
High-Solids, Solvent-Free Modification of Engineered Polysaccharides

 ,
,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
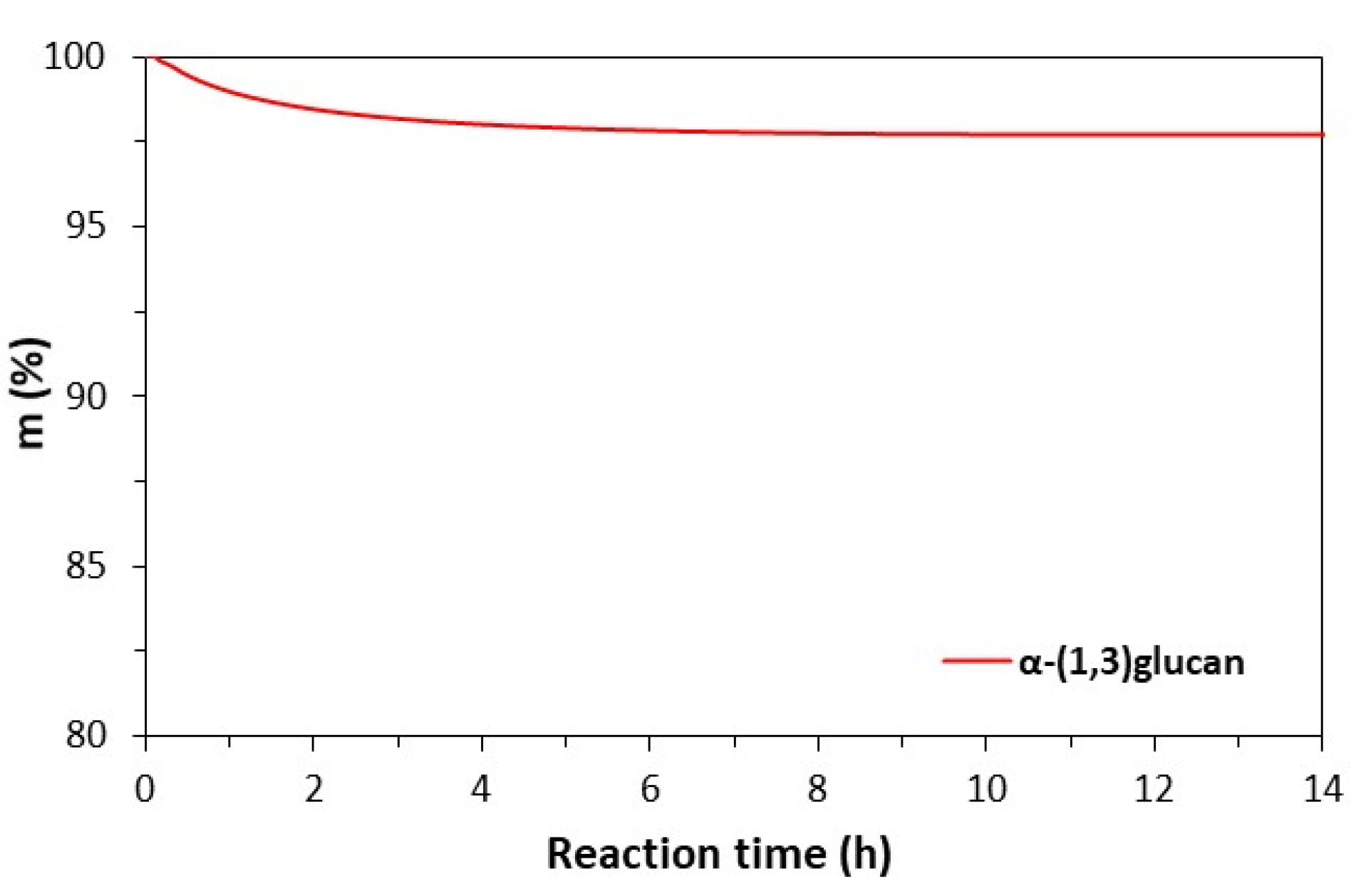
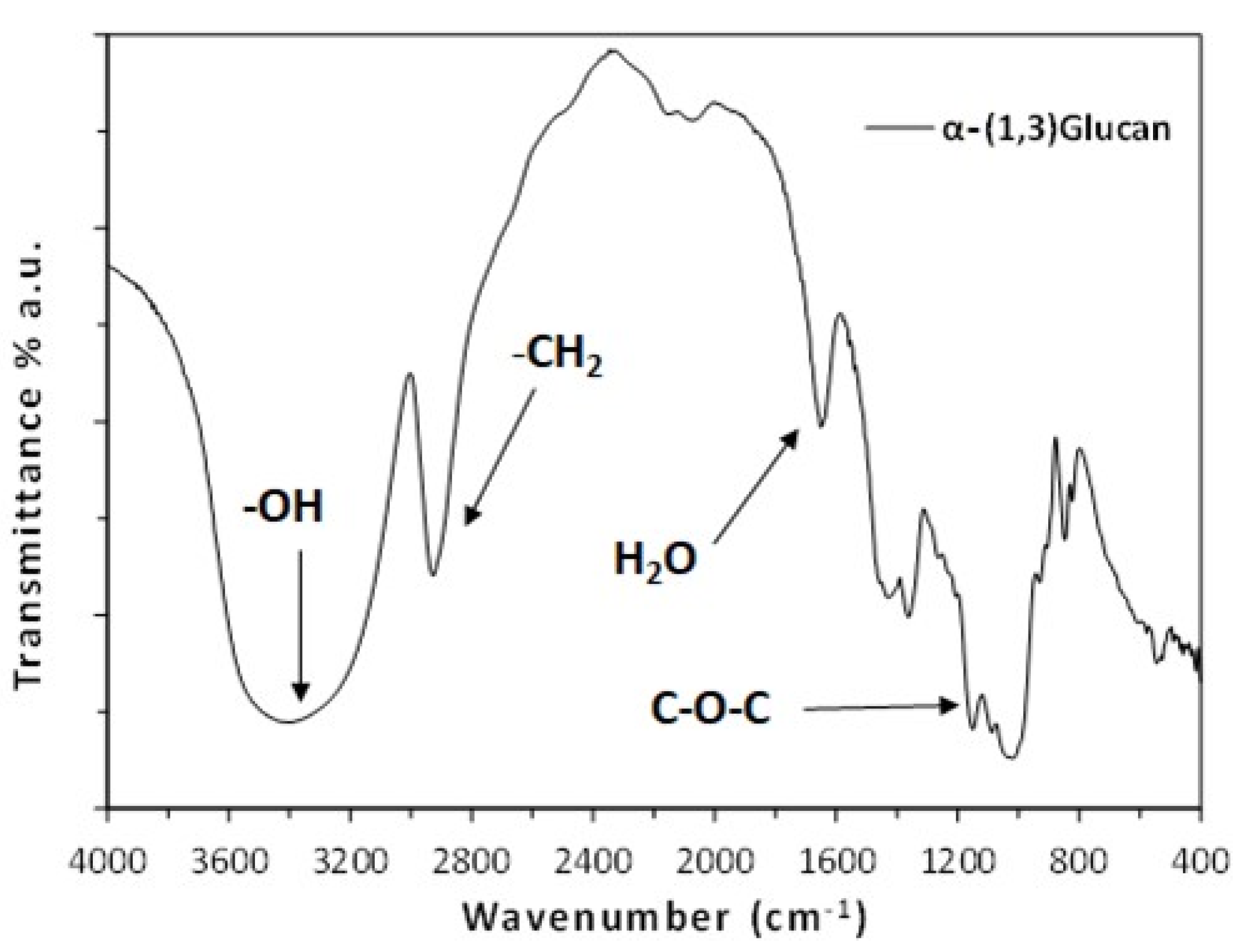
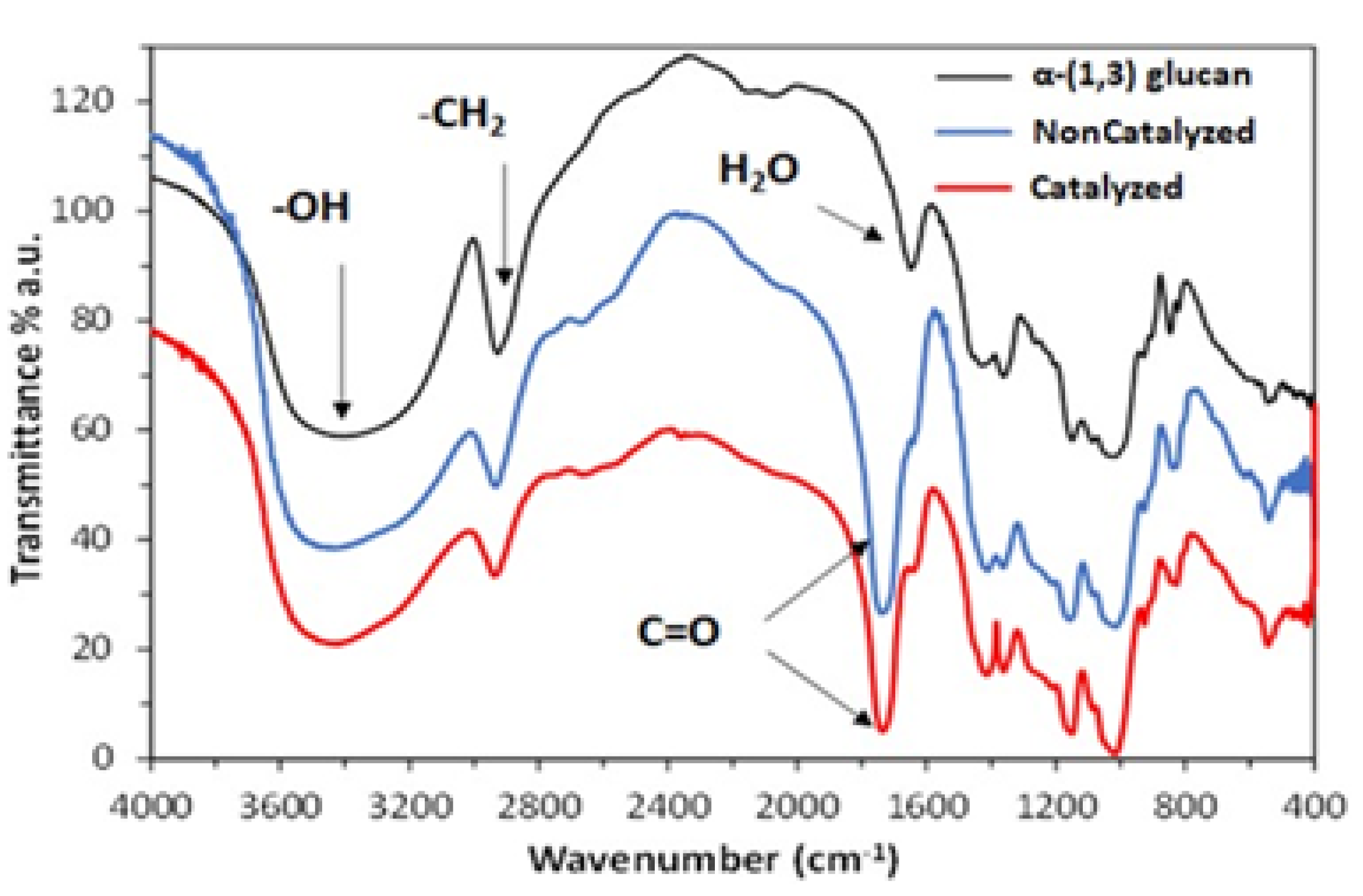
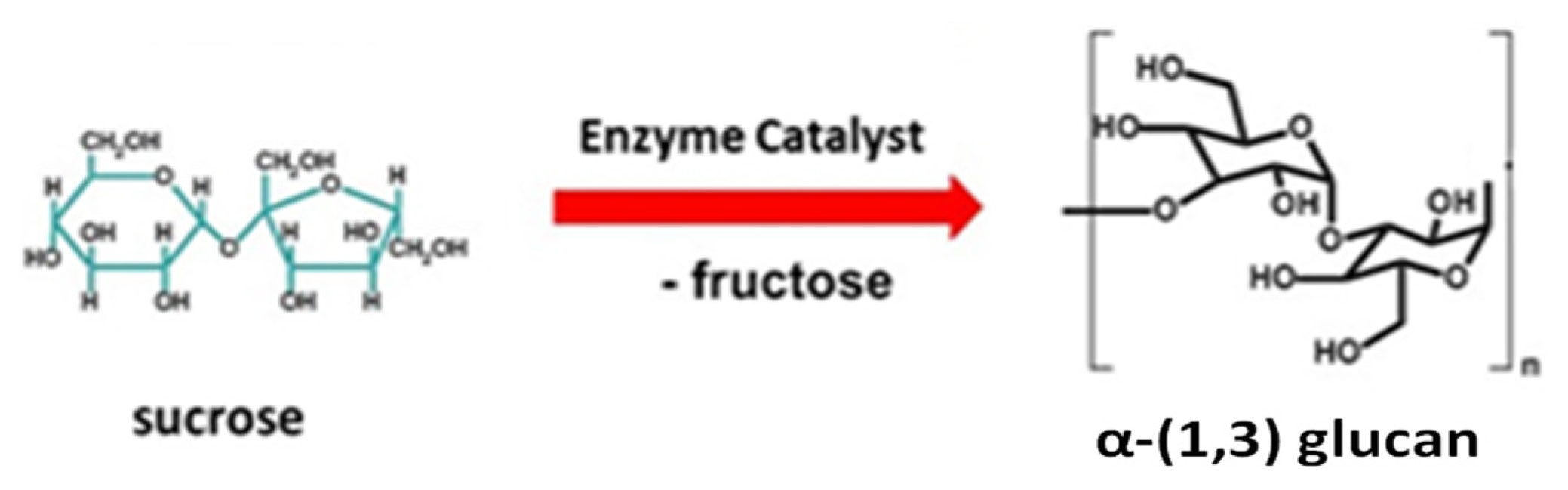
2.1. Characterization of α-(1,3) Glucan
2.2. Acylation of α-(1,3) Glucan with Succinic Anhydride
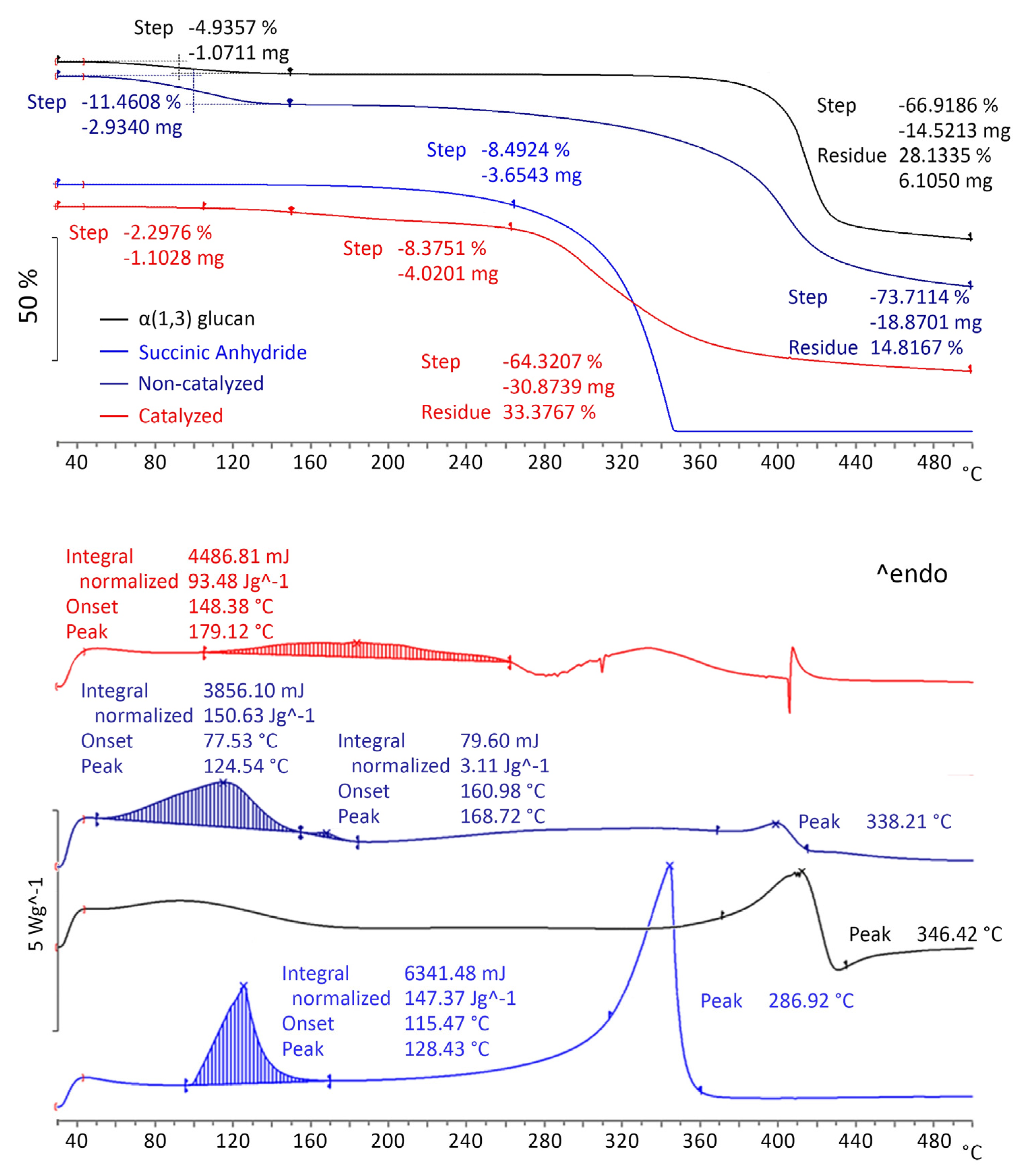
2.2.1. Acylation Reactions at the Micro Scale
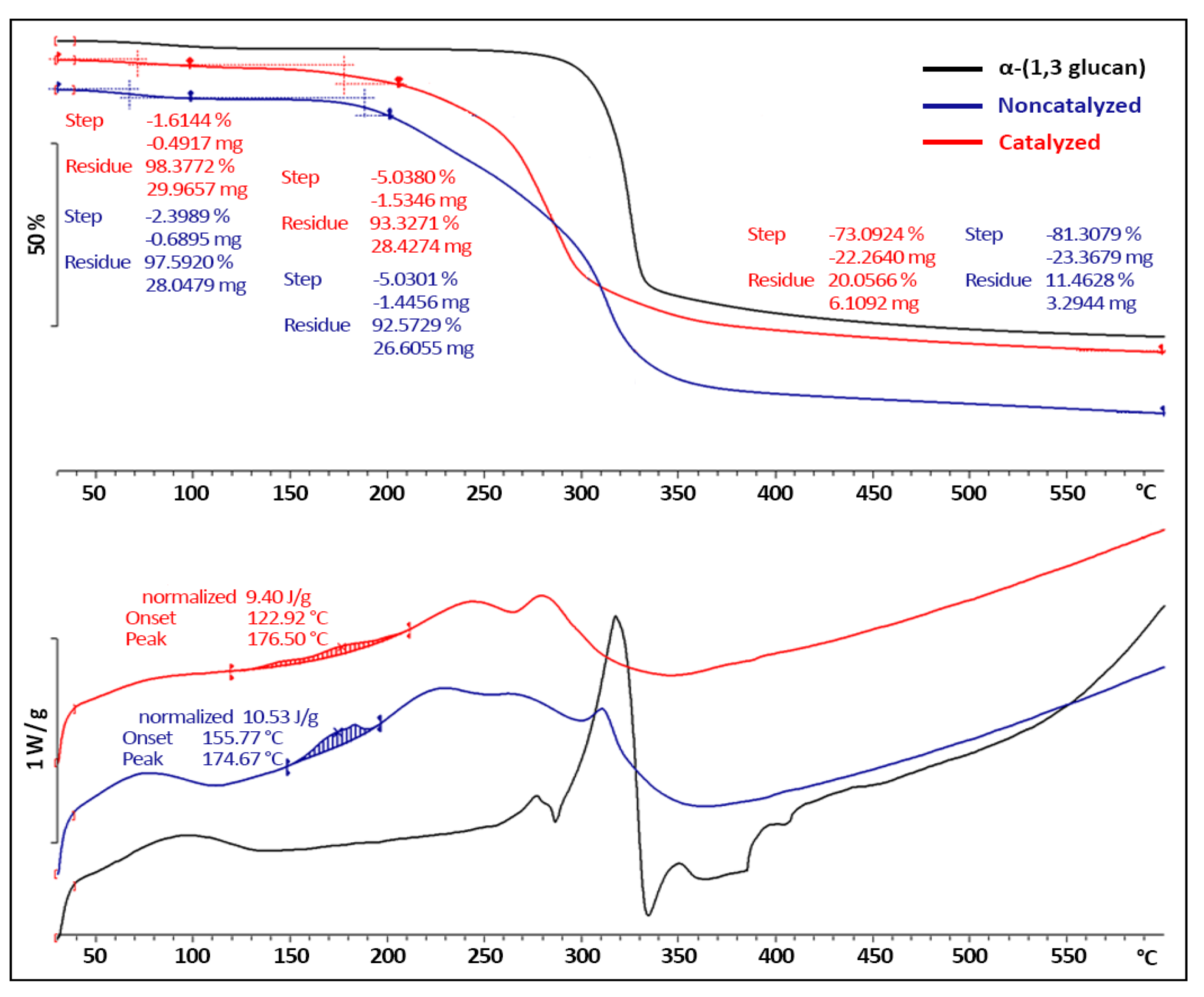
2.2.2. Acylation Reactions at Laboratory Scale
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Coma, V.; Freire, C.S.R.; Silvestre, A.J.D. Recent Advances on the Development of Antibacterial Polysaccharide-Based Materials. In Polysaccharides; Ramawat, K., Mérillon, J.M., Eds.; Springer International Publishing: Basel, Switzerland, 2015; pp. 1752–1791. [Google Scholar]
- Davidovic-Pinhas, M.; Barbut, S.; Marangoni, A.G. Physical structure and thermal behavior of ethylcellulose. Cellulose 2014, 21, 3243–3255. [Google Scholar] [CrossRef]
