
Natural Compounds as Metabolic Modulators of the Tumor Microenvironment




Abstract
:1. Introduction
2. Cells in the Tumor Microenvironment and Their Metabolic Plasticity
2.1. Malignant Cells
2.2. Immune Cells
2.2.1. Tumor-Associated Macrophages
2.2.2. T Lymphocytes
2.2.3. Natural Killer Cells
2.2.4. Dendritic Cells
2.3. Nonimmune Stromal Cells
2.3.1. Cancer-Associated Fibroblasts
2.3.2. Tumor Endothelial Cells
2.3.3. Cancer-Associated Adipocytes
3. Metabolic Modulation of TME Cells by Natural Compounds
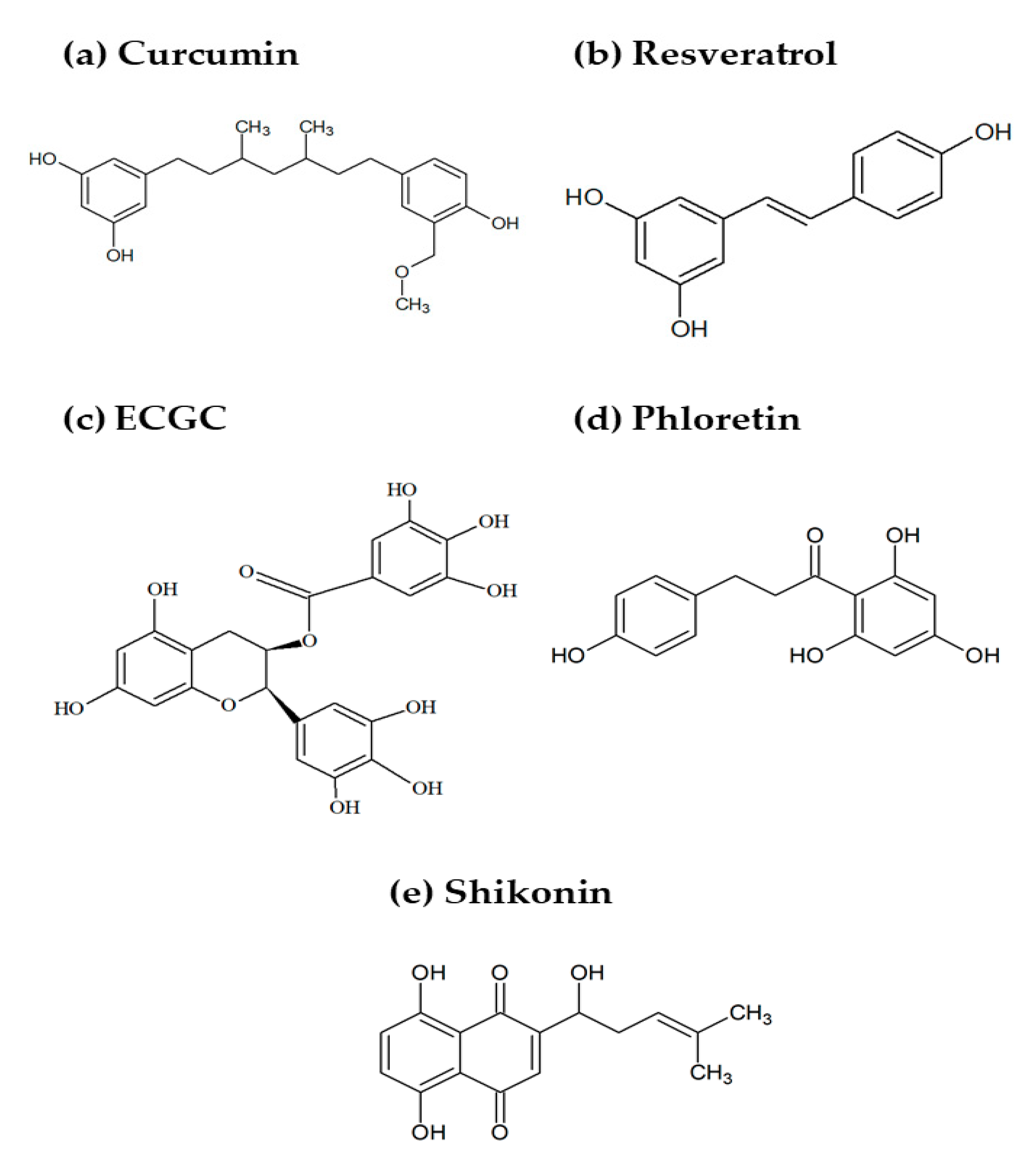
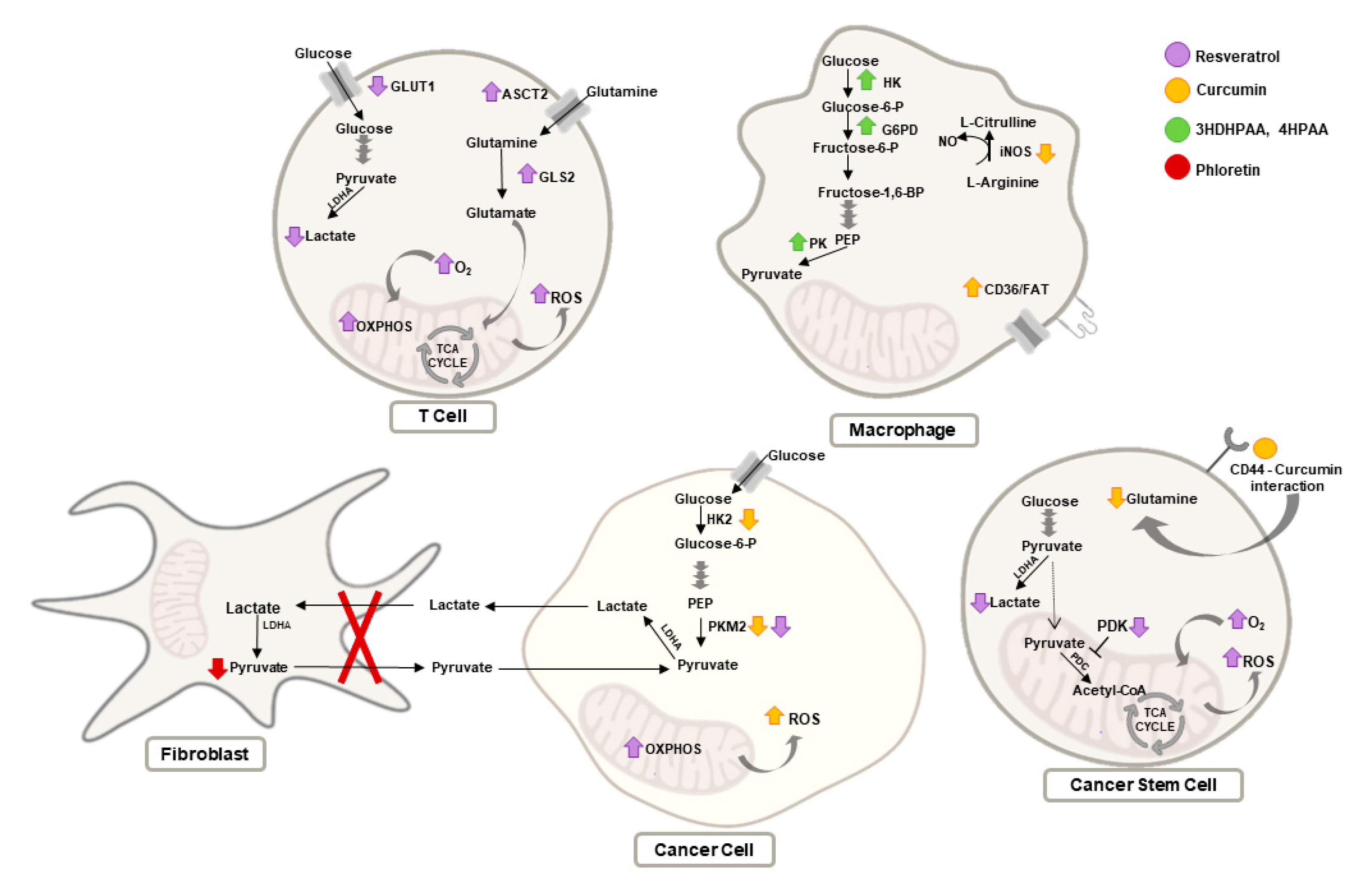
3.1. Curcumin
3.2. Resveratrol
3.3. Epigallocatechin Gallate
3.4. Phloretin
3.5. Shikonin
3.6. Other Natural Compounds
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar] [PubMed]
- Romero-Garcia, S.; Lopez-Gonzalez, J.S.; Báez-Viveros, J.L.; Aguilar-Cazares, D.; Prado-Garcia, H. Tumor cell metabolism: An integral view. Cancer Biol. Ther. 2011, 12, 939–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, A.S.; Almeida, C.R.; Helguero, L.; Duarte, I.F. Metabolic crosstalk in the breast cancer microenvironment. Eur. J. Cancer 2019, 121, 154–171. [Google Scholar] [CrossRef]
- De La Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic reprogramming of the tumour microenvironment. FEBS J. 2015, 282, 3892–3898. [Google Scholar] [CrossRef] [PubMed]
- Zyad, A.; Leouifoudi, I.; Tilaoui, M.; Mouse, H.A.; Khouchani, M.; Jaafari, A. Natural Products as Cytotoxic Agents in Chemotherapy against Cancer. Cytotoxicity 2018. [Google Scholar] [CrossRef] [Green Version]
- Liskova, A.; Samec, M.; Koklesova, L.; Brockmueller, A.; Zhai, K.; Abdellatif, B.; Siddiqui, M.; Biringer, K.; Kudela, E.; Pec, M.; et al. Flavonoids as an effective sensitizer for anti-cancer therapy: Insights into multi-faceted mechanisms and applicability towards individualized patient profiles. EPMA J. 2021, 17, 1–22. [Google Scholar] [CrossRef]
- Park, S.; Surh, Y. Modulation of tumor microenvironment by chemopreventive natural products. Ann. N. Y. Acad. Sci. 2017, 1401, 65–74. [Google Scholar] [CrossRef]
- Zubair, H.; Khan, M.A.; Anand, S.; Srivastava, S.K.; Singh, S.; Singh, A.P. Modulation of the tumor microenvironment by natural agents: Implications for cancer prevention and therapy. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Pan, P.; Huang, Y.-W.; Oshima, K.; Yearsley, M.; Zhang, J.; Arnold, M.; Yu, J.; Wang, L.-S. The immunomodulatory potential of natural compounds in tumor-bearing mice and humans. Crit. Rev. Food Sci. Nutr. 2019, 59, 992–1007. [Google Scholar] [CrossRef]
- Focaccetti, C.; Izzi, V.; Benvenuto, M.; Fazi, S.; Ciuffa, S.; Giganti, M.G.; Potenza, V.; Manzari, V.; Modesti, A.; Bei, R. Polyphenols as Immunomodulatory Compounds in the Tumor Microenvironment: Friends or Foes? Int. J. Mol. Sci. 2019, 20, 1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samec, M.; Liskova, A.; Koklesova, L.; Samuel, S.M.; Zhai, K.; Buhrmann, C.; Varghese, E.; Abotaleb, M.; Qaradakhi, T.; Zulli, A.; et al. Flavonoids against the Warburg phenotype—concepts of predictive, preventive and personalised medicine to cut the Gordian knot of cancer cell metabolism. EPMA J. 2020, 11, 377–398. [Google Scholar] [CrossRef]
- Guerra, A.R.; Duarte, M.F.; Duarte, I.F. Targeting Tumor Metabolism with Plant-Derived Natural Products: Emerging Trends in Cancer Therapy. J. Agric. Food Chem. 2018, 66, 10663–10685. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Heiden, M.G.V.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.-A.; Pan, S.-C.; Chu, I.; Lai, R.-Y.; Wei, Y.-H. Targeting cancer stem cells from a metabolic perspective. Exp. Biol. Med. 2020, 245, 465–476. [Google Scholar] [CrossRef] [Green Version]
