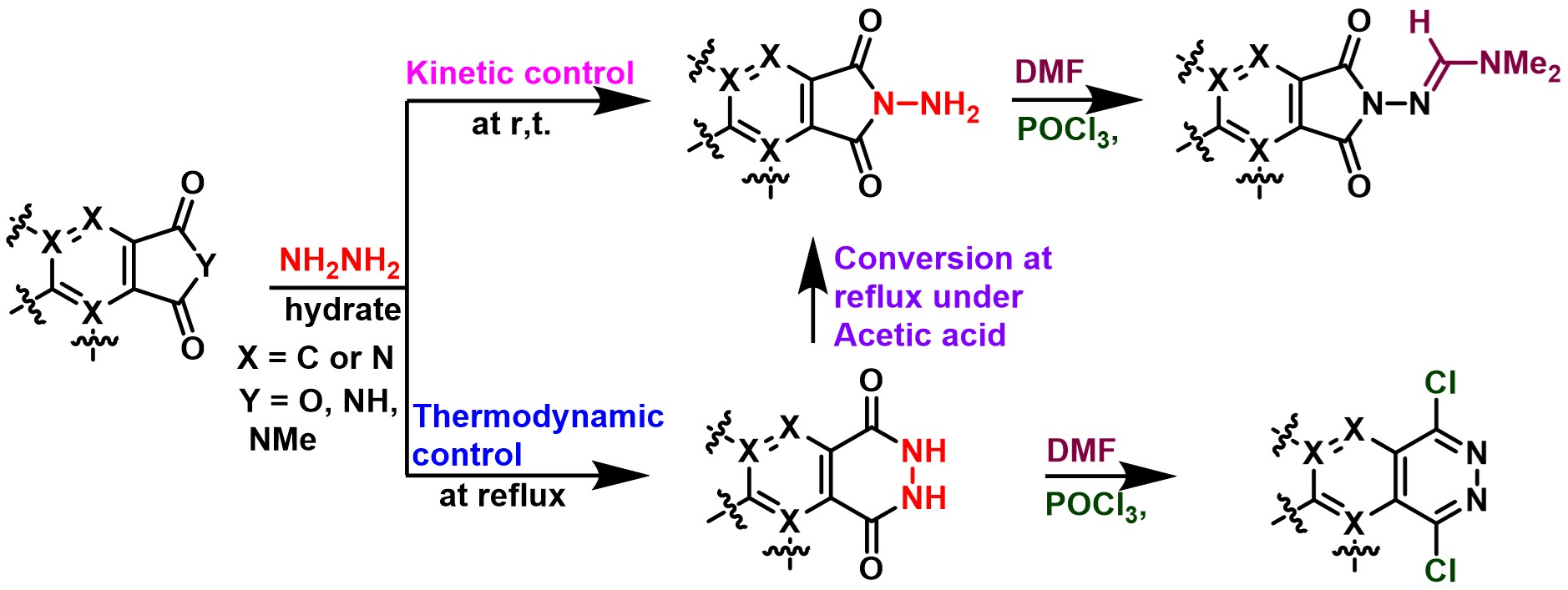
2. Results and Discussion
Initially, isobenzofuran-1,3-diones (
1a–
c), isoindoline-1,3-dione (
1d), furo [3,4-
b] pyrazine-5,7-dione (
1e), and 1
H-pyrrolo[3,4-
c] pyridine-1,3-dione (
1f) [
34,
35,
36] were purchased or prepared as the starting materials. Reacting compounds
1a–
f with monohydrate hydrazine in ethanol solution at low (at 0 °C or –20 °C) or room temperature led directly to the 5-exo cyclization
N-aminophthalimide products
2a–
f (81–94%) without the accumulation of hydrohydrazination products
3a–
f (
Table 1). On the other hand, the 6-exo cyclization of distal nitrogen instead of proximal one was exclusively observed at reflux for ~4 h, and the corresponding 6-endo phthalazine 1,4-dione products
3a–
f were formed (83–91%,
Table 1). Fortunately, compounds
2a–
f and
3a–
f can be selectively prepared via kinetic and thermodynamic control reaction with hydrazine hydrate. From the fundamental perspective, these symmetric molecules of
N-aminophthalimide products
2a–
f provide themselves for investigation of tautomeric conversion controlling processes as well as convenient platforms for the structural identity.
Additionally, phthalazine 1,4-diones
3a–
f were used as the reference standards. Although compounds
2 and
3 were tautomeric pairs, they significantly possessed the different polarity, such as, R
f = 0.33 for
N-aminophthalimide
2a and R
f = 0.41 for phthalazine 1,4-dione
3a (EA/MeOH = 9/1). All of the
2a–
f and
3a–
f compounds were fully characterized by spectroscopic methods. For example, compound
2a presented peaks at 3447 and 3482 cm
−1 for stretching of the –NH
2 group and at 1020 cm
−1 for stretching of the N–N group in FT-IR spectrum. For compound
3a, its IR absorption peaks were at 3461 cm
−1 for stretching of the –NH–NH– group and at 1051 cm
−1 for stretching of the N–N group. In
1H-NMR, compounds
2 and
3 presented similar chemical shift and coupling constants, resulting in difficult structural identification of each other. For example, the
1H-NMR spectra of compounds
2a and
3a were similar as shown in
Figure 1. This observation drove us to develop a novel identification method.
