
Novel 1,2,3-Triazole Derivatives as Mimics of Steroidal System—Synthesis, Crystal Structures Determination, Hirshfeld Surfaces Analysis and Molecular Docking

 , ,
, ,  ,
,
Abstract
:
1. Introduction
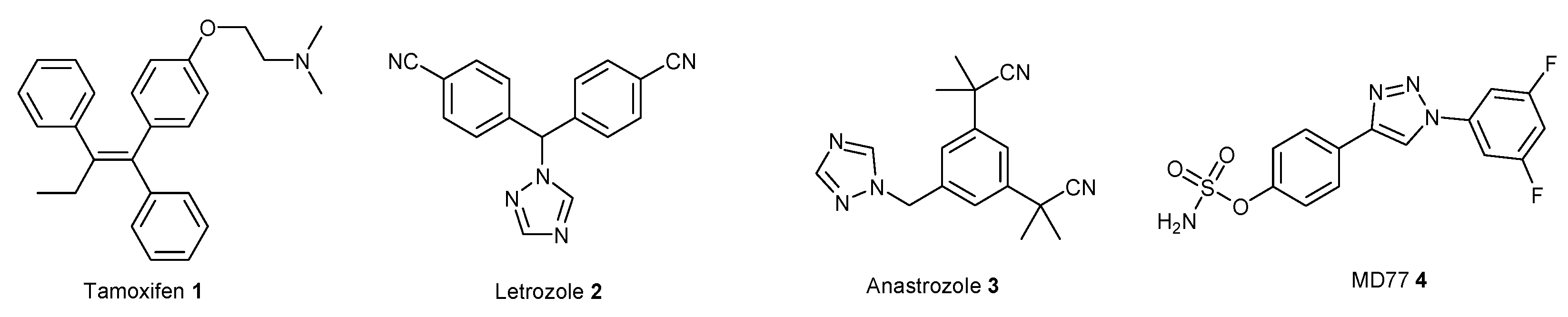
2. Results and Discussion
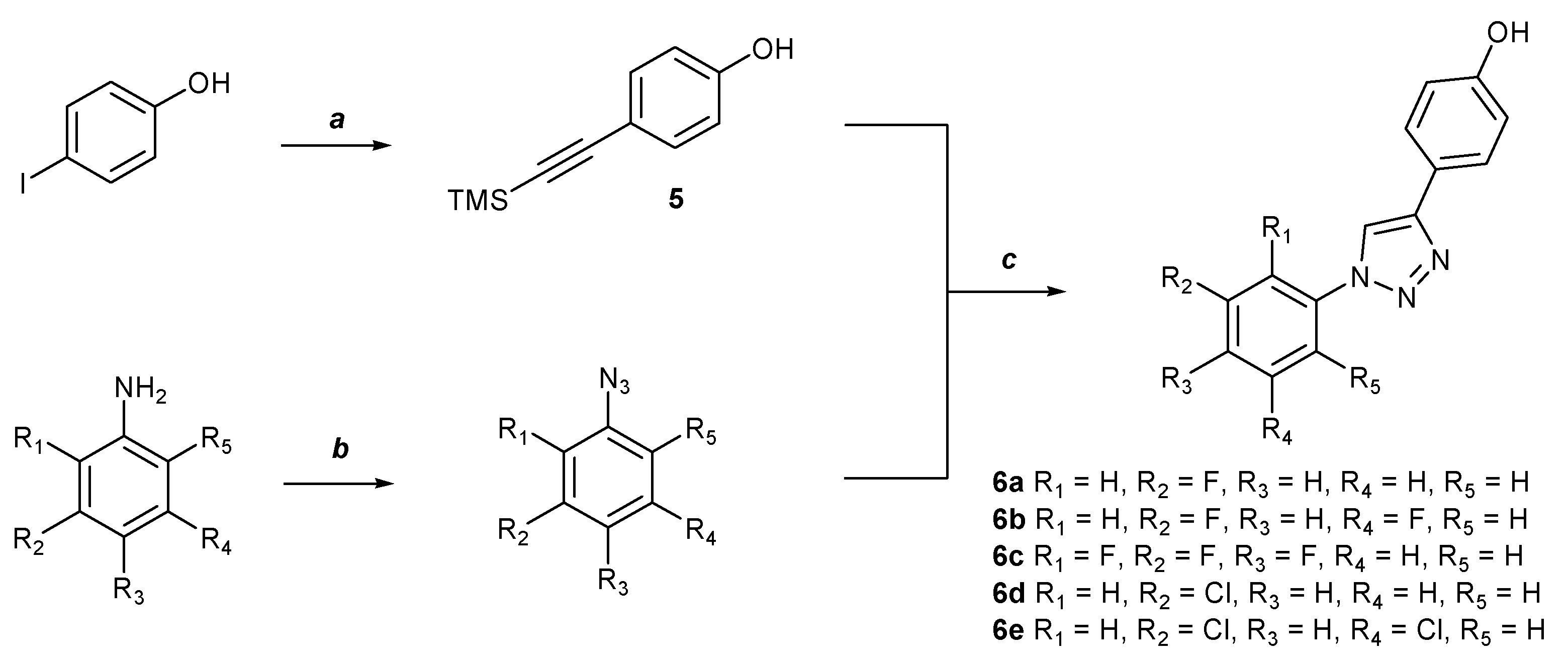
2.1. Chemistry
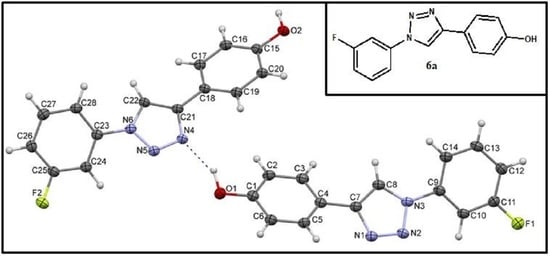
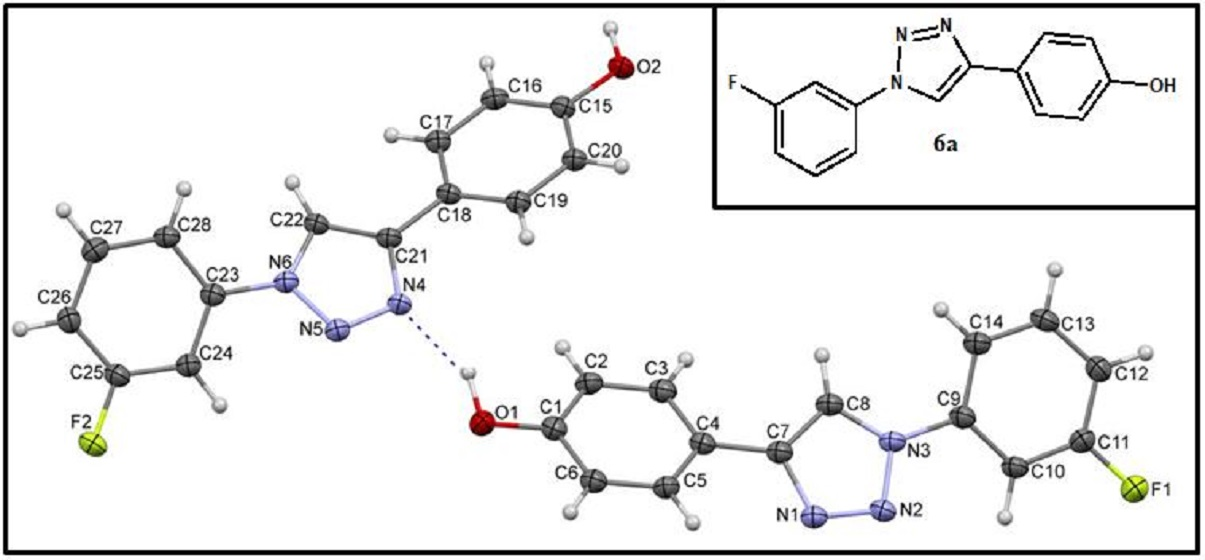
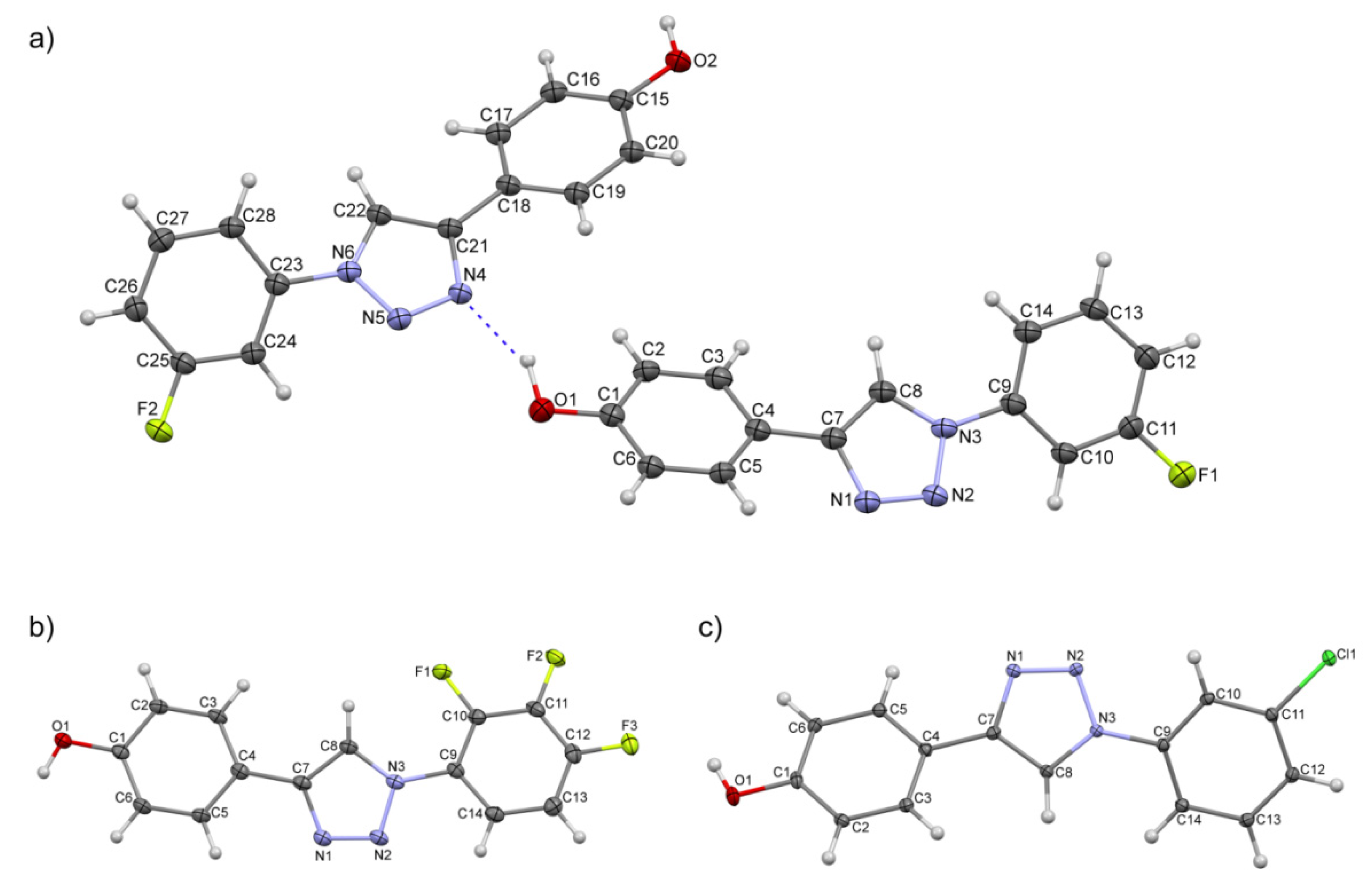
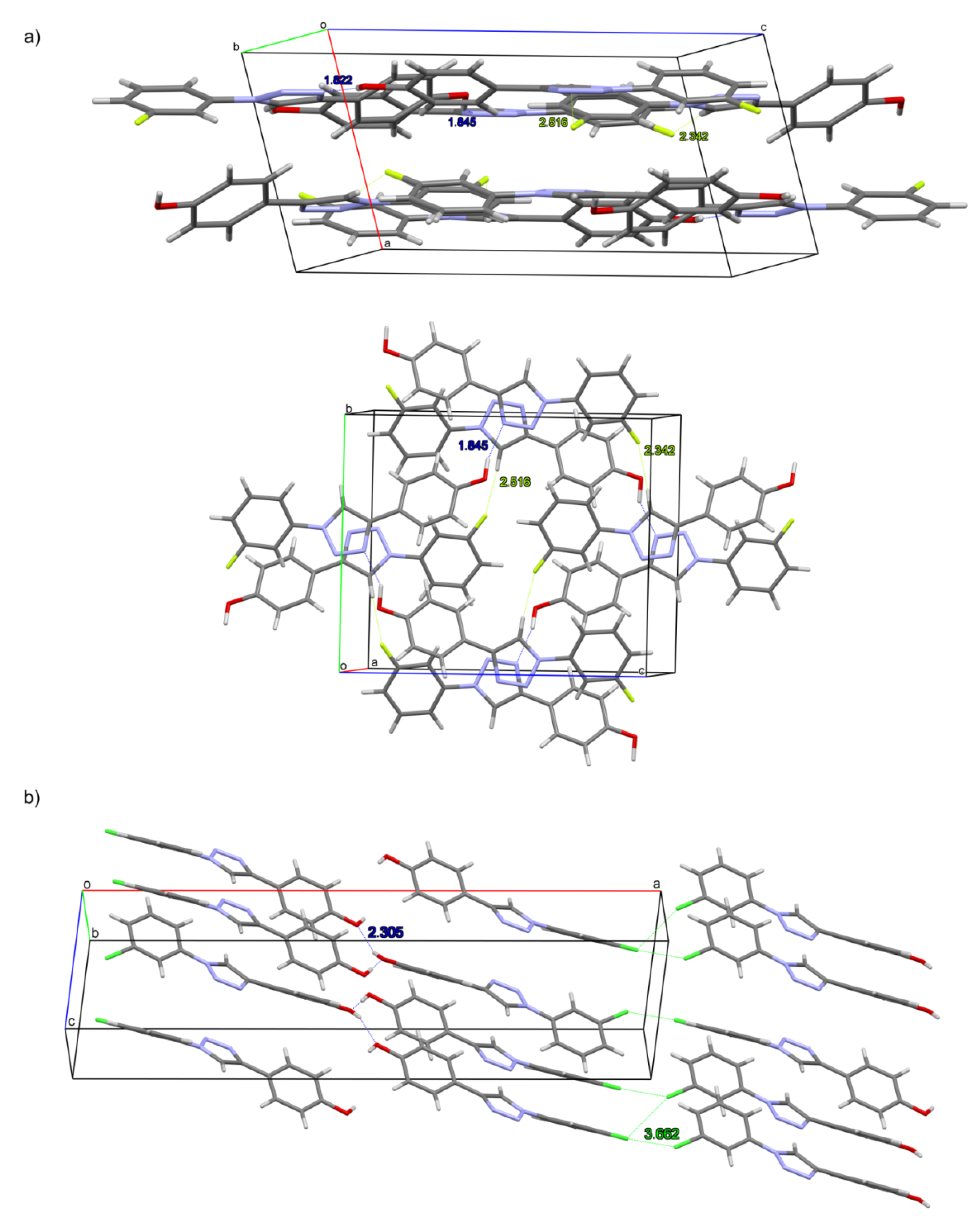
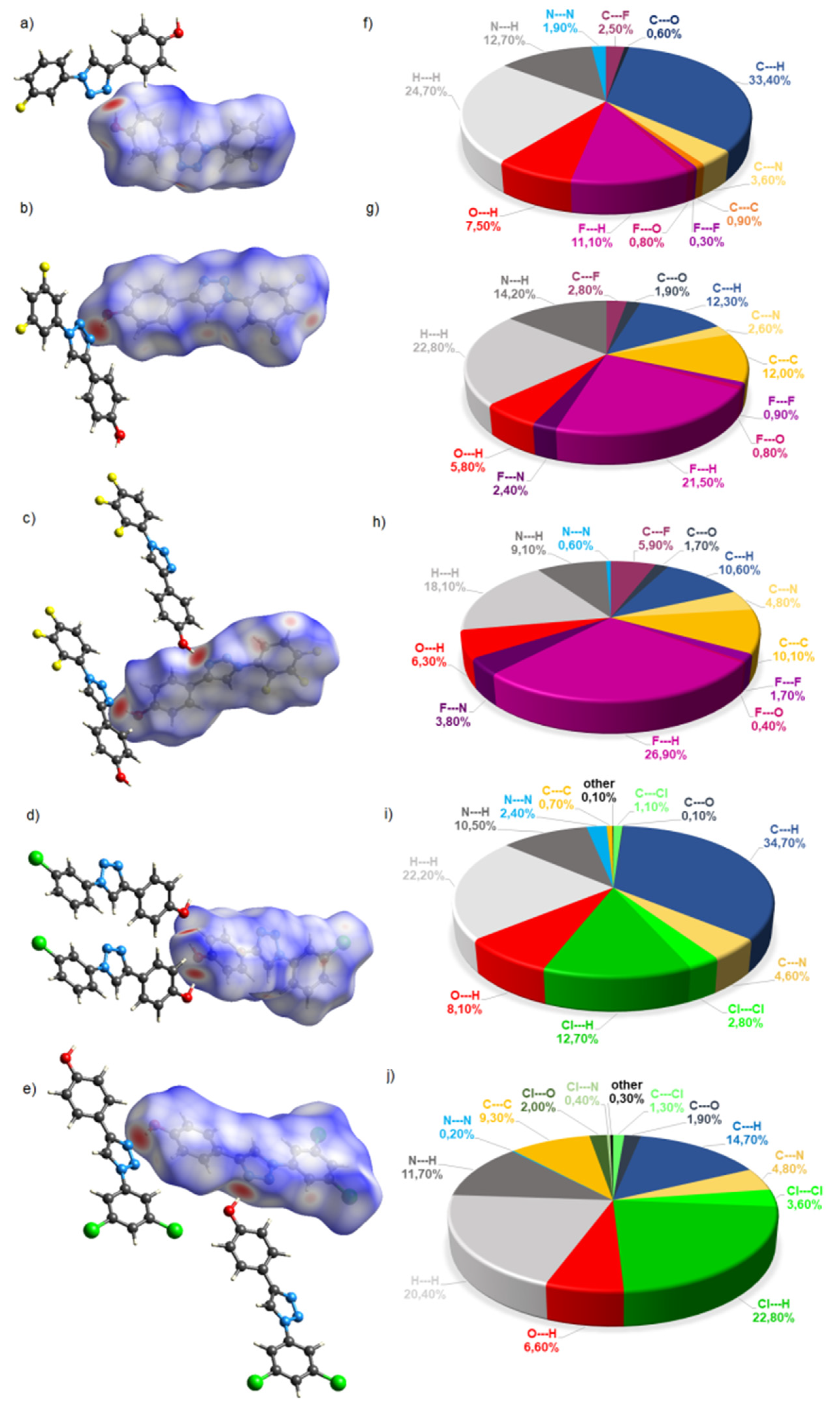
2.2. Crystal Structures and Hirshfeld Surfaces Analysis
2.3. Computational Studies
2.3.1. The Lipinski’s Rule of Five Calculations
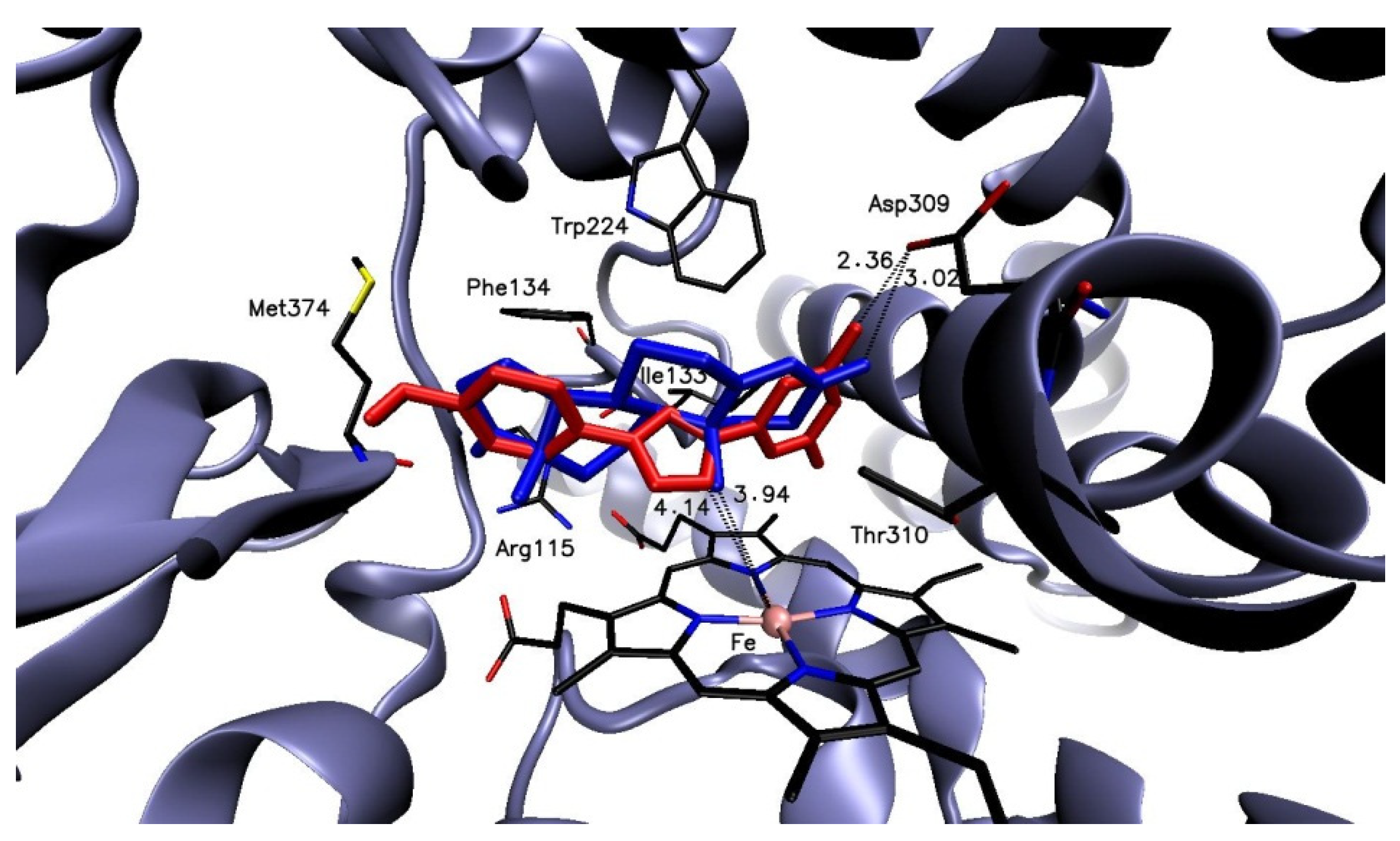
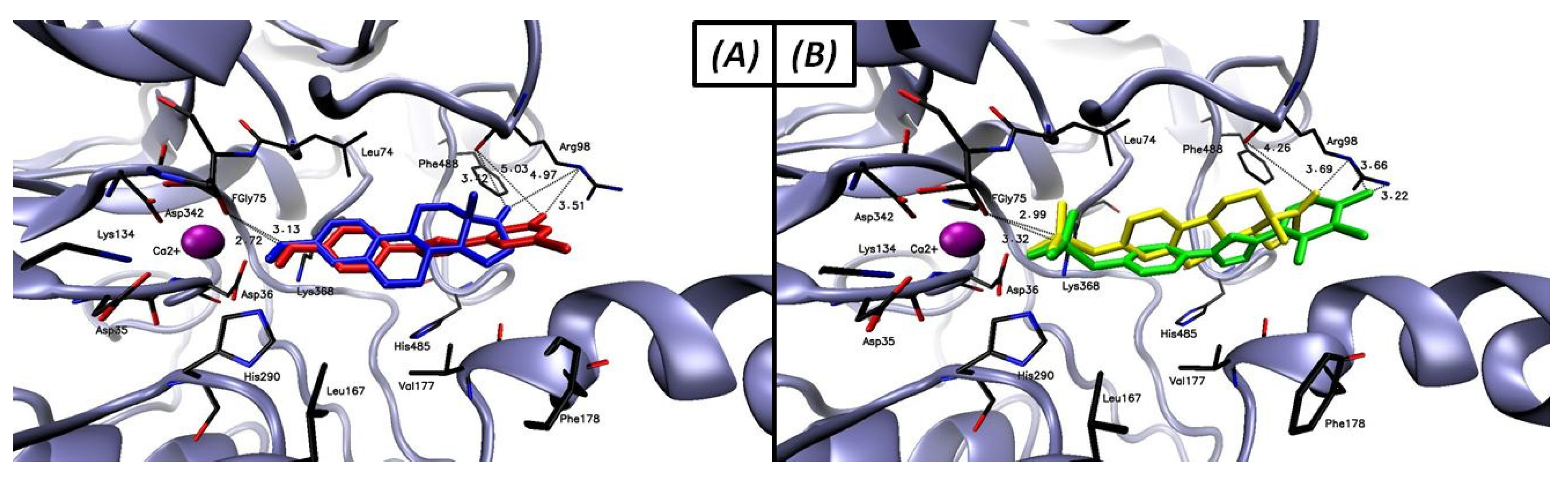
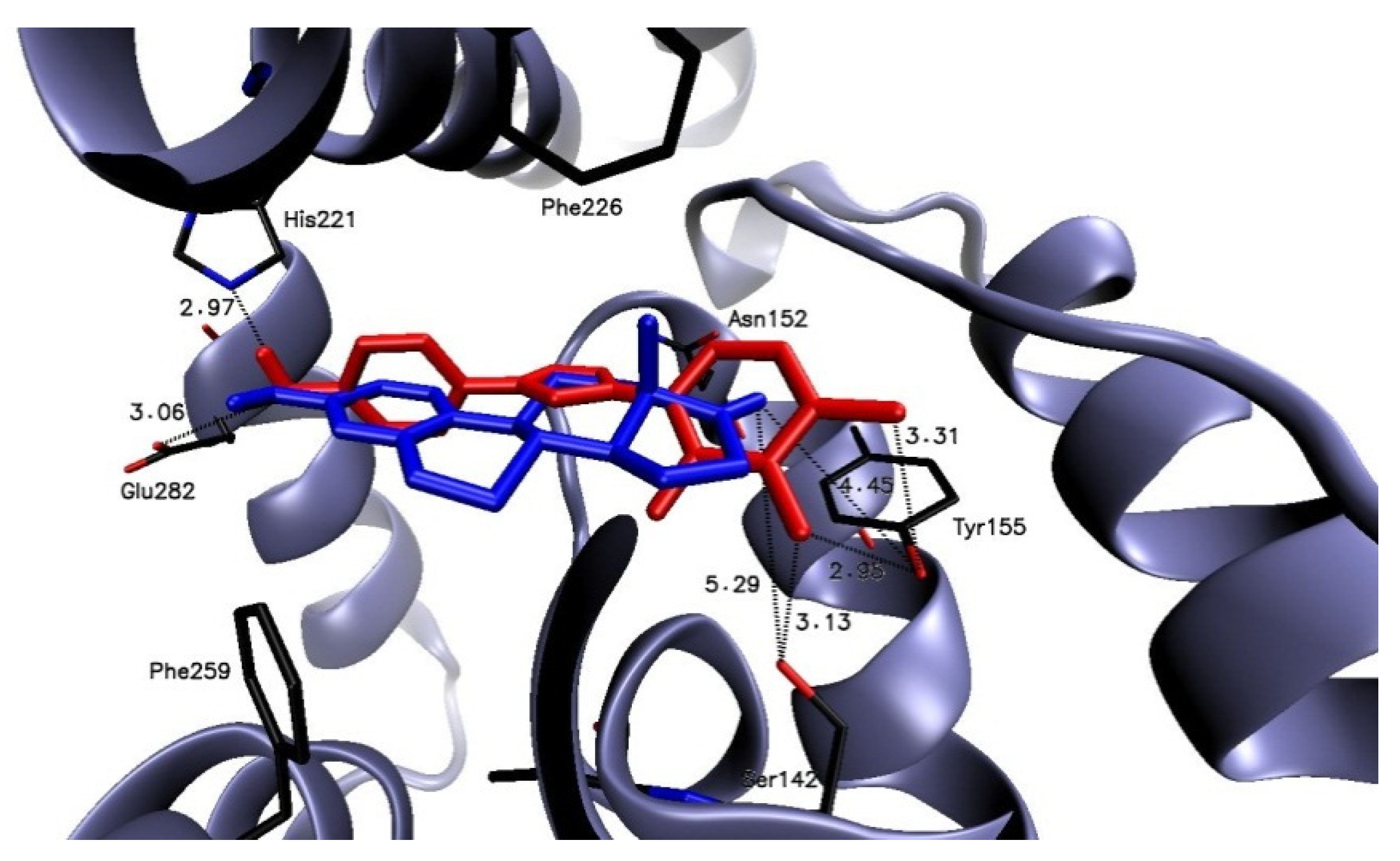
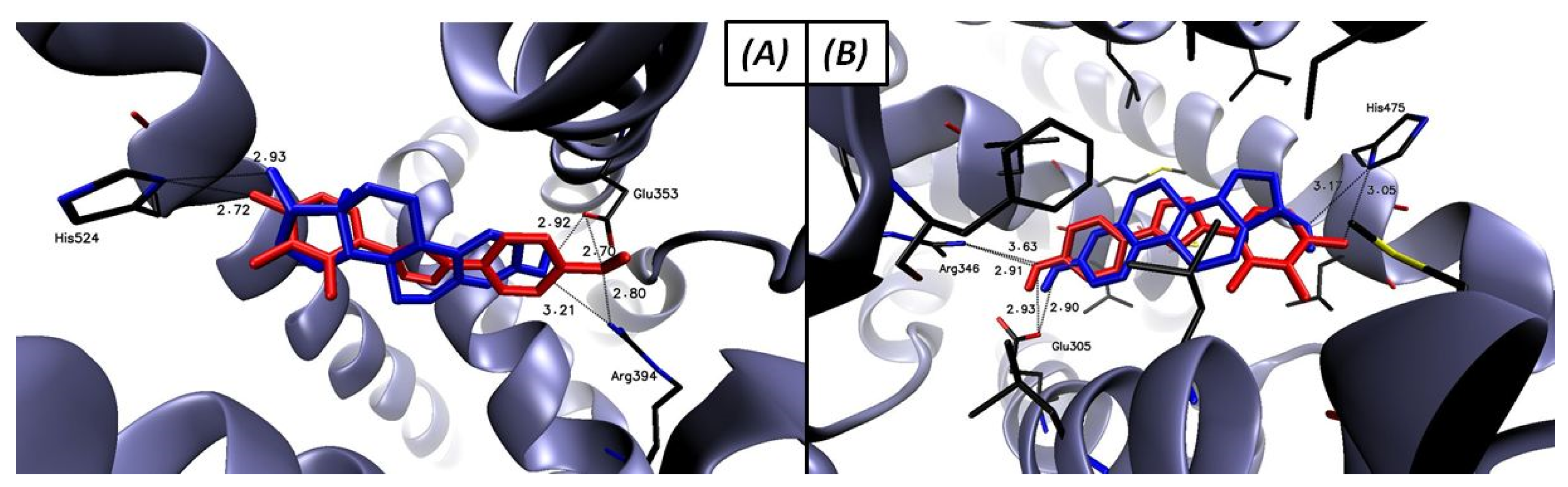
2.3.2. Molecular Docking
AROM
STS
17β-HSD1
ERα and ERβ
3. Materials and Methods
3.1. Synthesis
3.2. X-ray Diffraction Measurement
3.3. Computational Studies
3.3.1. The Lipinski’s Rule of Five Calculations
3.3.2. Ligands Preparation for Molecular Docking
3.3.3. Protein Preparation for Molecular Docking
