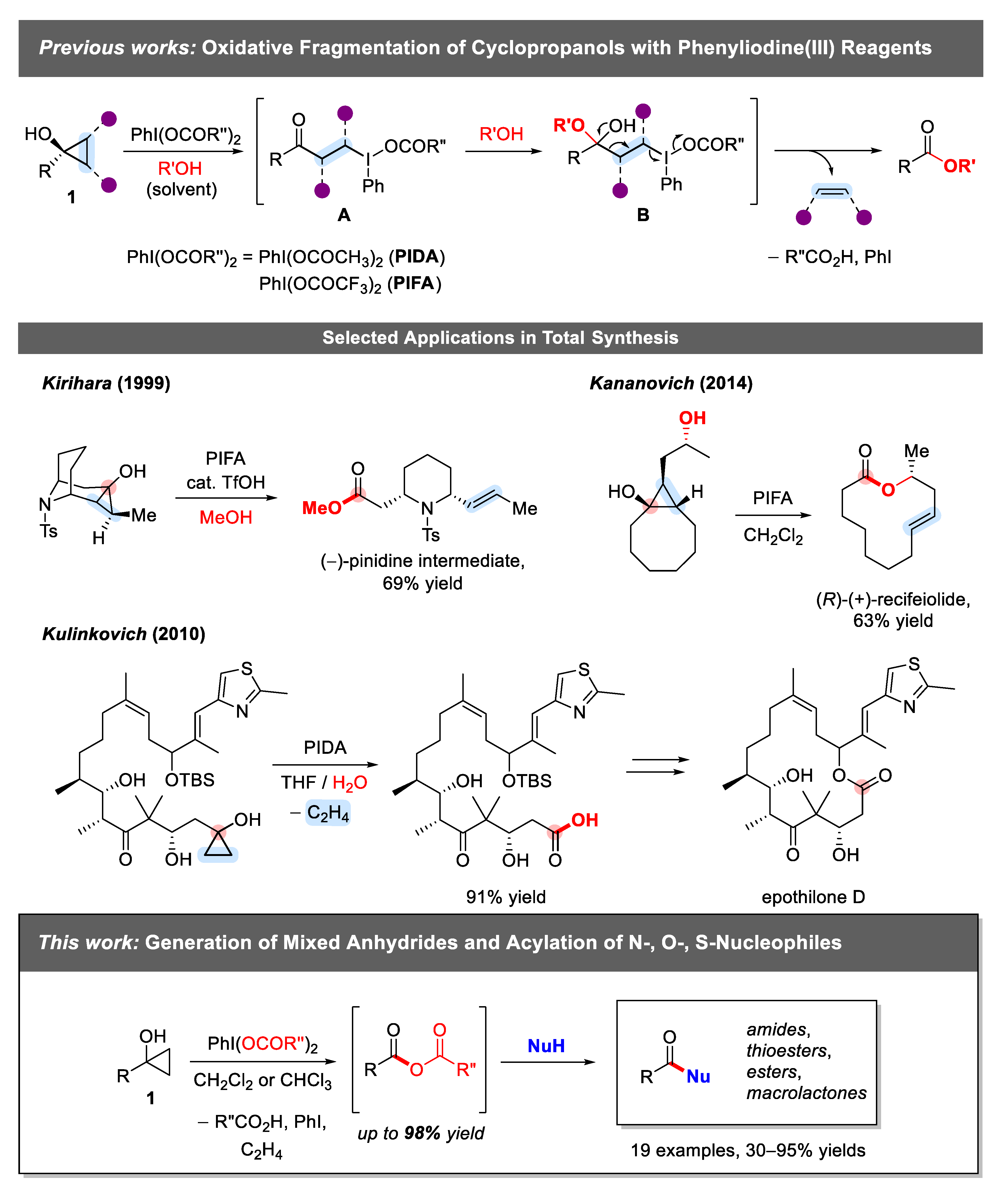
2. Results and Discussion
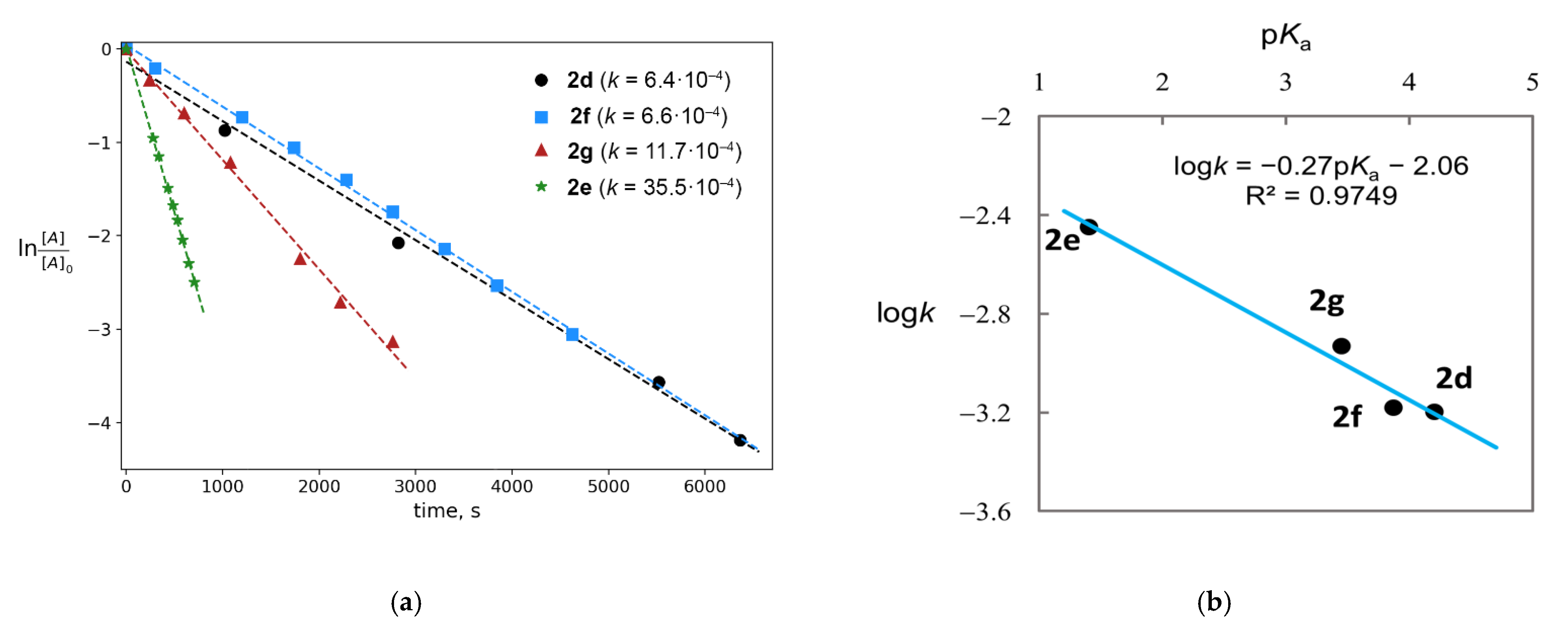
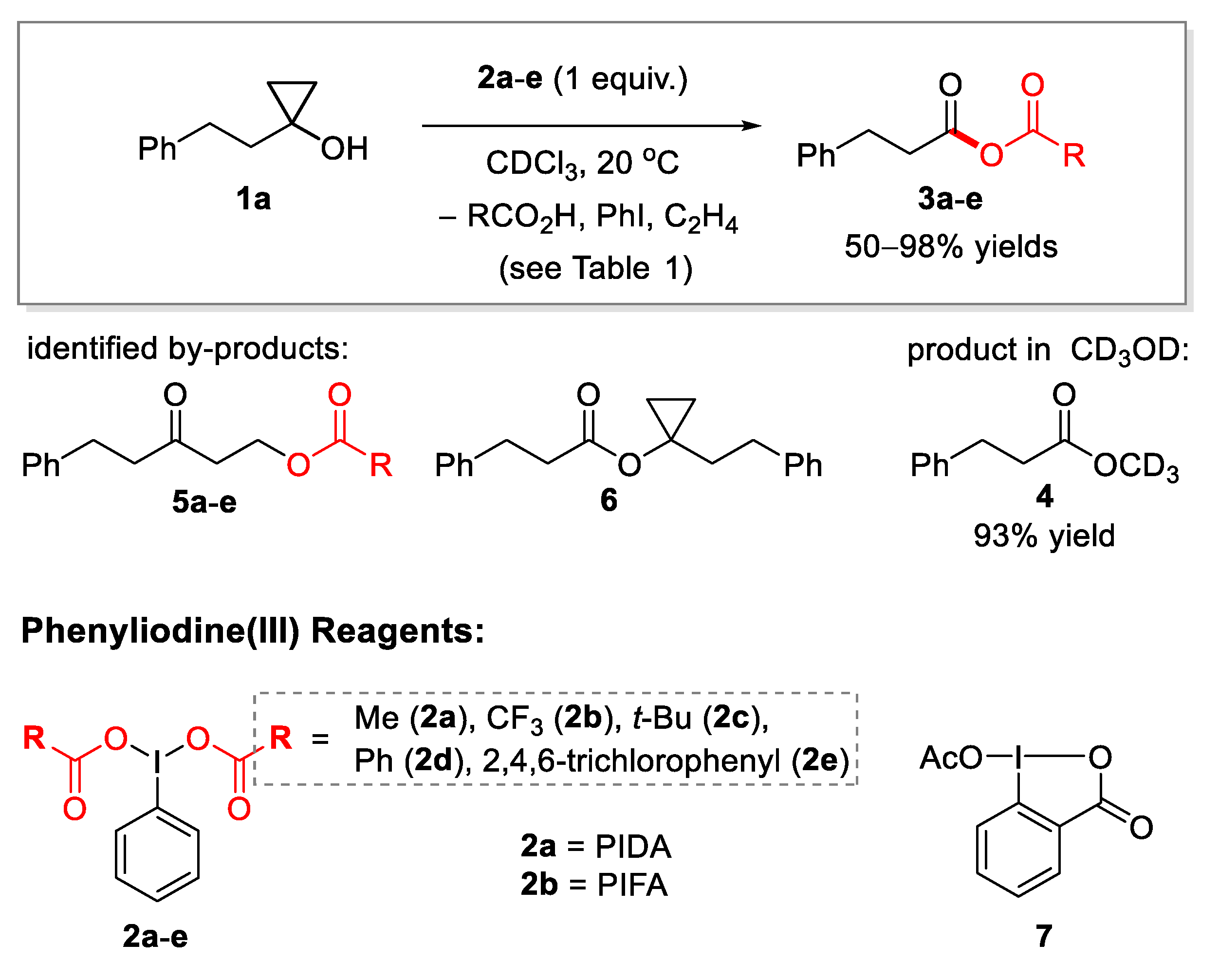
2.1. Generation of Mixed Anhydrides via Ring Scissoring of Tertiary Cyclopropanols with Phenyliodine(III) Dicarboxylates
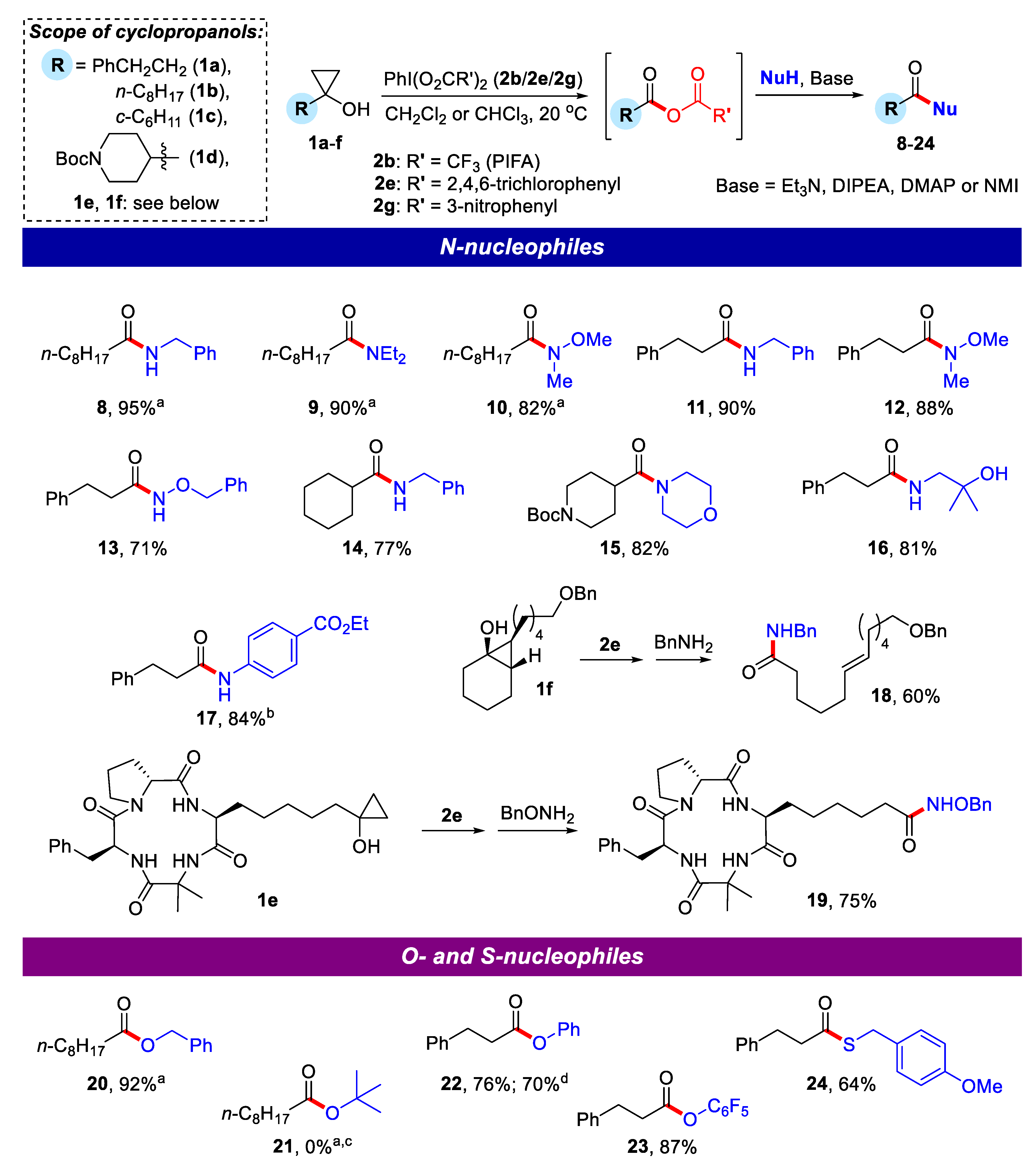
2.2. Application of Mixed Anhydrides for Acylation of N-, O-, S-Nucleophiles and in Macrolactonizations
3. Experimental Section
3.1. General Experimental Methods
3.2. Typical Procedure for Preparation of PhI(O2CAr)2 Reagents [for Ar = 2,4,6-trichlorophenyl, 2e]
3.3. General Procedure for Generation of Mixed Anhydrides from Cyclopropanols and Reagent 2e
3.3.1. N-Benzylnonanamide (8)
3.3.2. N,N-Diethylnonanamide (9)
3.3.3. N-Methoxy-N-methylnonanamide (10)
3.3.4. N-Benzyl-3-phenylpropanamide (11)
3.3.5. N-Methoxy-N-methyl-3-phenylpropanamide (12)
3.3.6. N-(Benzyloxy)-3-phenylpropanamide (13)
3.3.7. N-Benzylcyclohexanecarboxamide (14)
3.3.8. tert-Butyl 4-(morpholine-4-carbonyl)piperidine-1-carboxylate (15)
3.3.9. N-(2-Hydroxy-2-methylpropyl)-3-phenylpropanamide (16)
3.3.10. Ethyl 4-(3-phenylpropanamido)benzoate (17)
3.3.11. (E)-N-Benzyl-12-(benzyloxy)dodec-6-enamide (18)
3.3.12. 6-((3S,9S,14aR)-9-Benzyl-6,6-dimethyl-1,4,7,10-tetraoxotetradecahydropyrrolo[1,2-a][1,4,7,10]tetraazacyclododecin-3-yl)-N-(benzyloxy)hexanamide (cyclopeptide 19)
3.3.13. Benzyl nonanoate (20)
3.3.14. Phenyl 3-phenylpropanoate (22)
3.3.15. Pentafluorophenyl 3-phenylpropanoate (23)
3.3.16. S-(4-Methoxybenzyl) 3-phenylpropanethioate (24)
3.4. General Procedure for the Synthesis of Macrolactones 25a,b
3.4.1. Oxacycloundecan-2-one (25a)
3.4.2. Oxacyclotridecan-2-one (25b)
3.5. Preparation of (E)-oxacyclotridec-7-en-2-one (28)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kulinkovich, O.G. The Chemistry of Cyclopropanols. Chem. Rev. 2003, 103, 2597–2632. [Google Scholar] [CrossRef]
- Kulinkovich, O.G. Cyclopropanes in Organic Synthesis; Wiley: Hoboken, NJ, USA, 2015. [Google Scholar] [CrossRef]
- Haym, I.; Brimble, M.A. The Kulinkovich hydroxycyclopropanation reaction in natural product synthesis. Org. Biomol. Chem. 2012, 10, 7649–7665. [Google Scholar] [CrossRef]
- Cai, X.; Liang, W.; Dai, M. Total Syntheses via Cyclopropanols. Tetrahedron 2019, 75, 193–208. [Google Scholar] [CrossRef]
- Wolan, A.; Six, Y. Synthetic transformations mediated by the combination of titanium(iv) alkoxides and grignard reagents: Selectivity issues and recent applications. Part 1: Reactions of carbonyl derivatives and nitriles. Tetrahedron 2010, 66, 15–61. [Google Scholar] [CrossRef]
- McDonald, T.R.; Mills, L.R.; West, M.S.; Rousseaux, S.A.L. Selective Carbon–Carbon Bond Cleavage of Cyclopropanols. Chem. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pirenne, V.; Muriel, B.; Waser, J. Catalytic Enantioselective Ring-Opening Reactions of Cyclopropanes. Chem. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; You, B.; Xie, G.; Wang, X. Developments in the construction of cyclopropanols. Org. Biomol. Chem. 2020, 18, 191–204. [Google Scholar] [CrossRef]