- Das, M.; Law, S. Role of tumor microenvironment in cancer stem cell chemoresistance and recurrence. Int. J. Biochem. Cell Biol. 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Peiris-Pagès, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 1–10. [Google Scholar] [CrossRef]
- De Luca, A.; Fiorillo, M.; Peiris-Pagès, M.; Ozsvari, B.; Smith, D.L.; Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Cappello, A.R.; Pezzi, V.; Lisanti, M.P.; et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget 2015, 6, 14777–14795. [Google Scholar] [CrossRef] [Green Version]
- Farnie, G.; Sotgia, F.; Lisanti, M.P. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget 2015, 6, 30472–30486. [Google Scholar] [CrossRef] [Green Version]
- Patil, S. Metformin treatment decreases the expression of cancer stem cell marker CD44 and stemness related gene expression in primary oral cancer cells. Arch. Oral Biol. 2020, 113, 104710. [Google Scholar] [CrossRef] [PubMed]
- Song, C.W.; Lee, H.; Dings, R.; Williams, B.; Powers, J.; Dos Santos, T.; Choi, B.-H.; Park, H.J. Metformin kills and radiosensitizes cancer cells and preferentially kills cancer stem cells. Sci. Rep. 2012, 2, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef] [PubMed]
- Rhee, I. Diverse macrophages polarization in tumor microenvironment. Arch. Pharmacal Res. 2016, 39, 1588–1596. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Kurahara, H.; Shinchi, H.; Mataki, Y.; Maemura, K.; Noma, H.; Kubo, F.; Sakoda, M.; Ueno, S.; Natsugoe, S.; Takao, S. Significance of M2-Polarized Tumor-Associated Macrophage in Pancreatic. Cancer J. Surg. Res. 2011, 167, e211–e219. [Google Scholar] [CrossRef] [PubMed]
- Lan, C.; Huang, X.; Lin, S.; Huang, H.; Cai, Q.; Wan, T.; Lu, J.; Liu, J. Expression of M2-Polarized Macrophages is Associated with Poor Prognosis for Advanced Epithelial Ovarian Cancer. Technol. Cancer Res. Treat. 2013, 12, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Haque, A.S.M.R.; Moriyama, M.; Kubota, K.; Ishiguro, N.; Sakamoto, M.; Chinju, A.; Mochizuki, K.; Sakamoto, T.; Kaneko, N.; Munemura, R.; et al. CD206+ tumor-associated macrophages promote proliferation and invasion in oral squamous cell carcinoma via EGF production. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Benner, B.; Scarberry, L.; Suarez-Kelly, L.P.; Duggan, M.C.; Campbell, A.R.; Smith, E.; Lapurga, G.; Jiang, K.; Butchar, J.P.; Tridandapani, S.; et al. Generation of monocyte-derived tumor-associated macrophages using tumor-conditioned media provides a novel method to study tumor-associated macrophages in vitro. J. Immunother. Cancer 2019, 7, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.M.; Davies, L.C.; Karwan, M.; Ileva, L.; Ozaki, M.; Cheng, R.Y.; Ridnour, L.A.; Annunziata, C.M.; Wink, D.A.; McVicar, D.W. Itaconic acid mediates crosstalk between macrophage metabolism and peritoneal tumors. J. Clin. Investig. 2018, 128, 3794–3805. [Google Scholar] [CrossRef]
- Saha, S.; Shalova, I.N.; Biswas, S.K. Metabolic regulation of macrophage phenotype and function. Immunol. Rev. 2017, 280, 102–111. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunology 2019, 10, 1462. [Google Scholar] [CrossRef] [Green Version]
- Khan, F.H.; Dervan, E.; Bhattacharyya, D.D.; McAuliffe, J.D.; Miranda, K.M.; Glynn, S.A. The Role of Nitric Oxide in Cancer: Master Regulator or NOt? Int. J. Mol. Sci. 2020, 21, 9393. [Google Scholar] [CrossRef] [PubMed]
