General Procedure A: Synthesis of N-Alkyladenosine
DBU (1.5 mmol) was added dropwise to a stirred solution of inosine (1 mmol) and BOP (1.2 mmol) in DMF; the mixture was then heated at 40 °C. After the consumption of starting material (approximately 40 min, as assessed by TLC), the reaction was cooled to room temperature and the appropriate amine (5 mmol) was added dropwise and the reaction was stirred overnight. The crude product mixture was concentrated under reduced pressure, then diluted with ethyl acetate and was washed with water (3 × 10 mL). The organic layer was dried (anhydrous MgSO4) and concentrated under vacuum. The resulted solid was recrystallised twice from iso-propanol.
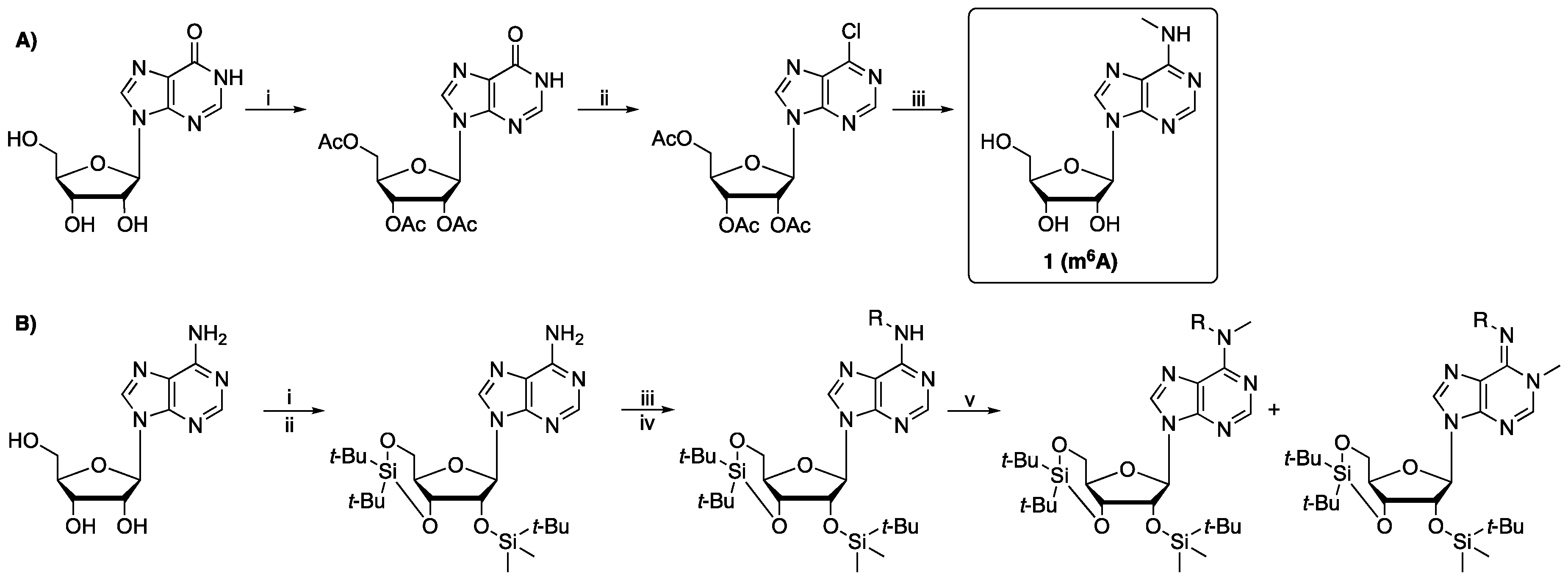
N6-Methyladenosine (1). The desired product was prepared according to General Procedure A; white solid (250 mg, 90%). m.p 214.5 °C; 1H-NMR (500 MHz, D2O) δ 2.93 (s, 3H), 3.74 (dd, J = 13.0, 3.5 Hz, 1H), 3.83 (dd, J = 13.0, 2.5 Hz, 1H), 4.20 (q, J = 3.0 Hz, 1H), 4.32 (dd, J = 5.0, 3.0 Hz, 1H), 4.66 (t, J = 5.5 Hz, 1H), 5.90 (d, J = 6.0 Hz, 1H), 8.02 (s, 1H), 8.11 (s, 1H); 13C-NMR (126 MHz, D2O) δ 27.5, 62.1, 71.1, 74.0, 88.36, 88.39, 120.0, 140.1, 152.9, 155.6. HRMS (ESI) m/z: calculated for C11H16O4N5 [M + H]+ 282.1197, observed: 282.1196.
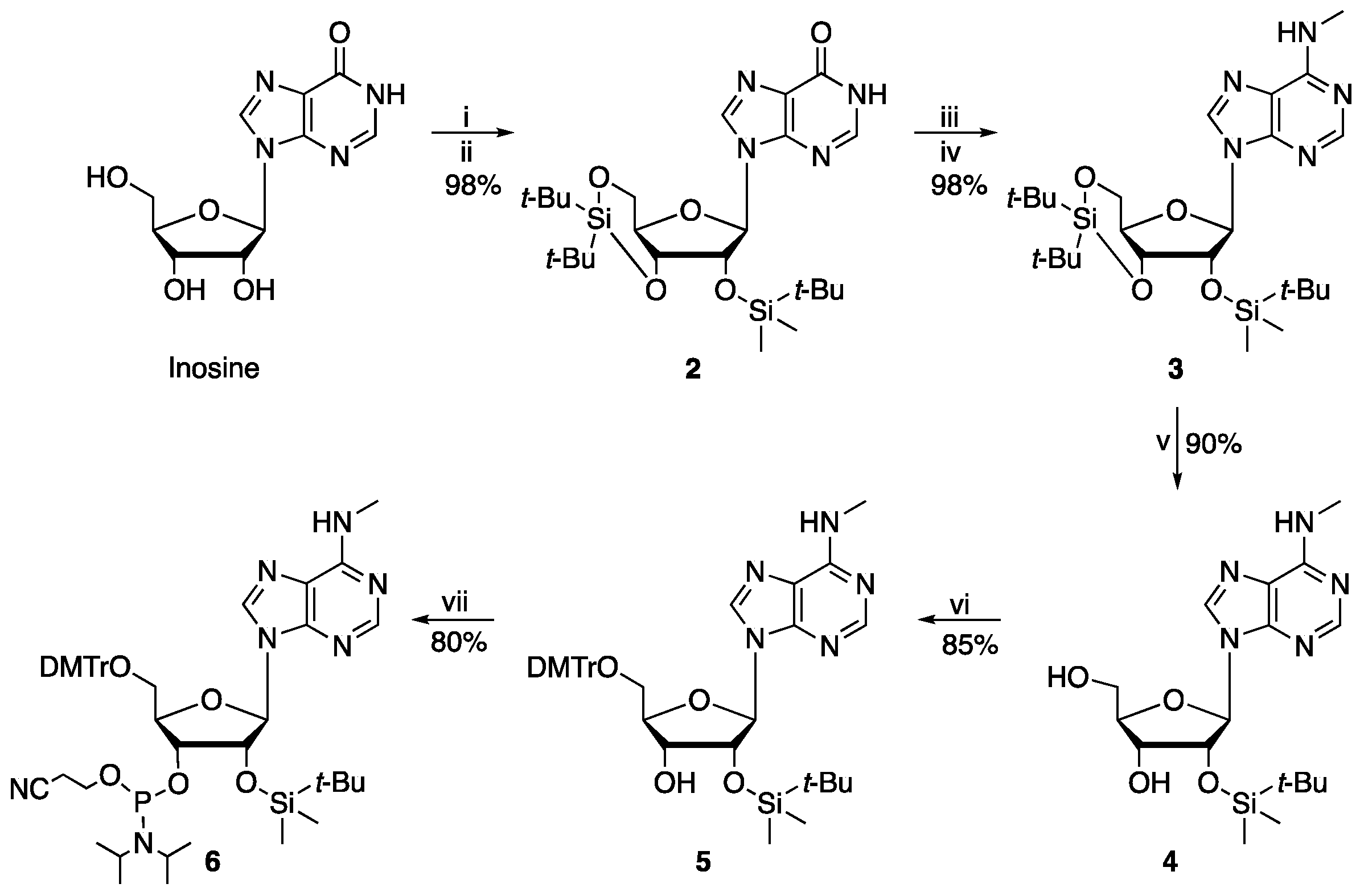
3′,5′-O-(Di-tert-butyl)silyl-2′-O-dimethyl(tert-butyl)silylinosine (
2). The desired compound was prepared according to a modified version of the reported procedure [
22]. To a stirred suspension of inosine (2.12 g, 8 mmol) in 40 mL anhydrous DMF at 0 °C, di-
t-butylsilyl ditrifluoromethanesulfonate (3.0 mL, 8.8 mmol) was added dropwise under an N
2 atmosphere. After consumption of starting material (30 min, as assessed by TLC), the reaction was quenched immediately with imidazole (2.7 g, 40 mmol) at 0 °C. After 5 min, the reaction was warmed to room temperature.