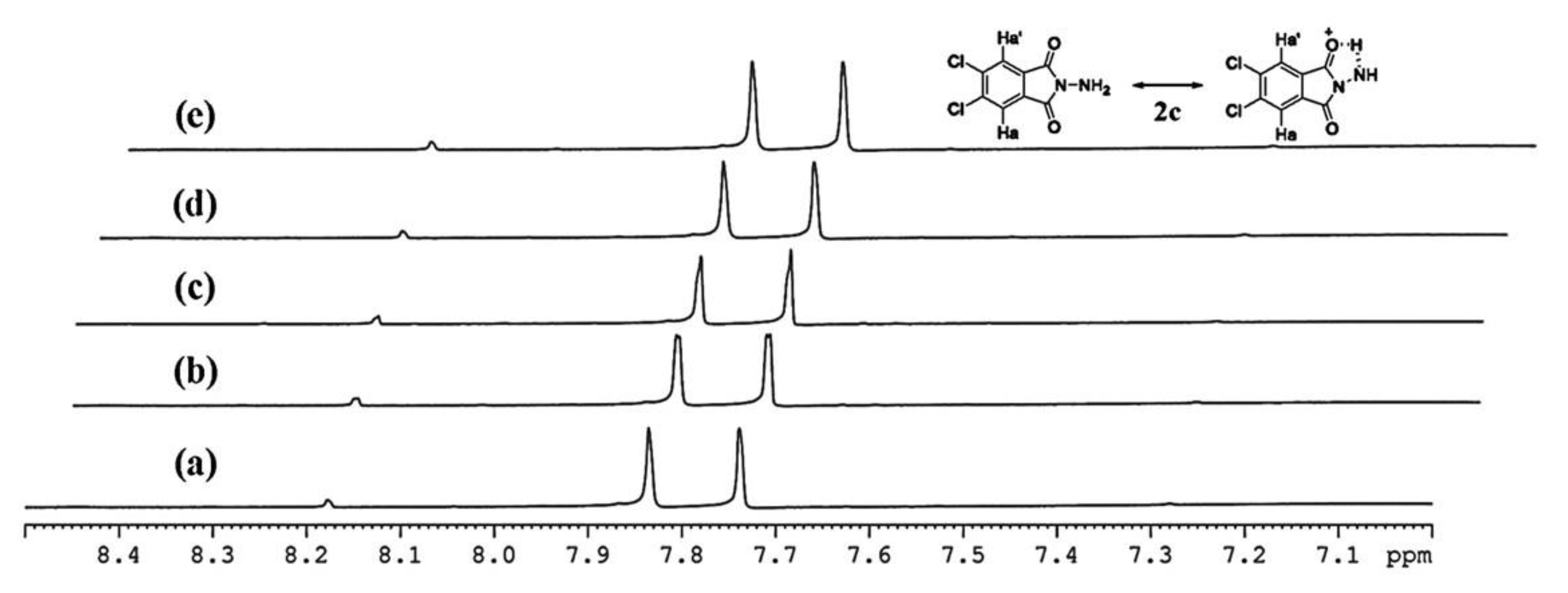
On the other hand,
N-aminophthalimides
2c and
2e reveal the existence of intramolecular hydrogen bonding phenomenon between carbonyl and amino group in
1H-NMR spectra. This phenomenon leads to the different chemical shift values between H
a and H
a’ in an aromatic ring (
Figure 1). However, compounds
3a–
f were favorable for the free base form in DMSO-
d6 solvent. For example, the
1H-NMR spectrum of compound
3e was presented in
Figure 1. Furthermore, Compound
2c was dissolved in DMSO-d6 and heated at ~100 °C by NMR technique. The sample was monitored in 0, 10, 20, 30, and 60 min, the timed programming result was shown in
Figure 2, we found that the intramolecular hydrogen bonding phenomenon was very clearly stable.
Vilsmeier amidination methodology was essentially examined for the applicable protected utilization of primary amines. The usual method was directly treating primary amines with dimethylformamide (DMF) and coupling agents including POCl
3, P
2O
5, PCl
5, (COCl)
2, PyBOP, SOCl
2, acyl chlorides, trifluoroacetic anhydride (TFAA), or sulfonyl chloride to give the corresponding amidine products [
37,
38,
39]. To further probe the structural divergence, pyrazolopyridopyridazine diones
2f and
N-aminopyrazolopyrrolopyridine-6,8-diones
3f were selected as model cases for the further control experiments [
28]. At first, we employed Vilsmeier reagent (halomethyleniminium salt) [
29,
30,
31,
32,
33] to compounds
2f and
3f (
Scheme 1). The reactions were individually monitored by TLC method. When compound
2f was completely consumed for 4 h at 65 °C, the corresponding acquired amidination product
4 was formed and obtained in 89% yield without producing chlorinated compound
5. The structure of compound
4 was fully characterized by spectroscopic methods and single-crystal X-ray diffraction study. Based on
1H NMR spectroscopic characterization, compound
4 possesses singlet signal of pyridine ring proton H
a around 9.03 ppm, and significant amidinyl moiety signals of iminium proton H
b around 7.70 ppm and two peaks of NMe
2 around 2.97 and 3.02 ppm (
Figure 3). These results showed the free primary amine group of compound
2f was successfully converted into the amidinyl substituent. On the other hand, chlorination of compound
3f was accomplished without amidination product
4 formation by Vilsmeier reagent at reflux for 4 h, affording the corresponding product
5 with down-field proton signal H
c of pyridine ring around 9.68 ppm in good yield (80%,
Scheme 1 and
Figure 3) [
27]. Based on the above derivatization study, Vilsmeier reaction was conceived as the significant derivatization agent to identify isomers between
2f and
3f.
