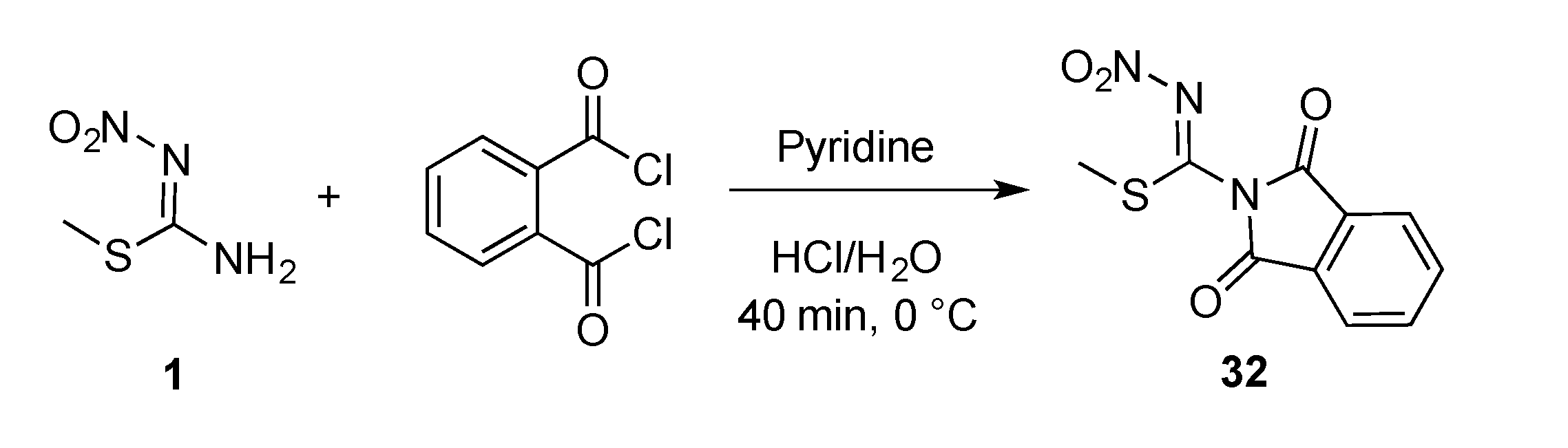
Appendix A
Data for 2-nitro-1-(pyridin-2-ylmethyl)guanidine 2. Gray solid, 82%, m.p. 175–177 °C (decomposes); TLC Rf 0.78 (MeOH); 1H NMR (500 MHz, DMSO-d6, 50 °C): δ 8.56 (ddd, J = 4.9, 1.8, 1.0 Hz, 1H), 7.93 (br s, 2H), 7.81 (td, J = 7.6, 1.8 Hz, 1H), 7.36–7.33 (m, 2H), 7.33–7.31 (m, 1H), 4.54 (s, 2H), 13C NMR (125 MHz, DMSO-d6, 50 °C): δ 149.4, 137.5, 123.0, 121.8, 46.0. HRMS (ESI): [M + Na]+ calculated for C7H9N5NaO2 [M + Na]+ 218.0648, found 218.0647 (5.0 ppm). IR (ATR) νmax: 3378, 3145, 3301, 1652.
Data for 2-nitro-1-(thiophen-2-ylmethyl)guanidine3. White solid, 74% yield, m.p. 129–131 °C; TLC Rf 0.21 (AcOEt/Hexane, 1:1); ¹H NMR (500 MHz, DMSO-d6, 50 °C): δ 7.93 (br s, 2H), 7.43–7.42 (m, 1H), 7.07–7.05 (m, 1H), 7.01–6.98 (m, 1H), 4.58 (br s, 2H), 13C NMR (125 MHz, DMSO-d6, 50 °C): δ 159.0, 127.0, 126.8, 126.1, 125.6, 38.4. HRMS (ESI): calculated for C6H9N4O2S [M + H]+ 201.0441, found 201.0441 (5.0 ppm). IR (ATR) νmax: 3371, 3160, 3312, 1644.
Data for 2-nitro-1-phenethylguanidine4. Yellow solid, 89% yield, m.p. 128–130 °C; TLC Rf 0.46 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, DMSO-d6, 50 °C): δ 7.35–7.29 (m, 2H), 7.29–7.20 (m, 3H), 3.48–3.40 (m, 2H), 2.83 (t, J = 7.4 Hz, 2H), 13C NMR (125 MHz, DMSO-d6, 50 °C): δ 159.7, 139.0, 129.2, 128.8, 126.8, 42.5, 34.4. HRMS (ESI): calculated for C9H13N4O2 [M + H]+ 209.1033, found 209.1030 (5.0 ppm). IR (ATR) νmax: 3367, 3170, 3302, 1644.
Data for (E)-1-(4-methoxybenzyl)-2-nitroguanidine5. White solid, 91% yield, m.p. 192–194 °C; TLC Rf 0.19 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, DMSO-d6, 50 °C): δ 7.86 (br s, 2H), 7.26 (d, J = 8.6 Hz, 1H), 6.93 (d, J = 8.6 Hz, 1H), 4.34 (s, 2H), 3.75 (s, 3H), 13C NMR (125 MHz, DMSO-d6, 50 °C): δ 159.0, 129.1, 114.3, 55.5, 43.7. HRMS (ESI): calculated for C9H13N403 [M + H]+ 225.0982, found 225.0988 (−2.2 ppm). IR (ATR) νmax: 3382, 3322, 3166.522, 1648.
Data for (E)-1-(4-methylbenzyl)-2-nitroguanidine6. White solid, 86% yield, m.p. 188–191 °C; TLC Rf 0.28 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, DMSO-d6, 50 °C): δ 7.87 (br s, 3H), 7.21 (d, J = 8.1 Hz, 2H), 7.17 (d, J = 8.1 Hz, 2H), 4.37 (br s, 2H), 2.30 (s, 3H), 13C NMR (125 MHz, DMSO-d6, 50 °C): δ 159.8, 136.9, 129.4, 127.7, 44.1, 21.0. HRMS (ESI): [M + H]+ calculated for C9H13N4O2 [M + H]+ 209.1033, found 209.1039 (−3.8 ppm). IR (ATR) νmax: 3374, 3305, 3155, 1648.
Data for (E)-1-(naphthalen-1-ylmethyl)-2-nitroguanidine7. White solid, 73% yield, m.p. 189–192 °C; TLC Rf 0.34 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, DMSO-d6, 50 °C): δ 8.05 (d, J = 8.2 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1H), 7.89 (d, J = 8.0 Hz, 1H), 7.63–7.54 (m, 2H), 7.54–7.45 (m, 2H), 4.89 (s, 2H), 13C NMR (125 MHz, DMSO-d6, 50 °C): δ 160.0, 133.8, 131.1, 129.0, 128.3, 126.8, 126.3, 125.8, 123.6, 42.6. HRMS (ESI): calculated for C12H13N4O2 [M + H]+ 245.1033 found 245.1039, (−2.9 ppm). IR (ATR) νmax: 3381, 3311, 3180, 1644.
Data for (E)-1-(4-chlorobenzyl)-2-nitroguanidine8. White solid, 72% yield, m.p. 190–192 °C; TLC Rf 0.22 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, DMSO-d6): δ 7.95 (s, 2H), 7.46–7.38 (m, 3H), 7.33 (d, J = 8.5 Hz, 2H), 4.40 (s, 2H), 13C NMR (125 MHz, DMSO-d6): δ 159.9, 132.3, 129.5, 128.8, 43.6. HRMS (ESI): calculated for C8H10ClN4O2 [M + H]+ 229.0487, found 229.0492 (−3.9 ppm). IR (ATR) νmax: 3370, 3312, 3180, 1645.
Data for (E)-2-nitro-1-(4-(trifluoromethyl)benzyl)guanidine9. White solid, 83% yield, m.p. 161–164 °C; TLC Rf 0.19 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, DMSO-d6, 50 °C): δ 7.97 (br s, 2H), 7.72 (d, J = 8.0 Hz, 2H), 7.52 (d, J = 7.9 Hz, 2H), 4.52 (s, 2H), 13C NMR (125 MHz, DMSO-d6, 50 °C): δ 159.8, 128.3, 128.1, 125.7 (q, J = 3.75 Hz), 125.3 (q, J = 270 Hz), 43.72; 19F NMR (470 MHz, DMSO-d6, 50 °C): δ −60.86. HRMS (ESI): calculated for C9H10F3N4O2 [M + H]+ 263.0750, found 263.0756 (−3.8 ppm). IR (ATR) νmax: 3378, 3305, 3178, 1648.
Data for (E)-1-(2-methoxybenzyl)-2-nitroguanidine10. White solid, 85% yield, m.p. 203–205 °C; TLC Rf 0.25 (AcOEt/Hexane, 6:4); ¹H NMR (500 MHz, DMSO-d6): δ 7.82 (br s, 2H), 7.33–7.28 (m, 1H), 7.23 (dd, J = 1.0, 7.4 Hz, 1H), 7.04 (d, J = 8.2 Hz, 1H), 7.04 (d, J = 8.2 Hz, 1H), 6.95 (t, J = 7.4 Hz, 1H), 4.37 (br s, 2H), 3.84 (s, 3H), 13C NMR (125 MHz, DMSO-d6): δ 157.2, 129.3, 128.3, 120.7, 111.2, 55.8. HRMS (ESI): calculated for C9H12N4O3Na [M + Na]+ 247.0802, found 247.0807 (−1.6 ppm). IR (ATR) νmax: 3382, 3322, 3166, 1648.