- Nath, N.; Kashfi, K. Tumor associated macrophages and ‘NO’. Biochem. Pharmacol. 2020, 176, 113899. [Google Scholar] [CrossRef]
- Kouidhi, S.; Elgaaied, A.B.; Chouaib, S. Impact of Metabolism in on T-Cell Differentiation and Function and Cross Talk with Tumor Microenvironment. Front. Immunol. 2017, 8, 270. [Google Scholar] [CrossRef] [Green Version]
- Saleh, R.; Elkord, E. FoxP3+ T regulatory cells in cancer: Prognostic biomarkers and therapeutic targets. Cancer Lett. 2020, 490, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Bai, L.; Li, W.; Zeng, T.; Tian, H.; Cui, J. Targeting T cell metabolism in the tumor microenvironment: An anti-cancer therapeutic strategy. J. Exp. Clin. Cancer Res. 2019, 38, 1–10. [Google Scholar] [CrossRef]
- Altman, B.J.; Dang, C.V. Normal and cancer cell metabolism: Lymphocytes and lymphoma. FEBS J. 2012, 279, 2598–2609. [Google Scholar] [CrossRef]
- Carr, E.L.; Kelman, A.; Wu, G.S.; Gopaul, R.; Senkevitch, E.; Aghvanyan, A.; Turay, A.M.; Frauwirth, K.A. Glutamine Uptake and Metabolism Are Coordinately Regulated by ERK/MAPK during T Lymphocyte Activation. J. Immunol. 2010, 185, 1037–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Liu, W.; Hu, B.; Wang, P.; Lv, X.; Chen, S.; Shao, Z. Prognostic Significance of Tumor-Infiltrating Natural Killer Cells in Solid Tumors: A Systematic Review and Meta-Analysis. Front. Immunol. 2020, 11, 1242. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, B.; Mandelboim, O. Sweet Killers: NK Cells Need Glycolysis to Kill Tumors. Cell Metab. 2018, 28, 183–184. [Google Scholar] [CrossRef]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-Derived Lactate Modifies Antitumor Immune Response: Effect on Myeloid-Derived Suppressor Cells and NK Cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Harmon, C.; Robinson, M.W.; Hand, F.; AlMuaili, D.; Mentor, K.; Houlihan, D.D.; Hoti, E.; Lynch, L.; Geoghegan, J.; O’Farrelly, C. Lactate-Mediated Acidification of Tumor Microenvironment Induces Apoptosis of Liver-Resident NK Cells in Colorectal Liver Metastasis. Cancer Immunol. Res. 2018, 7, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Plebanek, M.P.; Sturdivant, M.; DeVito, N.C.; Hanks, B.A. Role of dendritic cell metabolic reprogramming in tumor immune evasion. Int. Immunol. 2020, 32, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Sharma, M.D.; Lee, J.R.; Jhaver, K.G.; Johnson, T.S.; Keskin, D.B.; Marshall, B.; Chandler, P.; Antonia, S.J.; Burgess, R.; et al. Potential Regulatory Function of Human Dendritic Cells Expressing Indoleamine 2,3-Dioxygenase. Science 2002, 297, 1867–1870. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Investig. 2007, 117, 1147–1154. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.D.; Baban, B.; Chandler, P.; Hou, D.-Y.; Singh, N.; Yagita, H.; Azuma, M.; Blazar, B.R.; Mellor, A.L.; Munn, D.H. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J. Clin. Investig. 2007, 117, 2570–2582. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Malinarich, F.; Duan, K.; Hamid, R.A.; Bijin, A.; Lin, W.X.; Poidinger, M.; Fairhurst, A.-M.; Connolly, J.E. High Mitochondrial Respiration and Glycolytic Capacity Represent a Metabolic Phenotype of Human Tolerogenic Dendritic Cells. J. Immunol. 2015, 194, 5174–5186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Z.; Tan, Z.W.; Zhu, P.; Tan, N.S. Cancer-associated fibroblasts in tumor microenvironment–Accomplices in tumor malignancy. Cell. Immunol. 2019, 343, 103729. [Google Scholar] [CrossRef] [PubMed]
- Karta, J.; Bossicard, Y.; Kotzamanis, K.; Dolznig, H.; Letellier, E. Mapping the Metabolic Networks of Tumor Cells and Cancer-Associated Fibroblasts. Cells 2021, 10, 304. [Google Scholar] [CrossRef]