- Gonçalvesac, I.; Lopes, J.; Barra, A.; Hernadez, D.; Nunes, C.; Coimbra, M.A. Tailoring the surface properties and flexibility of starch-based films using oil and waxes recovered from potato chips byproducts. Int. J. Biol. Macromol. 2020, 163, 251–259. [Google Scholar] [CrossRef]
- Ferreira, W.H.; Carmo, M.I.B.; Lúcia, A.; Silva, N.; Andrade, C.T. Effect of structure and viscosity of the components on some properties of starch-rich hybrid blends. Carbohydr. Polym. 2015, 117, 988–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, D.; Pandey, J.; Raj, V.; Kumar, P. A Review on the Modification of Polysaccharide Through Graft Copolymerization for Various Potential Applications. Open J. Med. Chem. 2017, 11, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Nesic, A.; Cabrera-Barjas, G.; Dimitrijevic-Brankovic, S.; Davidovic, S.; Radovanovic, N.; Delattre, C. Prospect of Polysaccharide-Based Materials as Advanced Food Packaging. Molecules 2020, 25, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinze, T.; Liebert, T.; Koschella, A. Esterification of Polysaccharides; Springer Laboratory: Berlin, Germany, 2006. [Google Scholar]
- Jiang, T.; Duan, Q.; Zhu, J.; Liu, H.; Yu, L. Starch-based Biodegradable Materials: Challenges and Opportunities. Adv. Ind. Eng. Polym. Res. 2020, 3, 8–18. [Google Scholar] [CrossRef]
- Lenges, C. DuPont Biomaterials: Enzymatic Polymerization A new process for engineered polysaccharides. In Proceedings of the Bio Based Performance Materials Symposium, Wageningen, The Netherlands, 19 June 2017. [Google Scholar]
- Kassat, R.; Paullin, J. Preparation of Poly Alpha-1,3-Glucan Esters and Films Therefrom. U.S. Patent US2014/0187766, 3 July 2014. [Google Scholar]
- Carlisle, R. Scientific American Inventions and Discoveries; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004; p. 338. [Google Scholar]
- Vilivalam, V.D.; Illum, L.; Iqbal, K. Starch capsules: An alternative system for oral drug delivery. Pharmaceut. Sci. Tech. 2000, 3, 64–69. [Google Scholar] [CrossRef]
- Stepto, R.F.T. Understanding the processing of thermoplastic starch. Macromol. Symp. 2006, 245, 571–577. [Google Scholar] [CrossRef]
- Shanks, R.; Kong, I. Thermoplastic Starch. In Thermoplastic Elastomers; El-Sonbati, A., Ed.; InTech: Rijeka, Croatia, 2012; pp. 155–170. [Google Scholar]