3.3.4. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| 17β-HSD | 17β-hydroxysteroid dehydrogenase |
| ACN | acetonitrile |
| Adiol | androstenediol |
| AROM | aromatase |
| CuSO4·5H2O | copper(II) sulfate pentahydrate |
| DHES | dehydroepiandrosterone |
| DHEAS | dehydroepiandrosterone sulfate |
| E1 | estrone |
| E2 | estradiol |
| E1S | estrone sulfate |
| ER | estrogen receptor |
| HB | hydrogen bond |
| HS | Hirshfeld surface |
| PC | physicochemical |
| SERM | selective estrogen receptor modulator |
| STS | steroid sulfatase |
| TBAF | tetrabutylammonium fluoride |
| t-BuONO | tert-butyl nitrite |
| THF | tetrahydrofuran |
| TMSN3 | azidotrimethylsilane |
References
- Bonandi, E.; Christodoulou, M.S.; Fumagalli, G.; Perdicchia, D.; Rastelli, G.; Passarella, D. The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov. Today 2017, 22, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Saha, T.; Makar, S.; Swetha, R.; Gutti, G.; Singh, S.K. Estrogen signaling: An emanating therapeutic target for breast cancer treatment. Eur. J. Med. Chem. 2019, 177, 116–143. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Singh, J.; Singh, D.; Singh Jaggi, A.; Singh, N. Sulfatase inhibitors for recidivist breast cancer treatment: A chemical review. Eur. J. Med. Chem. 2016, 114, 170–190. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.W.; Gilligan, L.C.; Idkowial, J.; Arlt, W.; Foster, P.A. The regulation of steroid action by sulfation and desulfation. Endocr. Rev. 2015, 36, 526–563. [Google Scholar] [CrossRef] [PubMed]
- Daśko, M.; Demkowicz, S.; Rachon, J.; Biernacki, K.; Aszyk, J.; Kozak, W.; Masłyk, M.; Kubiński, K. New potent STS inhibitors based on fluorinated 4-(1-phenyl-1H-[1,2,3]triazol-4-yl)-phenyl sulfamates. J. Asian Nat. Prod. Res. 2019, 22, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Ciupak, O.; Daśko, M.; Biernacki, K.; Rachon, J.; Masłyk, M.; Kubiński, K.; Martyna, A.; Demkowicz, S. New potent steroid sulphatase inhibitors based on 6-(1-phenyl-1H-1,2,3-triazol-4-yl)naphthalen-2-yl sulphamate derivatives. Enzym. Inhib. Med. Chem. 2021, 36, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Sonogashira, K. Development of Pd-Cu catalyzed cross-coupling of terminal acetylenes with sp2-carbon halides. J. Organomet. Chem. 2002, 653, 46–49. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds. The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- X-Area 1.75, Software Package for Collecting Single-Crystal Data on STOE Area-Detector Diffractometers, for Image Processing, Scaling Reflection Intensities and for Outlier Rejection; STOE and Cie GmbH: Darmstadt, Germany, 2015.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXL-2014; University of Göttingen and Bruker AXS: Karlsruhe, Germany, 2014. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refirement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | R1 | R2 | R3 | R4 | R5 | Molecular Weight (g/mol) | Log P | Number of HB Donors | Number of HB Acceptors |
|---|---|---|---|---|---|---|---|---|---|
| 6a | H | F | H | H | H | 255.25 | 2.67 | 1 | 4 |
| 6b | H | F | H | F | H | 273.24 | 2.97 | 1 | 5 |
| 6c | F | F | F | H | H | 291.23 | 3.22 | 1 | 6 |
| 6d | H | Cl | H | H | H | 271.70 | 2.90 | 1 | 3 |
| 6e | H | Cl | H | Cl | H | 306.15 | 3.42 | 1 | 3 |
| No. | R1 | R2 | R3 | R4 | R5 | Binding Free Energy (kcal mol−1) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| AROM | STS | 17β-HSD1 | ERα | ERβ | ||||||
| 6a | H | F | H | H | H | −8.3 | −8.1/−7.1 * | −7.9 | −8.8 | −8.5 |