For further investigation into the reactivity of Vilsmeier amidination derivatization, Vilsmeier reaction was carried out using different substrates including
N-aminophthalimides
2a–
e at 50 °C for 0.5 h. Various substituted reactants
2a–
e were demonstrated to perform the reactions smoothly, regardless of whether electron-donating or electron-withdrawing substituents, and the corresponding amidination products
6−
10 were afforded in 74−88% yields (
Table 2). All products
6–
10 were fully characterized by spectroscopic methods, and they actually presented singlet peak for the significant amidinyl moiety signals of iminium proton H and two peaks of NMe
2 in
1H-NMR. Subsequently, a series of phthalazine 1,4-diones
3a–
e were treated with Vilsmeier reagent (POCl
3/DMF) at 65 °C or 80 °C for 2–4 h. The chlorination happened smoothly to afford the desired products
11–
15 in high yields (82−90%,
Table 2), except for
3d (31%). Owing to the electron-rich property of nitrogen atoms on the aromatic motif of compound
3d, the complicated aromatic substitution and polylization were proceeded. All chlorinated products
11–
15 were also fully characterized by spectroscopic methods, and two peaks for the significant dione moieties were converted into –N =
13C–Cl singlet signal at δ 153–157 ppm in
13C-NMR spectrum. Therefore, Vilsmeier reagent (POCl
3/DMF) was used as the derivatization reagent for the different reactive phenomenon to distinguish
N-aminophthalimides
2 and phthalazine-1,4-diones
3.
To explore the interconversion reactivity of the tautomerization, the solvent scope was first examined by using 7-aminopyrazolopyrrolopyridine-6,8-dione
2f. Compound
2f was screened and refluxed in the various solvents including CH
2Cl
2, THF, EtOH, MeCN, toluene, dioxane, and DMSO for 24 h. However, the reactions in CH
2Cl
2, EtOH, toluene recovered the starting material
2f without conversion happening (
Table 3 entries 1–3). The use of polar THF, MeCN, dioxane, and DMSO led to lower interconversion ratios of
2f/
3f from 93/7 to 88/12 (
Table 3, entries 4–7). Subsequently, Brønsted–Lowry acids including acetic acid (AcOH), methanesulfonic acid (TsOH), methanesulfonic chloride (TsCl), and trifluoroacetic acid (TFA) were studied for the interconversion reaction at reflux for 4 h (Entries 8–11,
Table 3). Several experimental observations are worthy to discuss: we firstly found that the conversion ratios were improved under acidic condition (Entries 8–11,
Table 3). Secondly, under the strong acid such as trifluoroacetic acid (pKa = 0.30),
p-toluenesulfonic acid (TsOH, pKa = −1.9), and methanesulfonic chloride (TsCl), the low conversion ratio and decomposed products were observed (Entries 8–10,
Table 3).
For further investigations, the timed programming of the thermodynamic conversion of compound
2f was carried out under acetic acid (AcOH) solution and shown in
Figure 4. The reaction mixture was sampled at 1.5, 2.3, 3.5, and 5 h and detected by the
1H-NMR spectroscopic method. This result showed that compound
2f was gradually converted to the thermodynamic stable product
3f (
Figure 4). Finally, transformation reaction was equilibrated at reflux for more than 5 h, and the conversion ratio was obtained proximately 6/94 (
2f/
3f, Entry 11 of
Table 4 and
Figure 4). Based on the above experimental result, acetic acid was conceived as the best acidic solvent with 6/94 conversion ratio. Fortunately,
2f can be successfully and smoothly transformed to more thermodynamically stable product
3f by refluxing in acidic medium [
40,
41]. To further demonstrate the reliable of conversion procedure,
N-aminophthalimides
2a–
e were also used as starting materials at reflux for 8–9 h. Fortunately, compounds
2a–
e can be smoothly transformed to give the corresponding thermodynamically pyrazolopyridopyridazine diones
3a–
e under acetic acid solvent, with the ratio of
2a–
e/
3a–
e from 6/94 to 1/99 (
Table 4).
3. Experimental Section
3.1. General Procedure
All reagents were purchased commercially. All reactions were carried out under argon or nitrogen atmosphere and monitored by TLC. Flash column chromatography was carried out on silica gel (230–400 mesh). Analytical thin-layer chromatography (TLC) was performed using pre-coated plates (silica gel 60 F-254) purchased from Merck Inc. Flash column chromatography purification was carried out by gradient elution using n-hexane in ethyl acetate (EtOAc) unless otherwise stated. 1H NMR spectra were recorded at 400 or 500 MHz and 13C NMR spectra were recorded at 100 or 125 MHz, respectively, in CDCl3, DMSO-d6, or D2O solvent. The standard abbreviations s, d, t, q, and m refer to the singlet, doublet, triplet, quartet, and multiplet, respectively. Coupling constant (J), whenever discernible, has been reported in Hz. Infrared spectra (IR) were recorded in neat solutions or solids; and mass spectra were recorded using electron impact or electrospray ionization techniques. The reported wavenumbers are referenced to the polystyrene 1601 cm−1 absorption. ESI-MS analyses were performed on an Applied Biosystems API 300 mass spectrometer. High-resolution mass spectra were obtained from a JEOL JMS-HX110 mass spectrometer.