Data for (S)-1-(1-cyclopropylethyl)-2-nitroguanidine S-11. White solid, 72%, m.p. 109–111 °C; TLC Rf 0.37 (AcOEt/Hexane, 7:3); [α]26 = +18.81 (c = 1.0, acetone); ¹H NMR (500 MHz, CDCl3): δ 7.78 (br s, 1H), 6.85 (br s, 2H), 3.10–3.06 (m, 1H), 1.38 (d, J = 6.5 Hz, 3H), 1.04–0.97 (m, 1H), 0.68–0.61 (m, 2H), 0.40–0.27 (m, 2H), 13C NMR (125 MHz, CDCl3): δ 158.6, 52.3, 20.0, 16.8, 3.8, 3.1. HRMS (ESI): calculated for C6H13N4O2 [M + H]+ 173.1033, found 173.1035 (5.0 ppm). IR (ATR) νmax: 3310, 3159, 3373, 1634.
Data for (S)-2-nitro-1-(1,2,3,4-tetrahydronaphthalen-1-yl)guanidine S-12. White solid, 59% yield, m.p. 180–182 °C; TLC Rf 0.40 (AcOEt/Hexane, 1:1); [α]26 = −49.29 (c = 0.1, isopropyl alcohol); 1H NMR (500 MHz, CD3OD, 50 °C): δ 7.29–7.11 (m, 4H), 5.00 (s, 1H), 2.91–2.74 (m, 2H), 2.13–2.05 (m, 1H), 1.97–1.81 (m, 3H). 13C NMR (126 MHz, CD3OD, 50 °C): δ 159.05, 137.35, 128.89, 128.02, 127.30, 125.94, 49.53, 29.62, 28.58, 19.48. HRMS (ESI): [M + Na]+ calculated for C11H14N4NaO2 [M + Na]+ 257.1009 found 257.1014 (5.0 ppm). IR (ATR) νmax: 3380, 3191, 3322, 1643.
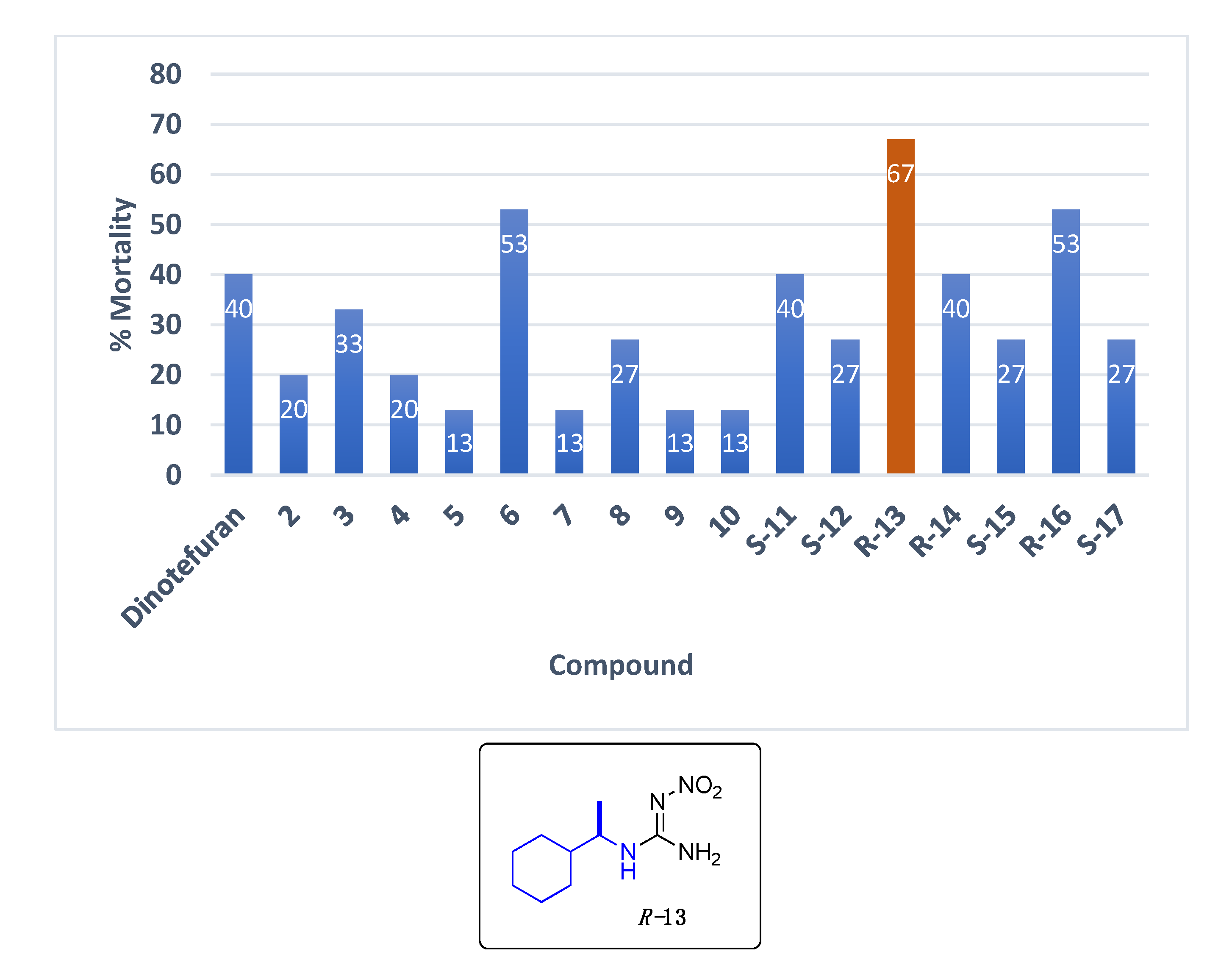
Data for (R)-1-(1-cyclohexylethyl)-2-nitroguanidine R-13. White solid, 61% yield, m.p. 139–140 °C; TLC Rf 0.38 (AcOEt/Hexane, 6:4); [α]26.3 = −30.71 (c = 0.5, acetone); ¹H NMR (500 MHz, CDCl3): δ 8.45 (br s, 1H), 6.63 (br s, 2H), 3.32 (br s, 1H), 1.82–1.68 (m, 6H), 1.48 (br s, 1H), 1.29–0.95 (m, 9H), 13C NMR (125 MHz, CDCl3): δ 158.6, 53.3, 42.9, 28.9, 26.1, 25.9, 17.6. HRMS (ESI): calculated for C9H19N4O2 [M + H]+ 215.1503, found 215.1508 (5.0 ppm). IR (ATR) νmax: 3355, 3310, 3170, 1644.
Data for (R)-1-(1-(naphthalen-1-yl)ethyl)-2-nitroguanidine R-14. Yellow solid, 30% yield, m.p. 175 °C (decomposes); TLC Rf 0.38 (AcOEt/Hexane, 6:4); [α]26.5 = −67.5 (c = 0.5, acetone,); 1H NMR (500 MHz, CDCl3): δ 8.30 (br s, 1H), 8.02 (d, J = 8.4 Hz, 1H), 7.95 (d, J = 7.5 Hz, 1H), 7.87 (d, J = 8.2 Hz, 1H), 7.65–7.60 (m, 1H), 7.60–7.56 (m, 3H), 7.54–7.50 (m, 1H), 6.15 (br s, 2H), 5.42 (s, 1H), 1.78 (d, J = 6.8 Hz, 3H), 13C NMR (125 MHz, CDCl3): δ 158.9, 135.4, 134.1, 129.6, 129.6, 129.3, 127.2, 126.3, 125.8, 122.8, 121.5, 49.2, 22.9. HRMS (ESI): calculated for C13H14N4NaO2 [M + Na]+ 281.1009, found 281.1014 (5.0 ppm). IR (ATR) νmax: 3415, 3222, 3283, 1637.
Data for (S)-1-(1-(naphthalen-1-yl)ethyl)-2-nitroguanidine S-15. Yellow solid, 46% yield, m.p 178 °C; TLC Rf 0.38 (AcOEt/Hexane, 6:4); [α]26.6 = +74.77 (c = 0.5, acetone), 1H NMR (500 MHz, CDCl3): δ 8.35 (s, 1H), 8.02 (d, J = 8.4 Hz, 1H), 7.97–7.93 (m, 1H), 7.87 (d, J = 8.1 Hz, 1H), 7.62 (ddd, J = 8.5, 6.8, 1.5 Hz, 1H), 7.60–7.56 (m, 2H), 7.52 (dd, J = 8.2, 7.2 Hz, 1H), 6.19 (s, 2H), 5.42 (s, 1H), 1.78 (d, J = 6.7 Hz, 3H), 13C NMR (125 MHz, CDCl3): δ 158.9, 135.3, 134.1, 129.6, 129.5, 129.3, 127.2, 126.3, 125.8, 122.8, 121.5, 49.1, 22.9. HRMS (ESI): [M + Na]+ calculated for C13H14N4NaO2 [M + Na]+ 281.1009, found 281.1014 (5.0 ppm). IR (ATR) νmax: 3415, 3222, 3283, 1636.
Data for (R)-2-nitro-1-(1-phenylethyl)guanidine R-16. Yellow solid, 79% yield, m.p. 126–127 °C; TLC Rf 0.29 (AcOEt/Hexane, 6:4) [α]24.4 = +37.4 (c = 0.5, acetone), ¹H NMR (500 MHz, CDCl3): δ 8.36 (br s, 1H), 7.43–7.40 (m, 2H), 7.37–7.33 (m, 3H), 6.46 (br s, 2H), 4.62 (br s, 1H), 1.64 (d, J = 6.8 Hz, 3H), 13C NMR (125 MHz, CDCl3): δ 159.0, 140.6, 129.6, 128.7, 125.5, 52.7, 24.0. HRMS (ESI): calculated for C9H13N4O2 [M + H]+ 209.1033, found 209.1039 (5.0 ppm). IR (ATR) νmax: 3392, 3210.