| 6b | H | F | H | F | H | −7.9 | −8.3/−6.6 * | −8.2 | −8.5 | −8.7 |
| 6c | F | F | F | H | H | −7.7 | −8.3/−7.9 * | −8.5 | −8.5 | −8.9 |
| 6d | H | Cl | H | H | H | −7.5 | −8.1/−6.9 * | −8.0 | −8.2 | −8.5 |
| 6e | H | Cl | H | Cl | H | −5.5 | −8.3/−6.9 * | −8.4 | −7.9 | −7.6 |
| Androstenedione | - | - | - | - | - | −12.4 | - | - | - | - |
| E1S | - | - | - | - | - | - | −6.3 | - | - | - |
| E1 | - | - | - | - | - | - | −8.9 | −8.9 | - | - |
| E2 | - | - | - | - | - | - | - | - | −10.7 | −11.1 |
| Identification Code | 6a | 6b | 6c | 6d | 6e |
|---|---|---|---|---|---|
| Empirical formula | C14H10FN3O | C14H9F2N3O | C14H8F3N3O | C14H10ClN3O | C14H9Cl2N3O |
| Formula weight (u) | 255.25 | 273.24 | 291.23 | 271.70 | 306.14 |
| Temperature (K) | 120(2) | 120(2) | 120(2) | 120(2) | 120(2) K |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 Ĺ |
| Crystal system | Triclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P-1 | P21/n | P21/c | P21/c | P21/c |
| Unit cell dimensions | |||||
| a (Å) | 7.138(4) | 8.2384(16) | 9.162(3) | 27.8104(14) | 13.590(8) |
| b (Å) | 11.869(4) | 11.772(2) | 10.218(4) | 5.6525(2) | 14.898(6) |
