
Ligand-Dependent Conformational Transitions in Molecular Dynamics Trajectories of GPCRs Revealed by a New Machine Learning Rare Event Detection Protocol


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Construction of the Rare Events Detection (RED) Protocol
2.2. Detection of Rare Events in the Dynamics of the Ligand-Bound 5-HT2AR
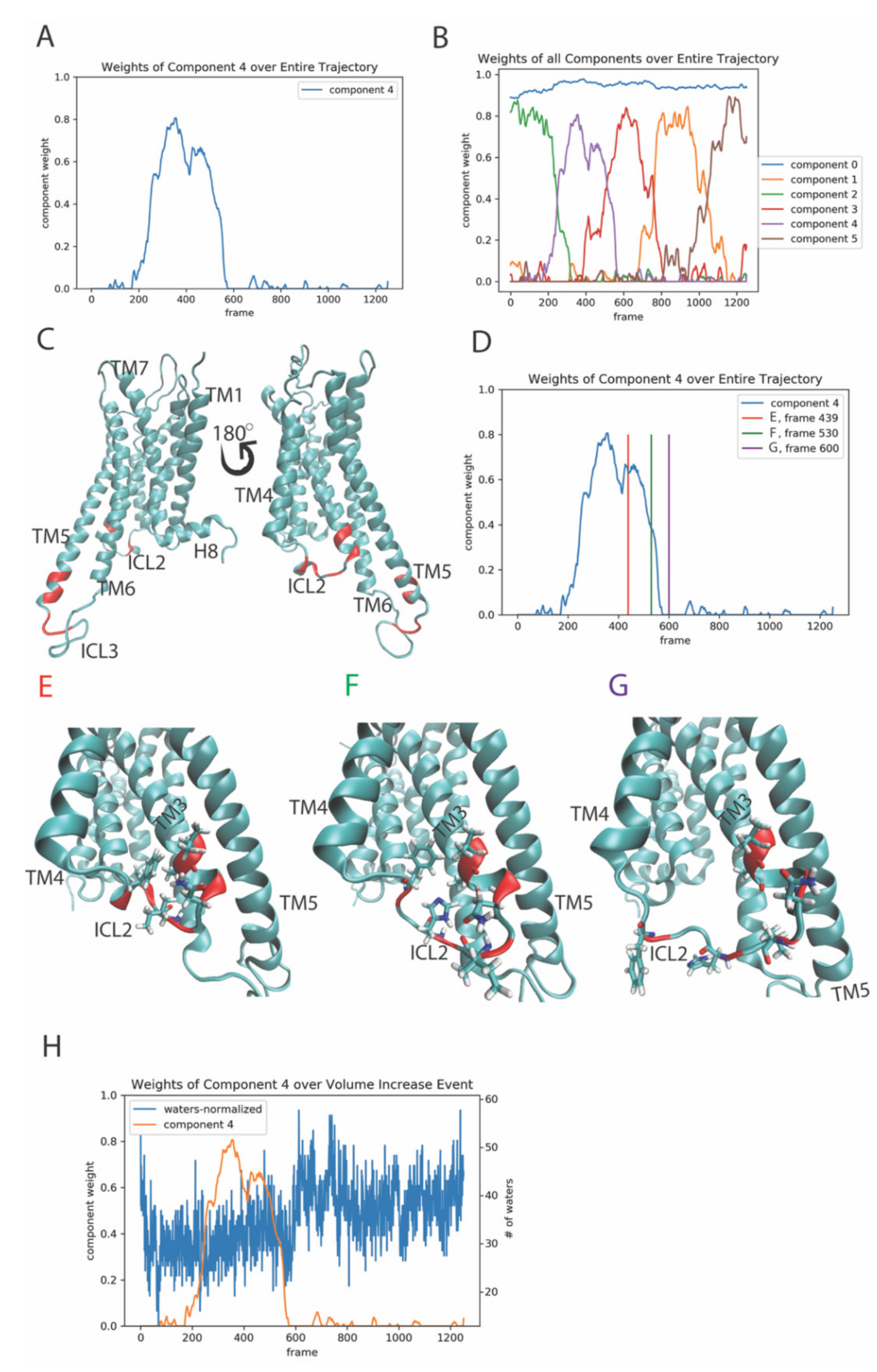
2.3. Function-Related Rare Events in the Dynamics of the 5-HT2AR Bound to Serotonin (5-HT)
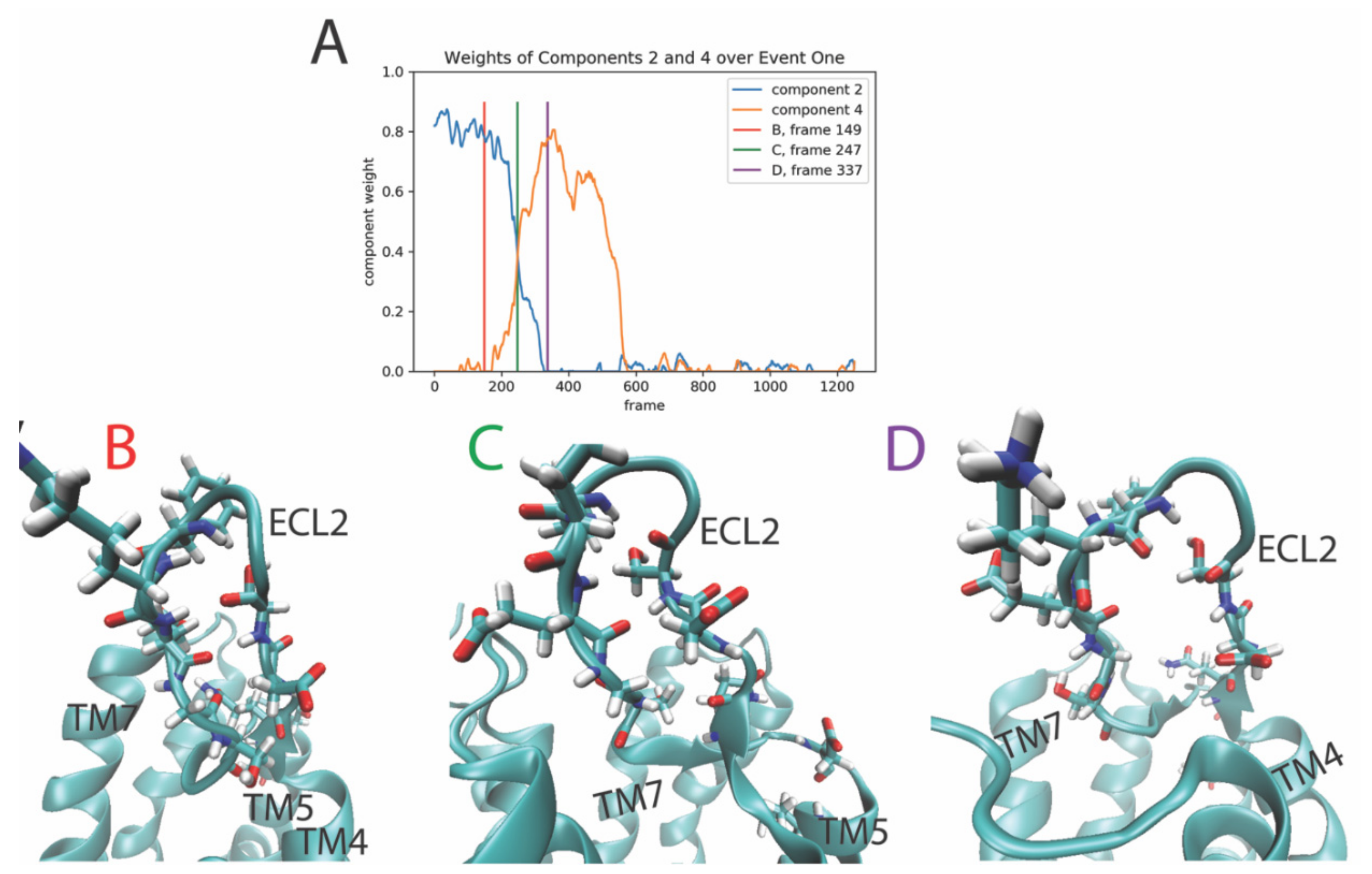
2.3.1. The First Event