t-Butyldimethylsilyl chloride (1.5 g, 9.6 mmol) was then added portionwise and the reaction was refluxed at 60 °C for 12 h. The suspension was then cooled to room temperature, water was added, and the precipitate was collected by suction filtration. The filtrate was discarded, and the white precipitate was washed with cold methanol. The methanol layer was evaporated under reduced pressure and the product was crystallised from CH
2Cl
2 to give a white solid (4.0 g, 98%). m.p 191–193.4 °C. TLC R
f 0.45 (3:2 cyclohexane/ethyl acetate);
1H NMR (600 MHz, CDCl
3) δ 0.17 (s, 3H), 0.18 (s, 3H), 0.96 (s, 9H), 1.07 (s, 9H), 1.10 (s, 9H), 4.02–4.09 (m, 1H), 4.25 (td,
J = 10.0, 5.0 Hz, 1H), 4.38 (dd,
J = 9.5, 4.5 Hz, 1H), 4.45–4.58 (m, 2H), 5.96 (s, 1H), 7.87 (s, 1H), 8.11 (s, 1H), 12.56 (s, 1H);
13C NMR (151 MHz, CDCl
3) δ −5.0, −4.3, 18.3, 20.4, 22.8, 25.9, 27.0, 27.5, 67.8, 74.8, 75.89, 75.94, 92.3, 125.5, 138.3, 144.7, 148.1, 158.9; HRMS (ESI)
m/
z: calculated for C
24H
43O
4N
528Si
2 [M + H]
+ 523.2767, observed: 523.2756.
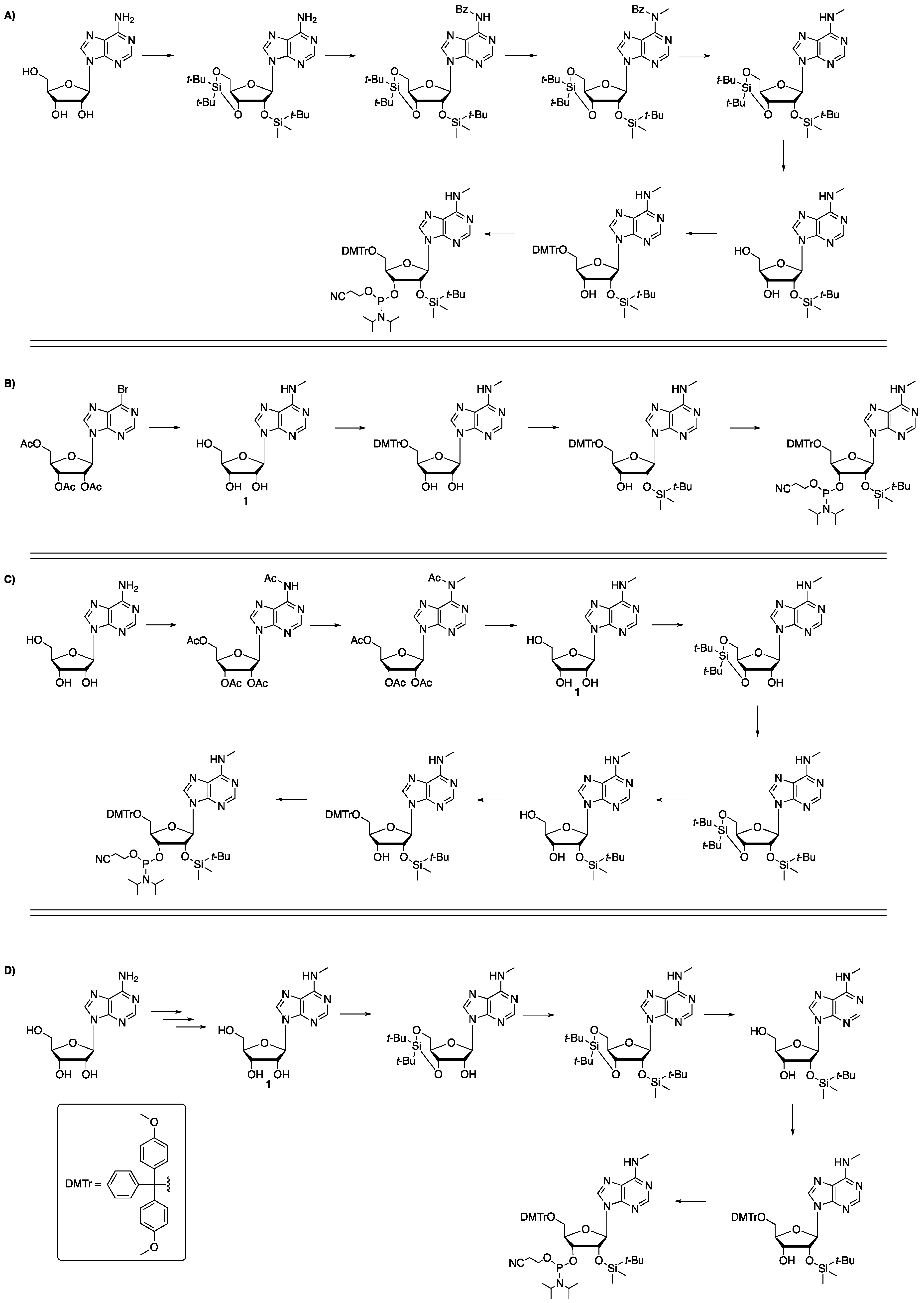
3′,5′-O-Bis(tert-butyl)silyl-2′-O-(tert-butyldimethyl)silyl-N6-methyladenosine (
3). The desired compound was prepared according to a modified version of the reported procedure [
10]. To a stirred solution of 3′,5′-
O-Bis(
tert-butylsilyl)-2′-
O-(
tert-butyldimethylsilyl)inosine (
2; 663 mg, 1.2 mmol) and BOP (0.64 g, 1.44 mmol) in 20 mL of THF, DBU (0.3 mL, 1.8 mmol) was added dropwise and the mixture was heated at 40 °C. After the consumption of the starting material (40 min, as assessed by TLC), the reaction was cooled to room temperature and methylamine (0.3 mL, 6.0 mmol) was added dropwise and the reaction was stirred overnight. The crude product mixture was concentrated under reduced pressure and diluted with ethyl acetate and was washed with water (3 × 10 mL). The organic layer was dried (anhydrous MgSO
4) and concentrated under vacuum. The residue was purified by column chromatography (9:1 to 3:2 cyclohexane/ethyl acetate) which resulted in an oil (665 mg, 98%). TLC R
f 0.20 (7:3 cyclohexane/ethyl acetate);
1H NMR (400 MHz, CDCl
3) δ 0.00 (s, 3H) 0.02 (s, 3H) 0.78 (s, 9H) 0.90 (s, 9H) 0.94 (s, 9H) 3.05 (d,
J = 1.0 Hz, 3H) 3.86–3.90 (m, 1H) 4.02–4.10 (m, 1H) 4.34 (dd,
J = 9.0, 5.0 Hz, 1 H) 4.38–4.44 (m, 1 H) 4.47 (d,
J = 4.5 Hz, 1 H) 5.76 (b.s, 2 H) 7.62 (s, 1 H) 8.22 (s, 1 H);
13C NMR (101 MHz, CDCl
3) δ −5.0, −4.3, 18.3, 20.4, 22.8, 25.9, 27.1, 27.5, 27.6, 67.9, 74.6, 75.5, 75.8, 92.4, 120.5, 125.0, 138.0, 153.4, 155.5; HRMS (ESI)
m/
z: calculated for C
25H
46O
4N
528Si
2 [M + H]
+ 536.3082, observed: 536.3078. Analytical data are consistent with those reported [
4].
2′-O-(tert-Butyldimethyl)silyl-N6-methyladenosine (
4). The desired compound was prepared according to the reported procedure [
4]. To a stirred solution of 3′,5′-
O-Bis(
tert-butylsilyl)-2′-
O-(