- Cha, J.K.; Kulinkovich, O.G. The Kulinkovich Cyclopropanation of Carboxylic Acid Derivatives. Org. React. 2012, 77, 1–160. [Google Scholar] [CrossRef]
- Kulinkovich, O.G.; de Meijere, A. 1,n-Dicarbanionic Titanium Intermediates from Monocarbanionic Organometallics and Their Application in Organic Synthesis. Chem. Rev. 2000, 100, 2789–2834. [Google Scholar] [CrossRef]
- Du, H.; Long, J.; Shi, Y. Catalytic Asymmetric Simmons−Smith Cyclopropanation of Silyl Enol Ethers. Efficient Synthesis of Optically Active Cyclopropanol Derivatives. Org. Lett. 2006, 8, 2827–2829. [Google Scholar] [CrossRef]
- Cheng, K.; Carroll, P.J.; Walsh, P.J. Diasteroselective Preparation of Cyclopropanols Using Methylene Bis(iodozinc). Org. Lett. 2011, 13, 2346–2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kananovich, D.G.; Zubrytski, D.M.; Kulinkovich, O.G. A Convenient Transformation of 2-Alkylidenecycloalkanones into Alkyl-Substituted Bicyclo[n.1.0]alkan-1-ols: Application to the Synthesis of Capsaicin. Synlett 2010, 7, 1043–1046. [Google Scholar] [CrossRef]
- Kananovich, D.G.; Kulinkovich, O.G. Studies on the origin of cis-diastereoselectivity of the titanium-mediated cyclopropanation of carboxylic esters with Grignard reagents. Stereochemistry of the intramolecular cyclization of β-metalloketones. Tetrahedron 2008, 64, 1536–1547. [Google Scholar] [CrossRef]
- Rivera, R.M.; Jang, Y.; Poteat, C.M.; Lindsay, V.N.G. General Synthesis of Cyclopropanols via Organometallic Addition to 1-Sulfonylcyclopropanols as Cyclopropanone Precursors. Org. Lett. 2020, 22, 6510–6515. [Google Scholar] [CrossRef]
- Yu, Z.; Mendoza, A. Enantioselective Assembly of Congested Cyclopropanes Using Redox-Active Aryldiazoacetates. ACS Catal. 2019, 9, 7870–7875. [Google Scholar] [CrossRef] [Green Version]
- Montesinos-Magraner, M.; Costantini, M.; Ramírez-Contreras, R.; Muratore, M.E.; Johansson, M.J.; Mendoza, A. General Cyclopropane Assembly via Enantioselective Transfer of a Redox-Active Carbene to Aliphatic Olefins. Angew. Chem. Int. Ed. 2019, 58, 5930–5935. [Google Scholar] [CrossRef] [Green Version]
- Simaan, M.; Marek, I. Asymmetric Catalytic Preparation of Polysubstituted Cyclopropanol and Cyclopropylamine Derivatives. Angew. Chem. Int. Ed. 2018, 57, 1543–1546. [Google Scholar] [CrossRef]
- Benoit, G.; Charette, A.B. Diastereoselective Borocyclopropanation of Allylic Ethers Using a Boromethylzinc Carbenoid. J. Am. Chem. Soc. 2017, 139, 1364–1367. [Google Scholar] [CrossRef]
- Mills, L.R.; Rousseaux, S.A.L. Modern Developments in the Chemistry of Homoenolates. Eur. J. Org. Chem. 2019, 2019, 8–26. [Google Scholar] [CrossRef]