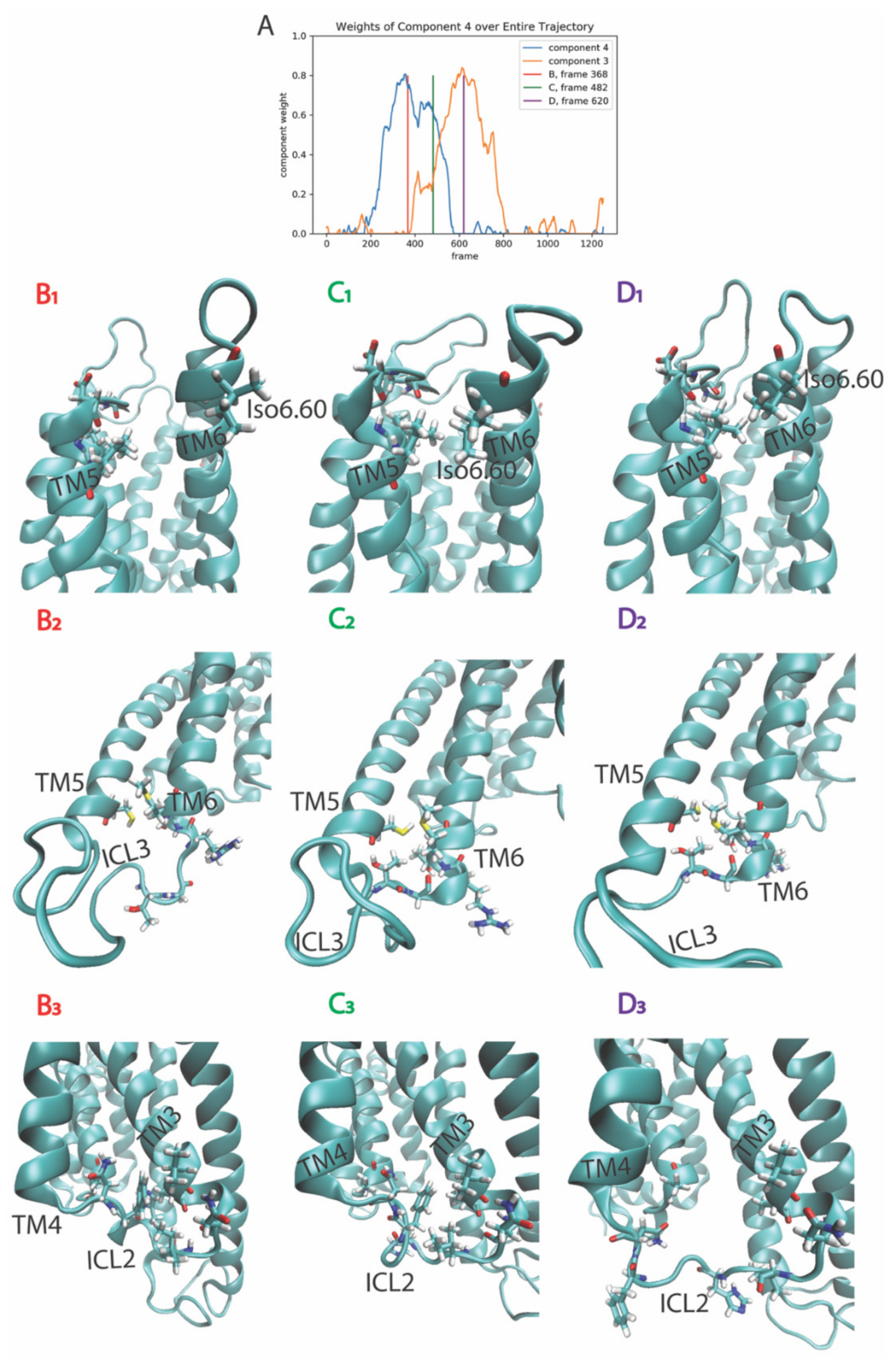
2.3.2. The Second Event
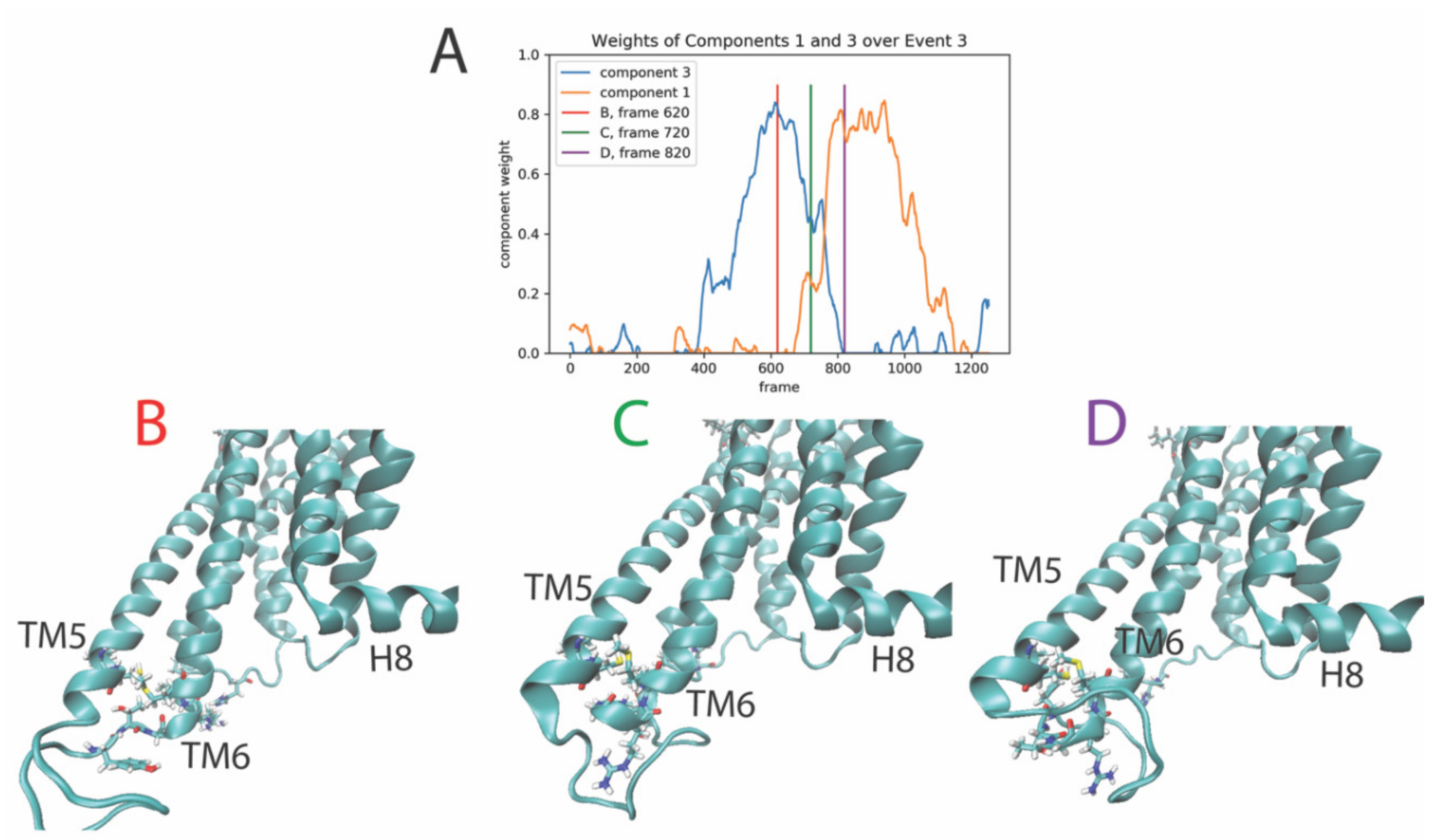
2.3.3. The Third Event
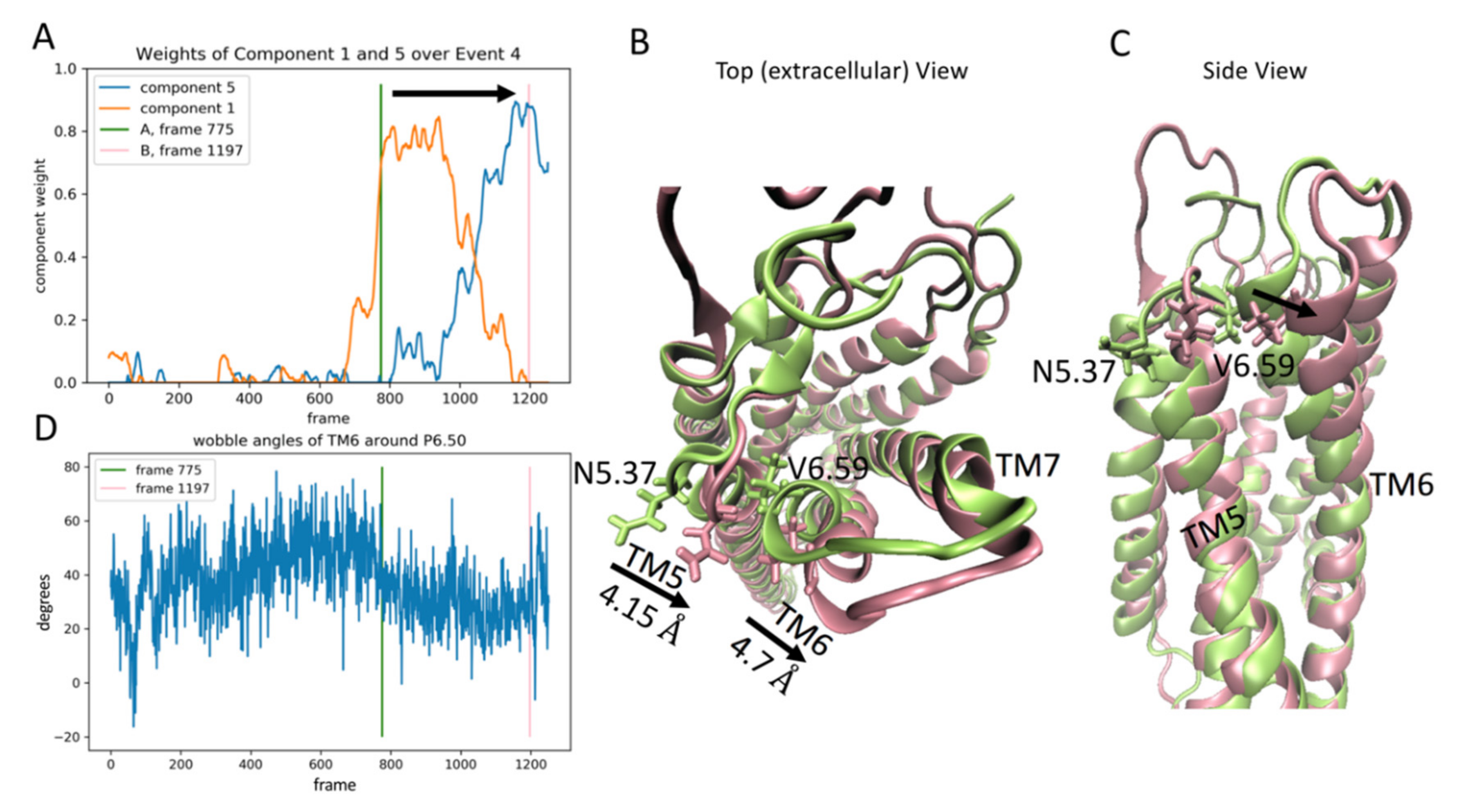
2.3.4. The Fourth Event
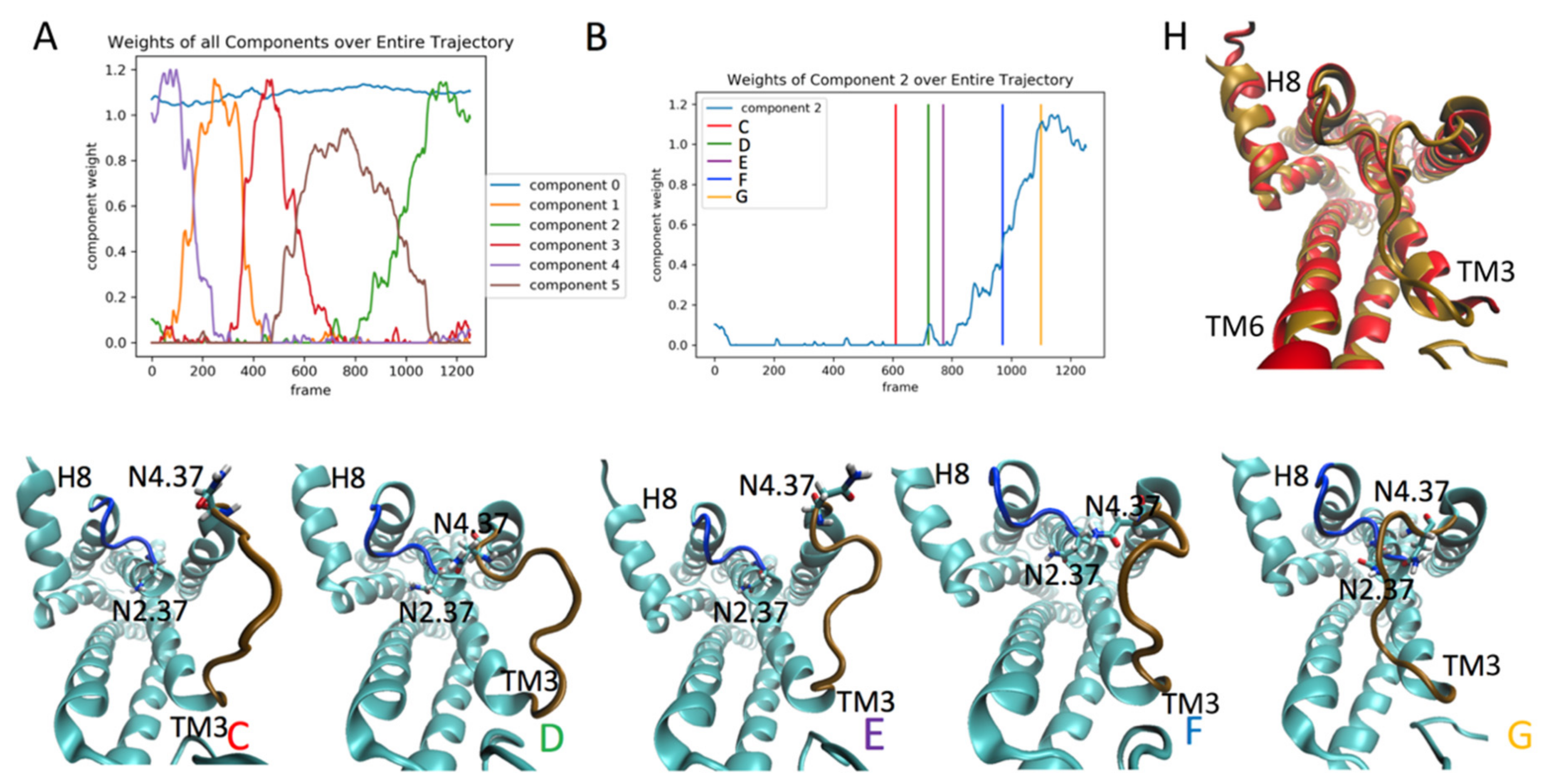
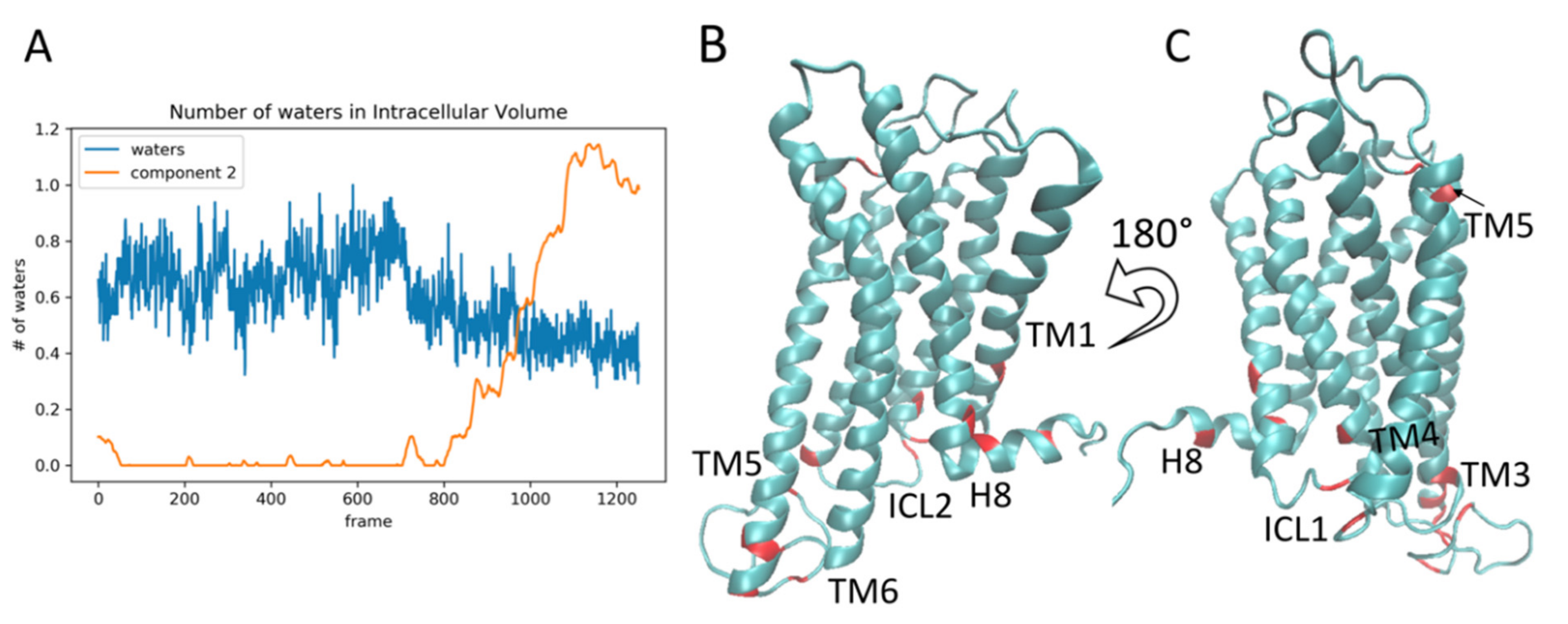
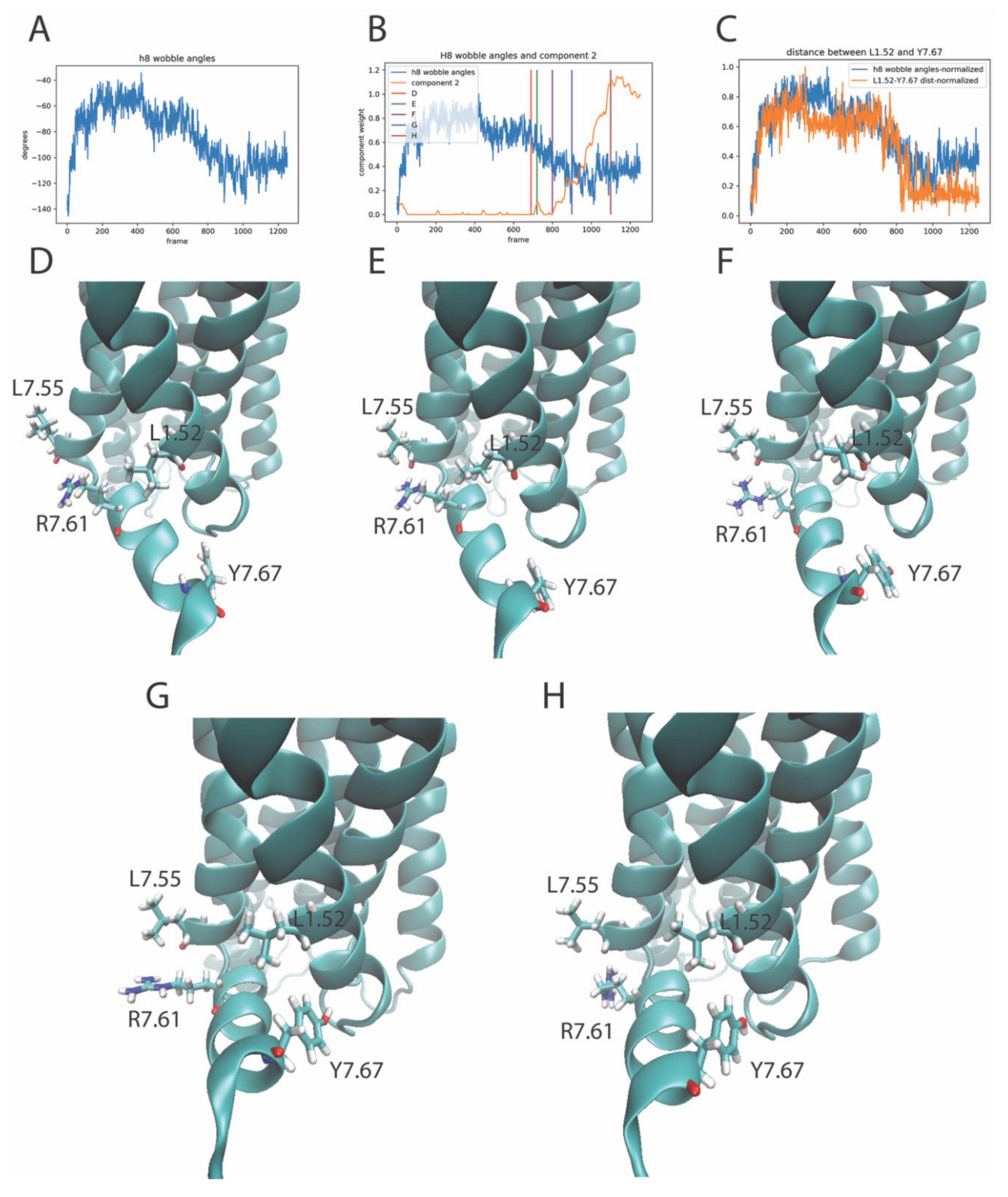
2.4. The Relation of RED-Identified Rare Events to the Functional Mechanism of the 5-HT2AR
2.4.1. Detection of Rare Events in The Dynamics of the Ketanserin (KET)-Bound 5-HT2AR
2.4.2. The RED Protocol Reveals Salient Structural Features of Simultaneous Conformational Changes in Different Structural Motifs
2.4.3. The Role of The Ligand in Transitions to Functional States
3. Discussion
4. Materials and Methods
4.1. Homology Model of the 5-HT2AR
- Set 1:
- consisted of two structures of the human 5-HT2BR (PDBID: 4ib4 and 5tvn);
- Set 2:
- included two structures of the human 5-HT2BR (PDBID: 4ib4 and 5tvn) and two structures of the human 5-HT1BR (PDBID: 4iaq and 4iar);
- Set 3:
- included all the structures in Set 2, augmented by 2 structures of the human β2-adrenergic receptor b2AR (PDBID: 4lde and 4ldl).