tert-butyldimethylsilyl)-
N6-methyladenosine (
3; 240 mg, 0.45 mmol) in 4 mL of CH
2Cl
2 at −15 °C, a cooled solution of (HF)
x·pyridine (0.06 mL, 2.3 mmol) in 365 μL pyridine was added. The reaction temperature was maintained at -15 °C and stirred for 12 h. The reaction was diluted with CH
2Cl
2, then washed first with sat. aq. NaHCO
3 solution, then with water (3 × 10 mL). The organic layer was dried (anhydrous MgSO
4) and concentrated under reduced pressure. The residue was purified by column chromatography (9:1 to 3:2 cyclohexane/ethyl acetate) which resulted in oil (160 mg, 90%). TLC R
f 0.15 (2:3 hexane/ethyl acetate);
1H-NMR (400 MHz, CDCl
3) δ 0.00 (s, 3H), 0.02 (s, 3H), 0.94 (s, 9H), 3.42 (d,
J = 1.0 Hz, 3H), 3.89 (dd,
J = 10.5, 9.0 Hz, 1H), 4.01–4.11 (m, 1H), 4.34 (dd,
J = 9.0, 5.0 Hz, 1H), 4.41 (dd,
J = 9.0, 5.0 Hz, 1H), 4.47 (d,
J = 5.0 Hz, 1H), 5.76 (s, 2H), 7.62 (s, 1H), 8.22 (s, 1H);
13C NMR (101 MHz, CDCl
3) δ −5.4, −5.3, 17.9, 25.6, 25.8, 27.5, 63.5, 73.1, 74.4, 87.8, 91.3, 119.7, 140.0, 140.1, 152.9, 155.8; HRMS (ESI)
m/
z: calculated for C
17H
30O
4N
528Si [M + H]
+ 396.2062, observed: 396.2068. Analytical data are consistent with those reported [
4].
5′-O-(4,4′-Dimethoxytrityl)−2′-O-dimethyl(tert-butyl)silyl-N6-methyladenosine (5). The desired compound was prepared according to the reported procedure [
4]. To a stirred solution of 2′-
O-dimethyl(
tert-butyl)silyl-
N6-methyladenosine (
4) (2.6 g, 6.6 mmol) in 4 mL anhydrous pyridine at 0 °C, DMTrCl (2.7 g, 8.0 mmol) was added portionwise at regular intervals for 12 h. The reaction was quenched by addition of an excess of anhydrous methanol (0.5 mL) at room temperature. After 1 h, the solution was concentrated under vacuum. The crude solid was first dissolved and fractioned between aqueous NaHCO
3 and ethyl acetate; the organic layer was then washed with water (3 × 10 mL). The organic layer was dried (MgSO
4) and concentrated under vacuum. The residue was purified by column chromatography (9:1 to 3:2 cyclohexane/ethyl acetate) resulted in a green oil (3.9 g, 85%). TLC R
f 0.45 (2:3 cyclohexane/ethyl acetate);
1H-NMR (400 MHz, CDCl
3) δ −0.13 (s, 3H) 0.00 (s, 3H) 0.86 (s, 9H) 2.77 (d,
J = 4.0 Hz, 1H) 3.17 (s, 3H) 3.36–3.43 (m, 1H) 3.54 (dd,
J = 10.5, 3.5 Hz, 1H) 3.80 (s, 6 H) 4.27 (d,
J = 3.5 Hz, 1H) 4.33–4.37 (m, 1H) 5.02 (t,
J = 5.5 Hz, 1H) 5.85 (d,
J = 4.5 Hz, 1H) 6.04 (b.s, 2H) 6.83 (d,
J = 9.0 Hz, 4H) 7.18–7.28 (m, 3H) 7.36 (d,
J = 8.0 Hz, 4H) 7.47 (dd,
J = 8.5 Hz, 1.5, 2H) 7.98 (s, 1H) 8.35 (s, 1H);
13C NMR (101 MHz, CDCl
3) δ −5.6, −5.5, 18.3, 25.8, 25.9, 55.2, 60.4, 63.0, 73.6, 75.5, 85.0, 87.5, 89.2, 113.4, 120.0, 127.3, 128.1, 128.3, 130.39, 130.45, 135.9, 138.0, 145.0, 153.0, 155.4, 158.89, 158.91; HRMS (ESI)
m/
z: calculated for C
38H
48O
6N
528Si [M + H]
+ 698.3368, observed: 698.3359. Analytical data are consistent with those reported [
4].
