
Heterotypic Supramolecular Hydrogels Formed by Noncovalent Interactions in Inflammasomes



Abstract
:1. Introduction
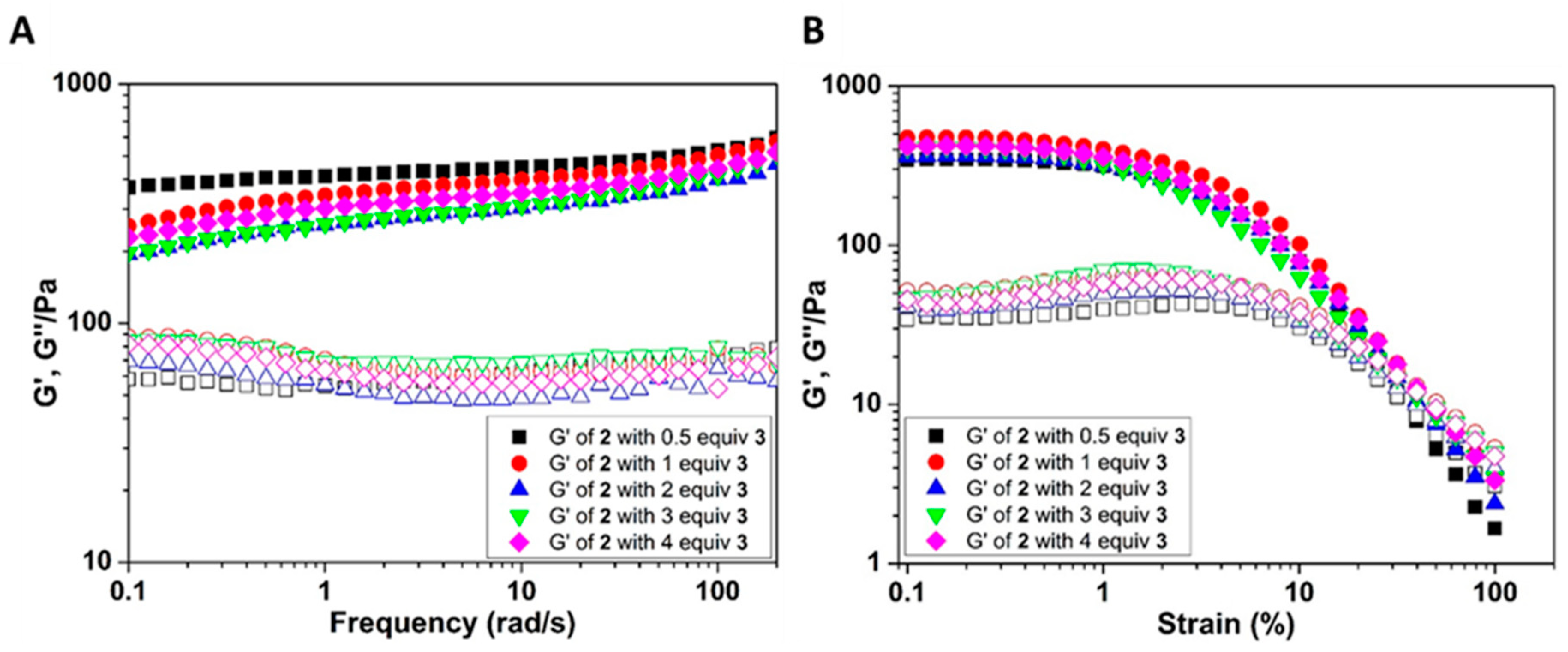
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.1.1. Procedure for Hydrogel Preparation
3.1.2. Circular Dichroism Measurement
3.1.3. Rheology Experiment
3.1.4. TEM Sample Preparation
3.2. Instruments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Yuan, C.; Ji, W.; Xing, R.; Li, J.; Gazit, E.; Yan, X. Hierarchically oriented organization in supramolecular peptide crystals. Nat. Rev. Chem. 2019, 3, 567–588. [Google Scholar]
- Edwards-Gayle, C.J.C.; Hamley, I.W. Self-assembly of bioactive peptides, peptide conjugates, and peptide mimetic materials. Org. Biomol. Chem. 2017, 15, 5867–5876. [Google Scholar]
- Levin, A.; Hakala, T.A.; Schnaider, L.; Bernardes, G.J.L.; Gazit, E.; Knowles, T.P.J. Biomimetic peptide self-assembly for functional materials. Nat. Rev. Chem. 2020, 4, 615–634. [Google Scholar]
- Ulijn, R.V.; Lampel, A. Order/Disorder in Protein and Peptide-Based Biomaterials. Isr. J. Chem. 2019, 60, 1129–1140. [Google Scholar]
- Beesley, J.L.; Woolfson, D.N. The de novo design of α-helical peptides for supramolecular self-assembly. Curr. Opin. Biotechnol. 2019, 58, 175–182. [Google Scholar]
- Kim, B.J.; Yang, D.; Xu, B. Emerging Applications of Supramolecular Peptide Assemblies. Trends Chem. 2020, 2, 71–83. [Google Scholar]
- Ji, W.; Zhang, S.; Yukawa, S.; Onomura, S.; Sasaki, T.; Miyazawa, K.; Zhang, Y. Regulating Higher-Order Organization through the Synergy of Two Self-Sorted Assemblies. Angew. Chem. Int. Ed. 2018, 57, 3636–3640. [Google Scholar] [CrossRef]
- Estroff, L.A.; Hamilton, A.D. Water gelation by small organic molecules. Chem. Rev. 2004, 104, 1201–1217. [Google Scholar] [PubMed]
- Du, X.; Zhou, J.; Shi, J.; Xu, B. Supramolecular Hydrogelators and Hydrogels: From Soft Matter to Molecular Biomaterials. Chem. Rev. 2015, 115, 13165–13307. [Google Scholar]
- Shy, A.N.; Kim, B.J.; Xu, B. Enzymatic Noncovalent Synthesis of Supramolecular Soft Matter for Biomedical Applications. Matter 2019, 1, 1127–1147. [Google Scholar]
- Chen, C.H.; Hsu, E.L.; Stupp, S.I. Supramolecular self-assembling peptides to deliver bone morphogenetic proteins for skeletal regeneration. Bone 2020, 141, 115565. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, X.; Horii, A.; Wang, X.; Qiao, L.; Zhang, S.; Cui, F.-Z. In vivo studies on angiogenic activity of two designer self-assembling peptide scaffold hydrogels in the chicken embryo chorioallantoic membrane. Nanoscale 2012, 4, 2720–2727. [Google Scholar] [CrossRef] [PubMed]
- Jayawarna, V.; Richardson, S.M.; Hirst, A.R.; Hodson, N.W.; Saiani, A.; Gough, J.E.; Ulijn, R.V. Introducing chemical functionality in Fmoc-peptide gels for cell culture. Acta Biomater. 2009, 5, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Lutolf, M.P.; Hubbell, J.A. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat. Biotechnol. 2005, 23, 47–55. [Google Scholar] [CrossRef]
- Kopeček, J. Hydrogel biomaterials: A smart future? Biomaterials 2007, 28, 5185–5192. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Xing, R.; Bai, S.; Yan, X. Recent advances of self-assembling peptide-based hydrogels for biomedical applications. Soft Matter 2019, 15, 1704–1715. [Google Scholar] [CrossRef]