4.2. Parametrization and Docking of the Molecular Models
4.3. Comparison of Modeled Starting Structures to 5-HT2AR Structures in the PDB
4.3.1. Comparison of Starting Structures
4.3.2. Structures Resulting from the MD Simulations: 5-HT2AR/KET vs. 5-HT2AR/Risperidone Binding Mode
4.3.3. Comparison of Functional Motifs, “Toggle Switch” W6.48
4.3.4. Comparison of Functional Motifs, Intracellular Orientation of TM6
4.3.5. Comparison of Functional Motifs, Intracellular Orientation of TM7
4.3.6. Comparison of Functional Motifs, the Ionic Lock
4.4. Molecular Dynamics Simulations
4.5. Calculation of the Intracellular Cavity Volume
4.6. Visualization of Intracellular Cavity Volume
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Khelashvili, G.; Stanley, N.; Sahai, M.A.; Medina, J.; LeVine, J.; Shi, L.; De Fabritiis, G.; Weinstein, H. Spontaneous inward opening of the dopamine transporter is triggered by PIP2-regulated dynamics of the N-terminus. ACS Chem. Neurosci. 2015, 6, 1825–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Arnittali, M.; Rissanou, A.N.; Harmandaris, V. Structure of Biomolecules through Molecular Dynamics Simulations. Procedia Comput. Sci. 2019, 156, 69–78. [Google Scholar] [CrossRef]
- Noe, F.; Clementi, C. Collective variables for the study of long-time kinetics from molecular trajectories: Theory and methods. Curr. Opin. Struct. Biol. 2017, 43, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Schütt, K.T.; Chmiela, S.; von Lilienfeld, O.A.; Tkatchenko, A.; Tsuda, K.; Müller, K.-R. (Eds.) Machine Learning for Molecular Dynamics on Long Timescales. In Machine Learning Meets Quantum Physics; Lecture Notes in Physics; Springer: Cham, Switzerland, 2020. [Google Scholar]
- Plante, A.; Shore, D.M.; Morra, G.; Khelashvili, G.; Weinstein, H. A Machine Learning Approach for the Discovery of Ligand-Specific Functional Mechanisms of GPCRs. Molecules 2019, 24, 2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urban, J.D.; Clarke, W.P.; von Zastrow, M.; Nichols, D.E.; Kobilka, B.; Weinstein, H.; Javitch, J.A.; Roth, B.L.; Christopoulos, A.; Sexton, P.M.; et al. Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther. 2007, 320, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gregorio, G.G.; Masureel, M.; Hilger, D.; Terry, D.S.; Juette, M.; Zhao, H.; Zhou, Z.; Perez-Aguilar, J.M.; Hauge, M.; Mathiasen, S.; et al. Single-molecule analysis of ligand efficacy in beta2AR-G-protein activation. Nature 2017, 547, 68–73. [Google Scholar] [CrossRef]
- LeVine, M.V.; Weinstein, H. AIM for Allostery: Using the Ising Model to Understand Information Processing and Transmission in Allosteric Biomolecular Systems. Entropy 2015, 17, 2895–2918. [Google Scholar] [CrossRef] [Green Version]
- Perez-Aguilar, J.M.; Shan, J.; LeVine, M.V.; Khelashvili, G.; Weinstein, H. A functional selectivity mechanism at the serotonin-2A GPCR involves ligand-dependent conformations of intracellular loop 2. J. Am. Chem. Soc. 2014, 136, 16044–16054. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.D.; Seung, H.S. Learning the parts of objects by non-negative matrix factorization. Nature 1999, 401, 788–791. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, K. Nonnegative matrix factorization: An analytical and interpretive tool in computational biology. PLoS Comput. Biol. 2008, 4, e1000029. [Google Scholar] [CrossRef] [PubMed]
- Cichocki, A.; Zdunek, R.; Phan, A.H.; Amari, S.-I. Nonnegative Matrix and Tensor Factorizations: Applications to Exploratory Multi-Way Data Analysis and Blind Source Separation; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Devarajan, K.; Cheung, V.C. On nonnegative matrix factorization algorithms for signal-dependent noise with application to electromyography data. Neural Comput. 2014, 26, 1128–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. JMLR 2011, 12, 2825–2830. [Google Scholar]
- Ballesteros, J.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- LeVine, M.V.; Weinstein, H. NbIT—A new information theory-based analysis of allosteric mechanisms reveals residues that underlie function in the leucine transporter LeuT. PLoS Comput. Biol. 2014, 10, e1003603. [Google Scholar] [CrossRef]
- Visiers, I.; Braunheim, B.B.; Weinstein, H. Prokink: A protocol for numerical evaluation of helix distortions by proline. Protein Eng. 2000, 13, 603–606. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Che, T.; Panova, O.; DiBerto, J.F.; Lyu, J.; Krumm, B.E.; Wacker, D.; Robertson, M.J.; Seven, A.B.; Nichols, D.E.; et al. Structure of a Hallucinogen-Activated Gq-Coupled 5-HT2A Serotonin Receptor. Cell 2020, 182, 1574–1588-e19. [Google Scholar] [CrossRef]
- Kimura, K.T.; Asada, H.; Inoue, A.; Kadji, F.M.N.; Im, D.; Mori, C.; Arakawa, T.; Hirata, K.; Nomura, Y.; Nomura, N.; et al. Structures of the 5-HT2A receptor in complex with the antipsychotics risperidone and zotepine. Nat. Struct. Mol. Biol. 2019, 26, 121–128. [Google Scholar] [CrossRef]
- Brogden, R.N.; Sorkin, E.M. Ketanserin. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in hypertension and peripheral vascular disease. Drugs 1990, 40, 903–949. [Google Scholar] [CrossRef]
- LeVine, M.V.; Terry, D.S.; Khelashvili, G.; Siegel, Z.S.; Quick, M.; Javitch, J.A.; Blanchard, S.C.; Weinstein, H. The allosteric mechanism of substrate-specific transport in SLC6 is mediated by a volumetric sensor. Proc. Natl. Acad. Sci. USA 2019, 116, 15947–15956. [Google Scholar] [CrossRef] [Green Version]
- LeVine, M.V.; Cuendet, M.A.; Razavi, A.M.; Khelashvili, G.; Weinstein, H. Thermodynamic Coupling Function Analysis of Allosteric Mechanisms in the Human Dopamine Transporter. Biophys. J. 2018, 114, 10–14. [Google Scholar] [CrossRef] [Green Version]
- Kobilka, B. Nobel Lecture-The Structural Basis of G Protein Coupled Receptor Signaling; Nobel Media AB: Stockholm, Sweden, 2021; Available online: https://www.nobelprize.org (accessed on 10 May 2021).
- Lefkowitz, R.J. Nobel Lecture-A Brief. History of G Protein Coupled Receptors; Nobel Media AB: Durham, NC, USA, 2021; Available online: https://www.nobelprize.org (accessed on 10 May 2021).