- Nagl, L.; Horvath, L.; Pircher, A.; Wolf, D. Tumor Endothelial Cells (TECs) as Potential Immune Directors of the Tumor Microenvironment–New Findings and Future Perspectives. Front. Cell Dev. Biol. 2020, 8, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Cantelmo, A.R.; Conradi, L.-C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.-A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef] [Green Version]
- Zecchin, A.; Kalucka, J.; Dubois, C.; Carmeliet, P. How Endothelial Cells Adapt Their Metabolism to Form Vessels in Tumors. Front. Immunol. 2017, 8, 1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diebold, L.P.; Gil, H.J.; Gao, P.; Martinez, C.A.; Weinberg, S.; Chandel, N.S. Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat. Metab. 2019, 1, 158–171. [Google Scholar] [CrossRef]
- Huang, H.; Vandekeere, S.; Kalucka, J.; Bierhansl, L.; Zecchin, A.; Brüning, U.; Visnagri, A.; Yuldasheva, N.Y.; Goveia, J.; Cruys, B.; et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017, 36, 2334–2352. [Google Scholar] [CrossRef]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty Acid Carbon Is Essential for Dntp Synthesis in Endothelial Cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y. Adipocyte and lipid metabolism in cancer drug resistance. J. Clin. Investig. 2019, 129, 3006–3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.-A.; Xing, X.; Harris, J.W.; Zaytseva, Y.Y.; I Mitov, M.; Napier, D.L.; Weiss, H.L.; Evers, B.M.; Gao, T. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. 2017, 8, e2593. [Google Scholar] [CrossRef]
- Wu, Q.; Li, J.; Li, Z.; Sun, S.; Zhu, S.; Wang, L.; Wu, J.; Yuan, J.; Zhang, Y.; Sun, S.; et al. Exosomes from the tumour-adipocyte interplay stimulate beige/brown differentiation and reprogram metabolism in stromal adipocytes to promote tumour progression. J. Exp. Clin. Cancer Res. 2019, 38, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Clement, E.; Lazar, I.; Attané, C.; Carrié, L.; Dauvillier, S.; Ducoux-Petit, M.; Esteve, D.; Menneteau, T.; Moutahir, M.; Le Gonidec, S.; et al. Adipocyte extracellular vesicles carry enzymes and fatty acids that stimulate mitochondrial metabolism and remodeling in tumor cells. EMBO J. 2020, 39, e102525. [Google Scholar] [CrossRef] [PubMed]
- Francescone, R.; Vendramini-Costa, D.B.; Franco-Barraza, J.; Wagner, J.; Muir, A.; Lau, A.N.; Gabitova, L.; Pazina, T.; Gupta, S.; Luong, T.; et al. Netrin G1 Promotes Pancreatic Tumorigenesis through Cancer-Associated Fibroblast–Driven Nutritional Support and Immunosuppression. Cancer Discov. 2021, 11, 446–479. [Google Scholar] [CrossRef]
- Yang, L.; Achreja, A.; Yeung, T.-L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, A.; Tommonaro, G. Curcumin and Cancer. Nutrients 2019, 11, 2376. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Du, H.; Zhang, G.; Wu, Y.; Qiu, P.; Liu, J.; Guo, J.; Liu, X.; Sun, L.; Du, B.; et al. Curcumin plays a synergistic role in combination with HSV-TK/GCV in inhibiting growth of murine B16 melanoma cells and melanoma xenografts. PeerJ 2019, 7, e7760. [Google Scholar] [CrossRef] [PubMed]
- Šudomová, M.; Hassan, S. Nutraceutical Curcumin with Promising Protection against Herpesvirus Infections and Their Associated Inflammation: Mechanisms and Pathways. Microorganisms 2021, 9, 292. [Google Scholar] [CrossRef] [PubMed]
- Vishvakarma, N.K. Novel antitumor mechanisms of curcumin: Implication of altered tumor metabolism, reconstituted tumor microenvironment and augmented myelopoiesis. Phytochem. Rev. 2014, 13, 717–724. [Google Scholar] [CrossRef]