3.2. Standard Procedure for Synthesis of N-Aminophthalimides 2a–f
Standard procedure for synthesis of
N-aminophthalimides
2a–
f. The reliable procedure involved the treatment of isobenzofuran-1,3-diones (
1a–
c), isoindoline-1,3-dione (
1d), furo[3,4-
b] pyrazine-5,7-dione (
1e), or 1
H-pyrrolo[3,4-
c] pyridine-1,3-dione (
1f, 1.0 equiv.) with monohydrate hydrazine (~5.0 equiv.) in EtOH/H
2O (2.0 mL/2.0 mL) at 0 °C or –20 °C to room temperature within 4 h. When the reaction was completed, the reaction mixture was added water (10 mL) for precipitation. The precipitate was filtered, washed with cold water (10 mL) and
n-hexane/EA (1/2, 15 mL) to give the corresponding crude
N-aminophthalimides
2a–f. The crude desired products
2a–f were recrystallized in acetone/THF (1/4) solution to obtain the pure
N-aminophthalimides
2a–f in 81–94% yields. The low solubility of the compounds
2a–f made the
13C-NMR characterization of quaternary and carbonyl carbons of these substrates unclear [
28].
N-Aminophthalimide (
2a) [
42]: White solid; 92% yield; mp 202–205 °C;
1H NMR (DMSO-
d6, 400 MHz) δ 7.83 (dd,
J = 5.8, 3.4 Hz, 2H, Ar
H), 8.05 (dd,
J = 5.8, 3.4 Hz, 2H, Ar
H);
13C NMR (DMSO-
d6, 100 MHz): δ 123.28 (2 × C), 130.53 (2 × C), 134.81 (2 × C), 167.36 (2 × C); FT-IR (KBr): 3482, 3341, 3262, 3093, 1778, 1719, 1605, 1468, 1409, 1291, 1197, 1091, 1075, 1020, 918, 800, 718, 526 cm
−1; EIMS
m/z: 162 (M
+, 100), 105 (12), 104 (74), 77 (12), 76 (26), 50 (11); HRMS (EI)
m/z: [M]
+ Calcd for C
8H
6N
2O
2: 162.0429; Found: 162.0425.
N-Amino-4,5-difluoropthalimide (2b): white solid; 92% yield; mp 288–290 °C; 1H NMR (D2O, 500 MHz): δ 7.40 (t, J = 9.38 Hz, 2H, ArH); 13C NMR (D2O, 125 MHz): δ 116.43 (dd, J = 12.98, 6.09 Hz, 2 × C), 134.91 (2 × C), 148.62 (d, J = 14.79 Hz), 150.61 (d, J = 14.78 Hz), 175.06 (2 × C). FT-IR (KBr): 3358, 3290, 3072, 2593, 1658, 1597, 1495, 1372, 1182, 1094, 1029, 910, 788, 743 cm−1; EIMS m/z: 198 (M+, 100), 141 (24), 140 (83), 113 (14), 112 (33); HRMS (EI) m/z: [M]+ Calcd for C8H4F2N2O2: 198.0241; Found: 198.0233.
N-Amino-4,5-dichlorophthalimide (2c): White solid; 81% yield; mp 302–305 °C; 1H NMR (DMSO-d6, 400 MHz) δ 7.72 (s, 1H, ArH), 7.84 (s, 1H, ArH); 13C NMR (DMSO-d6, 100 MHz) δ 130.36, 131.12, 132.08, 132.25, 132.57, 141.90, 164.74, 169.22; FT-IR (KBr): 3466, 3321, 3220, 3027, 2646, 1653, 1619, 1521, 1393, 1356, 1308, 1106, 1022, 930, 809, 782, 613 cm−1; EIMS m/z: 232 (M+ + 2, 64), 230 (M+, 100), 174 (46), 173 (12), 172 (72), 146 (13), 144 (19), 109 (19), 74 (17); HRMS m/z: [M]+calcd for C8H4Cl2N2O2: 229.9650; found: 229.9653.
N-Amino-2,3-pyrazinedicarboxylicphthalimide (
2d) [
42]: Brown solid; 82% yield; mp 220–222 °C;
1H NMR (D
2O, 400 MHz) δ 8.59 (s, 2H, Ar
H);
13C NMR (D
2O, 100 MHz) δ 143.28 (2 × C), 149.18 (2 × C), 172.29 (2 × C); FT-IR (KBr): 3430, 3282, 3164, 2936, 2750, 2636, 1679, 1621, 1353, 1161, 1107, 975, 825 cm
−1. EIMS
m/z: 164 (M
+, 36), 150 (25), 124 (40), 106 (76), 80 (100), 79 (30), 78 (56), 53 (69), 52 (95), 51 (52). HRMS
m/z: [M]
+calcd for C
6H
4N
4O
2: 164.0334; found: 164.0338.
Naphthalene-2,3-dicarboxylic hydrazide (2e): Yellow solid; 94% yield; mp 298–300 °C; 1H NMR (DMSO-d6, 400 MHz) δ 7.51–7.56 (m, 2H, ArH), 7.92–7.96 (m, 2H, ArH), 8.11 (s, 1H, ArH), 8.16 (s, 1H, ArH); 13C NMR (DMSO-d6, 100 MHz) δ 127.12, 127.52, 128.32, 128.48, 128.68, 128.87, 132.17, 132.32, 133.31, 137.68, 168.21, 171.85; FT-IR (KBr): 3466, 3429, 3304, 3190, 3054, 2653, 1673, 1636, 1606, 1390, 1339, 1177, 1140, 1099, 964, 809, 758, 481 cm−1; EIMS m/z: 213 (14), 212 (M+, 100), 155 (10), 154 (45), 127 (14), 126 (39); HRMS m/z: [M]+calcd for C12H8N2O2: 212.0586; found: 212.0578.