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Wootten, D.; Christopoulos, A.; Marti-Solano, M.; Babu, M.M.; Sexton, P.M. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell. Biol. 2018, 19, 638–653. [Google Scholar] [CrossRef]
- Smith, J.S.; Lefkowitz, R.J.; Rajagopal, S. Biased Signalling: From Simple Switches to Allosteric Microprocessors. Nat. Rev. Drug Discov. 2018, 17, 243–260. [Google Scholar] [CrossRef]
- Han, Y.; Moreira, I.S.; Urizar, E.; Weinstein, H.; Javitch, J.A. Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nat. Chem. Biol. 2009, 5, 688–695. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Shi, L.; Filizola, M.; Weinstein, H.; Javitch, J.A. Crosstalk in G protein-coupled receptors: Changes at the transmembrane homodimer interface determine activation. Proc. Natl. Acad. Sci. USA 2005, 102, 17495–17500. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Espigares, I.; Torrens-Fontanals, M.; Tiemann, J.K.S.; Aranda-García, D.; Ramírez-Anguita, J.M.; Stepniewski, T.M.; Worp, N.; Varela-Rial, A.; Morales-Pastor, A.; Medel-Lacruz, B.; et al. GPCRmd uncovers the dynamics of the 3D-GPCRome. Nat. Methods 2020, 17, 777–787. [Google Scholar] [CrossRef]
- Jones, E.M.; Lubock, N.B.; Venkatakrishnan, A.J.; Wang, J.; Tseng, A.M.; Paggi, J.M.; Latorraca, N.R.; Cancilla, D.; Satyadi, M.; Davis, J.E.; et al. Structural and functional characterization of G protein-coupled receptors with deep mutational scanning. Elife 2020, 9. [Google Scholar] [CrossRef]
- Suomivuori, C.-M.; Latorraca, N.R.; Wingler, L.M.; Eismann, S.; King, M.C.; Kleinhenz, A.L.W.; Skiba, M.A.; Staus, D.P.; Kruse, A.C.; Lefkowitz, R.J.; et al. Molecular mechanism of biased signaling in a prototypical G protein-coupled receptor. Science 2020, 367, 881–887. [Google Scholar] [CrossRef]
- Kohlhoff, K.J.; Shukla, D.; Lawrenz, M.; Bowman, G.R.; Konerding, D.E.; Belov, D.; Altman, R.B.; Pande, V.S. Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways. Nat. Chem. 2014, 6, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Noe, F.; Tkatchenko, A.; Muller, K.R.; Clementi, C. Machine Learning for Molecular Simulation. Annu. Rev. Phys. Chem. 2020, 71, 361–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ribeiro, J.M.L.; Tiwary, P. Machine learning approaches for analyzing and enhancing molecular dynamics simulations. Curr. Opin. Struct. Biol. 2020, 61, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yang, D.; Wu, M.; Guo, Y.; Guo, W.; Zhong, L.; Cai, X.; Dai, A.; Jang, W.; Shakhnovich, E.I.; et al. Common activation mechanism of class A GPCRs. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Zhou, X.E.; Gao, X.; He, Y.; Liu, W.; Ishchenko, A.; Barty, A.; White, T.A.; Yefanov, O.; Han, G.W.; et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 2015, 523, 561–567. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. Methods Mol. Biol. 2017, 1654, 39–54. [Google Scholar]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model 2012, 52, 1757–1768. [Google Scholar] [CrossRef]
- Sherman, W.; Beard, H.S.; Farid, R. Use of an induced fit receptor structure in virtual screening. Chem. Biol. Drug Des. 2006, 67, 83–84. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef]
- Razavi, A.M.; Khelashvili, G.; Weinstein, H. A Markov State-based Quantitative Kinetic Model of Sodium Release from the Dopamine Transporter. Sci. Rep. 2017, 7, 40076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehmeyer, C.; Noe, F. Time-lagged autoencoders: Deep learning of slow collective variables for molecular kinetics. J. Chem. Phys. 2018, 148, 241703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhowmik, D.; Gao, S.; Young, M.T.; Ramanathan, A. Deep clustering of protein folding simulations. BMC Bioinform. 2018, 19, 484. [Google Scholar] [CrossRef] [Green Version]
- Degiacomi, M.T. Coupling Molecular Dynamics and Deep Learning to Mine Protein Conformational Space. Structure 2019, 27, 1034–1040-e3. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plante, A.; Weinstein, H. Ligand-Dependent Conformational Transitions in Molecular Dynamics Trajectories of GPCRs Revealed by a New Machine Learning Rare Event Detection Protocol. Molecules 2021, 26, 3059. https://doi.org/10.3390/molecules26103059
Plante A, Weinstein H. Ligand-Dependent Conformational Transitions in Molecular Dynamics Trajectories of GPCRs Revealed by a New Machine Learning Rare Event Detection Protocol. Molecules. 2021; 26(10):3059. https://doi.org/10.3390/molecules26103059
Chicago/Turabian StylePlante, Ambrose, and Harel Weinstein. 2021. "Ligand-Dependent Conformational Transitions in Molecular Dynamics Trajectories of GPCRs Revealed by a New Machine Learning Rare Event Detection Protocol" Molecules 26, no. 10: 3059. https://doi.org/10.3390/molecules26103059
APA StylePlante, A., & Weinstein, H. (2021). Ligand-Dependent Conformational Transitions in Molecular Dynamics Trajectories of GPCRs Revealed by a New Machine Learning Rare Event Detection Protocol. Molecules, 26(10), 3059. https://doi.org/10.3390/molecules26103059