5′-
O-(4,4′-Dimethoxytrityl)-(3′-
O-[(2cyanoethyl)(
N,
N-diisopropylamino)phosphino]−2′-
O-dimethyl(
tert-butyl)silyl-
N6-methyladenosine (6). The desired compound was prepared according to the reported procedure [
4]. To a stirred solution of 5′-
O-(4,4′-dimethoxytrityl)−2′-
O-dimethyl(
tert-butyl)silyl-
N6-methyladenosine (5, 500 mg, 0.7 mmol) in anhydrous CH
2Cl
2 in an over-dried flask under argon, DIPEA (1.3 mL, 7.2 mmol) was added dropwise and the reaction mixture was allowed to stir at 0 °C for 10 min. (2-Cyanoethyl)-
N,N-diisopropylchlorophosphoramidite (0.40 mL, 1.8 mmol) was added to the reaction mixture dropwise at 0 °C under an argon atmosphere. The reaction was stirred at 0 °C for 30 min, then gradually (about 30 min) warmed to room temperature. After another five hours under an inert atmosphere, the reaction mixture was treated with a saturated aq. KCl solution, then evaporated by rotary evaporation. The desired product was separated by silica gel column chromatography (1:1:0.01 hexane/ethyl acetate/pyridine) resulting in a colourless oil (520 mg, 80%) yield. TLC R
f 0.40 (1:1:0.01 hexane/ethyl acetate/pyridine);
1H NMR (700 MHz, CD
2Cl
2-
d2) Major peaks are listed. δ −0.15 (s, 3H), −0.01 (s, 3H), 0.82 (s, 9H), 1.10 (s,3H), 1.11 (s, 3H), 1.22 (s, 3H), 1.22 (s, 3H), 1.65 (s, 2H), 2.62–2.74 (m, 2H), 3.19 (s, 3H), 3.36 (dd,
J = 10.5, 4.5 Hz, 1H), 3.54 (dd,
J = 10.5, 4.0 Hz, 1H), 3.82 (s, 6H), 3.85–3.93 (m, 1H), 3.95–4.10 (m, 1H), 4.41–4.49 (m, 1H), 5.12 (dd,
J = 6.1, 4.4 Hz, 1H), 5.33–5.40 (m, 2H), 5.79 (s, 1H), 5.99 (d,
J = 6.0 Hz, 1H), 6.78–6.90 (m, 4H), 7.23–7.29 (m, 1H), 7.28–7.34 (m, 2H), 7.34–7.40 (m, 4H), 7.47–7.52 (m, 2H), 7.94 (s, 1H), 8.25 (s, 1H);
13C NMR (176 MHz, CD
2Cl
2) Major peaks are listed. δ −5.4, −5.0, 0.8, 17.8, 20.4, 20.44, 21.1, 24.37, 24.4, 25.4, 25.44, 42.9, 43.0, 55.2, 58.8, 58.9, 63.5, 72.8, 72.9, 74.7, 74.7, 83.46, 83.48, 86.5, 88.4, 113.1, 117.8, 125.2, 126.8, 127.8, 128.1, 128.2, 129.0, 130.10, 130.14, 135.7, 139.0, 144.9, 153.0, 155.5, 158.6, 158.7;
31P-NMR (202 MHz, CD
2Cl
2) δ 148.0, 150.8.
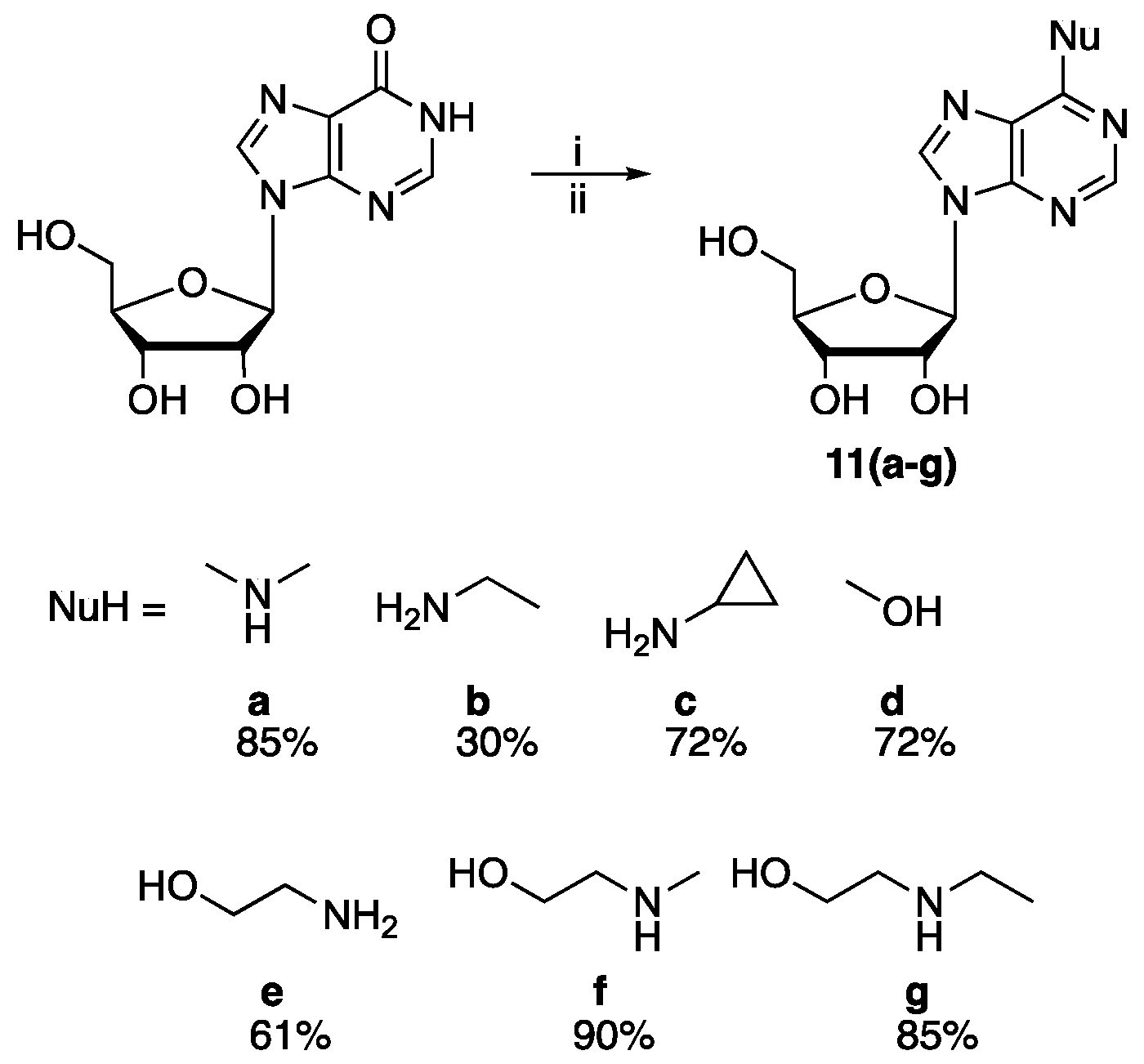
N6,N6-Dimethyladenosine (11a). The desired product was prepared according to General Procedure A; white solid (250 mg, 85%). 1H NMR (600 MHz, D2O) δ 3.06 (s, 6H), 3.75 (dd, J = 13.0, 3.5 Hz, 1H), 3.85 (dd, J = 13.0, 2.5 Hz, 1H), 4.19 (q, J = 3.0 Hz, 1H), 4.29–4.33 (t, J = 4.5 Hz, 1H), 4.59 (t, J = 5.5 Hz, 1H), 5.82 (d, J = 6.0 Hz, 1H), 7.78 (b.s, 1H), 8.00 (s, 1H); 13C-NMR (151 MHz, D2O) δ 38.7, 61.4, 70.5, 73.7, 85.5, 88.2, 118.9, 138.2, 148.2, 151.3, 153.6; HRMS (ESI) m/z: calculated for C12H18O4N5 [M + H]+ 296.1353, observed: 296.1352.