7-Amino-1,3-diphenylpyrazolo[3,4-b]pyrrolo[3,4-d]pyridine-6,8(3H,7H)-dione (
2f) [
28]: White solid; 83% yield; mp 217–219 °C;
1H NMR (DMSO-
d6, 400 MHz) δ 4.55 (br, 2H, N
H2), 7.42 (t,
J = 7.46 Hz, 1H, Ar
H), 7.49–7.55 (m, 3H, Ar
H), 7.60–7.65 (m, 4H, Ar
H), 8.25 (d,
J = 8.08 Hz, 2H, Ar
H), 8.82 (s, 1H, Ar
H).
3.3. Standard Procedure for Synthesis of Phthalazine 1,4-Diones 3a–f
The reliable procedure involved the treatment of isobenzofuran-1,3-diones (
1a–
c), isoindoline-1,3-dione (
1d), furo[3,4-
b] pyrazine-5,7-dione (
1e), or 1
H-pyrrolo[3,4-
c] pyridine-1,3-dione (
1f, 1.0 equiv.) with monohydrate hydrazine (~40 equiv.) in EtOH solution (2.0 mL) at room temperature or in neat at reflux for 4 h. When the reaction was completed, the reaction mixture was added water (10 mL) for precipitation. The precipitation was filtered, washed with cold water (10 mL) and
n-hexane/EA (1/2, 15 mL) to give the corresponding crude phthalazine 1,4-diones
3a–f. The crude desired products
3a–f were recrystallized in acetone/THF (1/4) solution to obtain the pure phthalazine 1,4-diones
3a–f in 83–91% yields. The low solubility of the compounds
3a–f made the
13C-NMR characterization of quaternary and carbonyl carbons of these substrates unclear [
28].
2,3-Dihydro-phthalazine-1,4-dione (
3a) [
43]: White solid; 89% yield; mp 227–229 °C;
1H NMR (DMSO-
d6, 400 MHz) δ 7.82 (dd,
J = 5.93, 3.30 Hz, 2H, Ar
H), 8.06 (dd,
J = 5.89, 3.28 Hz, 2H, Ar
H);
13C NMR (DMSO-
d6, 100 MHz) δ 125.78 (2 × C), 128.68 (2 × C), 132.35 (2 × C), 156.41 (2 × C); FT-IR (KBr): 3461, 3207, 2715, 1773, 1749, 1609, 1468, 1386, 1309, 1051, 715 cm
−1.
6,7-Fluoroo-2,3-dihydrophthalazine-1,4-dione (3b): White solid; 85% yield; mp 220–222 °C; 1H NMR (DMSO-d6, 400 MHz) δ 7.99 (t, J = 9.11 Hz, 2H, ArH); 13C NMR (DMSO-d6, 100 MHz) δ 114.38 (2 × C), 126.58 (2 × C), 151.48 (d, J = 16.11 Hz), 154.02 (d, J = 16.10 Hz), 154.61 (2 × C); FT-IR (KBr): 3490, 3180, 3070, 2610, 1660, 1590, 1511, 1460, 1354, 1303, 1189, 1067, 899, 804, 565 cm−1; EIMS m/z: 199 (13), 198 (M+, 100), 141 (23), 140 (75), 113 (17), 112 (30), 63 (13); HRMS m/z: [M]+calcd for C8H4F2N2O2: 198.0241; found: 198.0234.
6,7-Dichloro-2,3-dihydrophthalazine-1,4-dione (3c): White solid; 87% yield; mp 237–239 °C; 1H NMR (DMSO-d6, 400 MHz) δ 8.15 (s, 2H, ArH); 13C NMR (DMSO-d6, 100 MHz) δ 128.16 (2 × C), 129.64 (2 × C), 134.98 (2 × C), 156.12 (2 × C); FT-IR (KBr): 3314, 3193, 2984, 2926, 2879, 1666, 1565, 1467, 1450, 1373, 1075, 822 cm−1; EIMS m/z: 232 (M+ + 2, 64), 231 (M+ + 1, 17) 230 (M+, 100), 174 (46), 173 (12), 172 (72), 146 (13), 144 (19), 109 (19), 74 (17); HRMS m/z: [M]+calcd for C8H4Cl2N2O2: 229.9650; found:229.9643.
7-Dihydropyrazino[2,3-d] pyridazine-5,8-dione (
3d) [
44]: Brown solid; 83% yield; mp 325–327 °C;
1H NMR (D
2O, 500 MHz) δ 8.82 (s, 2H, ArH);
13C NMR (D
2O, 125 MHz) δ 145.14 (2 × C), 146.29 (2 × C), 169.64 (2 × C). FT-IR (KBr): 3343, 3204, 1661, 1632, 1543, 1314, 1293, 1100 cm
−1; EIMS
m/z: 164 (M
+, 1), 150 (26) 140 (16), 106 (77), 80 (100), 78 (46), 52 (2).
