
Syntheses and Study of a Pyrroline Nitroxide Condensed Phospholene Oxide and a Pyrroline Nitroxide Attached Diphenylphosphine

 ,
,  ,
,
Abstract
:
1. Introduction
2. Results and Discussions
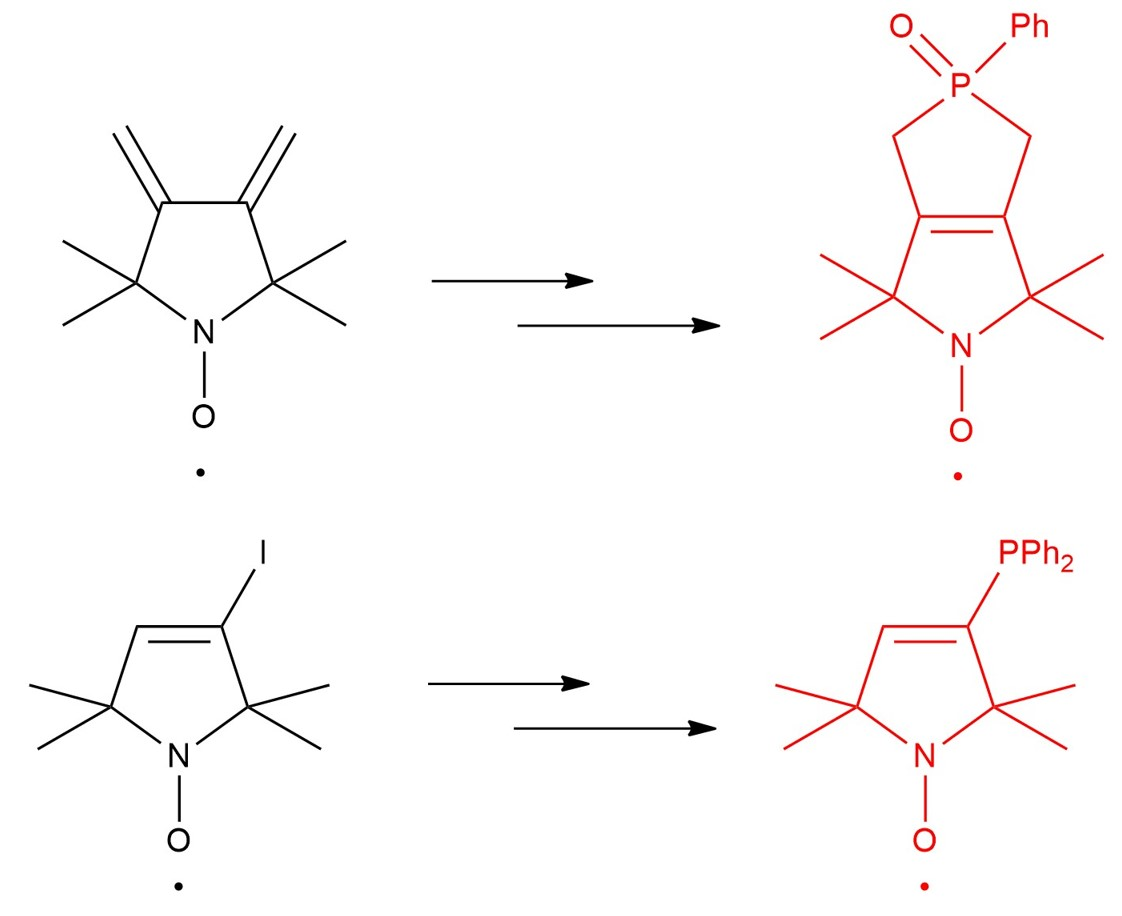
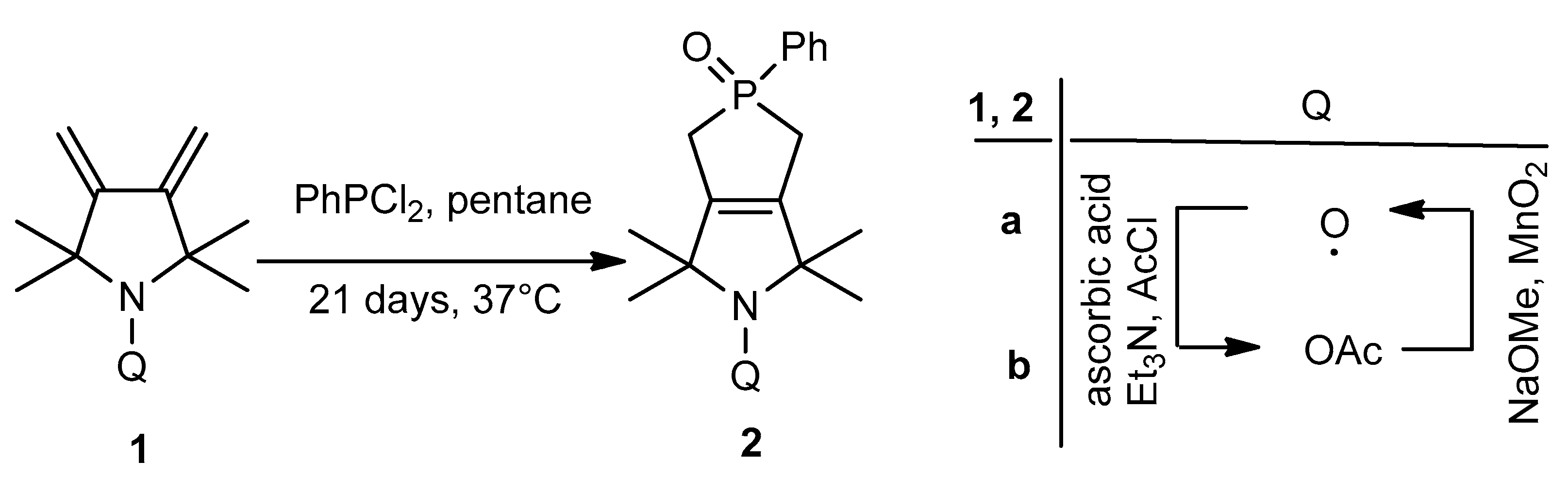
2.1. Synthesis of Paramagnetic Phospholene Oxide
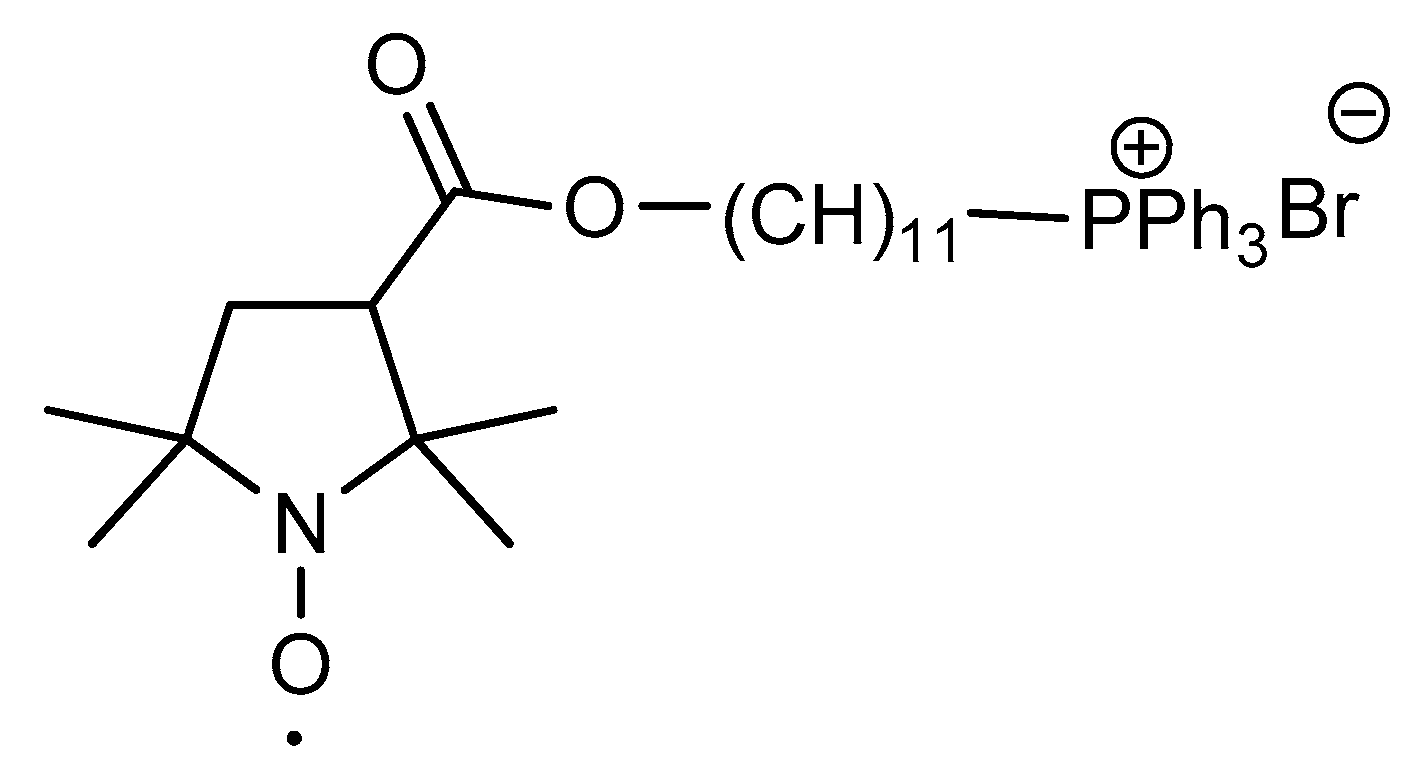
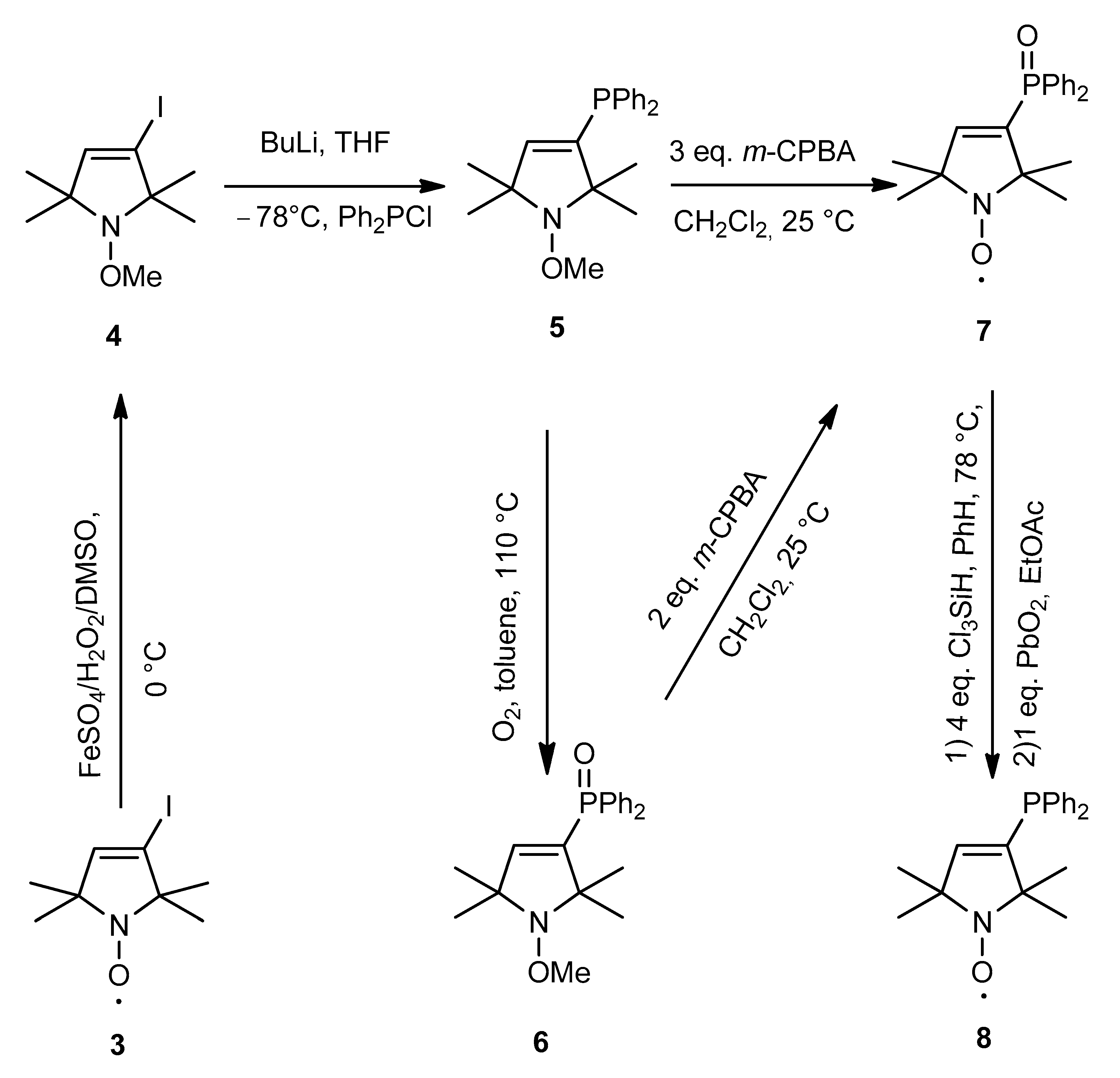
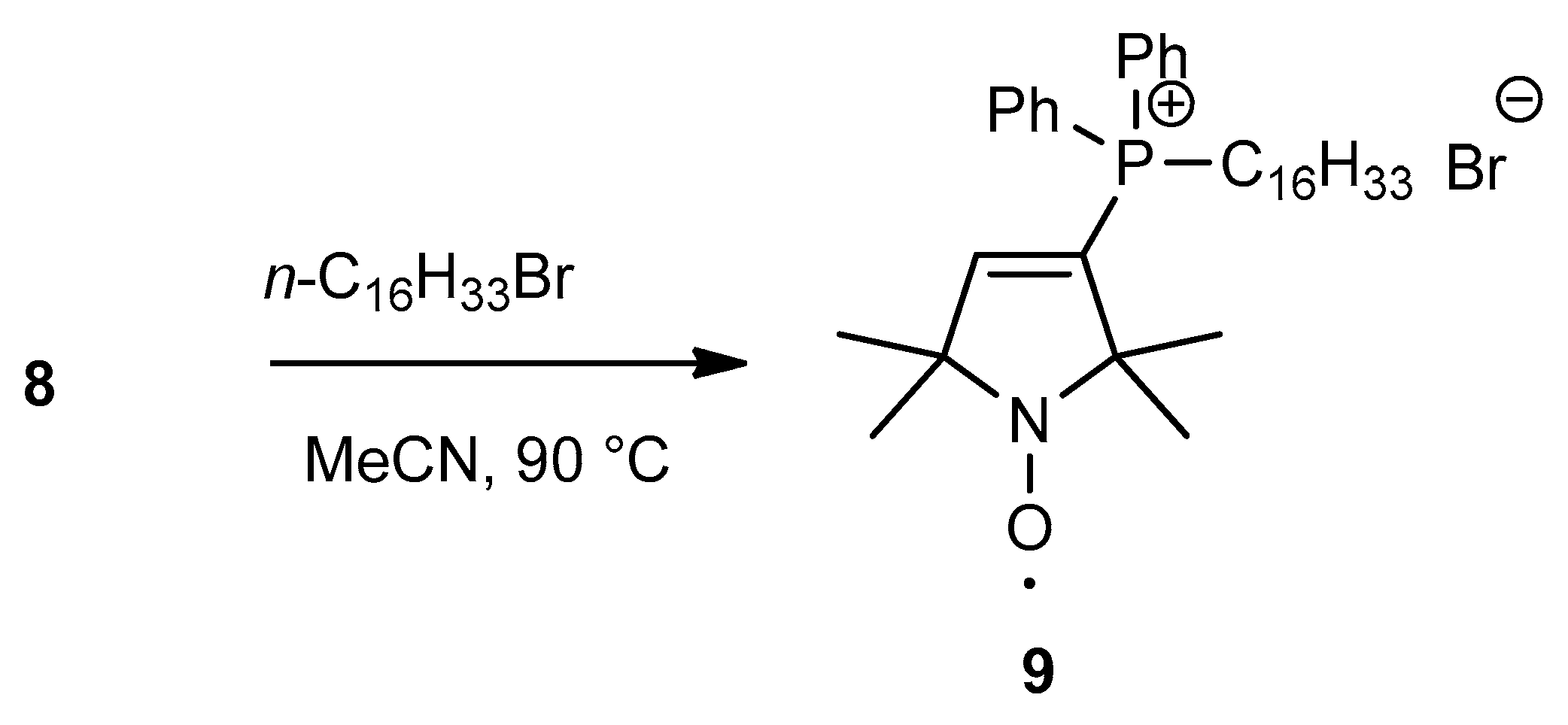
2.2. Synthesis of Pyrroline Nitroxide Diphenylphosphine and Its Phosphonium Salt
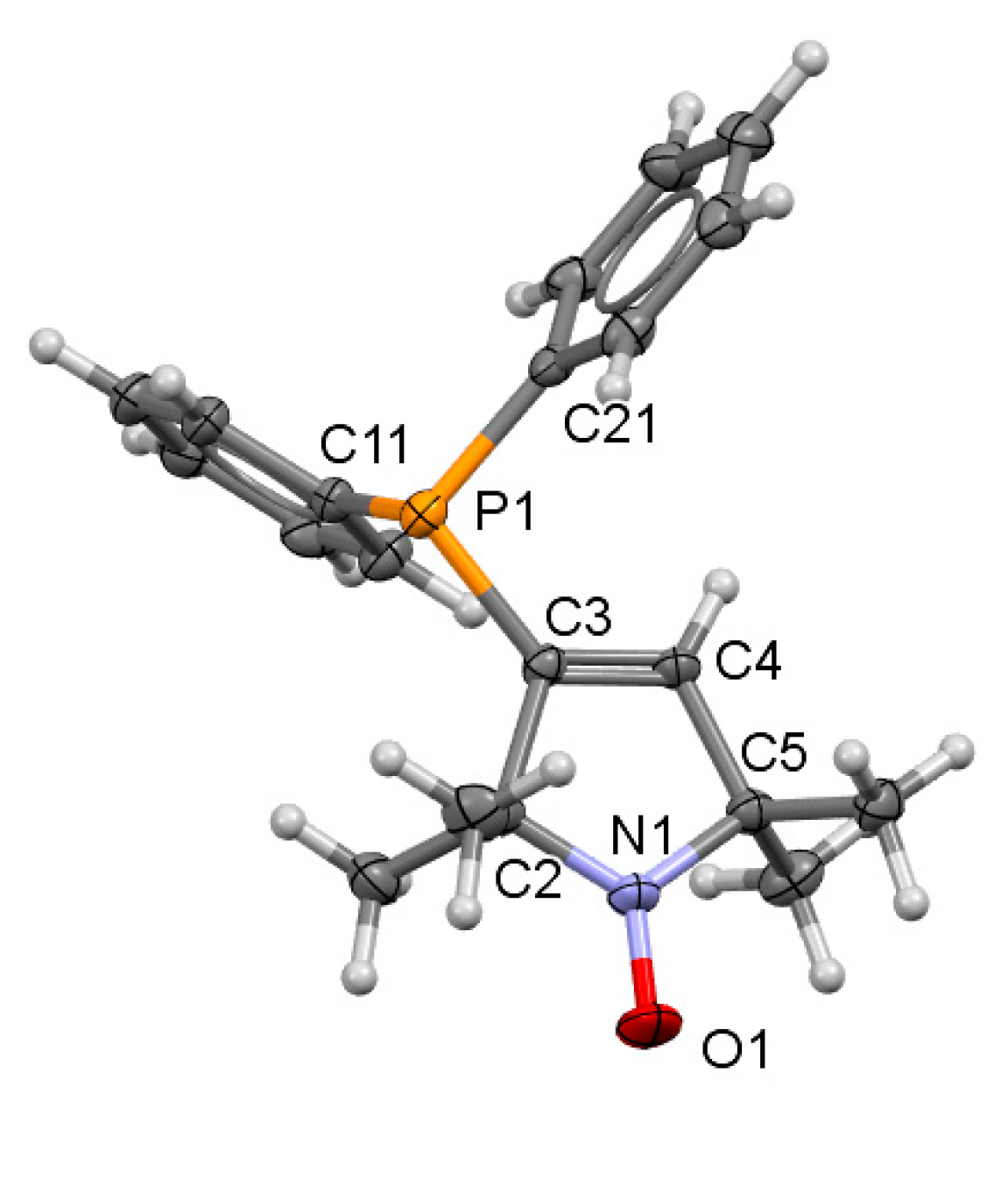
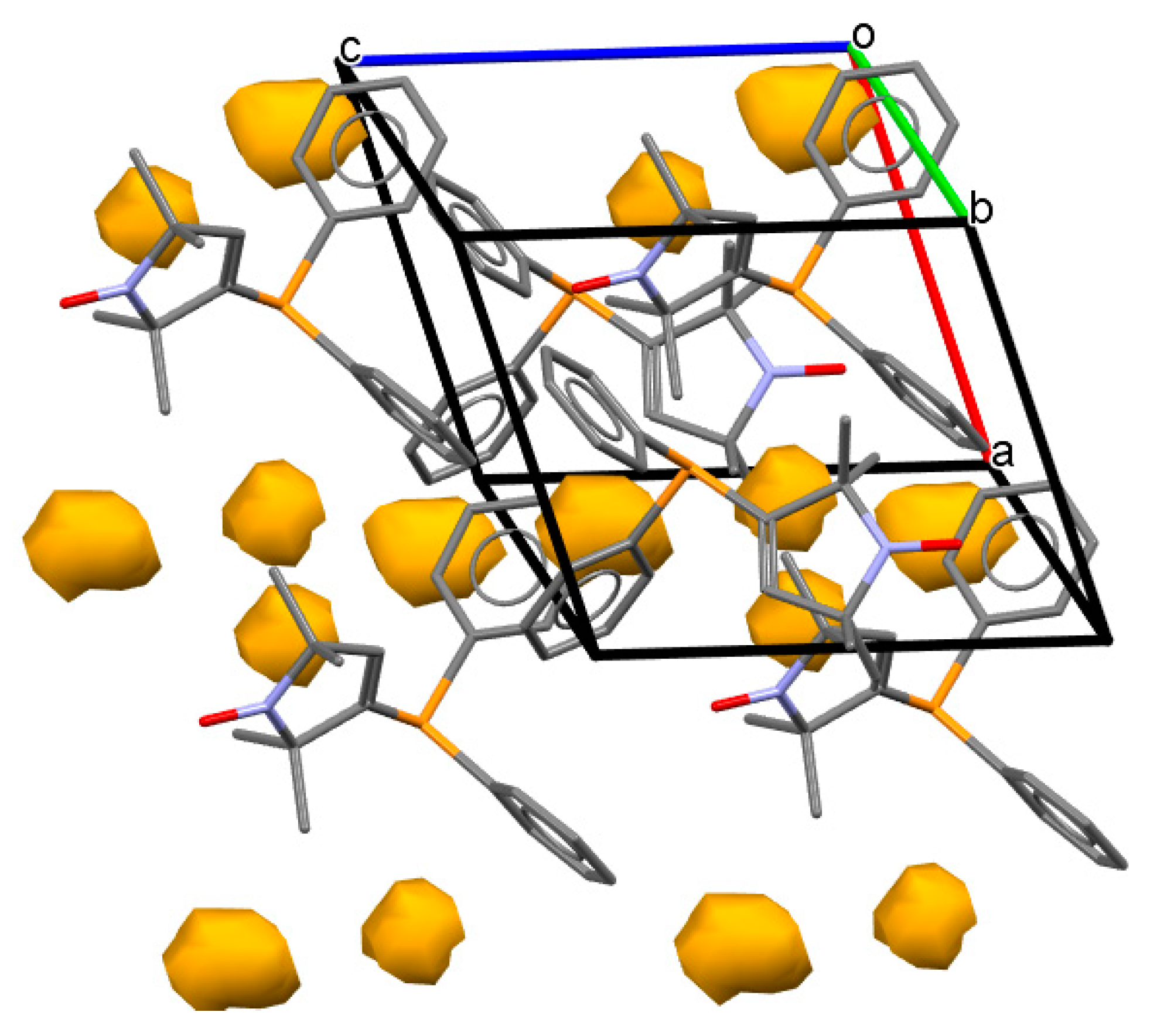
2.3. X-ray Crystallographic Study of Pyrroline Nitroxide-Diphenylphosphine
3. Materials and Methods
3.1. General Methods and Reagents
3.2. Synthesis of 2,2,5,5-Tetramethyl-3,4-dimethylenepyrrolidin-1-yl Acetate (1b)
3.3. Synthesis of 1,1,3,3-Tetramethyl-5-oxido-5-phenyl-5,6-dihydrophospholo[3,4-c]pyrrol-2(1H,3H,4H)-yl Acetate (2b)
3.4. Synthesis of 1,1,3,3-Tetramethyl-5-phenyl-5-oxido-1,2,3,4,5,6-hexahydrophospholo[3,4-c]pyrrole-2-yloxyl Radical (2a)