N6-Ethyladenosine (11b). The desired product was prepared according to General Procedure A; Ethylamine was prepared in-situ. To a stirred solution of ethylamine hydrochloride (1.1 g, 13.7 mmol) in 10 mL of ethanol in 50 mL round bottom flask, Ag2O (3.8 g, 16.4 mmol) was added and the mixture was stirred at room temperature under N2 for 1 h. The precipitate was collected by suction filtration; the resultant solution was then added to the stirred mixture of inosine, BOP and DBU. Yellowish solid (90 mg, 30%). 1H NMR (600 MHz, D2O + DMSO-d6) δ 2.63 (t, J = 2.0 Hz, 3H), 3.53 (b.s, 2H), 3.77 (dd, J = 13.0, 3.5 Hz, 1H), 3.85 (dd, J = 13.0, 3.0 Hz, 1H), 4.23 (q, J = 3.0 Hz, 1H), 4.35 (dd, J = 5.0, 3.0 Hz, 1H), 5.98 (d, J = 6.5 Hz, 1H), 8.18 (s, 1H), 8.24 (s, 1H), (1H under solvent peak); 13C-NMR (151 MHz, D2O + DMSO-d6) δ 13.9, 61.6, 70.8, 73.7, 86.0, 88.3, 117.6, 130.0, 140.1, 152.7, 154.6, (1C under solvent peak); HRMS (ESI) m/z: calculated for C12H18O4N5 [M + H]+ 296.1308, observed: 296.1353.
N6-Cyclopropyladenosine (11c). The desired product was prepared according to General Procedure A; white solid (222 mg, 72%). 1H-NMR (700 MHz, DMSO-d6 + D2O) δ 0.62–0.71 (m, 2H), 0.88 (dd, J = 7.0, 2.0 Hz, 2H), 3.00 (bs, 1H), 3.67 (dd, J = 12.0, 3.5 Hz, 1H), 3.77 (dd, J = 12.0, 3.5 Hz, 1H), 4.21–4.28 (m, 1H), 4.67 (t, J = 5.5 Hz, 1H), 5.97 (d, J = 6.0 Hz, 1H), 8.32 (s, 1H), 8.41 (s, 1H), (1 proton under solvent); 13C NMR (176 MHz, DMSO-d6 + D2O) δ 7.0, 61.9, 70.8, 70.9, 73.9, 86.2, 86.3, 88.4, 119.8, 140.3, 140.4, 152.7, 155.9; HRMS (ESI) m/z: calculated for C13H18O4N5 [M + H]+ 308.1353, observed: 308.1351.
O6-Methylinosine (11d). DBU (1.5 mmol) was added dropwise to a stirred solution of inosine (1 mmol), BOP (1.2 mmol) in THF; the mixture was heated at 40 °C. After the consumption of starting material (40 min, as assessed by TLC), the reaction mixture was concentrated under reduced pressure and an excess of MeOH was added to the flask and the reaction was stirred at 40 °C overnight. The crude product mixture was concentrated under reduced pressure and diluted with ethyl acetate, then washed with water (3 × 10 mL). The organic layer was dried (MgSO4) and concentrated under vacuum. The crude mixture was purified (99:1 to 9:1 ethyl acetate/methanol) by column chromatography which resulted in a white solid (0.2 g, 72%). TLC Rf 0.3 (9:1 CH2Cl2/MeOH); 1H-NMR (600 MHz, DMSO-d6) δ 3.58 (ddd, J = 12.0, 6.0, 4.0 Hz, 1H), 3.69 (dt, J = 12.0, 4.5 Hz, 1H), 3.98 (q, J = 4.0 Hz, 1H), 4.11 (s, 3H), 4.17 (q, J = 4.5 Hz, 1H), 4.60 (q, J = 5.5 Hz, 1H), 5.13 (t, J = 5.5 Hz, 1H), 5.22 (d, J = 5.0 Hz, 1H), 5.50 (d, J = 6.0 Hz, 1H), 6.00 (d, J = 5.5 Hz, 1H), 8.57 (s, 1H), 8.63 (s, 1H); 13C-NMR (151 MHz, DMSO-d6) δ 54.5, 61.8, 70.8, 74.2, 86.2, 88.2, 121.6, 142.9, 152.2, 152.24, 160.9; calculated for C11H13O5N4 [M + H]+ 283.0934, observed: 283.0932.
N6-(2-Hydroxyethyl)adenosine (11e). The desired product was prepared according to General Procedure A; white solid (190 mg, 61%). 1H-NMR (600 MHz, DMSO-d6) δ 3.57 (dd, J = 12.0, 4.0 Hz, 1H), 3.67 (dd, J = 12.0, 3.5 Hz, 1H), 4.13–4.21 (m, 1H), 4.51–4.64 (m, 3H), 5.96 (d, J = 6.0 Hz, 1H), 8.49 (s, 1H), 8.52 (s, 1H) (4 protons under the residual water peak); 13C NMR (151 MHz, DMSO-d6) δ 61.6, 63.4, 70.7, 74.0, 86.1, 88.2, 121.5, 142.7, 152.0, 152.2, 160.6; HRMS (ESI) m/z: calculated for C12H18O5N5 [M + H]+312.1302, observed: 312.1297.
N6,N6-Methyl(2-hydroxyethyl)adenosine (11f). The desired product was prepared according to General Procedure A; white solid (293 mg, 90%). 1H-NMR (700 MHz, DMSO-d6) δ 3.56 (ddd, J = 12.0, 7.0, 3.5 Hz, 1H), 3.61–4.43 (m, 6H), 4.59 (q, J = 6.0 Hz, 1H), 4.75 (t, J = 5.5 Hz, 1H), 5.18 (d, J = 5.0 Hz, 1H), 5.37 (dd, J = 7.0, 4.5 Hz, 1H), 5.45 (d, J = 6.0 Hz, 1H), 5.91 (d, J = 6.0 Hz, 1H), 8.22 (s, 1H), 8.37 (s, 1H) (3 methyl protons and one hydroxyl group under the residual water peak in DMSO); 13C NMR (176 MHz, DMSO-d6) δ 37.3, 52.7, 60.0, 62.0, 71.0, 73.9, 86.2, 88.3, 120.1, 139.2, 150.4, 152.2, 154.6; HRMS (ESI) m/z: calculated for C13H20O5N5 [M + H]+ 326.1459, observed: 326.1459.