Data for (S)-2-nitro-1-(1-phenylethyl)guanidine S-17. Yellow solid, 95% yield, m.p. 127–128 °C; TLC Rf 0.29 (AcOEt/Hexane, 6:4) [α]23.9 = −40.32 (c = 0.5, acetone,), ¹H NMR (500 MHz, CDCl3): δ 8.41 (br s, 1H), 7.43–7.40 (m, 2H), 7.36–7.33 (m, 3H), 6.52 (br s, 2H), 4.62 (br s, 1H), 1.63 (3H, d, J = 6.8 Hz), 13C NMR (125 MHz, CDCl3): δ 159.0, 140.6, 129.6, 128.6, 125.5, 52.6, 23.9. HRMS (ESI): calculated for C9H13N4O2 [M + H]+ 209.1033, found 209.1039 (2.86 ppm). IR (ATR) νmax: 3392, 3211.
Appendix B
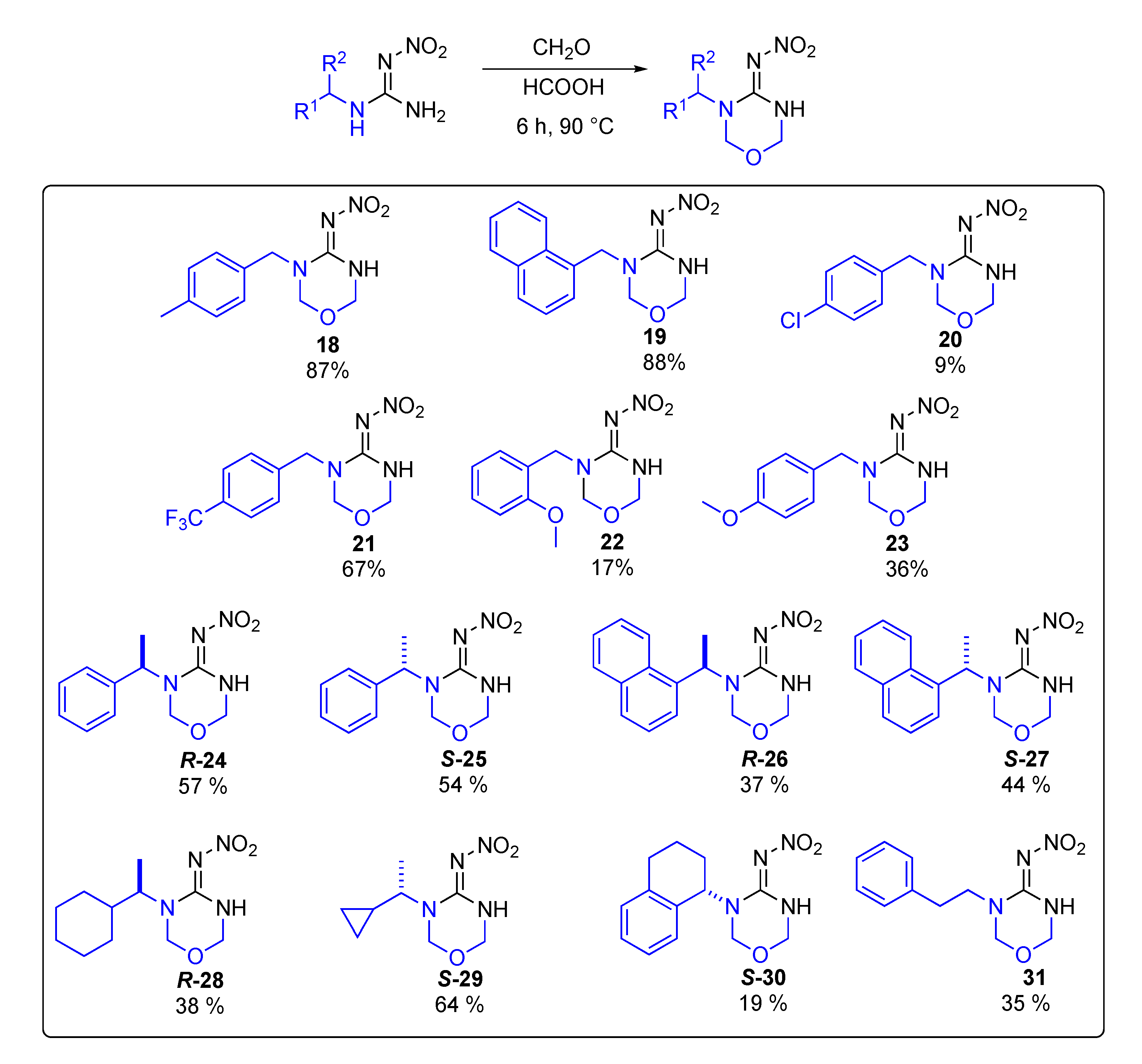
Data for (E)-N-(3-(4-methylbenzyl)-1,3,5-oxadiazinan-4-ylidene)nitramide18. White solid, 87% yield, m.p. 110–113 °C; TLC Rf 0.44 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, DMSO-d6, 50 °C): δ 9.73 (br s, 1H), 7.21 (d, J = 8.2 Hz, 2H), 7.18 (d, J = 8.1 Hz, 2H), 4.97 (s, 2H), 4.93 (s, 2H), 4.56 (s, 2H), 2.30 (s, 3H), 13C NMR (125 MHz, DMSO-d6, 50 °C): δ 154.6, 137.2, 133.6, 129.6, 127.8, 77.3, 73.8, 48.5, 21.0. HRMS (ESI): calculated for C11H14N4O3Na [M + Na]+ 273.0958, found 273.0964 (−3.7 ppm). IR (ATR) νmax: 3275, 1545.
Data for (E)-N-(3-(naphthalen-2-ylmethyl)-1,3,5-oxadiazinan-4-ylidene)nitramide19. White solid, 88% yield, m.p. 170–174 °C; TLC Rf 0.47 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, DMSO-d6, 50 °C): δ 9.85 (s, 1H), 8.12–8.07 (m, 1H), 8.01–7.95 (m, 1H), 7.90 (d, J = 8.2 Hz, 1H), 7.63–7.56 (m, 2H), 7.52 (dd, J = 8.2, 7.1 Hz, 1H), 7.43–7.39 (m, 1H), 5.13 (s, 2H), 5.04 (s, 2H), 4.97 (s, 2H), 13C NMR (125 MHz, DMSO-d6, 50 °C): δ 154.7, 133.9, 131.8, 131.1, 129.1, 128.4, 126.9, 126.5, 125.9, 124.6, 123.4, 78.1, 73.9, 47.1 ppm. HRMS (ESI): calculated for C14H14N4O3Na [M + Na]+ 309.0958, found 309.0964 (−2.6 ppm). IR (ATR) νmax: 3297, 1546.
Data for (E)-N-(3-(4-chlorobenzyl)-1,3,5-oxadiazinan-4-ylidene)nitramide20. White solid, 9% yield, m.p. 123–127 °C; TLC Rf 0.34 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, DMSO-d6): δ 9.82 (s, 1H), 7.44 (d, J = 8.1 Hz, 3H), 7.33 (d, J = 8.1 Hz, 3H), 5.00–4.94 (m, 4H), 4.59 (s, 2H), 13C NMR (125 MHz, DMSO-d6): δ 154.4, 135.9, 132.5, 129.6, 129.1, 78.1, 73.86, 48.1. HRMS (ESI): calculated for C10H11CIN4O3Na [M + Na]+ 293.0412, found 293.0417 (−3.4 ppm). IR (ATR) νmax: 3308, 1589.
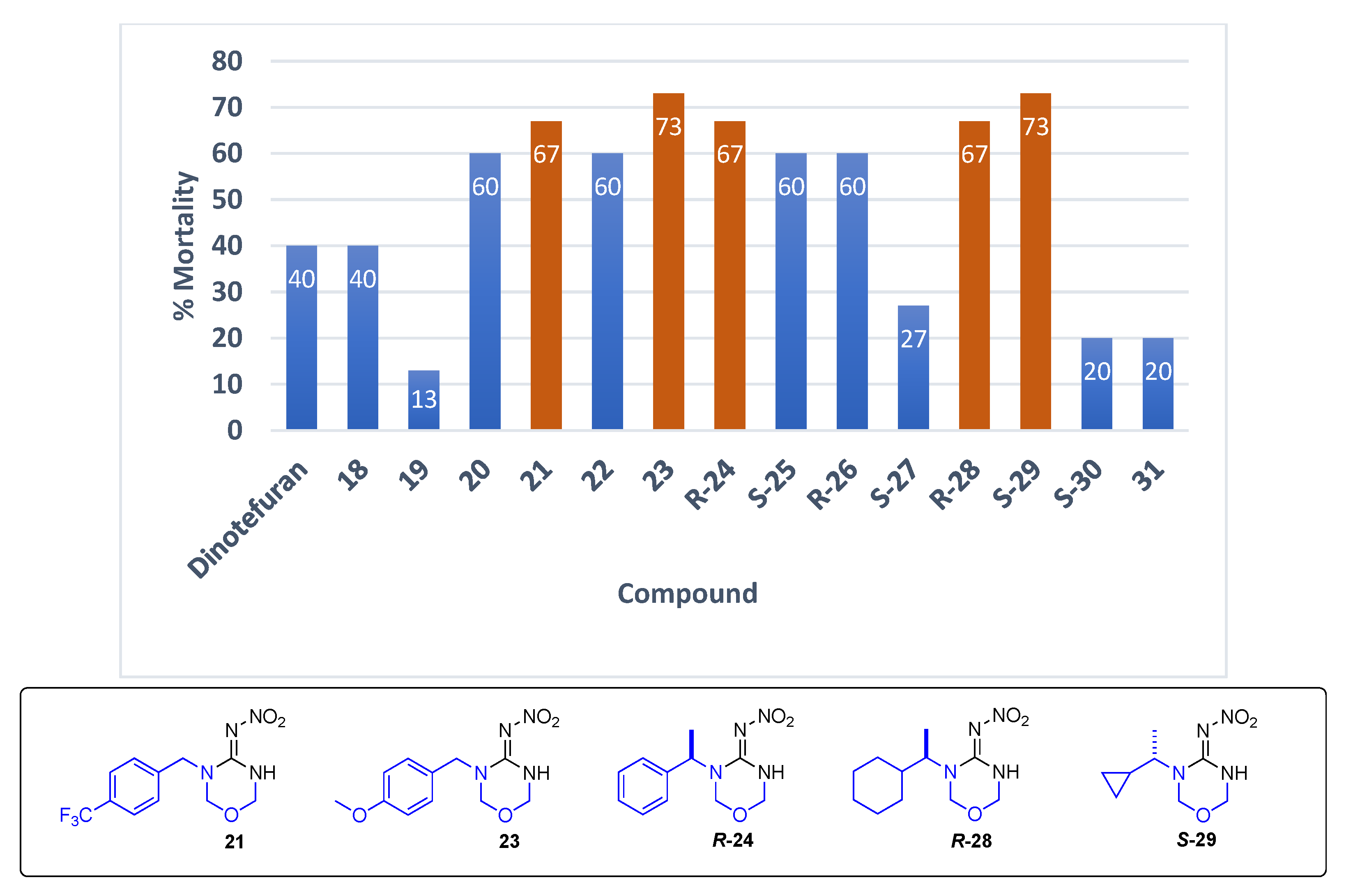
Data for (E)-N-(3-(4-(trifluoromethyl)benzyl)-1,3,5-oxadiazinan-4-ylidene)nitramide21. White solid, 67% yield, m.p. 176–178 °C; TLC Rf 0.31 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, DMSO-d6): δ 9.86 (s, 1H), 7.75 (d, J = 8.1 Hz, 2H), 7.51 (d, J = 8.0 Hz, 2H), 5.00 (s, 2H), 4.99 (s, 2H), 4.69 (s, 2H), 13C NMR (125 MHz, DMSO-d6): δ 154.9, 138.9, 130.6 (q, J = 36.25 Hz), 128.0, 126.02 (q, J = 3.75 Hz), 123.8 (q, J = 270 Hz), 77.6, 73.5, 48.8, 19F NMR (470 MHz, DMSO-d6): δ −60.86. HRMS (ESI): calculated for C11H12F3N4O3+ [M + H]+ 305.0856, found 305.0861 (−4.9 ppm). IR (ATR) νmax: 3316, 1550.