| c (Å) | 14.148(4) | 12.358(3) | 12.658(4) | 7.3445(4) | 14.307(8) |
| α (°) | 89.54(2) | 90 | 90 | 90 | 90 |
| β (°) | 75.50(3) | 102.211(17) | 93.66(3) | 94.728(4) | 116.70(4) |
| γ (°) | 88.42(3) | 90 | 90 | 90 | 90 |
| Volume (Å3) | 1160.0(8) | 1171.4(4) | 1182.5(7) | 1150.61(9) | 2588(3) |
| Z | 4 | 4 | 4 | 4 | 8 |
| Z’ | 2 | 1 | 1 | 1 | 2 |
| Density (calculated) (Mg/m3) | 1.462 | 1.549 | 1.636 | 1.568 | 1.571 |
| Absorption coefficient (mm−1) | 0.107 | 0.123 | 0.139 | 0.326 | 0.499 |
| F(000) | 528 | 560 | 592 | 560 | 1248 |
| Crystal size (mm) | 0.154 × 0.116 × 0.064 | 0.47 × 0.24 × 0.105 | 0.646 × 0.383 × 0.238 | 0.31 × 0.174 × 0.092 | 0.269 × 0.209 × 0.151 |
| Theta range for data collection (°) | 2.270 to 25.995. | 2.416 to 25.997. | 2.564 to 25.986 | 2.204 to 25.997 | 2.099 to 26.000 |
| Index ranges | −8 ≤ h ≤ 8, −14 ≤ k ≤ 14, −17 ≤ l ≤ 17 | −9 ≤ h ≤ 10, −14 ≤ k ≤ 14, −15 ≤ l ≤ 15 | −10 ≤ h ≤ 11, −12 ≤ k ≤ 12, −15 ≤ l ≤ 15 | −34 ≤ h ≤ 34, −6 ≤ k ≤ 5, −8 ≤ l ≤ 9 | −16 ≤ h ≤ 16, −18 ≤ k ≤ 18, −17 ≤ l ≤ 17 |
| Reflections collected | 12,538 | 14,025 | 14,455 | 5871 | 16,627 |
| Independent reflections | 4556 [R(int) = 0.0543] | 2297 [R(int) = 0.0282] | 2325 [R(int) = 0.0268] | 2223 [R(int) = 0.0330] | 5074 [R(int) = 0.0311] |