- Wang, K.; Fan, H.; Chen, Q.; Ma, G.; Zhu, M.; Zhang, X.; Zhang, Y.; Yu, J. Curcumin inhibits aerobic glycolysis and induces mitochondrial-mediated apoptosis through hexokinase II in human colorectal cancer cells in vitro. Anti-Cancer Drugs 2015, 26, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, F.A.; Prakasam, G.; Chattopadhyay, S.; Rehman, A.U.; Padder, R.A.; Ansari, M.A.; Irshad, R.; Mangalhara, K.; Bamezai, R.N.K.; Husain, M.; et al. Curcumin decreases Warburg effect in cancer cells by down-regulating pyruvate kinase M2 via mTOR-HIF1α inhibition. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Fang, X.-L.; Zhen, Q.; Chen, Q.-Y.; Feng, C. Mitochondrial targeting nano-curcumin for attenuation on PKM2 and FASN. Colloids Surf. B Biointerfaces 2019, 182, 110405. [Google Scholar] [CrossRef]
- Soni, V.K.; Shukla, D.; Kumar, A.; Vishvakarma, N.K. Curcumin circumvent lactate-induced chemoresistance in hepatic cancer cells through modulation of hydroxycarboxylic acid receptor-1. Int. J. Biochem. Cell Biol. 2020, 123, 105752. [Google Scholar] [CrossRef]
- Fan, H.; Tian, W.; Ma, X. Curcumin induces apoptosis of HepG2 cells via inhibiting fatty acid synthase. Target. Oncol. 2014, 9, 279–286. [Google Scholar] [CrossRef]
- Younesian, O.; Kazerouni, F.; Dehghan-Nayeri, N.; Omrani, D.; Rahimipour, A.; Shanaki, M.; Kalkhoran, M.R.; Cheshmi, F. Effect of Curcumin on Fatty Acid Synthase Expression and Enzyme Activity in Breast Cancer Cell Line SKBR3. Int. J. Cancer Manag. 2017, 10. [Google Scholar] [CrossRef]
- Bianchi, G.; Ravera, S.; Traverso, C.; Amaro, A.; Piaggio, F.; Emionite, L.; Bachetti, T.; Pfeffer, U.; Raffaghello, L. Curcumin induces a fatal energetic impairment in tumor cells in vitro and in vivo by inhibiting ATP-synthase activity. Carcinogenesis 2018, 39, 1141–1150. [Google Scholar] [CrossRef]
- Sordillo, P.P.; Helson, L. Curcumin and cancer stem cells: Curcumin has asymmetrical effects on cancer and normal stem cells. Anticancer Res. 2015, 35, 599–614. [Google Scholar]
- Huang, Y.-T.; Lin, Y.-W.; Chiu, H.-M.; Chiang, B.-H. Curcumin Induces Apoptosis of Colorectal Cancer Stem Cells by Coupling with CD44 Marker. J. Agric. Food Chem. 2016, 64, 2247–2253. [Google Scholar] [CrossRef]
- Nakagawa, K.; Zingg, J.-M.; Kim, S.H.; Thomas, M.J.; Dolnikowski, G.G.; Azzi, A.; Miyazawa, T.; Meydani, M. Differential cellular uptake and metabolism of curcuminoids in monocytes/macrophages: Regulatory effects on lipid accumulation. Br. J. Nutr. 2014, 112, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingg, J.-M.; Hasan, S.T.; Cowan, D.; Ricciarelli, R.; Azzi, A.; Meydani, M. Regulatory effects of curcumin on lipid accumulation in monocytes/macrophages. J. Cell. Biochem. 2012, 113, 833–840. [Google Scholar] [CrossRef]
- Han, Y.; Jo, H.; Cho, J.H.; Dhanasekaran, D.N.; Song, Y.S. Resveratrol as a Tumor-Suppressive Nutraceutical Modulating Tumor Microenvironment and Malignant Behaviors of Cancer. Int. J. Mol. Sci. 2019, 20, 925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Chen, K.; Cheng, L.; Yan, B.; Qian, W.; Cao, J.; Liang, C.; Wu, E.; Ma, Q.; Yang, W. Resveratrol and cancer treatment: Updates. Ann. N. Y. Acad. Sci. 2017, 1403, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Brockmueller, A.; Sameri, S.; Liskova, A.; Zhai, K.; Varghese, E.; Samuel, S.M.; Büsselberg, D.; Kubatka, P.; Shakibaei, M. Resveratrol’s Anti-Cancer Effects through the Modulation of Tumor Glucose Metabolism. Cancers 2021, 13, 188. [Google Scholar] [CrossRef]