2,3-Dihydrobenzo[g]phthalazine-1,4-dione (3e): White solid; 91% yield; mp 301–303 °C; 1H NMR (DMSO-d6, 400 MHz) δ 7.71(dd, J = 6.29, 3.27 Hz, 2H, ArH), 8.25 (dd, J = 6.28, 3.28 Hz, 2H, ArH), 8.72 (s, 2H, ArH); 13C NMR (DMSO-d6, 100 MHz) δ 125.13 (2 × C), 126.69 (2 × C), 128.84 (2 × C), 129.66 (2 × C), 134.54 (2 × C), 156.22 (2 × C); FT-IR (KBr): 3416, 1663, 1626, 1501, 1467, 1440, 1366, 1072, 815 cm−1; EIMS m/z: 213 (14), 212 (M+, 100), 155 (12), 154 (43), 237 (17), 126 (44); HRMS m/z: [M]+calcd for C12H8N2O2: 212.0586; found: 212.0588.
1,3-Diphenyl-7,8-dihydro-3H-pyrazolo [4’,3’:5,6] pyrido[3,4-d] pyridazine-6,9-dione(3f) [
28]: white solid; 84% yield; mp 292–295 °C;
1H NMR (DMSO-
d6, 600 MHz) δ 7.43–7.47 (m, 4H, Ar
H), 7.60–7.64 (m, 4H, Ar
H), 8.20 (d,
J = 7.88 Hz, 2H, Ar
H), 9.43 (s, 1H, Ar
H).
3.4. Standard Procedure for Preparation of Amidination Products 4 and 6–10 from N-Aminophthalimides 2a–f with Vilsmeier Reagent (POCl3/DMF)
The reliable procedure that involved
N-aminophthalimides
2a–
f (1.0 equiv..) was individually treated with ~3.0 equivalent amount of POCl
3 in
N,
N-dimethylformamide solution (DMF, 2.0 mL) at 50 °C or 65 °C for 0.5–4 h. When the reaction was completed, the reaction mixture was added to saturate sodium bicarbonate (15 mL) and extracted with dichloromethane (15 mL). The organic extracts were dried over MgSO
4, filtered, and concentrated under reduced pressure. The residues were purified by column chromatography on silica gel to give the corresponding amidination products
4 in 89% yield and
6–
10 in 74–88% yields [
37,
38,
39].
N’-(6,8-Dioxo-1,3-diphenyl-6,8-dihydropyrazolo[3,4-b]pyrrolo[3,4-d]pyridin-7(3H)-yl)-N,N-dimethylformimidamide (4): Brown solid; 89% yield; mp 225–227 °C; 1H NMR (CDCl3, 400 MHz) δ 2.97 (s, 3H, NMe), 3.02 (s, 3H, NMe), 7.37 (t, J = 7.36 Hz, 1H, ArH), 7.48–7.50 (m, 3H, ArH), 7.54 (t, J = 7.74 Hz, 2H, ArH), 7.70 (s, 1H, ArH), 7.87 (d, J = 4.40 Hz, 2H, ArH), 8.24 (d, J = 8.00 Hz, 2H, ArH), 9.03 (s, 1H, ArH); 13C NMR (CDCl3, 100 MHz) δ 33.83, 41.06, 108.85, 119.70, 122.21 (2 × C), 127.20, 127.90 (2 × C), 129.14 (2 × C), 129.37, 129.82 (2 × C), 131.52, 133.65, 138.40, 143.04, 146.08, 153.77, 161.77, 163.79, 165.45; FT-IR (KBr): 3064, 2923, 2852, 1714, 1626, 1500, 1413, 846 cm−1; EIMS m/z: 411 (25), 410 (M+, 100), 341 (29), 340 (87), 339 (52), 268 (14), 77 (31); HRMS calcd. For C23H18N6O2: 410.1491; found: 410.1482.
N’-(1,3-Dioxo-1,3-dihydro-2H-isoindol-2-yl)-N,N-dimethyliminoformamide hydrochloride (6): Yellow solid; 88% yield; mp 177–179 °C; 1H NMR (CDCl3, 400 MHz) δ 3.02 (s, 6H, N(CH3)2), 7.66 (dd, J = 5.37, 3.03, 2H, ArH), 7.75 (s, 1H), 7.79 (dd, J = 5.42, 3.09, 2H, ArH); 13C NMR (CDCl3, 100 MHz) δ 34.84, 41.10, 123.01 (2 × C), 123.58 (2 × C), 130.77 (2 × C), 133.77 (2 × C), 161.52, 166.38 (2 × C); FT-IR (KBr): 3446, 2933, 1699, 1620, 1321, 1138, 706 cm−1; EIMS m/z: 218 (12), 217 (M+, 100), 148 (19), 130 (27), 105 (17), 104 (22), 90 (11), 76 (29), 71 (41), 70 (21); HRMS m/z: [M]+calcd for C11H11N3O2: 217.0851; found: 217.0842.
N’-(1,3-Dioxo-5,6-difluoro-2H-isoindolin-2-yl)-N,N-dimethylformimidamide (7): Light yellow solid; 74% yield; mp 183–185 °C; 1H NMR (CDCl3, 400 MHz) δ 2.88 (s, 3H, NMe), 2.96 (s, 3H, NMe), 7.69 (t, J = 8.96 Hz, 2H, ArH), 8.04 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 31.83, 36.98, 119.75 (dd, J = 13.4, 7.63 Hz, 2 × C), 129.46 (t, J = 4.77 Hz, 2 × C), 150.11 (d, J = 14.45 Hz), 152.68 (d, J =14.31 Hz), 163.68, 168.39 (2 × C); FT-IR (KBr): 3048, 2919, 1695, 1620, 1450, 1355, 1148, 818 cm−1; EIMS m/z: 254 (13), 253 (M+, 100), 166 (20), 141 (25), 140 (40), 139 (16), 126 (22), 125 (12), 113 (13), 112 (52), 111 (20), 109 (16), 97 (23), 95 (17), 85 (16), 83 (21), 81 (17); HRMS m/z: [M]+calcd for C11H9F2N3O2: 253.0663; found: 253.0657.