| Completeness to theta = 25.242° | 99.9% | 100.0% | 99.9% | 98.6% | 99.7% |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 4556/0/352 | 2297/0/185 | 2325/0/194 | 2223/0/176 | 5074/0/369 |
| Goodness-of-fit on F2 | 1.050 | 1.054 | 1.063 | 1.111 | 1.058 |
| Final R indices (I > 2sigma(I)) | R1 = 0.0708, wR2 = 0.1924 | R1 = 0.0303, wR2 = 0.0830 | R1 = 0.0315, wR2 = 0.0825 | R1 = 0.0369, wR2 = 0.0869 | R1 = 0.0409, wR2 = 0.1051 |
| R indices (all data) | R1 = 0.0975, wR2 = 0.2167 | R1 = 0.0339, wR2 = 0.0848 | R1 = 0.0379, wR2 = 0.0858 | R1 = 0.0513, wR2 = 0.0948 | R1 = 0.0543, wR2 = 0.1121 |
| Extinction coefficient | 0.064(10) | n/a | n/a | n/a | n/a |
| Largest diff. peak and hole (e⋅Å−3) | 0.384 and −0.318 | 0.266 and −0.202 | 0.239 and −0.195 | 0.299 and −0.378 | 0.308 and −0.386 |
| CCDC number | 2,063,930 | 2,063,931 | 2,063,934 | 2,063,932 | 2,063,933 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daśko, M.; Dołęga, A.; Siedzielnik, M.; Biernacki, K.; Ciupak, O.; Rachon, J.; Demkowicz, S. Novel 1,2,3-Triazole Derivatives as Mimics of Steroidal System—Synthesis, Crystal Structures Determination, Hirshfeld Surfaces Analysis and Molecular Docking. Molecules 2021, 26, 4059. https://doi.org/10.3390/molecules26134059