Data for (E)-N-(3-(2-methoxybenzyl)-1,3,5-oxadiazinan-4-ylidene)nitramide22. White solid, 17% yield, m.p. 143–145 °C; TLC Rf 0.44 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, DMSO-d6, 50 °C): δ 9.71 (s, 1H), 7.29 (td, J = 8.3, 7.4, 1.7 Hz, 1H), 7.21 (dd, J = 7.5, 1.7 Hz, 1H), 7.03 (dd, J = 8.2, 1.1 Hz, 1H), 6.95 (td, J = 7.5, 1.1 Hz, 1H), 4.98 (s, 2H), 4.96 (s, 2H), 4.54 (s, 2H), 3.83 (s, 3H), 13C NMR (125 MHz, DMSO-d6, 50 °C): δ 157.3, 154.7, 129.2, 128.3, 124.4, 120.8, 111.4, 78.5, 73.9, 55.9, 44.7. HRMS (ESI): calculated for C11H14N404Na [M + Na]+ 289.0907, found 289.0913 (−2.4 ppm). IR (ATR) νmax: 3284, 1551.
Data for (E)-N-(3-(4-methoxybenzyl)-1,3,5-oxadiazinan-4-ylidene)nitramide23. White solid, 36% yield, m.p. 162–167 °C; TLC Rf 0.34 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, DMSO-d6, 50 °C): δ 9.72 (s, 1H), 7.29–7.23 (m, 2H), 6.93 (d, J = 8.7 Hz, 2H), 4.96 (d, J = 2.4 Hz, 2H), 4.93 (s, 2H), 4.54 (s, 2H), 3.76 (s, 3H), 13C NMR (125 MHz, DMSO-d6, 50 °C): δ 159.3, 154.5, 129.3, 128.6, 114.6, 77.8, 73.8, 55.6, 48.2. HRMS (ESI): calculated for C11H14N4O4Na [M + Na]+ 289.0907, found 289.0913 (−3.5 ppm). IR (ATR) νmax: 3266, 1547.
Data for (R)-N-(3-(1-phenylethyl)-1,3,5-oxadiazinan-4-ylidene)nitramide R-24. White solid, 57% yield, m.p. 108–109 °C; TLC Rf 0.44 (AcOEt/Hexane, 6:4) [α]25.5 = +169.78 (c = 0.5, acetone), 1H NMR (500 MHz, CDCl3): δ 10.00 (s, 1H), 7.41–7.36 (m, 3H), 7.35–7.30 (m, 4H), 6.06 (q, J = 7.1 Hz, 1H), 5.00 (dd, J = 8.6, 2.4 Hz, 1H), 4.95 (dd, J = 8.6, 2.2 Hz, 1H), 4.76 (d, J = 9.4 Hz, 1H), 4.51 (d, J = 9.4 Hz, 1H), 13C NMR (125 MHz, CDCl3): δ 154.4, 137.8, 128.9, 128.2, 127.2, 73.8, 73.7, 52.2, 16.1. HRMS (ESI): calculated for C11H14N4NaO3+ [M + Na]+ 273.0958, found 273.0964 (5.0 ppm). IR (ATR) νmax: 3280.
Data for (S)-N-(3-(1-phenylethyl)-1,3,5-oxadiazinan-4-ylidene)nitramide S-25. White solid, 54% yield, m.p. 104–105 °C; TLC Rf 0.44 (AcOEt/Hexane, 6:4) [α]25.9 = −165.12 (c = 0.5, acetone,), 1H NMR (500 MHz, CDCl3): δ 9.98 (s, 1H), 7.41–7.35 (m, 2H), 7.35–7.30 (m, 3H), 6.05 (q, J = 7.1 Hz, 1H), 5.01 (dd, J = 8.6, 2.6 Hz, 1H), 4.95 (dd, J = 8.6, 2.5 Hz, 1H), 4.76 (d, J = 9.4 Hz, 1H), 4.51 (d, J = 9.4 Hz, 1H), 1.59 (d, J = 7.2 Hz, 3H), 13C NMR (125 MHz, CDCl3): δ 154.4, 137.8, 128.9, 128.2, 127.2, 73.8, 73.7, 52.1, 16.1. HRMS (ESI): calculated for C11H14N4NaO3 [M + Na]+ 273.0958, found 273.0964 (5.0 ppm). IR (ATR) νmax: 3282, 1578.
Data for (R)-N-(3-(1-(naphthalen-1-yl)ethyl)-1,3,5-oxadiazinan-4-ylidene)nitramide R-26. White solid, 37% yield, m.p. 211–214 °C (decomposes); TLC Rf 0.55 (AcOEt/Hexane, 6:4) [α]26.7 = +174.44 (c = 0.5, acetone), 1H NMR (500 MHz, CDCl3): δ 10.00 (s, 1H), 8.06 (dd, J = 8.4, 1.1 Hz, 1H), 7.93–7.87 (m, 2H), 7.64–7.60 (m, 1H), 7.59–7.55 (m, 2H), 7.52–7.48 (m, 1H), 6.58 (q, J = 6.8 Hz, 1H), 4.98 (ddd, J = 8.7, 3.5, 1.2 Hz, 1H), 4.72 (ddd, J = 9.6, 7.7, 1.6 Hz, 2H), 3.97 (d, J = 9.6 Hz, 1H), 1.79 (d, J = 6.8 Hz, 3H), 13C NMR (125 MHz, CDCl3): δ 153.9, 133.8, 132.4, 131.5, 129.8, 128.9, 127.5, 126.4, 125.0, 124.8, 123.2, 73.7, 73.4, 49.9, 16.3. HRMS (ESI): [M + Na]+ calculated for C15H16N4NaO3 [M + Na]+ 323.1115, found 323.1120 (5.0 ppm). IR (ATR) νmax: 3269, 1572.
Data for (S)-N-(3-(1-(naphthalen-1-yl)ethyl)-1,3,5-oxadiazinan-4-ylidene)nitramide S-27. White solid, m.p. 210–213 °C; TLC Rf 0.55 (AcOEt/Hexane, 6:4) [α]26.6 = −198.29 (c = 0.5, acetone), 1H NMR (500 MHz, CDCl3): δ 9.98 (s, 1H), 8.04 (d, J = 8.3 Hz, 1H), 7.89 (t, J = 7.8 Hz, 2H), 7.60 (t, J = 6.9 Hz, 1H), 7.55 (dd, J = 7.0, 3.0 Hz, 2H), 7.50–7.46 (m, 1H), 6.56 (q, J = 6.8 Hz, 1H), 4.96 (ddd, J = 8.6, 3.5, 1.0 Hz, 1H), 4.73–4.66 (m, 2H), 3.95 (d, J = 9.6 Hz, 1H), 1.77 (d, J = 6.8 Hz, 3H), 13C NMR (125 MHz, CDCl3): δ 153.9, 133.8, 132.5, 131.5, 129.8, 128.9, 127.5, 126.4, 125.0, 124.8, 123.2, 73.7, 73.4, 49.9, 16.3. HRMS (ESI): calculated for C15H16N4NaO3+ [M + Na]+ 323.1115, found 323.1120 (5.0 ppm). IR (ATR) νmax: 3269, 1573.
Data for (R)-N-(3-(1-cyclohexylethyl)-1,3,5-oxadiazinan-4-ylidene)nitramide R-28. White solid, 38% yield, m.p. 139–140 °C; TLC Rf 0.51 (AcOEt/Hexane, 6:4) [α]26.3 = −22.39 (c = 0.5, acetone), ¹H NMR (500 MHz, CDCl3): δ 9.99 (br s, 1H), 5.01-5.00 (d, J = 2.4 Hz, 2H), 4.88–4.86 (d, J = 9.5 Hz, 1H), 4.82–4.80 (d, J = 9.4 Hz, 1H), 4.48–4.43 (m, 1H), 1.78–1.66 (m, 5H), 1.37–1.30 (m, 1H), 1.25–0.95 (m, 9H), 13C NMR (125 MHz, CDCl3): δ 154.90, 73.72, 73.31, 55.50, 40.82, 29.81, 29.73, 25.93, 25.80, 16.06. HRMS (ESI): calculated for C11H21N4O3 [M + H]+ 257.1608, found 257.1614 (5.0 ppm). IR (ATR) νmax: 3729, 1567.