N’-(1,3-Dioxo-5,6-dichloro-2H-isoindolin-2-yl)-N,N-dimethylformimidamide (8): Light orange solid; 85% yield; mp 192–194 °C; 1H NMR (CDCl3, 400 MHz) δ 3.04 (s, 6H, N(CH3)2), 7.74 (s, 1H), 7.87 (s, 2H, ArH); 13C NMR (CDCl3, 100 MHz) δ 34.80, 41.07, 125.09 (2 × C), 129.90 (2 × C), 138.54 (2 × C), 161.35, 164.42 (2 × C). FT-IR (KBr): 3092, 3024, 2926. 1773, 1705, 1624, 1345, 1145, 774 cm−1; EIMS m/z: 287 (M+ + 2, 61), 286 (M+ +1, 12), 285 (M+, 100), 198 (13), 175 (12), 174 (14), 173 (22), 172 (20), 146 (16), 144 (24), 109 (11), 71 (98), 70 (36), 69 (10); HRMS m/z: [M]+calcd for C11H9Cl2N3O2: 285.0072; found: 285.0064.
N’-(1,3-Dioxo-5,7-dihydro-6H-pyrrolo[3,4-b]pyrazin-6-yl)-N,N-dimethylformimidamide (9): Light yellow solid; 88% yield, mp 219–221 °C; 1H NMR (CDCl3, 400 MHz) δ 3.03 (s, 3H, NMe), 3.05 (s, 3H, NMe), 7.81 (s, 1H), 8.84 (s, 2H, ArH); 13C NMR (CDCl3, 100 MHz) δ 34.83, 41.18, 146.17 (2 × C), 148.52 (2 × C), 161.28, 162.31 (2 × C); FT-IR (KBr): 1654, 1289, 1545, 1293, 1104, 825 cm−1; EIMS m/z: 220 (12), 219 (M+, 56), 193 (10), 179 (13), 178 (13), 169 (12), 168 (12), 167 (14), 165 (11), 155 (11), 152 (11), 151 (26), 150 (14), 149 (23), 147 (11), 141 (12), 139 (15), 137 (13), 135 (11), 127 (12), 125 (21), 123 (19), 121 (11), 119 (11), 115 (14), 113 (14), 112 (14), 111 (36), 110 (11), 109 (27), 107 (15), 106 (16), 105 (16), 99 (17), 98 (13), 97 (52), 96 (16), 95 (36), 93 (12), 91 (27); HRMS m/z: [M]+calcd for C9H9N5O2: 219.0756; found: 219.0748.
N’-(1,3-Dioxo-5,6-dihydro-2H-benzo[f]isoindol-2-yl)-N,N-dimethylformimidamide (10): Light yellow solid; 77% yield; mp 199–201 °C; 1H NMR (CDCl3, 400 MHz) δ 3.05 (s, 3H, NMe), 3.07 (s, 3H, NMe), 7.64–7.69 (m, 2H, ArH), 7.84 (s, 1H), 7.99–8.02 (m, 2H, ArH), 8.28 (s, 2H, ArH); 13C NMR (CDCl3, 100 MHz) δ 34.89, 41.10, 124.33 (2 × C), 126.68 (2 × C), 128.99 (2 × C), 130.20 (2 × C), 135.45 (2 × C), 161.23, 166.00 (2 × C) cm−1; FT-IR (KBr): 2918, 2807, 1757, 1693, 1621, 1521, 1425, 1411, 1321, 1154, 1118, 1004, 900, 754, 479 cm−1; EIMS m/z: 268 (19), 267 (M+, 100), 225 (13), 210 (10), 198 (26), 197 (49), 180 (28), 155 (64), 154 (24), 153 (20), 152 (13), 140 (21), 127 (26), 126 (74), 71 (17), 57 (11); HRMS m/z: [M]+calcd for C15H13N3O2: 267.1008; found: 267.1015.
3.5. Standard Procedure for Preparation of Chlorination Products 5, and 11–15 from Phthalazine 1,4-Diones 3a–f with Vilsmeier Reagent (POCl3/DMF)
The reliable procedure that involved phthalazine 1,4-diones
3a–
f (1.0 equiv.) was individually treated with ~3.0 equivalent amount of POCl
3 in
N,
N-dimethylformamide solution (DMF, 2.0 mL) at 65 °C or 80 °C for 2–4 h. When the reaction was completed, the reaction mixture was added to saturate sodium bicarbonate (15 mL) and extracted with dichloromethane (15 mL). The organic extracts were dried over MgSO
4, filtered, and concentrated under reduced pressure. The residues were purified by column chromatography on silica gel to give the corresponding chlorinated products
5 in 80% yield and
11–
15 in 31–90% yields [
29].