- Nikolaev, A.; Orellana, A. Transition-Metal-Catalyzed C–C and C–X Bond-Forming Reactions Using Cyclopropanols. Synthesis 2016, 48, 1741–1768. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Q.-L.; Chen, Z.; Zhou, C.-S.; Xiong, B.-Q.; Zhang, P.-L.; Yang, C.-A.; Zhou, Q. Oxidative radical ring-opening/cyclization of cyclopropane derivatives. Beilstein J. Org. Chem. 2019, 15, 256–278. [Google Scholar] [CrossRef] [PubMed]
- Rubottom, G.M.; Marrero, R.; Krueger, D.S.; Schreiner, J.L. The lead (IV) acetate oxidation of cyclopropane derivatives: A novel fragmentation reaction. Tetrahedron Lett. 1977, 18, 4013–4015. [Google Scholar] [CrossRef]
- Rubottom, G.M.; Beedle, E.C.; Kim, C.-W.; Mott, R.C. Stereochemistry of the Lead(IV) Acetate Fragmentation of 1-(Trimethylsiloxy)bicyclo[n.1.0]Alkanes. J. Am. Chem. Soc. 1985, 107, 4230–4233. [Google Scholar] [CrossRef]
- Kirihara, M.; Yokoyama, S.; Kakuda, H.; Momose, T. Efficient Fragmentation of the Tertiary Cyclopropanol System: Oxidation of 1-(Trimethylsiloxy)bicyclo[n.1.0]alkanes and Analogues by Using Phenyliodine(III) Diacetate. Tetrahedron Lett. 1995, 36, 6907–6910. [Google Scholar] [CrossRef]
- Kirihara, M.; Yokoyama, S.; Kakuda, H.; Momose, T. Hypervalent λn-Iodane-Mediated Fragmentation of Tertiary Cyclopropanol Systems. Tetrahedron 1998, 54, 13943–13954. [Google Scholar] [CrossRef]
- Zubrytski, D.M.; Kananovich, D.G.; Matiushenkov, E.A. Preparation of Stereochemically Pure E- and Z-Alkenoic Acids and Their Methyl Esters from Bicyclo[n.1.0]alkan-1-ols. Application in the Synthesis of Insect Pheromones. Russ. J. Org. Chem. 2017, 53, 813–823. [Google Scholar] [CrossRef]
- Momose, T.; Nishio, T.; Kirihara, M. Efficient Synthesis of an Enantiomeric Pair of Pinidine: An Illustration of Organochemical Carving on the Rigid Bridged System as the Stereochemical Tactics. Tetrahedron Lett. 1996, 37, 4987–4990. [Google Scholar] [CrossRef]
- Kirihara, M.; Nishio, T.; Yokoyama, S.; Kakuda, H.; Momose, T. Hypervalent λn-Iodane-Mediated Fragmentation of Tertiary Cyclopropanol Systems II: Application to Asymmetric Syntheses of Piperidine and Indolizidine Alkaloids. Tetrahedron 1999, 55, 2911–2926. [Google Scholar] [CrossRef]
- Zubrytski, D.M.; Kananovich, D.G.; Kulinkovich, O.G. A highly stereoselective route to medium-ring-sized trans-alkenolides via oxidative fragmentation of bicyclic oxycyclopropane precursors: Application to the synthesis of (+)-recifeiolide. Tetrahedron 2014, 70, 2944–2950. [Google Scholar] [CrossRef]
- Bekish, A.V.; Kulinkovich, O.G. Differentiation between the ethoxycarbonyl groups in diethyl malate via their titanium-catalyzed reductive cyclopropanation with ethylmagnesium bromide and subsequent site-selective three-carbon ring cleavage. Tetrahedron Lett. 2005, 46, 6975–6978. [Google Scholar] [CrossRef]
- Bekish, A.V.; Isakov, V.E.; Kulinkovich, O.G. A cyclopropanol approach to the synthesis of the C13–C21 fragment of epothilones from diethyl (S)-malate. Tetrahedron Lett. 2005, 46, 6979–6981. [Google Scholar] [CrossRef]