- Roosdiana, A.; Mardiana, D.; Indahyanti, E.; Oktavianie, D.A. Enzymatic Cellulose Palmitate Synthesis Using Immobilized Lipase. J. Life Sci. Res. 2017, 14, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Sustainable Packaging Conference Highlights New Materials, Applications. Available online: https://www.ptonline.com/articles/sustainable-packaging-conference-highlights-new-materials-applications (accessed on 26 November 2020).
- Adibi, A.; Kim, J.; Mok, J.; Lenges, C.; Simon, L.; Mekonnen, T.H. Enzymatic polymerization designed alpha-1,3 glucan particle morphology as reinforcing fillers of dipped and casted rubber films. Carbohydr. Polym. 2021, 267, 118234. [Google Scholar] [CrossRef]
- Bezerra, D.S.; Teixeira, P.R.S.; Teixeira, A.S.N.M.; Eiras, C.; Osajima, J.A.; Silva Filho, E.C. Chemical Functionalization of Cellulosic Materials—Main Reactions and Applications in the Contaminants Removal of Aqueous Medium. In Cellulose—Fundamental Aspects and Current Trends; Poletto, M., Ed.; InTechOpen: Rijeka, Croatia, 2015; pp. 93–113. [Google Scholar]
- Heinze, T.; Liebert, T.; Pfeiffer, K.; Hussain, M. Unconventional Cellulose Esters: Synthesis, Characterization and Structure–Property Relations. Cellulose 2003, 10, 283–296. [Google Scholar] [CrossRef]
- Cumpstey, I. Chemical Modification of Polysaccharides. ISRN Org. Chem. 2013, 2013, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Puanglek, S.; Kimura, S.; Enomoto-Rogers, Y.; Kabe, T.; Yoshida, M.; Wada, M.; Iwata, T. In vitro synthesis of linear α-1,3-glucan and chemical modification to ester derivatives exhibiting outstanding thermal properties. Sci. Rep. 2016, 6, 30479. [Google Scholar] [CrossRef] [PubMed]
- Puanglek, S.; Kimura, S.; Iwata, T. Thermal and mechanical properties of tailor-made unbranched α-1,3-glucan esters with various carboxylic acid chain length. Carbohydr. Polym. 2017, 169, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Vitas, S.; Keplinger, T.; Reichholf, N.; Figi, R.; Cabanne, E. Functional lignocellulosic material for the remediation of copper(II) ions from water: Towards the design of a wood filter. J. Hazard. Mater. 2018, 355, 119–127. [Google Scholar] [CrossRef]