3.5. Synthesis of 3-Iodo-1-methoxy-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrole (4)
3.6. Synthesis of 3-(Diphenylphosphino)-1-methoxy-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrole (5)
3.7. Synthesis of 1-Methoxy-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)diphenylphosphine Oxide (6)
3.8. Synthesis of 3-(Diphenylphosphinoxido)-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-1-yloxyl Radical (7)
3.9. Synthesis of 3-(Diphenylphosphino)-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-1-yloxyl Radical (8)
3.10. Synthesis of Hexadecyl (1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)diphenylphosphonium Bromide Radical (9)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Likhtenshtein, G.I. Nitroxides: Brief History, Fundamentals, and Recent Developments; Springer: Cham, Switzerland, 2020. [Google Scholar]
- Ouari, O.; Gigmes, D. (Eds.) Nitroxides: Synthesis, Properties and Applications; The Royal Society of Chemistry: Cambridge, UK, 2021. [Google Scholar]
- Neiman, M.B.; Rozantsev, E.G.; Mamedova, Y.G. Free radical reactions involving no unpaired electrons. Nature 1962, 196, 472–474. [Google Scholar] [CrossRef]
- Hideg, K.; Sár, P.C.; Hankovszky, H.O.; Tamás, T.; Jerkovich, G. Synthesis of new 3,4-disubstituted 2,5-dihydro-1h-pyrrol-1-yloxyl spin-label reagents via allylic rearrangements. Synthesis 1993, 25, 390–394. [Google Scholar] [CrossRef]
- Chalmers, B.A.; Morris, J.C.; Fairfull-Smith, K.E.; Grainger, R.S.; Bottle, S.E. A novel protecting group methodology for syntheses using nitroxides. Chem. Commun. 2013, 49, 10382–10384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isbera, M.; Bognár, B.; Jekő, J.; Sár, C.; Hideg, K.; Kálai, T. Syntheses and reactions of pyrroline, piperidine nitroxide phosphonates. Molecules 2020, 25, 2430. [Google Scholar] [CrossRef] [PubMed]
- Isbera, M.; Bognár, B.; Sár, C.; Jekő, J.; Kálai, T. Syntheses and utilizations of pyrroline-nitroxide- and tetrahydropyridine-nitroxide-based α-ketophosphonates, β-ketophosphonates, and a bisphosphonate. Synth. Commun. 2021, 51, 1353–1362. [Google Scholar] [CrossRef]