- Kovalenko, V.N.; Masalov, N.V.; Kulinkovich, O.G. Synthesis of (+)-Disparlure from Diethyl (−)-Malate via Opening and Fragmentation of the Three-Membered Ring in Tertiary Cyclopropanols. Russ. J. Org. Chem. 2009, 45, 1318–1324. [Google Scholar] [CrossRef]
- Shklyaruck, D.G.; Fedarkevich, A.N.; Kozyrkov, Y.Y. The First Synthesis of Spirocyclic Sulfates from Tertiary Cyclopropanols and Their Reaction with Normant Homocuprates. Synlett 2014, 25, 1855–1858. [Google Scholar] [CrossRef] [Green Version]
- Shklyaruck, D.G. A cyclopropanol-based approach to synthesis of Unit A of the cryptophycins. Tetrahedron Asymmetry 2014, 25, 644–649. [Google Scholar] [CrossRef]
- Shklyaruck, D.G. New Synthesis of syn-Stereodiad Building Block for Polyketides. Formal Synthesis of Arenamides A and C. Russ. J. Org. Chem. 2015, 51, 582–590. [Google Scholar] [CrossRef]
- Hurski, A.L.; Kulinkovich, O.G. Total synthesis of epothilone D by sixfold ring cleavage of cyclopropanol intermediates. Tetrahedron Lett. 2010, 51, 3497–3500. [Google Scholar] [CrossRef]
- Hurski, A.L.; Kulinkovich, O.G. Synthesis of Epothilone D with the Forced Application of Oxycyclopropane Intermediates. Russ. J. Org. Chem. 2011, 47, 1653–1674. [Google Scholar] [CrossRef]
- Elek, G.Z.; Koppel, K.; Zubrytski, D.M.; Konrad, N.; Järving, I.; Lopp, M.; Kananovich, D.G. Divergent Access to Histone Deacetylase Inhibitory Cyclopeptides via a Late-Stage Cyclopropane Ring Cleavage Strategy. Short Synthesis of Chlamydocin. Org. Lett. 2019, 21, 8473–8478. [Google Scholar] [CrossRef]
- Nishino, N.; Jose, B.; Shinta, R.; Kato, T.; Komatsu, Y.; Yoshida, M. Chlamydocin–hydroxamic acid analogues as histone deacetylase inhibitors. Bioorg. Med. Chem. 2004, 12, 5777–5784. [Google Scholar] [CrossRef]
- Wang, S.; Li, X.; Wei, Y.; Xiu, Z.; Nishino, N. Discovery of Potent HDAC Inhibitors Based on Chlamydocin with Inhibitory Effects on Cell Migration. ChemMedChem 2014, 9, 627–637. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Kitir, B.; Maolanon, A.R.; Ohm, R.G.; Colaço, A.R.; Fristrup, P.; Madsen, A.S.; Olsen, C.A. Chemical Editing of Macrocyclic Natural Products and Kinetic Profiling Reveal Slow, Tight-Binding HDAC Inhibitors with Picomolar Affinities. Biochemistry 2017, 56, 5134–5146. [Google Scholar] [CrossRef] [PubMed]
- Boström, J.; Brown, D.G.; Young, R.J.; Keserü, G.M. Expanding the medicinal chemistry synthetic toolbox. Nat. Rev. Drug Discov. 2018, 17, 709–727. [Google Scholar] [CrossRef] [PubMed]
- Roughley, S.D.; Jordan, A.M. The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem. 2011, 54, 3451–3479. [Google Scholar] [CrossRef]
- Massolo, E.; Pirola, M.; Benaglia, M. Amide bond formation strategies: Latest advances on a dateless transformation. Eur. J. Org. Chem. 2020, 2020, 4641–4651. [Google Scholar] [CrossRef]
- de Figueiredo, R.M.; Suppo, J.-S.; Campagne, J.-M. Nonclassical Routes for Amide Bond Formation. Chem. Rev. 2016, 116, 12029–12122. [Google Scholar] [CrossRef]