Data for (S)-N-(3-(1-cyclopropylethyl)-1,3,5-oxadiazinan-4-ylidene)nitramide S-29. White solid, 64% yield, m.p. 111–113 °C; TLC Rf 0.63 (AcOEt/Hexane, 7:3) [α]26 = +12.15 (c = 1, acetone), 1H NMR (500 MHz, DMSO-d6, 50 °C): δ 9.67 (s, 1H), 5.09 (s, 2H), 4.93 (s, 2H), 3.71 (dq, J = 9.3, 6.9 Hz, 1H), 1.21 (d, J = 6.9 Hz, 3H), 1.10–1.01 (m, 1H), 0.61–0.53 (m, 1H), 0.49–0.42 (m, 1H), 0.36–0.30 (m, 1H), 0.29–0.23 (m, 1H), 13C NMR (125 MHz, DMSO-d6, 50 °C): δ 153.3, 73.5, 73.1, 55.2, 16.8, 13.7, 4.2, 3.0. HRMS (ESI): calculated for C8H14N4NaO3 [M + Na]+ 237.0958 found 237.0954 (5.0 ppm). IR (ATR) νmax: 3295, 1578.
Data for (S)-N-(3-(1,2,3,4-tetrahydronaphthalen-1-yl)-1,3,5-oxadiazinan-4-ylidene)nitramide S-30. White solid, 19% yield, m.p. 167–169 °C; TLC Rf 0.25 (AcOEt/Hexane, 6:4) [α]24 = −47.14 (c = 1, acetone). 1H NMR (500 MHz, CDCl3): δ 10.05 (s, 1H), 7.21–7.11 (m, 4H), 5.94 (dd, J = 10.2, 6.1 Hz, 1H), 5.04 (d, J = 2.4 Hz, 2H), 4.69 (d, J = 9.6 Hz, 1H), 4.60 (d, J = 9.5 Hz, 1H), 2.86–2.73 (m, 2H), 2.24–2.17 (m, 1H), 1.99–1.91 (m, 1H), 1.89–1.79 (m, 1H), 1.72 (tdd, J = 12.2, 10.0, 2.9 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ 155.19, 138.67, 132.49, 129.64, 127.76, 127.36, 126.59, 74.76, 74.03, 54.17, 29.19, 28.21, 21.43. HRMS (ESI): calculated for C13H16N4NaO3 [M + Na]+ 299.1115, found 299.1111 (5.0 ppm). IR (ATR) νmax: 3303, 1557.
Data for N-(3-phenethyl-1,3,5-oxadiazinan-4-ylidene)nitramide R-31. White solid, 35% yield, m.p. 134–136 °C; TLC Rf 0.45 (AcOEt/Hexane, 6:4) 1H NMR (500 MHz, CDCl3): δ 9.77 (br s, 1H), 7.35–7.31 (m, 2H), 7.28–7.25 (m, 1H), 7.23–7.19 (m, 2H), 4.89 (d, J = 2.7 Hz, 2H), 4.46 (s, 2H), 3.67 (t, J = 6.8 Hz, 2H), 2.96 (t, J = 6.8 Hz, 2H), 13C NMR (125 MHz, CDCl3): δ 154.5, 138.1, 128.9, 128.8, 126.9, 78.8, 73.2, 48.9, 34.4. HRMS (ESI): [M + Na]+ calculated for C11H14N4NaO3 [M + Na]+ 273.0958, found 273.0956. (5.0 ppm). IR (ATR) νmax: 3324, 1587.
Appendix C
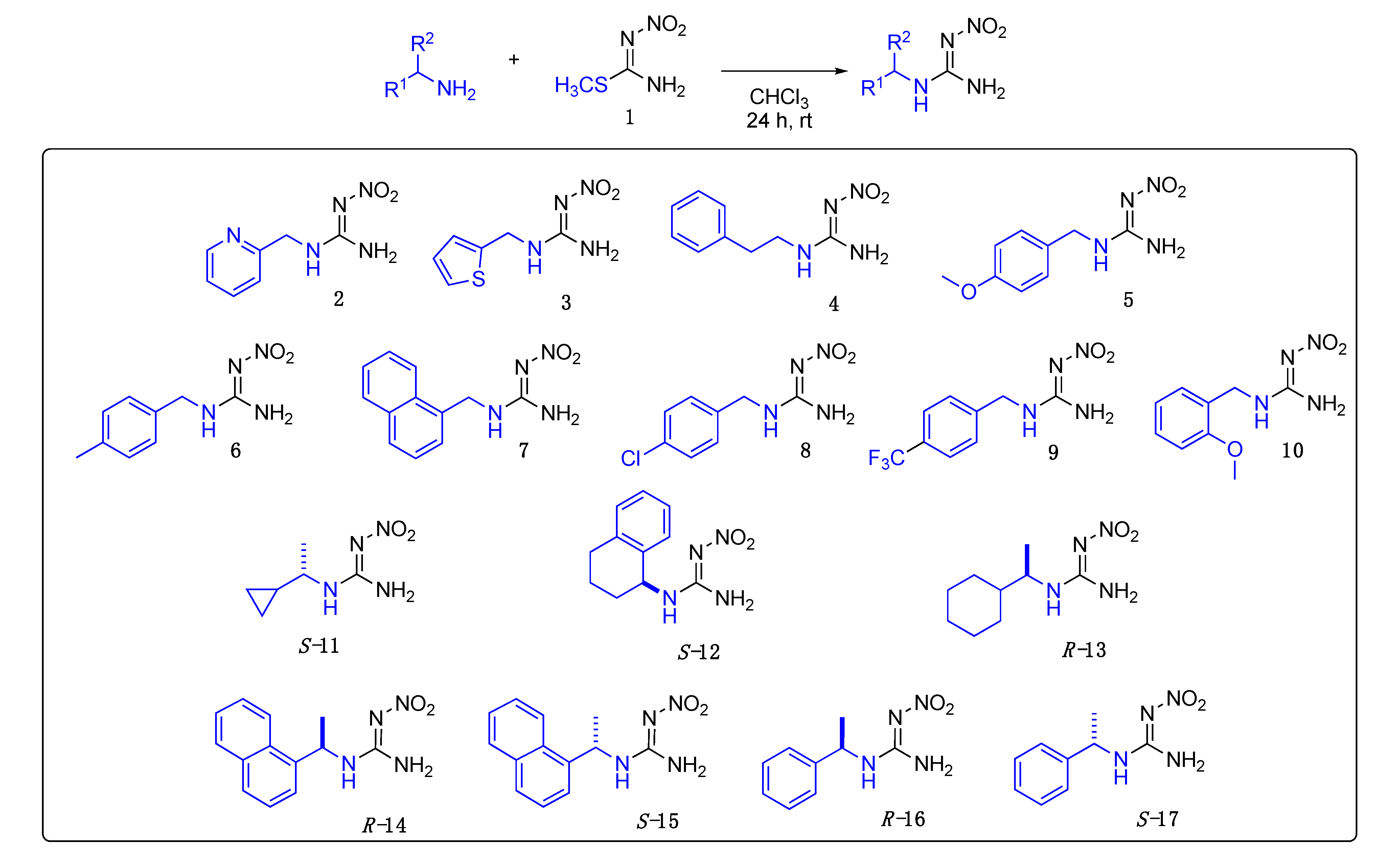
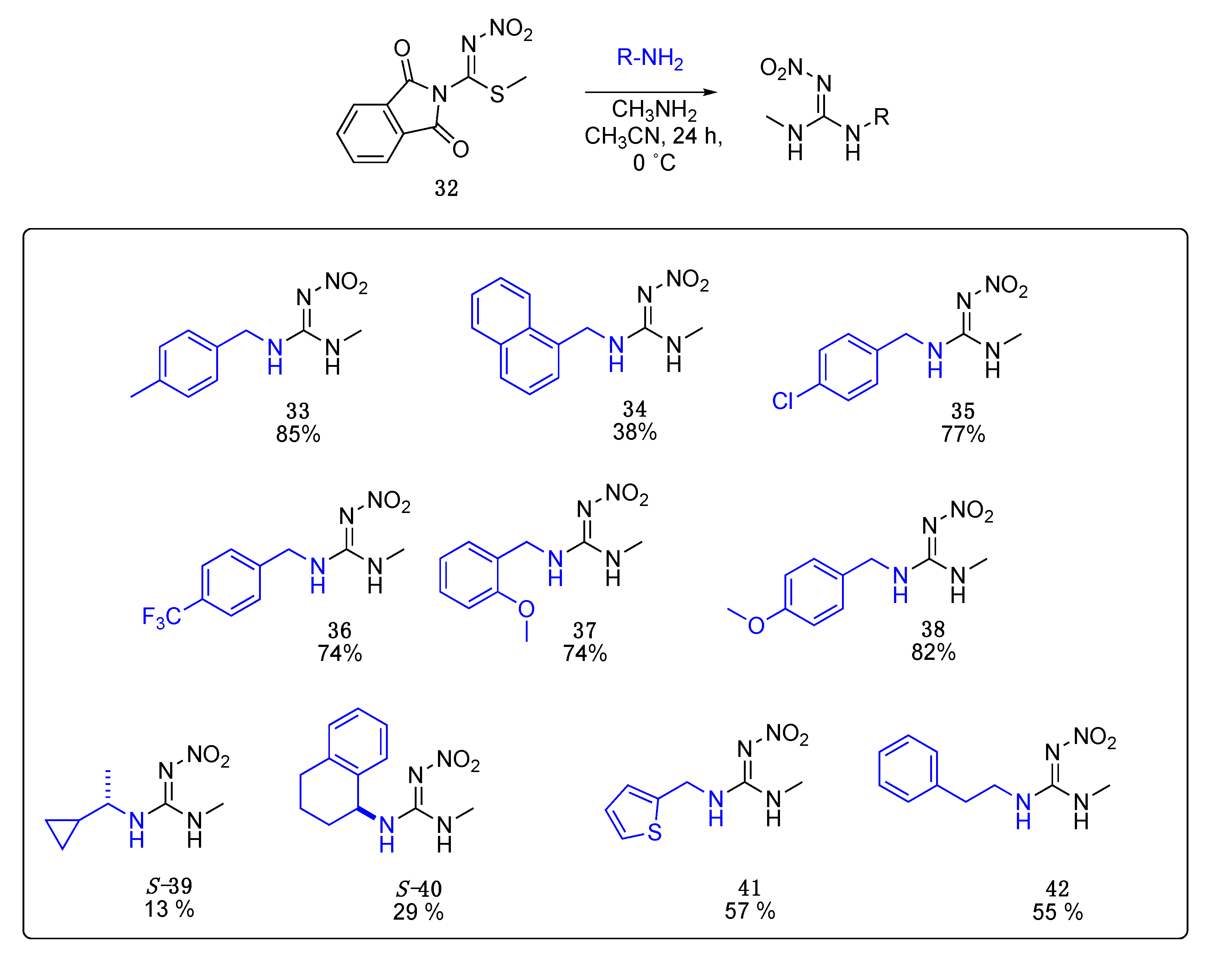
Data for (E)-1-methyl-3-(4-methylbenzyl)-2-nitroguanidine33. White solid, 85%, m.p. 166–168 °C; TLC Rf 0.33 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, DMSO-d6): δ 9.17 (br s, 1H), 7.79 (br s, 1H), 7.19 (d, J = 7.8 Hz, 2H), 7.15 (d, J = 7.8 Hz, 2H), 4.38 (s, 2H), 2.84 (br s, 3H), 2.28 (s, 3H), 13C NMR (125 MHz, DMSO-d6): δ 158.3, 136.6, 129.4, 127.4, 44.2, 28.7, 21.1. HRMS (ESI): calculated for C10H14N4O2Na [M + Na]+ 245.1009, found 245.1014 (−2.9 ppm). IR (ATR) νmax: 3285, 1627.