- Rancurel, C.; Heise, H.; Köhler, F.H.; Schatzschneider, U.; Rentschler, E.; Vidal-Gancedo, J.; Veciana, J.; Sutter, J.-P. Spin transfer and magnetic interaction via phosphorus in nitronyl nitroxide radical-substituted triphenylphosphine derivatives. J. Phys. Chem. A 2004, 108, 5903–5914. [Google Scholar] [CrossRef] [Green Version]
- Stipa, P.; Finet, J.P.; Le Moigne, F.; Tordo, P. β-Phosphorylated five-membered ring nitroxides: Synthesis and EPR study of 2-phosphonyl-4-(hydroxymethyl)pyrrolidine aminoxyl radicals. J. Org. Chem. 1993, 58, 4465–4468. [Google Scholar] [CrossRef]
- Kálai, T.; Balog, M.; Jekő, J.; Hideg, K. Synthesis and reactions of symmetric paramagnetic pyrrolidine diene. Synthesis 1999, 31, 973–980. [Google Scholar] [CrossRef]
- McCormack, W.B. Substituted Phosphacyclopentene Oxides and Process of Preparing Them. U.S. Patent 2,663,737, 22 December 1953. [Google Scholar]
- Quin, L.D.; Middlemas, E.D.; Rao, S.N.; Miller, R.W.; McPhail, A.T. Synthesis and conformational properties of 3,8-phosphanedione 1-oxides. J. Am. Chem. Soc. 1982, 104, 1893–1900. [Google Scholar] [CrossRef]
- Wang, J.Y.; Li, Q.J.; Xiao, Y.M.; Fu, B.; Qin, Z.H. Triphenylphosphonium (TPP)-based antioxidants: A new perspective on antioxidant Design. ChemMedChem 2020, 15, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-targated triphenylphosphonium-based compounds: Syntheses, mechanism of action, and therapeutic and diagnostic applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, A.; Kotamraju, S.; Karunakaran, C.; Kalivendi, S.V.; Thomas, S.; Joseph, J.; Kalyanaraman, B. Mitochondria superoxide dismutase mimetic inhibits peroxide-induced oxidative damage and apoptosis: Role of mitochondrial Superoxide. Free Rad. Biol. Med. 2005, 39, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Kálai, T.; Bognár, B.; Jekő, J.; Hideg, K. Synthesis of new, paramagnetically modified heterocycles. Synthesis 2006, 38, 2573–2579. [Google Scholar]