- Papadopoulos, G.N.; Kokotos, C.G. Photoorganocatalytic One-Pot Synthesis of Hydroxamic Acids from Aldehydes. Chem. Eur. J. 2016, 22, 6964–6967. [Google Scholar] [CrossRef]
- Montalbetti, C.A.G.N.; Falque, V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar] [CrossRef]
- Kirihara, M.; Kakuda, H. Hypervalent Iodine-Mediated Fragmentation of Tertiary Cyclopropanol Systems. J. Synth. Org. Chem. Jpn. 2004, 62, 919–928. [Google Scholar] [CrossRef]
- Kirihara, M.; Yokoyama, S.; Momose, T. A Significant Effect of Triflic Acid on the Hypervalent λn-Iodane-Mediated Fragmentation of the Tertiary Cyclopropanol System. Synth. Commun. 1998, 28, 1947–1956. [Google Scholar] [CrossRef]
- Zhdankin, V.V.; Stang, P.J. Chemistry of Polyvalent Iodine. Chem. Rev. 2008, 108, 5299–5358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zefirov, N.S.; Zhdankin, V.V.; Dan’kov, Y.V.; Sorokin, V.D.; Semerikov, V.N.; Koz’min, A.S.; Caple, R.; Berglund, B.A. Novel Reagents Containing Hypervalent Iodine and Their Use for Electrophilic Additions to Olefins. Tetrahedron Lett. 1986, 27, 3971–3974. [Google Scholar] [CrossRef]
- Tóth, B.L.; Béke, F.; Egyed, O.; Bényei, A.; Stirling, A.; Novák, Z. Synthesis of Multifunctional Aryl(Trifloxyalkenyl)iodonium Triflate Salts. ACS Omega 2019, 4, 9188–9197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Guo, L.-N.; Wang, H.; Duan, X.-H. Alkynylation of Tertiary Cycloalkanols via Radical C–C Bond Cleavage: A Route to Distal Alkynylated Ketones. Org. Lett. 2015, 17, 4798–4801. [Google Scholar] [CrossRef]
- Jia, K.; Zhang, F.; Huang, H.; Chen, Y. Visible-Light-Induced Alkoxyl Radical Generation Enables Selective C(sp3)–C(sp3) Bond Cleavage and Functionalizations. J. Am. Chem. Soc. 2016, 138, 1514–1517. [Google Scholar] [CrossRef]
- Feng, Y.-S.; Shu, Y.-J.; Cao, P.; Xu, T.; Xu, H.-J. Copper(I)-catalyzed ring-opening cyanation of cyclopropanols. Org. Biomol. Chem. 2017, 15, 3590–3593. [Google Scholar] [CrossRef]
- Li, Y.; Ye, Z.; Bellman, T.M.; Chi, T.; Dai, M. Efficient Synthesis of β-CF3/SCF3-Substituted Carbonyls via Copper-Catalyzed Electrophilic Ring-Opening Cross-Coupling of Cyclopropanols. Org. Lett. 2015, 17, 2186–2189. [Google Scholar] [CrossRef] [Green Version]
- Kananovich, D.G.; Konik, Y.A.; Zubrytski, D.M.; Järving, I.; Lopp, M. A Simple Access to β-Trifluoromethyl-Substituted Ketones via Copper-Catalyzed Ring-Opening Trifluoromethylation of Substituted Cyclopropanols. Chem. Commun. 2015, 51, 8349–8352. [Google Scholar] [CrossRef]
- Stang, P.J.; Boehshar, M.; Wingert, H.; Kitamura, T. Acetylenic Esters. Preparation and Characterization of Alkynyl Carboxylates via Polyvalent Iodonium Species. J. Am. Chem. Soc. 1988, 110, 3272–3278. [Google Scholar] [CrossRef]
- Koposov, A.Y.; Boyarskikh, V.V.; Zhdankin, V.V. Amino Acid-Derived Iodobenzene Dicarboxylates: Reagents for Oxidative Conversion of Alkenes to Amino Acid Esters. Org. Lett. 2004, 6, 3613–3615. [Google Scholar] [CrossRef]