N6,N6-Ethyl(2-hydroxyethyl)adenosine (11g). The desired product was prepared according to General Procedure A; white solid (275 mg, 85%). 1H-NMR (700 MHz, D2O) δ 1.18 (t, J = 7.0 Hz, 3H), 2.53 (d, J = 9 Hz, 1H)3.75 (dd, J = 13.0, 3.5 Hz, 1H), 3.78–4.09 (m, 7H), 4.21 (q, J = 3.5 Hz, 1H), 4.34 (dd, J = 5.0, 3.5 Hz, 1H), 5.98 (d, J = 6.0 Hz, 1H), 8.13 (s, 1H), 8.17 (s, 1H) 13C-NMR (176 MHz, D2O) δ 15.4, 44.6, 59.6, 61.1, 61.5, 70.6, 73.5, 85.8, 88.1, 119.3, 138.7, 149.2, 152.0, 154.1; HRMS (ESI) m/z: calculated for C14H22O5N5 [M + H]+ 340.1617, observed: 340.1615.
2′,3′,5′-O-Tris(tert-butyldimethyl)silylinosine (12). To a stirred solution of inosine (3.75 g, 13.24 mmol) and imidazole (3.6 g, 53.0 mmol) in anhydrous DMF in a 50 mL round bottom flask, TBDMSCl (6.6 g, 43.7 mmol) was added portionwise. The reaction was heated at 60 °C for 12 h. The suspension was cooled to room temperature, water was added and the precipitate was collected by suction filtration. The filtrate was discarded, and the white precipitate was washed with cold methanol. The methanol layer was evaporated under vacuum; the product was crystallised as a white solid from CH2C12 (7.8 g, 94%). TLC Rf 0.6 (1:9 MeOH/CH2Cl2); 1H-NMR (400 MHz, CDCl3) δ −0.31 (s, 3H), −0.16 (s, 3H), −0.04 (s, 3H), −0.03 (s, 3H), 0.00 (s, 3H), 0.01 (s, 3H), 0.68 (s, 9H), 0.79 (s, 9H), 0.82 (s, 9H), 3.66 (dd, J = 11.5, 2.5 Hz, 1H), 3.86 (dd, J = 11.5, 4.0 Hz, 1H), 4.00 (q, J = 3.5 Hz, 1H), 4.17 (t, J = 4.0 Hz, 1H), 4.38 (t, J = 4.5 Hz, 1H), 5.88 (d, J = 5.0 Hz, 1H), 7.97 (s, 1H), 8.09 (s, 1H), 12.83 (s, 1H); 13C NMR (101 MHz, CDCl3) δ −5.4, −5.0, −4.70, −4.66, −4.4, 17.9, 18.1, 18.6, 25.7, 25.8, 26.1, 62.4, 71.8, 85.5, 88.3, 125.0, 139.1, 144.5, 148.9. 159.2; HRMS (ESI) m/z: calculated for C28H55O5N428Si3 [M + H]+ 611.3474, observed: 611.3468.
2′,3′,5′-O-Tris[dimethyl(tert-butyl)silyl]-N6-(2-hydroxyethyl)adenosine (13a). To a stirred solution of 2′,3′,5′-O-tris(tert-butyldimethyl)silylinosine (12; 0.1 g, 0.16 mmol) and PyBOP (0.1 g, 0.2 mmol) in 10 mL of THF in a 50 mL round bottom flask, DIPEA (42 μL, 0.24 mmol) was added dropwise and the mixture was heated at 40 °C. After the consumption of the starting material (40 min, as assessed by TLC), the reaction was cooled to room temperature and ethanolamine (0.2 mL, 0.35 mmol) was added dropwise; the reaction was then stirred overnight. The crude product mixture was concentrated under reduced pressure and then diluted with ethyl acetate and was washed with water (3 × 10 mL). The organic layer was dried (MgSO4) and concentrated under reduced pressure. The residue was purified by column chromatography (99:1 to 94:6 CH2Cl2/MeOH) which resulted in oil (95 mg, 90%). 1H NMR (500 MHz, CD3OD) δ −0.28 (s, 3H), −0.02 (s, 3H), 0.17 (s, 6H), 0.18 (s, 6H), 0.79 (s, 9H), 0.98 (s, 9H), 0.99 (s, 9H), 3.71–3.84 (m, 4H), 3.85 (dd, J = 11.5, 3.0 Hz, 1H), 4.06 (dd, J = 11.0, 4.5 Hz, 1H), 4.15 (dt, J = 5.0, 3.0 Hz, 1H), 4.40 (dd, J = 4.5, 2.5 Hz, 1H), 4.83 (dd, J = 6.0, 4.5 Hz, 1H), 4.89 (s, 5H), 6.07 (d, J = 6.0 Hz, 1H), 8.26 (s, 1H), 8.31 (s, 1H), (-OH peak under solvent peak); 13C-NMR (126 MHz, CD3OD) δ −6.6, −6.5, −6.3, −5.6, −5.6, −5.5, 17.4, 17.6, 18.0, 24.9, 25.1, 25.2, 45.97, 46.0, 60.4, 62.7, 72.7, 76.0, 86.2, 87.7, 119.5, 139.3, 148.6, 152.5, 155.0; HRMS (ESI) m/z: calculated for C30H60O5N528Si3 [M + H]+ 635.3713, observed: 635.3797.
2′,3′,5′-O-Tris[dimethyl(tert-butyl)silyl]-N6,N6-methyl(2-hydroxyethyl)adenosine (13b). To a stirred solution of 2′,3′,5′-O-tris(tert-butyldimethyl)silylinosine (12; 1 g, 1.64 mmol) and BOP (0.9 g, 1.96 mmol) in 25 mL of EtOH in a 50 mL round bottom flask, DBU (0.3 mL, 1.97 mmol) was added dropwise; the mixture was heated at 40 °C. After the consumption of the starting material (40 min, TLC), the reaction was cooled to room temperature and methylethanolamine (0.65 mL, 8.2 mmol) was added dropwise; the reaction was then stirred overnight. The crude product mixture was concentrated under reduced pressure, then diluted with ethyl acetate and was washed with water (3 × 10 mL). The organic layer was dried (anhydrous MgSO4) and concentrated under reduced pressure. The residue was purified by column chromatography (99:1 to 94:6 CH2Cl2/MeOH) which resulted in oil (0.82 g, 80%). TLC Rf 0.6 (6:94 MeOH/CH2Cl2). 1H-NMR (600 MHz, CDCl3) δ −0.29 (s, 3H), −0.14 (s, 3H), −0.01 (s, 3H), 0.00 (s, 3H), 0.02 (s, 3H), 0.03 (s, 3H), 0.71 (s, 9H), 0.83 (s, 9H), 0.85 (s, 9H), 3.42 (s, 3H), 3.67 (dd, J = 11.5, 3.0 Hz, 1H), 3.87 (t, J = 5.0 Hz, 2H), 3.92 (dd, J = 11.4, 4.0 Hz, 1H), 3.94–4.09 (m, 4H), 4.21 (t, J = 4.0 Hz, 1H), 4.58 (t, J = 4.5 Hz, 1H), 5.92 (d, J = 5.0 Hz, 1H), 7.97 (s, 1H), 8.20 (s, 1H); 13C-NMR (151 MHz, CDCl3) δ −5.38, −5.36, −5.0, −4.72, −4.70, −4.4, 17.9, 18.1, 18.5, 25.7, 25.9, 26.1, 53.8, 61.6, 62.5, 71.9, 75.6, 85.3, 88.3, 120.4, 137.7, 150.5, 152.2, 155.6; HRMS (ESI) m/z: calculated for C31H62O5N528Si3 [M + H]+ 668.4053, observed: 668.4042.