- Vaidya, A.; Gaugler, M.; Smith, D. Green route to modification of wood waste, cellulose and hemicellulose using reactive extrusion. Carbohydr. Polym. 2016, 136, 1238–1250. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Duan, Y.M.; Li, F.S.; Wang, X.; Tong Cui, X.; Bacha, U.; Zhu, M.P.; Xiao, Z.; Zhao, Z. Preparation and characterization of modified and functional starch (hexadecyl corboxymethyl starch) ether using reactive extrusion. Starke 2016, 68, 1–9. [Google Scholar] [CrossRef]
- Volokhina, A.V.; Kudryavtsev, G.I.; Skuratov, S.M.; Bonetskaya, A.K. The polyamidation process in the solid state. J. Polym. Sci. 1961, 53, 289–294. [Google Scholar] [CrossRef]
- Papaspyrides, C.; Vouyiouka, S.; Bletsos, I. New aspects on the mechanism of the solid state polyamidation of PA 6,6 salt. Polymer 2006, 47, 1020–1027. [Google Scholar] [CrossRef]
- Papaspyrides, C.D.; Porfyris, A.D.; Rulkens, R.; Grolman, E.; Kolkman, A.J. The effect of diamine length on the direct solid state polycondensation of semi-aromatic nylon salts. J. Polym. Sci. A Polym. Chem. 2016, 54, 2493–2506. [Google Scholar] [CrossRef]
- Papaspyrides, C.D. Solid State Polyamidation. In The Polymeric Materials Encyclopedia, 1st ed.; Salamone, J.C., Ed.; CRC Press, Inc.: Boca Raton, FL, USA, 1996; pp. 7819–7831. [Google Scholar]