- Jia, L.; Gao, Y.; Zhou, T.; Zhao, X.-L.; Hu, H.-Y.; Chen, D.-W.; Qiao, M.-X. Enhanced response to PD-L1 silencing by modulation of TME via balancing glucose metabolism and robust co-delivery of siRNA/Resveratrol with dual-responsive polyplexes. Biomaterials 2021, 271, 120711. [Google Scholar] [CrossRef]
- Shen, Y.-A.; Lin, C.-H.; Chi, W.-H.; Wang, C.-Y.; Hsieh, Y.-T.; Wei, Y.-H.; Chen, Y.-J. Resveratrol Impedes the Stemness, Epithelial-Mesenchymal Transition, and Metabolic Reprogramming of Cancer Stem Cells in Nasopharyngeal Carcinoma through p53 Activation. Evid.-Based Complement. Altern. Med. 2013, 2013, 590393. [Google Scholar] [CrossRef]
- Craveiro, M.; Cretenet, G.; Mongellaz, C.; Matias, M.I.; Caron, O.; De Lima, M.C.P.; Zimmermann, V.S.; Solary, E.; Dardalhon, V.; Dulić, V.; et al. Resveratrol stimulates the metabolic reprogramming of human CD4+T cells to enhance effector function. Sci. Signal. 2017, 10, eaal3024. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.-H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.-C. Inflammatory T Cell Responses Rely on Amino Acid Transporter ASCT2 Facilitation of Glutamine Uptake and mTORC1 Kinase Activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Zhang, Z.; Han, Y.; Wang, J.; Wang, Y.; Chen, X.; Shao, Y.; Cheng, Y.; Zhou, W.; Lu, X.; et al. A review on anti-cancer effect of green tea catechins. J. Funct. Foods 2020, 74, 104172. [Google Scholar] [CrossRef]
- Wei, R.; Mao, L.; Xu, P.; Zheng, X.; Hackman, R.M.; Mackenzie, G.G.; Wang, Y. Suppressing glucose metabolism with epigallocatechin-3-gallate (EGCG) reduces breast cancer cell growth in preclinical models. Food Funct. 2018, 9, 5682–5696. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, S.; Yang, J.; Yi, P.; Xu, P.; Yi, M.; Peng, W. Integrated transcriptomic and metabolomic analyses to characterize the anti-cancer effects of (−)-epigallocatechin-3-gallate in human colon cancer cells. Toxicol. Appl. Pharmacol. 2020, 401, 115100. [Google Scholar] [CrossRef]
- Chu, K.O.; Chan, K.P.; Chan, S.O.; Ng, T.K.; Jhanji, V.; Wang, C.-C.; Pang, C.P. Metabolomics of Green-Tea Catechins on Vascular-Endothelial-Growth-Factor-Stimulated Human-Endothelial-Cell Survival. J. Agric. Food Chem. 2018, 66, 12866–12875. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.-H.; Ho, C.-T.; Chen, Z.-F.; Chen, L.-C.; Whang-Peng, J.; Lin, T.-N.; Ho, Y.-S. The apple polyphenol phloretin inhibits breast cancer cell migration and proliferation via inhibition of signals by type 2 glucose transporter. J. Food Drug Anal. 2018, 26, 221–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-T.; Tu, S.-H.; Yang, P.-S.; Hsu, S.-P.; Lee, W.-H.; Ho, C.-T.; Wu, C.-H.; Lai, Y.-H.; Chen, M.-Y.; Chen, L.-C. Apple Polyphenol Phloretin Inhibits Colorectal Cancer Cell Growth via Inhibition of the Type 2 Glucose Transporter and Activation of p53-Mediated Signaling. J. Agric. Food Chem. 2016, 64, 6826–6837. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.B.; Ackerstaff, E.; Serganova, I.S.; Kerrigan, J.E.; Blasberg, R.G.; Koutcher, J.A.; Banerjee, D. Tumor stroma interaction is mediated by monocarboxylate metabolism. Exp. Cell Res. 2017, 352, 20–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Přenosil, J.E.; Kut, Ö.M.; Dunn, I.J.; Heinzle, E. Biocatalysis, 2. Immobilized Biocatalysts. Ullmann’s Encycl. Ind. Chem. 2009, 5, 477–527. [Google Scholar] [CrossRef]
- Guo, C.; He, J.; Song, X.; Tan, L.; Wang, M.; Jiang, P.; Li, Y.; Cao, Z.; Peng, C. Pharmacological properties and derivatives of shikonin—A review in recent years. Pharmacol. Res. 2019, 149. [Google Scholar] [CrossRef]