- Lundblad, R.L.; Macdonald, F.M. Handbook of Biochemistry and Molecular Biology, 4th ed.; CRC Press: Boca Raton, FL, USA, 2010; pp. 1–1098. [Google Scholar]
- Beutner, G.L.; Young, I.S.; Davies, M.L.; Hickey, M.R.; Park, H.; Stevens, J.M.; Ye, Q. TCFH–NMI: Direct Access to N-Acyl Imidazoliums for Challenging Amide Bond Formations. Org. Lett. 2018, 20, 4218–4222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.-A.; Han, G.; Kim, H.-J.; Kim, H.-M.; Cho, S.-D. Chemopreventive and chemotherapeutic effect of a novel histone deacetylase inhibitor, by specificity protein 1 in MDA-MB-231 human breast cancer cells. Eur. J. Cancer Prev. 2014, 23, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Montiel-Smith, S.; Meza-Reyes, S.; Viñas-Bravo, O.; Fernández-Herrera, M.A.; Martínez-Pascual, R.; Sandoval-Ramírez, J.; Fuente, A.; Reyes, M.; Ruiz, J.A. In situ preparation of mixed anhydrides containing the trifluoroacetyl moiety. Application to the esterification of cholesterol and phenol. Arkivoc 2005, 2005, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Shiina, I. Total Synthesis of Natural 8- and 9-Membered Lactones: Recent Advancements in Medium-Sized Ring Formation. Chem. Rev. 2007, 107, 239–273. [Google Scholar] [CrossRef]
- Parenty, A.; Moreau, X.; Niel, G.; Campagne, J.-M. Update 1 of: Macrolactonizations in the Total Synthesis of Natural Products. Chem. Rev. 2013, 113, PR1–PR40. [Google Scholar] [CrossRef] [PubMed]
- Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. A Rapid Esterification by Means of Mixed Anhydride and Its Application to Large-ring Lactonization. Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. [Google Scholar] [CrossRef] [Green Version]
- Shiina, I.; Mukaiyama, T. A Novel Method for the Preparation of Macrolides from ω-Hydroxycarboxylic Acids. Chem. Lett. 1994, 23, 677–680. [Google Scholar] [CrossRef]
- Shiina, I.; Kubota, M.; Oshiumi, H.; Hashizume, M. An Effective Use of Benzoic Anhydride and Its Derivatives for the Synthesis of Carboxylic Esters and Lactones: A Powerful and Convenient Mixed Anhydride Method Promoted by Basic Catalysts. J. Org. Chem. 2004, 69, 1822–1830. [Google Scholar] [CrossRef]
- Ishihara, K.; Kubota, M.; Kurihara, H.; Yamamoto, H. Scandium Trifluoromethanesulfonate as an Extremely Active Lewis Acid Catalyst in Acylation of Alcohols with Acid Anhydrides and Mixed Anhydrides. J. Org. Chem. 1996, 61, 4560–4567. [Google Scholar] [CrossRef]
- Shiina, I. An effective method for the synthesis of carboxylic esters and lactones using substituted benzoic anhydrides with Lewis acid catalysts. Tetrahedron 2004, 60, 1587–1599. [Google Scholar] [CrossRef]
- de Léséleuc, M.; Collins, S.K. Direct synthesis of macrodiolides via hafnium(IV) catalysis. Chem. Commun. 2015, 51, 10471–10474. [Google Scholar] [CrossRef] [PubMed]
- de Léséleuc, M.; Collins, S.K. Direct Macrolactonization of Seco Acids via Hafnium(IV) Catalysis. ACS Catal. 2015, 5, 1462–1467. [Google Scholar] [CrossRef]