Daśko M, Dołęga A, Siedzielnik M, Biernacki K, Ciupak O, Rachon J, Demkowicz S. Novel 1,2,3-Triazole Derivatives as Mimics of Steroidal System—Synthesis, Crystal Structures Determination, Hirshfeld Surfaces Analysis and Molecular Docking. Molecules. 2021; 26(13):4059. https://doi.org/10.3390/molecules26134059
Chicago/Turabian StyleDaśko, Mateusz, Anna Dołęga, Magdalena Siedzielnik, Karol Biernacki, Olga Ciupak, Janusz Rachon, and Sebastian Demkowicz. 2021. "Novel 1,2,3-Triazole Derivatives as Mimics of Steroidal System—Synthesis, Crystal Structures Determination, Hirshfeld Surfaces Analysis and Molecular Docking" Molecules 26, no. 13: 4059. https://doi.org/10.3390/molecules26134059
APA StyleDaśko, M., Dołęga, A., Siedzielnik, M., Biernacki, K., Ciupak, O., Rachon, J., & Demkowicz, S. (2021). Novel 1,2,3-Triazole Derivatives as Mimics of Steroidal System—Synthesis, Crystal Structures Determination, Hirshfeld Surfaces Analysis and Molecular Docking. Molecules, 26(13), 4059. https://doi.org/10.3390/molecules26134059