- Boulos, J.C.; Rahama, M.; Hegazy, M.-E.F.; Efferth, T. Shikonin derivatives for cancer prevention and therapy. Cancer Lett. 2019, 459, 248–267. [Google Scholar] [CrossRef]
- Zhao, X.; Zhu, Y.; Hu, J.; Jiang, L.; Li, L.; Jia, S.; Zen, K. Shikonin Inhibits Tumor Growth in Mice by Suppressing Pyruvate Kinase M2-mediated Aerobic Glycolysis. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef]
- Liu, B.; Jin, J.; Zhang, Z.; Zuo, L.; Jiang, M.; Xie, C. Shikonin exerts antitumor activity by causing mitochondrial dysfunction in hepatocellular carcinoma through PKM2–AMPK–PGC1α signaling pathway. Biochem. Cell Biol. 2019, 97, 397–405. [Google Scholar] [CrossRef]
- Tao, T.; Su, Q.; Xu, S.; Deng, J.; Zhou, S.; Zhuang, Y.; Huang, Y.; He, C.; He, S.; Peng, M.; et al. Down-regulation of PKM2 decreases FASN expression in bladder cancer cells through AKT/mTOR/SREBP-1c axis. J. Cell. Physiol. 2019, 234, 3088–3104. [Google Scholar] [CrossRef]
- Wang, H.; Tang, Y.; Fang, Y.; Zhang, M.; Wang, H.; He, Z.; Wang, B.; Xu, Q.; Huang, Y. Reprogramming Tumor Immune Microenvironment (TIME) and Metabolism via Biomimetic Targeting Codelivery of Shikonin/JQ1. Nano Lett. 2019, 19, 2935–2944. [Google Scholar] [CrossRef] [PubMed]
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J.; et al. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and rec-ommendations for a universal nomenlature. Nat. Prod. Rep. 2013, 30, 108–160. [Google Scholar] [CrossRef]
- Dahlem, C.; Siow, W.X.; Lopatniuk, M.; Tse, W.K.F.; Kessler, S.M.; Kirsch, S.H.; Hoppstädter, J.; Vollmar, A.M.; Müller, R.; Luzhetskyy, A.; et al. Thioholgamide A, a New Anti-Proliferative Anti-Tumor Agent, Modulates Macrophage Polarization and Metabolism. Cancers 2020, 12, 1288. [Google Scholar] [CrossRef]
- Liu, M.; Luo, F.; Ding, C.; Albeituni, S.; Hu, X.; Ma, Y.; Cai, Y.; McNally, L.R.; Sanders, M.A.; Jain, D.; et al. Dectin-1 Activation by a Natural Product β-Glucan Converts Immunosuppressive Macrophages into an M1-like Phenotype. J. Immunol. 2015, 195, 5055–5065. [Google Scholar] [CrossRef]
- Carrasco-Pozo, C.; Ni Tan, K.; Avery, V.M. Hemin Prevents Increased Glycolysis in Macrophages upon Activation: Protection by Microbiota-Derived Metabolites of Polyphenols. Antioxidants 2020, 9, 1109. [Google Scholar] [CrossRef]
- Ma, J.-L.; Yang, P.-Y.; Rui, Y.-C.; Lu, L.; Kang, H.; Zhang, J. Hemin modulates cytokine expressions in macrophage-derived foam cells via heme oxygenase-1 induction. J. Pharmacol. Sci. 2007, 103, 261–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.; Song, H.; Cai, W.; Shen, X. Real Time Monitoring of Inhibition of Adipogenesis and Angiogenesis by (−)-Epigallocatechin-3-Gallate in 3T3-L1 Adipocytes and Human Umbilical Vein Endothelial Cells. Nutrients 2015, 7, 8871–8886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, T.; Huang, C.; Yang, C.; Jiang, T.; Su, J.; Chen, M.; Fatima, S.; Gong, R.; Hu, X.; Bian, Z.; et al. Apigenin inhibits STAT3/CD36 signaling axis and reduces visceral obesity. Pharmacol. Res. 2020, 152, 104586. [Google Scholar] [CrossRef]
- Pérez-Jiménez, A.; Rufino-Palomares, E.E.; Fernández-Gallego, N.; Ortuño-Costela, M.C.; Reyes-Zurita, F.J.; Peragón, J.; Salguero, E.L.G.; Mokhtari, K.; Medina, P.P.; Lupiáñez, J.A. Target molecules in 3T3-L1 adipocytes differentiation are regulated by maslinic acid, a natural triterpene from Olea europaea. Phytomedicine 2016, 23, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, J.; Heinrich, M.A.; Teixeira, L.M.; Prakash, J. 3D In Vitro Model (R)evolution: Unveiling Tumor–Stroma Interactions. Trends Cancer 2021, 7, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Gurav, P. PhytoNanotechnology: Enhancing Delivery of Plant Based Anti-cancer Drugs. Front. Pharmacol. 2018, 8, 1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Cells in the TME | Phenotypic Features | Metabolic Features | Ref. |