- Mocci, F.; Uccheddu, G.; Frongia, A.; Cerioni, G. Solution Structure of Some λ3 Iodanes: An 17O NMR and DFT Study. J. Org. Chem. 2007, 72, 4163–4168. [Google Scholar] [CrossRef] [PubMed]
- Shibahara, F.; Sugiura, R.; Murai, T. Direct Thionation and Selenation of Amides Using Elemental Sulfur and Selenium and Hydrochlorosilanes in the Presence of Amines. Org. Lett. 2009, 11, 3064–3067. [Google Scholar] [CrossRef]
- Fukuyama, T.; Nishitani, S.; Inouye, T.; Morimoto, K.; Ryu, I. Effective Acceleration of Atom Transfer Carbonylation of Alkyl Iodides by Metal Complexes. Application to the Synthesis of the Hinokinin Precursor and Dihydrocapsaicin. Org. Lett. 2006, 8, 1383–1386. [Google Scholar] [CrossRef]
- Lee, J.I. A Novel Synthesis of N-Methoxy-N-Methylamides from 4,6-Pyrimidyl Urethane and Grignard Reagents. Bull. Korean Chem. Soc. 2007, 28, 695–697. [Google Scholar] [CrossRef]
- Allen, C.L.; Davulcu, S.; Williams, J.M.J. Catalytic Acylation of Amines with Aldehydes or Aldoximes. Org. Lett. 2010, 12, 5096–5099. [Google Scholar] [CrossRef]
- Murphy, J.A.; Commeureuc, A.G.J.; Snaddon, T.N.; McGuire, T.M.; Khan, T.A.; Hisler, K.; Dewis, M.L.; Carling, R. Direct Conversion of N-Methoxy-N-Methylamides (Weinreb Amides) to Ketones via a Nonclassical Wittig Reaction. Org. Lett. 2005, 7, 1427–1429. [Google Scholar] [CrossRef]
- Gissot, A.; Volonterio, A.; Zanda, M. One-Step Synthesis of O-Benzyl Hydroxamates from Unactivated Aliphatic and Aromatic Esters. J. Org. Chem. 2005, 70, 6925–6928. [Google Scholar] [CrossRef]
- Maki, T.; Ishihara, K.; Yamamoto, H. 4,5,6,7-Tetrachlorobenzo[d][1,3,2]Dioxaborol-2-ol as an Effective Catalyst for the Amide Condensation of Sterically Demanding Carboxylic Acids. Org. Lett. 2006, 8, 1431–1434. [Google Scholar] [CrossRef]
- Besbes, N. Regiospecific ring opening of N-acylaziridines by neutral hydrolysis. Tetrahedron Lett. 1999, 40, 6569–6570. [Google Scholar] [CrossRef]
- Sheikh, M.C.; Takagi, S.; Ogasawara, A.; Ohira, M.; Miyatake, R.; Abe, H.; Yoshimura, T.; Morita, H. Studies on the Lossen-Type rearrangement of N-(3-phenylpropionyloxy)phthalimide and N-tosyloxy derivatives with several nucleophiles. Tetrahedron 2010, 66, 2132–2140. [Google Scholar] [CrossRef]
- Hon, Y.-S.; Wong, Y.-C.; Chang, C.-P.; Hsieh, C.-H. Tishchenko reactions of aldehydes promoted by diisobutylaluminum hydride and its application to the macrocyclic lactone formation. Tetrahedron 2007, 63, 11325–11340. [Google Scholar] [CrossRef]
- Muller, T.; Coowar, D.; Hanbali, M.; Heuschling, P.; Luu, B. Improved synthesis of tocopherol fatty alcohols and analogs: Microglial activation modulators. Tetrahedron 2006, 62, 12025–12040. [Google Scholar] [CrossRef]
- Chen, H.; Sun, S.; Liao, X. Nickel-Catalyzed Decarboxylative Alkenylation of Anhydrides with Vinyl Triflates or Halides. Org. Lett. 2019, 21, 3625–3630. [Google Scholar] [CrossRef]