- Fritzsche, H.; Hasserodt, U.; Korte, F. Reduktion organischer verbindungen des fünfwertigen phosphors zu phosphinen, II. reduktion tertiärer phosphinoxyde zu tertiären phosphinen mit trichlorsilan. Chem. Ber. 1965, 98, 171–174. [Google Scholar] [CrossRef]
- Kovács, T.; Keglevich, G. The reduction of tertiary phosphine oxides by silanes. Curr. Org. Chem. 2017, 21, 569–585. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westrip, S.P. publCIF: Software for editing, validating and formatting crystallograph-ic information files. J. Appl. Crystallogr. 2010, 43, 920–925. [Google Scholar] [CrossRef] [Green Version]
- Mercury, C.F.; Macrae, P.R.; Edgington, P.M.E.; Pidcock, G.P.; Shields, R.; Taylor, M.T.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The cambridge structural database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Formula | C20H23NOP |
|---|---|
| Mr | 324.36 |
| Crystal system, space group | Monoclinic, P21 |
| Temperature (K) | 200 |
| a, b, c (Å) | 8.3032 (3), 12.1786 (4), 9.3477 (3) |
| β (°) | 108.792 (2) |
| V (Å3) | 894.86 (5) |
| Z | 2 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.16 |
| Crystal size (mm) | 0.45 × 0.44 × 0.28 |
| Data collection | |
| Diffractometer | Bruker D8 VENTURE |
| Absorption correction | Multiscan SADABS2016/2-Bruker AXS area detector scaling and absorption correction |
| Tmin, Tmax | 0.85, 0.96 |
| No. of measured, independent, and observed (I > 2σ(I)) reflections | 13,054, 3528, 3427 |
| Rint | 0.037 |
| (sin θ/λ)max (Å−1) | 0.619 |
| Refinement | |
| R(F2 > 2σ(F2)), wR(F2), S | 0.031, 0.096, 1.21 |
| No. of reflections | 3528 |
| No. of parameters | 213 |
| No. of restraints | 1 |
| H-atom treatment | H-atom parameters constrained |
| Δ>max, Δ>min (e Å−3) | 0.51, −0.50 |
| Absolute structure | Flack x determined using 1581 quotients [(I+) − (I−)]/[(I+) + (I−)] (Parsons, Flack and Wagner, Acta Cryst. B69 (2013) 249–259). |
| Absolute structure parameter | –0.05 (3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isbera, M.; Bognár, B.; Gallyas, F.; Bényei, A.; Jekő, J.; Kálai, T. Syntheses and