- Huettner, N.; Dargaville, T.R.; Forget, A. Discovering Cell-Adhesion Peptides in Tissue Engineering: Beyond RGD. Trends Biotechnol. 2018, 36, 372–383. [Google Scholar] [CrossRef]
- Gao, Y.; Kuang, Y.; Guo, Z.-F.; Guo, Z.; Krauss, I.J.; Xu, B. Enzyme-Instructed Molecular Self-assembly Confers Nanofibers and a Supramolecular Hydrogel of Taxol Derivative. J. Am. Chem. Soc. 2009, 131, 13576–13577. [Google Scholar] [CrossRef]
- Du, X.; Zhou, J.; Wang, H.; Shi, J.; Kuang, Y.; Zeng, W.; Yang, Z.; Xu, B. In situ generated D-peptidic nanofibrils as multifaceted apoptotic inducers to target cancer cells. Cell Death Dis. 2017, 8, e2614. [Google Scholar] [CrossRef]
- He, H.; Wang, J.; Wang, H.; Zhou, N.; Yang, D.; Green, D.R.; Xu, B. Enzymatic Cleavage of Branched Peptides for Targeting Mitochondria. J. Am. Chem. Soc. 2018, 140, 1215–1218. [Google Scholar] [CrossRef]
- Feng, Z.; Wang, H.; Wang, S.; Zhang, Q.; Zhang, X.; Rodal, A.A.; Xu, B. Enzymatic Assemblies Disrupt the Membrane and Target Endoplasmic Reticulum for Selective Cancer Cell Death. J. Am. Chem. Soc. 2018, 140, 9566–9573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, L.; He, S.; Liu, Z.; Xu, K.; Zhong, W. Co-assembled supramolecular hydrogels of doxorubicin and indomethacin-derived peptide conjugates for synergistic inhibition of cancer cell growth. Chem. Commun. 2019, 55, 4411–4414. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Feng, Z.; Qin, Y.; Wang, J.; Xu, B. Nucleopeptide Assemblies Selectively Sequester ATP in Cancer Cells to Increase the Efficacy of Doxorubicin. Angew. Chem.-Int. Ed. 2018, 57, 4931–4935. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Feng, Z.; Wang, Y.; Zhou, R.; Yang, Z.; Xu, B. Integrating Enzymatic Self-Assembly and Mitochondria Targeting for Selectively Killing Cancer Cells without Acquired Drug Resistance. J. Am. Chem. Soc. 2016, 138, 16046–16055. [Google Scholar] [CrossRef]
- Feng, Z.; Wang, H.; Chen, X.; Xu, B. Self-Assembling Ability Determines the Activity of Enzyme-Instructed Self-Assembly for Inhibiting Cancer Cells. J. Am. Chem. Soc. 2017, 139, 15377–15384. [Google Scholar] [CrossRef]
- Lyu, L.; Liu, F.; Wang, X.; Hu, M.; Mu, J.; Cheong, H.; Liu, G.; Xing, B. Stimulus-Responsive Short Peptide Nanogels for Controlled Intracellular Drug Release and for Overcoming Tumor Resistance. Chem. Asian J. 2017, 12, 744–752. [Google Scholar] [CrossRef]
- Tang, W.; Zhao, Z.; Chong, Y.; Wu, C.; Liu, Q.; Yang, J.; Zhou, R.; Lian, Z.-X.; Liang, G. Tandem Enzymatic Self-Assembly and Slow Release of Dexamethasone Enhances Its Antihepatic Fibrosis Effect. ACS Nano 2018, 12, 9966–9973. [Google Scholar] [CrossRef]
- Dasgupta, A.; Mondal, J.H.; Das, D. Peptide hydrogels. RSC Adv. 2013, 3, 9117–9149. [Google Scholar] [CrossRef]
- Toledano, S.; Williams, R.J.; Jayawarna, V.; Ulijn, R.V. Enzyme-Triggered Self-Assembly of Peptide Hydrogels via Reversed Hydrolysis. J. Am. Chem. Soc. 2006, 128, 1070–1071. [Google Scholar] [CrossRef]