6,9-Dichloro-1,3-diphenyl-3H-pyrazolo[4’,3’:5,6]pyrido[3,4-d]pyridazine (5): Light yellow solid; 80% yield; mp 196–197 °C; 1H NMR (CDCl3, 500 MHz) δ 7.46 (t, J = 7.43 Hz, 1H, ArH), 7.49–7.51 (m, 3H, ArH), 7.57–7.60 (m, 4H, ArH), 8.17 (dd, J = 8.60, 1.09 Hz, 2H, ArH), 9.68 (s, 1H, ArH); 13C NMR (CDCl3, 125 MHz) δ 103.92, 118.46, 123.39 (2 × C), 127.91, 127.95 (2 × C), 128.19, 129.13, 129.33 (2 × C), 130.01 (2 × C), 135.22, 137.87, 147.79, 149.88, 151.00, 152.05, 153.49; FT-IR (KBr): 3064, 2923, 2846, 1571, 1501, 1413, 1243, 1119, 863 cm−1; EIMS m/z: 395 (12), 394 (17), 393 (M+ + 2, 65), 392 (36), 391 (M+, 99), 390 (20), 356 (20), 321 (35), 320 (60), 288 (14), 263 (12), 244 (33), 218 (17), 91 (19), 77 (100); HRMS calcd. For C20H11Cl2N5: 391.0392; found: 391.0397.
1,4-Dichlorophthalazine (11): Yellow solid; 90% yield; mp 162–164 °C; 1H NMR (CDCl3, 400 MHz) δ 7.74–7.76 (m, 2H, ArH), 7.85–7.7.87 (m, 2H, ArH); 13C NMR (CDCl3, 100 MHz) δ 125.86 (2 × C), 127.21 (2 × C), 134.49 (2 × C), 155.03 (2 × C); IR (KBr): 3157, 3000, 2896, 2875, 1671, 1346, 1289, 1157, 993, 775, 664 cm−1; EIMS m/z: 202 (M+ + 4, 10), 200 (M+ + 2, 63), 198 (M+, 100), 182 (25), 180 (77), 172 (17), 170 (26), 151 (17), 135 (20), 128 (14), 125 (11), 123 (29), 102 (17), 101 (11), 99 (20), 90 (11). HRMS calcd. For C8H4Cl2N2: 197.9752; found: 197.9746.
1,4-Dichloro-2,3-difluorophthalazine (12): White solid; 82% yield; mp 75–76 °C; 1H NMR (CDCl3, 400 MHz) δ 8.09 (t, J = 8.34 Hz, 2H, ArH); 13C NMR (CDCl3, 100 MHz) δ 113.97 (dd, J = 14.25, 7.66 Hz, 2 × C), 125.19 (t, J = 5.23 Hz, 2 × C), 153.50 (d, J = 16.46 Hz), 153.68 153.50 (2 × C), 156.15 (d, J = 16.39 Hz); IR (KBr): 3143, 3068, 2836, 2782, 2718, 2611, 1625, 1571, 1539, 1511, 1389, 1218, 1161, 1104, 893, 814 cm−1; EIMS m/z: 238 (M+ + 4, 11), 236 (M+ + 2, 78), 234 (M+, 100), 234 (30), 218 (12), 216 (36), 208 (24), 206 (38), 171 (27), 164 (30), 159 (18), 138 (11), 136 (16), 124 (10), 88 (15), 75 (11); HRMS calcd. For C8H2Cl2F2N2: 233.9563; found: 233.9568.
1,4,6,7-Tetrachlorophthalazine (13): Light orange solid; 84% yield; mp 135–136 °C; 1H NMR (CDCl3, 400 MHz) δ 8.40 (s, 2H, ArH); 13C NMR (CDCl3, 100 MHz) δ 126.07 (2 × C), 127.39 (2 × C), 140.2 (2 × C), 153.45 (2 × C); IR (KBr): 3094, 1596, 1535, 1453, 1501, 1380, 1268, 1242, 1130, 1035, 702, 663 cm−1; EIMS m/z: 270 (M+ + 2, 26), 270 (M+ + 2, 16), 268 (M+, 100), 266 (77), 240 (19), 238 (14), 205 (14), 203 (14), 196 (13), 84 (10); HRMS calcd. For C8H2Cl4N2: 265.8972; found: 265.8979.
6,7-Dichloropyrazino[2,3-d] pyridazine (
14) [
28]: Black solid; 31% yield; mp 204–206 °C;
1H NMR (CDCl
3, 400 MHz) δ 9.14 (s, 1H, Ar
H), 9.17 (s, 1H, Ar
H) [
28].
1,4-Dichlorobenzo[g]phthalazine (15): Gray solid; 85% yield; mp 212–214 °C; 1H NMR (CDCl3, 400 MHz) δ 7.78–7.81 (m, 2H, ArH), 8.18–8.21 (m, 2H, ArH), 8.82 (s, 2H, ArH); 13C NMR (CDCl3, 100 MHz) δ 123.23 (2 × C), 126.83 (2 × C), 129.22 (2 × C), 129.79 (2 × C), 135.46 (2 × C), 155.39 (2 × C); IR (KBr): 2961, 2932, 2857, 1739,1725, 1461, 1282, 1264, 1121, 1075, 739 cm−1; EIMS m/z: 250 (M+ + 2, 59), 249 (M+ + 1, 11), 248 (M+, 100), 178 (31), 152 (19), 151 (14), 150 (18); HRMS calcd. For C23H18N6O2: 247.9908; found: 247.9906.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


