- Papaspyrides, C.D.; Porfyris, A.D.; Vouyiouka, S.; Rulkens, R.; Grolman, E.; Vanden Poel, G. Solid state polymerization in a micro-reactor: The case of poly(tetramethylene terephthalamide). J. Appl. Polym. Sci. 2016, 133, 42371. [Google Scholar] [CrossRef]
- Hameed, S.; Hussain, M.A.; Masood, R.; Haseeb, M.T. Cross-linking of cotton fabric using maleic anhydride and sodium hypophosphite. Cellul. Chem. Technol. 2016, 50, 321–328. [Google Scholar]
- Billmeyer, F.W. Methods for estimating intrinsic viscosity. J. Polym. Sci. 1949, 4, 83–86. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reactants | MW (g/mol) | Reacting Groups (meq/kg) | Mass Ratio (GL:SA) |
|---|---|---|---|
| α-(1,3) glucan | 129,600 | 18,519 | - |
| succinic anhydride | 100.1 | 19,980/2 = 9990 | 1:0.618 1 |
| Initial Composition (Mass Ratio GL:SA = 1:0.62 for Target DS = 1) | ||||
| Experiments | Glucan Neat (g) | SA (g) | NaPO2H2.H2O (g) | |
| Noncatalyzed | 6.2 | 3.8 | 0 | |
| Catalyzed (10 wt.% NaPO2H2.H2O) | 10.54 | 6.23 | 1.62 | |
| Received Products after Reaction | ||||