Study of a Pyrroline Nitroxide Condensed Phospholene Oxide and a Pyrroline Nitroxide Attached Diphenylphosphine. Molecules 2021, 26, 4366. https://doi.org/10.3390/molecules26144366
Isbera M, Bognár B, Gallyas F, Bényei A, Jekő J, Kálai T. Syntheses and Study of a Pyrroline Nitroxide Condensed Phospholene Oxide and a Pyrroline Nitroxide Attached Diphenylphosphine. Molecules. 2021; 26(14):4366. https://doi.org/10.3390/molecules26144366
Chicago/Turabian StyleIsbera, Mostafa, Balázs Bognár, Ferenc Gallyas, Attila Bényei, József Jekő, and Tamás Kálai. 2021. "Syntheses and Study of a Pyrroline Nitroxide Condensed Phospholene Oxide and a Pyrroline Nitroxide Attached Diphenylphosphine" Molecules 26, no. 14: 4366. https://doi.org/10.3390/molecules26144366
APA StyleIsbera, M., Bognár, B., Gallyas, F., Bényei, A., Jekő, J., & Kálai, T. (2021). Syntheses and Study of a Pyrroline Nitroxide Condensed Phospholene Oxide and a Pyrroline Nitroxide Attached Diphenylphosphine. Molecules, 26(14), 4366. https://doi.org/10.3390/molecules26144366