Data for (E)-1-methyl-3-(naphthalen-2-ylmethyl)-2-nitroguanidine34. White solid, 38% yield, m.p. 159–162 °C; TLC Rf 0.37 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, DMSO-d6, 50 °C): δ 8.11 (d, J = 8.2 Hz, 1H), 7.99–7.95 (m, 1H), 7.87 (d, J = 8.1 Hz, 1H), 7.62–7.54 (m, 2H), 7.50 (dd, J = 8.1, 7.1 Hz, 1H), 7.46 (dd, J = 7.0, 1.3 Hz, 1H), 7.33 (s, 2H), 4.93 (d, J = 4.7 Hz, 2H), 2.88 (s, 3H), 13C NMR (125 MHz, DMSO-d6, 50 °C): δ 158.5, 133.7, 130.9, 128.1, 124.6, 42.8, 28.8. HRMS (ESI): calculated for C13H14N4O2Na [M + Na]+ 281.1009, found 281.1014 (−1.8 ppm). IR (ATR) νmax: 3352, 1623.
Data for (E)-1-(4-chlorobenzyl)-3-methyl-2-nitroguanidine35. White solid, 77% yield, m.p. 191–194 °C; TLC Rf 0.23 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, DMSO-d6): δ 9.19 (br s, 1H), 7.85 (br s, 1H), 7.41 (d, J = 8.1 Hz, 2H), 7.32 (d, J = 8.0 Hz, 2H), 4.41 (s, 2H), 2.85 (br s, 3H), 13C NMR (125 MHz, DMSO-d6): δ 158.3, 132.0, 129.3, 128.7, 43.8, 28.8. HRMS (ESI): calculated for C9H11CIN4O2Na [M + Na]+ 265.0463, found 265.0468 (−4.2 ppm). IR (ATR) νmax: 3282, 1625.
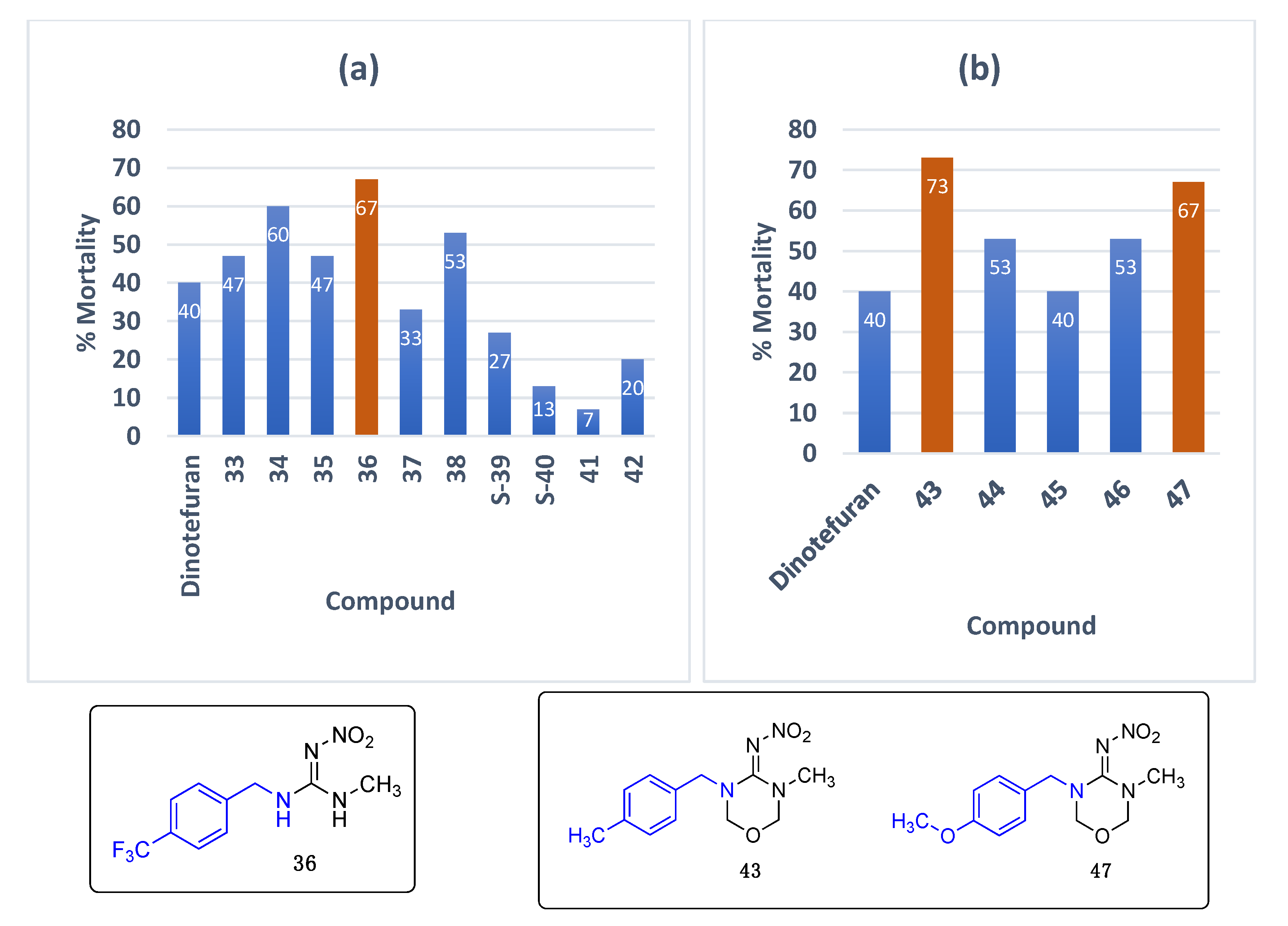
Data for (E)-1-methyl-2-nitro-3-(4-(trifluoromethyl)benzyl)guanidine36. White solid, 74% yield, m.p. 170–173 °C; TLC Rf 0.20 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, DMSO-d6): δ 9.22 (br s, 1H), 7.88 (br s, 1H), 7.72 (d, J = 8.0 Hz, 2H), 7.51 (d, J = 8.0 Hz, 2H), 4.51 (s, 2H), 2.88 (br s, 3H), 13C NMR (125 MHz, DMSO-d6): δ 158.4, 131.2, 128.0, 125.7 (q, J = 3.5 Hz), 124.7 (q, J = 261.25 Hz), 123.7, 44.1, 28.8, 19F NMR (470 MHz, DMSO-d6): δ −60.77. HRMS (ESI): calculated for C10H12F3N4O2 [M + H]+ 277.0907, found 277.0912 (−2.2 ppm). IR (ATR) νmax: 3235, 1625.
Data for (E)-1-(2-methoxybenzyl)-3-methyl-2-nitroguanidine37. White solid, 74% yield, m.p. 148–152 °C; TLC Rf 0.33 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, DMSO-d6, 50 °C): δ 8.20 (d, J = 164.9 Hz, 2H), 7.29 (td, J = 7.9, 1.7 Hz, 1H), 7.20 (d, J = 7.4 Hz, 1H), 7.03 (d, J = 8.2 Hz, 1H), 6.95 (td, J = 7.4, 1.0 Hz, 1H), 4.42 (d, J = 5.5 Hz, 2H), 3.84 (s, 3H), 2.85 (s, 3H), 13C NMR (125 MHz, DMSO-d6): δ 169.0, 136.8, 129.6, 127.9, 120.7, 111.0, 55.8, 28.7, 26.5. HRMS (ESI): [M + Na]+ calculated for C10H14N4O3Na [M + Na]+ 261.0958, found 261.0964 (−1.9 ppm). IR (ATR) νmax: 3328, 1621.