2′,3′,5′-O-Tris[dimethyl(tert-butyl)silyl]-N6,N6-ethyl(2-hydroxyethyl)adenosine (13c). To a stirred solution of 2′,3′,5′-O-tris(tert-butyldimethyl)silylinosine (12; 1 g, 1.64 mmol) and BOP (0.87 g, 1.96 mmol) in 25 mL of EtOH in a 50 mL round bottom flask, DBU (0.3 mL, 2.0 mmol) was added dropwise and the mixture was heated at 40 °C. After the consumption of the starting material (40 min, as assessed by TLC), the reaction was cooled to room temperature and ethylethanolamine (0.65 mL, 8.2 mmol) was added dropwise; the reaction was then stirred overnight. The crude product mixture was concentrated under reduced pressure, then diluted with ethyl acetate and washed with water (3 × 10 mL). The organic layer was dried (anhydrous MgSO4) and concentrated under reduced pressure. The residue was purified by column chromatography (99:1 to 94:6 CH2Cl2/MeOH) which resulted in an oil (960 mg, 86%). TLC Rf 0.5 (6:94 MeOH/CH2Cl2); 1H NMR (600 MHz, CDCl3) δ −0.30 (s, 3H), −0.15 (s, 3H), −0.02 (s, 3H), −0.01 (s, 3H), 0.00 (s, 3H), 0.01 (s, 3H), 0.69 (s, 9H), 0.82 (s, 9H), 0.83 (s, 9H), 1.18 (t, J = 7.0 Hz, 3H), 3.66 (dd, J = 11.0, 3.0 Hz, 1H), 3.72–4.15 (m, 8H), 4.21 (t, J = 4.0 Hz, 1H), 4.59 (t, J = 5.0 Hz, 1H), 4.67–5.17 (m, 1H), 5.89 (d, J = 5.0 Hz, 1H), 8.05 (s, 1H), 8.16 (s, 1H); 13C NMR (151 MHz, CDCl3) δ −5.39, −5.38, −5.0, −4.72, −4.70, −4.4, 13.3, 17.9, 18.1, 18.5, 25.7, 25.8, 26.0, 44.6, 51.6, 62.5, 62.6, 71.9, 75.5, 85.2, 88.3, 120.1, 137.8, 150.5, 152.1, 155.0. HRMS (ESI) m/z: calculated for C32H64O5N528Si3 [M + H]+ 682.4209, observed: 682.4201.
2′,3′,5′-O-Tris[dimethyl(tert-butyl)silyl]-N1,N6-ethanoadenosine (15a). To a stirred solution of 2′,3′,5′-O-tris[dimethyl(tert-butyl)silyl]-N1,N6-(2-hydroxyethyl)adenosine (13a; 1.3 g, 2 mmol) and Et3N (1.4 mL, 10 mmol) in 30 mL of anhydrous DMF in a 50 mL round bottom flask, methyltriphenoxyphosphonium iodide (1 g, 2.4 mmol) was added and the mixture was stirred at room temperature for 1 h. Anhydrous methanol was added and the crude product mixture was concentrated under reduced pressure, then diluted with ethyl acetate and washed with NaHCO3 and water (3 × 10 mL). The organic layer was dried (MgSO4) and concentrated under reduced pressure. The residue was purified by column chromatography (99:1 to 85:15 CH2Cl2/MeOH) resulting in an oil (0.75 g, 60%). 1H-NMR (600 MHz, CDCl3) δ −0.18 (s, 3H), −0.07 (s, 3H), 0.00 (s, 3H), 0.01 (s, 3H), 0.05 (s, 3H), 0.06 (s, 3H), 0.75 (s, 9H), 0.83 (s, 9H), 0.87 (s, 9H), 3.71 (dd, J = 11.5, 2.5 Hz, 1H), 3.93 (dd, J = 11.5, 3.0 Hz, 1H), 4.06 (dt, J = 5.5, 3.0 Hz, 1H), 4.18 (t, J = 4.5 Hz, 1H), 4.26–4.40 (m, 3H), 4.90–4.95 (m, 2H), 5.92 (d, J = 4.0 Hz, 1H), 8.38 (s, 1H), 8.51 (s, 1H); 13C NMR (151 MHz, CDCl3) δ −5.3, −5.27, −4.8, −4.6, −4.4, −4.2, 17.9, 18.0, 18.5, 25.7, 25.8, 26.1, 26.4, 26.5, 46.28, 46.33, 46.4, 49.1, 62.0, 71.1, 76.7, 85.2, 89.0, 117.6, 142.3, 143.4, 149.0, 151.3; HRMS (ESI) m/z: calculated for C30H58O4N528Si3 [M + H]+ 636.3767, observed: 636.3791.
N1,N6-Ethanoadenosine (16a). To a stirred solution of 2′,3′,5′-O-tris[dimethyl(tert-butyl)silyl]-N6-ethanoadenosine (15a, 470 mg, 0.74 mmol) in CH2Cl2, 3HF.Et3N (361 µL, 2.2 mmol) was added dropwise. The solution was left to stir for 48 h. The mixture was reduced under pressure and was purified by column chromatography (99:1 to 9:1 ethyl acetate/methanol) which resulted in a white solid (150 mg, 70%). 1H NMR (700 MHz, DMSO-d6) δ 3.49–3.57 (m, 2H), 3.60–3.68 (m, 1H), 3.86–3.97 (m, 3H), 4.08–4.16 (m, 3H), 4.44 (t, J = 5.0 Hz, 1H), 5.11–5.21 (m, 2H), 5.46 (s, 1H), 5.78 (d, J = 6.0 Hz, 1H), 8.09 (s, 1H), 8.17 (s, 1H); 13C-NMR (176 MHz, DMSO-d6) δ 46.5, 53.3, 61.9, 70.8, 74.5, 86.1, 88.0, 120.0, 138.5, 145.2, 145.4, 150.2; HRMS (ESI) m/z: calculated for C12H16O4N5 [M + H]+ 294.1197, observed: 294.1191.