- Li, J.; Zhan, Z.; Du, X.; Wang, J.; Hong, B.; Xu, B. Selection of Secondary Structures of Heterotypic Supramolecular Peptide Assemblies by an Enzymatic Reaction. Angew. Chem.-Int. Ed. 2018, 57, 11716–11721. [Google Scholar] [CrossRef]
- King, P.J.S.; Lizio, M.G.; Booth, A.; Collins, R.F.; Gough, J.E.; Miller, A.F.; Webb, S.J. A modular self-assembly approach to functionalised β-sheet peptide hydrogel biomaterials. Soft Matter 2016, 12, 1915–1923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Xu, K.; Wang, L.; Gu, H.; Wei, H.; Zhang, M.; Xu, B. Self-assembly of small molecules affords multifunctional supramolecular hydrogels for topically treating simulated uranium wounds. Chem. Commun. 2005, 21, 4414–4416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kuang, Y.; Gao, Y.; Xu, B. Versatile Small-Molecule Motifs for Self-Assembly in Water and the Formation of Biofunctional Supramolecular Hydrogels. Langmuir 2011, 27, 529–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Du, X.; Hashim, S.; Shy, A.; Xu, B. Aromatic-Aromatic Interactions Enable α-Helix to beta-Sheet Transition of Peptides to Form Supramolecular Hydrogels. J. Am. Chem. Soc. 2017, 139, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Xu, B. Heterotypic Supramolecular Hydrogels. J. Mater. Chem. B 2016, 4, 5638–5649. [Google Scholar] [CrossRef] [Green Version]
- Wester, J.R.; Lewis, J.A.; Freeman, R.; Sai, H.; Palmer, L.C.; Henrich, S.E.; Stupp, S.I. Supramolecular Exchange among Assemblies of Opposite Charge Leads to Hierarchical Structures. J. Am. Chem. Soc. 2020, 142, 12216–12225. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nat. Cell Biol. 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Park, H.H.; Lo, Y.-C.; Lin, S.-C.; Wang, L.; Yang, J.K.; Wu, H. The Death Domain Superfamily in Intracellular Signaling of Apoptosis and Inflammation. Annu. Rev. Immunol. 2007, 25, 561–586. [Google Scholar] [CrossRef] [Green Version]
- Lu, A.; Magupalli Venkat, G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.; Schröder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified Polymerization Mechanism for the Assembly of ASC-Dependent Inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| 1 | sol | sol | sol | precipitate/gel a |
| 2 | sol | sol | gel | precipitate/sol |
| 3 | sol | gel | sol | sol |
| 4 | precipitate/gel a | precipitate/sol | sol | sol |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shy, A.N.; Wang, H.; Feng, Z.; Xu, B. Heterotypic Supramolecular Hydrogels Formed by Noncovalent Interactions in Inflammasomes. Molecules 2021, 26, 77. https://doi.org/10.3390/molecules26010077
Shy AN, Wang H, Feng Z, Xu B. Heterotypic Supramolecular Hydrogels Formed by Noncovalent Interactions in Inflammasomes. Molecules. 2021; 26(1):77. https://doi.org/10.3390/molecules26010077
Chicago/Turabian StyleShy, Adrianna N., Huaimin Wang, Zhaoqianqi Feng, and Bing Xu. 2021. "Heterotypic Supramolecular Hydrogels Formed by Noncovalent Interactions in Inflammasomes" Molecules 26, no. 1: 77. https://doi.org/10.3390/molecules26010077
APA StyleShy, A. N., Wang, H., Feng, Z., & Xu, B. (2021). Heterotypic Supramolecular Hydrogels Formed by Noncovalent Interactions in Inflammasomes. Molecules, 26(1), 77. https://doi.org/10.3390/molecules26010077