Data for (E)-1-(4-methoxybenzyl)-3-methyl-2-nitroguanidine38. White solid, 82% yield, m.p. 189–192 °C; TLC Rf 0.23 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, DMSO-d6): δ 9.16 (s, 1H), 7.77 (s, 1H), 7.24 (d, J = 8.1 Hz, 2H), 6.91 (d, J = 8.2 Hz, 2H), 4.35 (d, J = 5.2 Hz, 2H), 3.73 (s, 3H), 2.83 (s, 3H), 13C NMR (125 MHz, DMSO-d6): δ 158.9, 158.2, 131.0, 129.0, 114.2, 55.5, 43.9, 28.7. HRMS (ESI): calculated for C10H14N4O3Na+ [M + Na]+ 261.0958, found 261.0964 (−1.5 ppm). IR (ATR) νmax: 3264, 1627.
Data for (S)-1-(1-cyclopropylethyl)-3-methyl-2-nitroguanidine S-39. White solid, 13% yield, m.p. 148–150 °C; TLC Rf 0.31 (AcOEt/Hexane, 6:4) [α]25 = +31.89 (c = 1, acetone), 1H NMR (500 MHz, DMSO-d6, 50 °C): δ 8.27 (s, 2H), 3.36–3.26 (m, 1H), 2.81 (d, J = 4.9 Hz, 3H), 1.21 (d, J = 6.6 Hz, 3H), 1.08–0.99 (m, 1H), 0.53–0.46 (m, 1H), 0.46–0.39 (m, 1H), 0.34–0.28 (m, 1H), 0.24–0.17 (m, 1H), 13C NMR (125 MHz, DMSO-d6, 50 °C): δ 157.25, 51.01, 28.01, 19.89, 16.68, 2.91, 2.70. HRMS (ESI): calculated for C7H14N4NaO2 [M + Na]+ 209.1009, found 209.1006 (5.0 ppm). IR (ATR) νmax: 3334, 3290, 1622.
Data for (S)-1-methyl-2-nitro-3-(1,2,3,4-tetrahydronaphthalen-1-yl)guanidine S-40. White solid, 29% yield, m.p. 192–194 °C; TLC Rf 0.43 (AcOEt/Hexane, 6:4) [α]26 = −2.98 (c = 1, acetone), 1H NMR (500 MHz, DMSO-d6, 50 °C): δ 8.28 (br s, 2H), 7.28–7.09 (m, 4H), 5.05 (s, 1H), 2.86 (s, 3H), 2.83–2.68 (m, 2H), 2.06–1.97 (m, 1H), 1.91 (d, J = 6.8 Hz, 1H), 1.86-1.80 (m, 1H), 1.79–1.70 (m, 1H), 13C NMR (125 MHz, DMSO-d6, 50°C): δ 157.6, 137.1, 128.6, 127.4, 126.9, 125.8, 49.3, 29.5, 28.4, 28.1, 19.9. HRMS (ESI): calculated for C12H17N4O2 [M + H]+ 249.1346, found 249.1340 (5.0 ppm) IR (ATR) νmax: 3337, 3302, 1623.
Data for 1-methyl-2-nitro-3-(thiophen-2-ylmethyl)guanidine41. Yellow solid, 57%, m.p. 100–102 °C; TLC Rf 0.29 (AcOEt/Hexane, 65:35) 1H NMR (500 MHz, DMSO-d6): δ 9.18 (s, 1H), 8.10–7.82 (m, 1H), 7.42 (d, J = 5.1 Hz, 1H), 7.03 (s, 1H), 7.00–6.95 (m, 1H), 4.57 (s, 2H), 2.81 (s, 3H), 13C NMR (125 MHz, DMSO-d6): δ 157.78, 126.86, 126.10, 125.68, 39.48, 28.51. HRMS (ESI): calculated for C7H10N4NaO2S [M + Na]+ 237.0417, found 237.0413 (5.0 ppm). IR (ATR) νmax: 3286, 3210.
Data for 1-methyl-2-nitro-3-phenethylguanidine42. White solid, 55% yield, m.p. 151–153 °C; TLC Rf 0.18 (AcOEt/Hexane, 6:4) 1H NMR (500 MHz, DMSO-d6): δ 7.31 (t, J = 7.5 Hz, 2H), 7.27–7.19 (m, 3H), 3.49–3.41 (m, 2H), 2.86 (t, J = 7.4 Hz, 2H), 2.78 (d, J = 2.8 Hz, 3H), 13C NMR (125 MHz, DMSO-d6): δ 157.83, 138.53, 128.51, 128.20, 126.13, 42.22, 34.59, 27.97. HRMS (ESI): calculated for C10H14N4NaO2 [M + Na]+ 245.1009, found 245.1009 (5.0 ppm). IR (ATR) νmax: 3336, 3284, 1619.
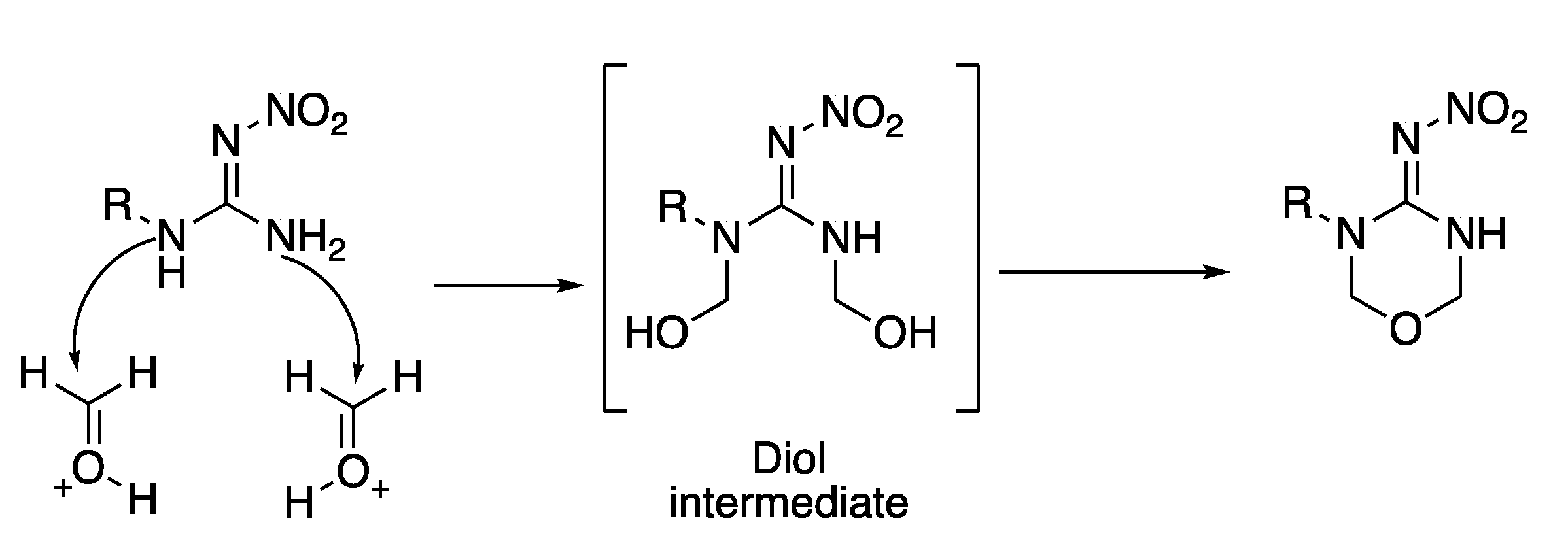
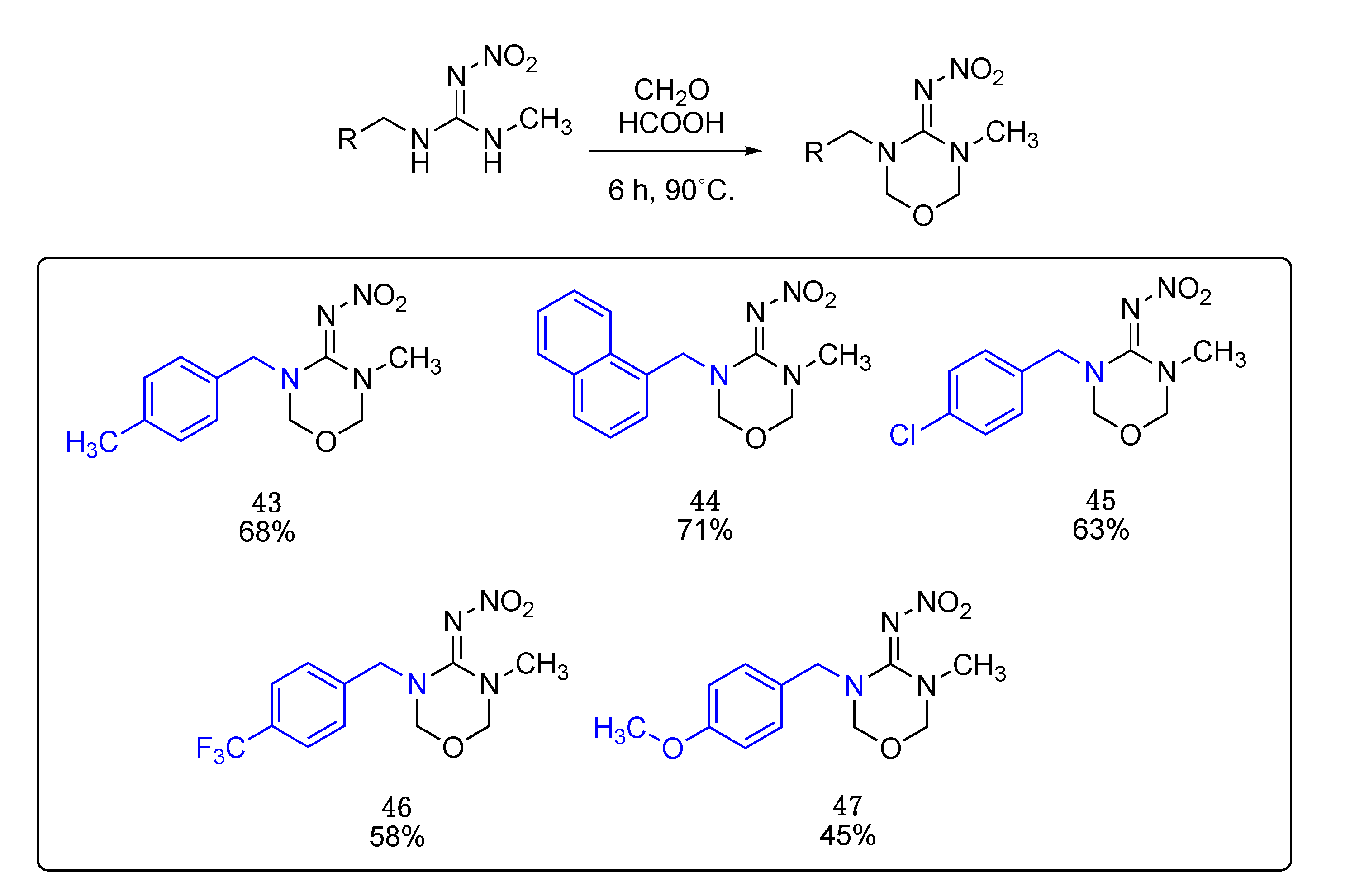
These compounds were prepared following the same procedure used for the cyclization of non-methylated nitroguanidines.