2′,3′,5′-O-Tris[dimethyl(tert-butyl)silyl]-N6,N1,N6-methylethanoadenosine (15b). To a stirred solution of 2′,3′,5′-O-tris[dimethyl(tert-butyl)silyl]-N6,N6-methyl(2-hydroxyethyl)adenosine (13b; 0.5 g, 0.75 mmol) and Et3N (0.54 mL, 3.75 mmol) in 30 mL of anhydrous DMF in a 50 mL round bottom flask, methyltriphenoxyphosphonium iodide (0.85 g, 1.9 mmol) was added and the mixture was stirred at room temperature for 1 h. Anhydrous methanol was added and the crude product mixture was concentrated under reduced pressure, then diluted with ethyl acetate and washed with NaHCO3 and water (3 × 10 mL). The organic layer was dried (anhydrous MgSO4), then concentrated under reduced pressure. The residue was purified by alumina column chromatography (99:1 to 90:10 CH3Cl/MeOH) which resulted in an oil. 1H NMR (600 MHz, CDCl3) δ −0.20 (s, 3H), −0.12 (s, 3H), −0.08 (s, 3H), −0.07 (s, 3H), 0.00 (s, 3H), 0.01 (s, 3H), 0.70 (s, 9H), 0.75 (s, 9H), 0.81 (s, 9H), 3.57 (s, 3H), 3.65 (dd, J = 11.5, 2.0 Hz, 1H), 3.88 (dd, J = 11.5, 3.0 Hz, 1H), 4.00 (dt, J = 5.0, 2.5 Hz, 1H), 4.12 (dd, J = 5.5, 4.0 Hz, 1H), 4.17–4.26 (m, 2H), 4.35–4.42 (m, 1H), 5.05–5.3 (m, 2H), 5.87 (d, J = 3.5 Hz, 1H), 8.43 (s, 1H), 8.50 (s, 1H); 13C NMR (151 MHz, CDCl3) δ −5.4, −5.2, −4.8, −4.6, −4.5, −4.2, 17.9, 18.1, 18.6, 25.7, 25.8, 26.2, 34.6, 45.9, 49.1, 51.6, 61.7, 70.7, 85.0, 89.3, 115.4, 117.0, 129.5, 142.8, 143.7, 149.9, 150.1; HRMS (ESI) m/z: calculated for C31H60O4N528Si3 [M] 650.3947, observed: 650.3924.
2′,3′,5′-O-Tris[dimethyl(tert-butyl)silyl]-N6,N1,N6-ethylethanoadenosine (15c). To a stirred solution of 2′,3′,5′-O-tris[dimethyl(tert-butyl)silyl]-N6,N6-ethyl(2-hydroxyethyl)adenosine (15c; 0.4 g, 0.6 mmol) and Et3N ( 0.4 mL, 3 mmol) in 20 mL of anhydrous DMF in a 50 mL round bottom flask, methyltriphenoxyphosphonium iodide (0.6 g, 1.2 mmol) was added; the mixture was stirred at room temperature for 1 h. Anhydrous methanol was added and the crude product mixture was concentrated under reduced pressure and then diluted with ethyl acetate and was washed subsequently with NaHCO3 and water (3 × 10 mL). The organic layer was dried (anhydrous MgSO4) and concentrated under reduced pressure. The residue was purified by alumina column chromatography (99:1 to 95:5 CH2Cl2/MeOH) which resulted in a solid (0.26 g, 36%). mp 195 °C; 1H-NMR (600 MHz, CDCl3) δ −0.25 (s, 3H), −0.18 (s, 3H), −0.03 (s, 3H), −0.01 (s, 3H), 0.00 (s, 3H), 0.01 (s, 3H), 0.70 (s, 9H), 0.74 (s, 9H), 0.83 (s, 9H), 1.33 (t, J = 7.0 Hz, 3H), 3.2 (q, J = 7.0 Hz, 2H), 3.65 (dd, J = 12.0, 2.0 Hz, 1H), 3.88 (dd, J = 12.0, 3.0 Hz, 1H), 4.00 (dt, J = 5.0, 2.5 Hz, 1H), 4.23–4.28 (m, 2H), 4.59 (t, J = 5.0 Hz, 1H), 4.69 (t, J = 5.0 Hz, 1H) 5.0–5.18 (m, 2H), 5.83 (d, J = 5.0 Hz, 1H), 8.34 (s, 1H), 8.46 (s, 1H); 13C-NMR (151 MHz, CDCl3) δ −5.3, −5.0, −5.0, −4.6, −4.6, −4.2, 13.0, 17.9, 18.1, 18.5, 25.7, 25.8, 26.2, 35.6, 48.4, 51.6, 61.7, 70.7, 73.4, 85.0, 89.3, 115.4, 144.8, 149.9, 150.0, 152.0; HRMS (ESI) m/z: calculated for C31H60O4N528Si3 [M] 664.3886, observed: 664.3896.
N6,N1,N6-Methylethanoadenosine (16b). To a stirred solution of 2′,3′,5′-O-tris[dimethyl(tert-butyl)silyl]-N6,N6-methylethanoadenosine (15b; 500 mg, 0.8 mmol) in CH2Cl2, 3HF.Et3N (0.4 mL, 2.3 mmol) was added dropwise. The solution was stirred for 48 h. The mixture was reduced under pressure, then purified by alumina column chromatography (99:1 to 9:1 ethyl acetate/methanol) which resulted in a white solid (54 mg, 22%). mp 202.5 °C; 1H-NMR (600 MHz, DMSO-d6) δ 3.01 (s, 3H), 3.29 (m, 1H), 3.38 (dt, J = 12.5, 4.5 Hz, 1H), 3.68 (q, J = 4.0 Hz, 1H), 3.76–3.84 (m, 2H), 3.87 (b.s, 1H), 4.18 (q, J = 5.0 Hz, 1H), 4.43 (t, J = 9.5 Hz, 1H), 4.79 (t, J = 5.5 Hz, 1H), 5.01 (s, 1H), 5.28–5.37 (m, 2H), 5.68 (d, J = 5.0 Hz, 1H), 8.50 (s, 1H), 8.52 (s, 1H); 13C-NMR (151 MHz, D2O) δ 36.4, 50.5, 53.4, 63.6, 72.6, 77.1, 88.6, 90.8, 119.0, 145.7, 148.0, 152.2, 152.3; HRMS (ESI) m/z: calculated for C13H18O4N5 [M]+ 308.1353, observed: 308.1353.
N6,N1,N6-Ethylethanoadenosine (16c). To a stirred solution of 2′,3′,5′-O-tris[dimethyl(tert-butyl)silyl]-N6,N6-methylethanoadenosine (15c; 470 mg, 0.74 mmol) in CH2Cl2, 3HF.Et3N (360 µL, 2.2 mmol)was added dropwise. The solution was left to stir for 48 h. The mixture was reduced under pressure and was purified by alumina column chromatography (99:1 to 9:1 ethyl acetate/methanol) which resulted in a white solid (65 mg, 30%). mp 210 °C; 1H-NMR (600 MHz, DMSO-d6) δ 1.15 (t, J = 7.0 Hz, 3H), 2.88 (q, J = 7.0 Hz, 2H), δ 3.49–3.57 (m, 2H), 3.36 (dt, J = 12.5 Hz, 4.0 Hz, 1H), 3.54 (q, J = 4.0 Hz, 1H), 3.76–3.84 (m, 2H), 3.95 (b.s, 1H), 4.15–4.27 (m, 1H), 4.34–4.43 (m, 1H), 5.1 (b.s, 1H), 5.43–5.57 (m, 2H), 6.0 (d, J = 5.0 Hz, 1H), 8.48 (s, 1H), 8.63 (s, 1H); 13C-NMR (151 MHz, DMSO-d6) δ 13.0, 32.4, 48.3, 52.3, 62.4, 70.4, 75.3, 85.4, 90.8, 119.0, 146.4, 148.2, 151.9, 152.0; HRMS (ESI) m/z: calculated for C13H18O4N5 [M]+ 322.1518, observed: 322.1515.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