|---|---|---|---|
| Malignant Cells | |||
| Cancer Cells | Proangiogenic Invasion and metastasis Immune evasion Immunosuppression | ↑ Aerobic glycolysis (lactate secretion) ↑ Glutaminolysis ↑ PPP ↑ One-carbon metabolism ↑ de novo lipid synthesis | [7,17,18] |
| Cancer Stem Cells (CSCs) | Expression of surface markers (CD44, CD133 or ALDH1) Stemness potential Prometastatic Protumorigenic Resistance to chemo/-radiation | Mitochondrial respiration (↑ mitochondrial mass, ↑ oxygen consumption) | [19,20,21,22,23] |
| Immune Cells | |||
| Tumor-Associated Macrophages | M1-like phenotype: - Proinflammatory - Tumoricidal functions | ↑ Glycolysis Citrate and succinate accumulation | [27,28,29,30,31,32,33] |
| M2-like phenotype: - Anti-inflammatory - Protumorigenic - Positive correlation with poor prognosis in cancer patients | ↑ TCA cycle and OXPHOS Itaconate production | ||
| T Lymphocytes | Cytotoxic T cells (Tc): - Antitumoral activities | Aerobic glycolysis (↑ glucose uptake, ↑ PPP, ↑ glutamine metabolism) | [38,39,40,41,42,43] |
| Regulatory T cells (Treg): - Inhibition of Tc activity - Positive correlation with poor prognosis in cancer patients | ↑ OXPHOS | ||
| Natural Killer Cells (NK cells) | Antitumoral activity Positive correlation with favorable prognosis in cancer patients | ↑ Glycolysis and OXPHOS to enhance cytotoxic capacity | [44,45,46,47] |
| Dendritic Cells (DCs) | Mature/activated DCs: - Activation of T lymphocytes - Induction of adaptive immune response | ↑ Glycolysis during activation | [48,49,50,51,52,53] |
| Tolerogenic DCs: - Inhibition of immune response - ↑ Expression of IDO - Promotion of Treg - Suppression of Tc | ↑ TCA cycle | ||
| Nonimmune Stromal Cells | |||
| Cancer-Associated Fibroblasts (CAFs) | Protumoral activity: - Modulation of cancer metastasis - ECM remodeling - Therapy resistance | Reverse Warburg effect (aerobic glycolysis) ↑ Glutamate and glutamine secretion (↑ glutamine synthetase) | [54,55,66,67] |
| Tumor Endothelial Cells (TECs) | Promotion of angiogenesis: - Highly proliferative - Self-renewal potential Can act as APC | ↑ Glycolysis ↑ Mitochondrial respiration (mitochondrial complex III), glutaminolysis and FAO | [56,57,58,59] |
| Cancer-Associated Adipocytes (CAA) | Promotion of tumor growth and invasion: - Release of adipokines, growth factors and hormones - Recruitment of immune cells - Production of proteases | ↑ Lipolysis and lipid release | [62,63,64,65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dias, A.S.; Helguero, L.; Almeida, C.R.; Duarte, I.F. Natural Compounds as Metabolic Modulators of the Tumor Microenvironment. Molecules 2021, 26, 3494. https://doi.org/10.3390/molecules26123494
Dias AS, Helguero L, Almeida CR, Duarte IF. Natural Compounds as Metabolic Modulators of the Tumor Microenvironment. Molecules. 2021; 26(12):3494. https://doi.org/10.3390/molecules26123494
Chicago/Turabian StyleDias, Ana S., Luisa Helguero, Catarina R. Almeida, and Iola F. Duarte. 2021. "Natural Compounds as Metabolic Modulators of the Tumor Microenvironment" Molecules 26, no. 12: 3494. https://doi.org/10.3390/molecules26123494
APA StyleDias, A. S., Helguero, L., Almeida, C. R., & Duarte, I. F. (2021). Natural Compounds as Metabolic Modulators of the Tumor Microenvironment. Molecules, 26(12), 3494. https://doi.org/10.3390/molecules26123494