N-(3-methyl-5-(4-methylbenzyl)-1,3,5-oxadiazinan-4-ylidene)nitramide43. White solid, 68% yield, m.p. 146–151 °C; TLC Rf 0.06 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, DMSO-d6): δ 7.22–7.15 (m, 4H), 5.00 (s, 2H), 4.95 (s, 2H), 4.57 (s, 2H), 2.87 (s, 3H), 2.29 (s, 3H), 13C NMR (125 MHz, DMSO-d6): δ 156.8, 137.5, 132.9, 129.6, 128.1, 79.6, 77.4, 50.7, 33.9, 21.1. HRMS (ESI): calculated for C12H17N4O3 [M + H]+ 265.1295, found 265.1301 (−0.8 ppm). IR (ATR) νmax: 1601.
Data for N-(3-methyl-5-(naphthalen-2-ylmethyl)-1,3,5-oxadiazinan-4-ylidene)nitramide44. White solid, 71% yield, m.p. 151–153 °C; TLC Rf 0.06 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, CDCl3): δ 7.99 (dd, J = 8.4, 1.1 Hz, 1H), 7.92–7.85 (m, 2H), 7.62–7.57 (m, 1H), 7.56–7.51 (m, 1H), 7.48–7.41 (m, 2H), 5.19 (s, 2H), 4.81 (s, 2H), 4.65 (s, 2H), 3.12 (s, 3H), 13C NMR (125 MHz, CDCl3): δ 157.8, 133.9, 131.2, 129.8, 129.0, 128.9, 127.7, 127.3, 126.4, 125.2, 123.0, 79.6, 50.0, 34.8. HRMS (ESI): calculated for C15H17N4O3 [M + H]+ 301.1295, found 301.1301 (0.3 ppm). IR (ATR) νmax: 1609.
Data for (E)-N-(3-(4-chlorobenzyl)-5-methyl-1,3,5-oxadiazinan-4-ylidene)nitramide 45. White solid, 63% yield, m.p. 163–166 °C; TLC Rf 0.03 (AcOEt/Hexane, 6:4); ¹H NMR (500 MHz, DMSO-d6): δ 7.50–7.40 (m, 2H), 7.36–7.29 (m, 2H), 5.02 (s, 2H), 5.0 (s, 2H), 4.62 (s, 2H), 2.87 (s, 3H), 13C NMR (125 MHz, DMSO-d6): δ 156.9, 135.3, 132.8, 129.9, 129.0, 79.6, 77.6, 50.3, 34.0. HRMS (ESI): calculated for C11H14ClN4O3 [M + H]+ 285.0749, found 285.0754 (0.7 ppm). IR (ATR) νmax: 1608.
Data for N-(3-methyl-5-(4-(trifluoromethyl)benzyl)-1,3,5-oxadiazinan-4-ylidene)nitramide46. White solid, 58% yield, m.p. 126–129 °C; TLC Rf 0.03 (AcOEt/Hexane, 6:4); ¹H NMR (500 MHz, DMSO-d6): δ 7.72 (d, J = 7.9 Hz, 2H), 7.50 (d, J = 8.0 Hz, 2H), 5.02 (s, 4H), 4.71 (s, 2H), 2.87 (s, 3H), 13C NMR (125 MHz, DMSO-d6): δ 157.0, 141.1, 128.7 (q, J= 31.25 Hz), 128.5, 125.9 (q, J = 3.75 Hz), 124.6 (q, J = 271.25 Hz), 79.6, 77.9, 50.6, 33.9, 19F (470 MHz, DMSO-d6): δ −60.77. HRMS (ESI): calculated for C12H13F3N4O3Na [M + Na]+ 341.0837, found 341.0837 (0.9 ppm). IR (ATR) νmax: 1609.
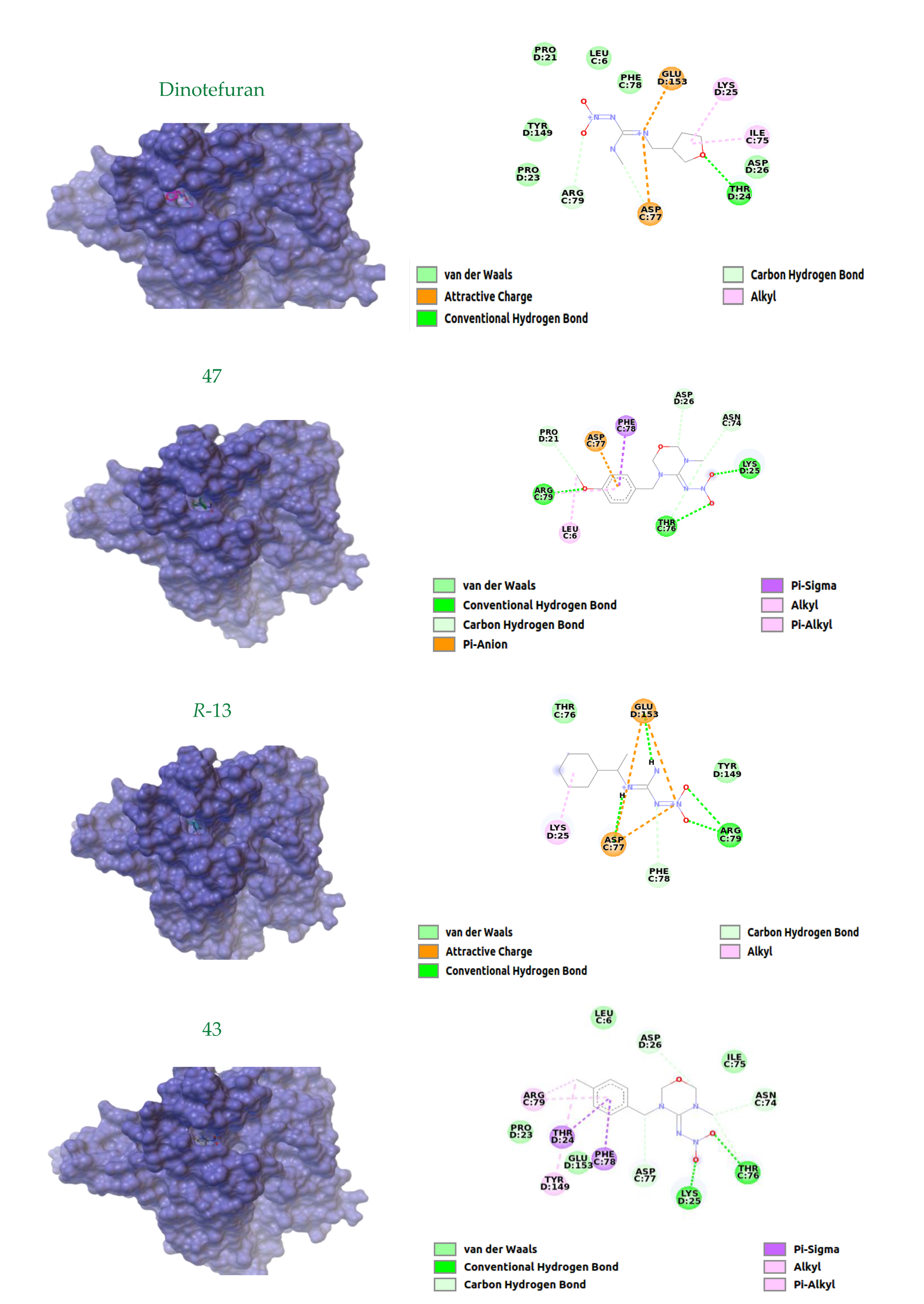
Data for N-(3-(4-methoxybenzyl)-5-methyl-1,3,5-oxadiazinan-4-ylidene)nitramide47. White solid, 45% yield, m.p. 139–142 °C; TLC Rf 0.03 (AcOEt/Hexane, 6:4); 1H NMR (500 MHz, DMSO-d6): δ 7.24 (d, J = 8.4 Hz, 2H), 6.92 (d, J = 8.6 Hz, 1H), 4.99 (s, 2H), 4.94 (s, 2H), 4.54 (s, 2H), 3.74 (s, 2H), 2.88 (s, 3H), 13C NMR (125 MHz, DMSO-d6): δ 159.4, 156.8, 129.7, 127.7, 114.5, 79.5, 77.3, 55.5, 50.4, 33.9. HRMS (ESI): calculated for C12H17N4O4+ [M + H]+ 281.1244, found 281.1250 (−1.4 ppm). IR (ATR) νmax: 1587.


 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