| Modified Glucan (g) | Received SA (g) | DS | [η] (dL/g) | |
| Noncatalyzed | 7.6 | 2.4 | 0.37 | Insoluble |
| Catalyzed (10 wt.% NaPO2H2.H2O) | 15.1 | 3.3 | 0.7 | Insoluble |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porfyris, A.; Papaspyrides, C.D.; Behabtu, N.; Lenges, C.; Kopatsis, A. High-Solids, Solvent-Free Modification of Engineered Polysaccharides. Molecules 2021, 26, 4058. https://doi.org/10.3390/molecules26134058
Porfyris A, Papaspyrides CD, Behabtu N, Lenges C, Kopatsis A. High-Solids, Solvent-Free Modification of Engineered Polysaccharides. Molecules. 2021; 26(13):4058. https://doi.org/10.3390/molecules26134058
Chicago/Turabian StylePorfyris, Athanasios, Constantine D. Papaspyrides, Natnael Behabtu, Cristian Lenges, and Alexander Kopatsis. 2021. "High-Solids, Solvent-Free Modification of Engineered Polysaccharides" Molecules 26, no. 13: 4058. https://doi.org/10.3390/molecules26134058
APA StylePorfyris, A., Papaspyrides, C. D., Behabtu, N., Lenges, C., & Kopatsis, A. (2021). High-Solids, Solvent-Free Modification of Engineered Polysaccharides. Molecules, 26(13), 4058. https://doi.org/10.3390/molecules26134058