- Pulikottil, F.T.; Pilli, R.; Suku, R.V.; Rasappan, R. Nickel-Catalyzed Cross-Coupling of Alkyl Carboxylic Acid Derivatives with Pyridinium Salts via C–N Bond Cleavage. Org. Lett. 2020, 22, 2902–2907. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
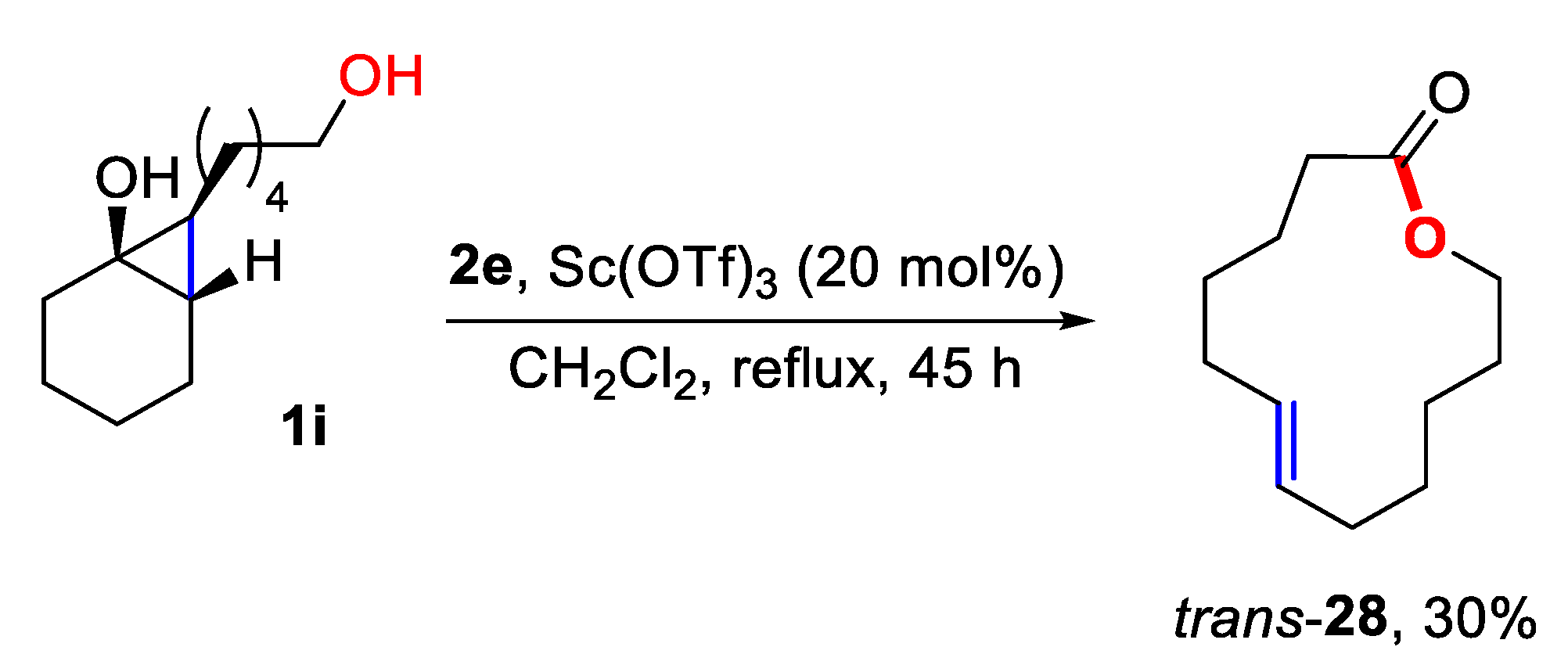{kind=link}
| Entry | Reagent | Solvent | Additive or Catalyst | Reaction Time, Min b | Main Product | Yield % c | Yield of 5, % c |
|---|---|---|---|---|---|---|---|
| 1 | 2a | CD3OD | – | <15 | 4 | 93 | – |
| 2 | 2a | CDCl3 | BnNH2 (3 equiv.) | 300 | no reaction | ||
| 3 | 2a | CDCl3 | – | 600 d | 3a | 50 | 12 |
| 4 | 2a | CDCl3 | TfOH (1 mol%) | <5 | 3a | 90 | 10 e |
| 5 | 2b | CDCl3 | – | 7 | 3b | 95 | Trace f |
| 6 | 2c | CDCl3 | – | 1500 | – g | – g | – g |
| 7 | 2c | CDCl3 | TfOH (1 mol%) | 1500 | – g | – g | – g |
| 8 | 2d | CDCl3 | – | 500 d | 3d | 46 | 8 |
| 9 | 2e | CDCl3 | – | 100 | 3e | 98 | 2 |
| 10 | 2e | CDCl3 | TfOH (1 mol%) | <5 | 3e | 70 | 30 e |
| 11 | 2e | CDCl3 | MsOH (1 mol%) | 30 | 3e | 95 | 4 e |
| 12 | 2e | CDCl3 | CF3CO2H (1 mol%) | 90 | 3e | 94 | 5 e |
| 13 | 2e | toluene-d8 | – | <15 | 3e | 90 | 4 |
| 14 | 7 | CD3ODor CDCl3 | – | 1500 | no reaction | ||

| Entry | Reagent 2a-k | R | Time, min b | Yield of 8, % c |
|---|---|---|---|---|
| 1 | 2a | Me | 120 | 22 e,d |
| 2 | 2h |  | 95 | 91 |
| 3 | 2e |  | 30 | 94 |
| 4 | 2i |  | <14 | 88 |
| 5 | 2g |  | 90 | 95 |
| 6 | 2j |  | – f | 58 |
| 7 | 2k |  | – f | – f |

| Entry | Substrate | Reagent | Catalyst | Solvent | Time, h | Yield of 25, % a | Yield of 26, % a |
|---|---|---|---|---|---|---|---|
| 1 | 1g (n = 9) | 2g | Sc(OTf)3 | CH3CN/THF | 20 | 0 b | 0 |
| 2 | 2g | Sc(OTf)3 | CH2Cl2/CH3CN c | 17 | 62 | 14 | |
| 3 | 1h (n = 11) | 2g | Sc(OTf)3 | CH2Cl2/CH3CN | 189 | 82 | 10 |
| 4 | 2e | Sc(OTf)3 | CH2Cl2/CH3CN | 187 | 91 | 8 | |
| 5 | 2g | Hf(OTf)4 | CH2Cl2/CH3CN | 178 | 93 | >7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zubrytski, D.M.; Elek, G.Z.; Lopp, M.; Kananovich, D.G. Generation of Mixed Anhydrides via Oxidative Fragmentation of Tertiary Cyclopropanols with Phenyliodine(III) Dicarboxylates. Molecules 2021, 26, 140. https://doi.org/10.3390/molecules26010140
Zubrytski DM, Elek GZ, Lopp M, Kananovich DG. Generation of Mixed Anhydrides via Oxidative Fragmentation of Tertiary Cyclopropanols with Phenyliodine(III) Dicarboxylates. Molecules. 2021; 26(1):140. https://doi.org/10.3390/molecules26010140
Chicago/Turabian StyleZubrytski, Dzmitry M., Gábor Zoltán Elek, Margus Lopp, and Dzmitry G. Kananovich. 2021. "Generation of Mixed Anhydrides via Oxidative Fragmentation of Tertiary Cyclopropanols with Phenyliodine(III) Dicarboxylates" Molecules 26, no. 1: 140. https://doi.org/10.3390/molecules26010140
APA StyleZubrytski, D. M., Elek, G. Z., Lopp, M., & Kananovich, D. G. (2021). Generation of Mixed Anhydrides via Oxidative Fragmentation of Tertiary Cyclopropanols with Phenyliodine(III) Dicarboxylates. Molecules, 26(1), 140. https://doi.org/10.3390/molecules26010140