
Prediction of Molecular Properties Using Molecular Topographic Map


Abstract
:1. Introduction
2. Results
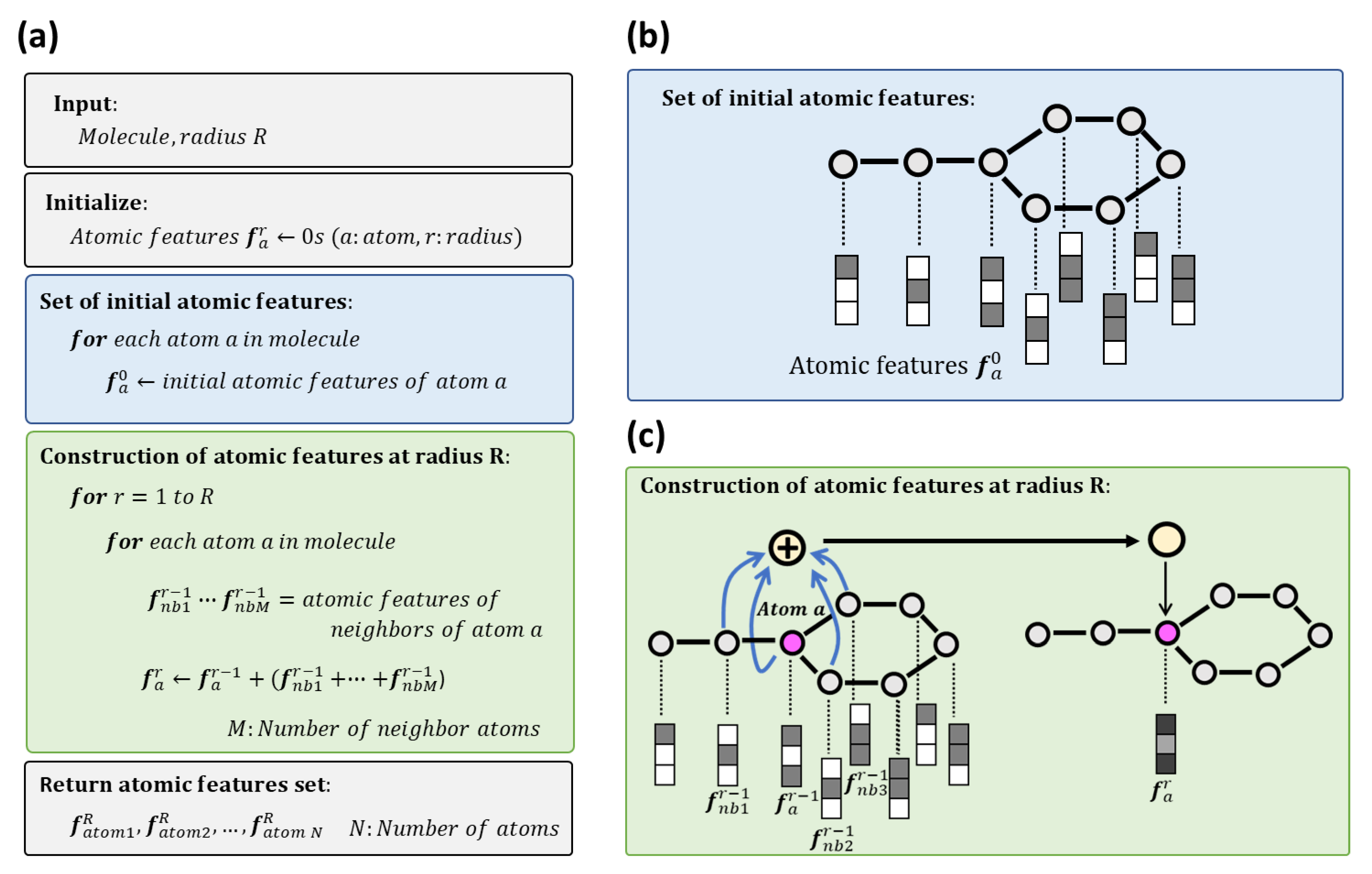
2.1. Atomic Features Set
2.2. Molecular Topographic Map
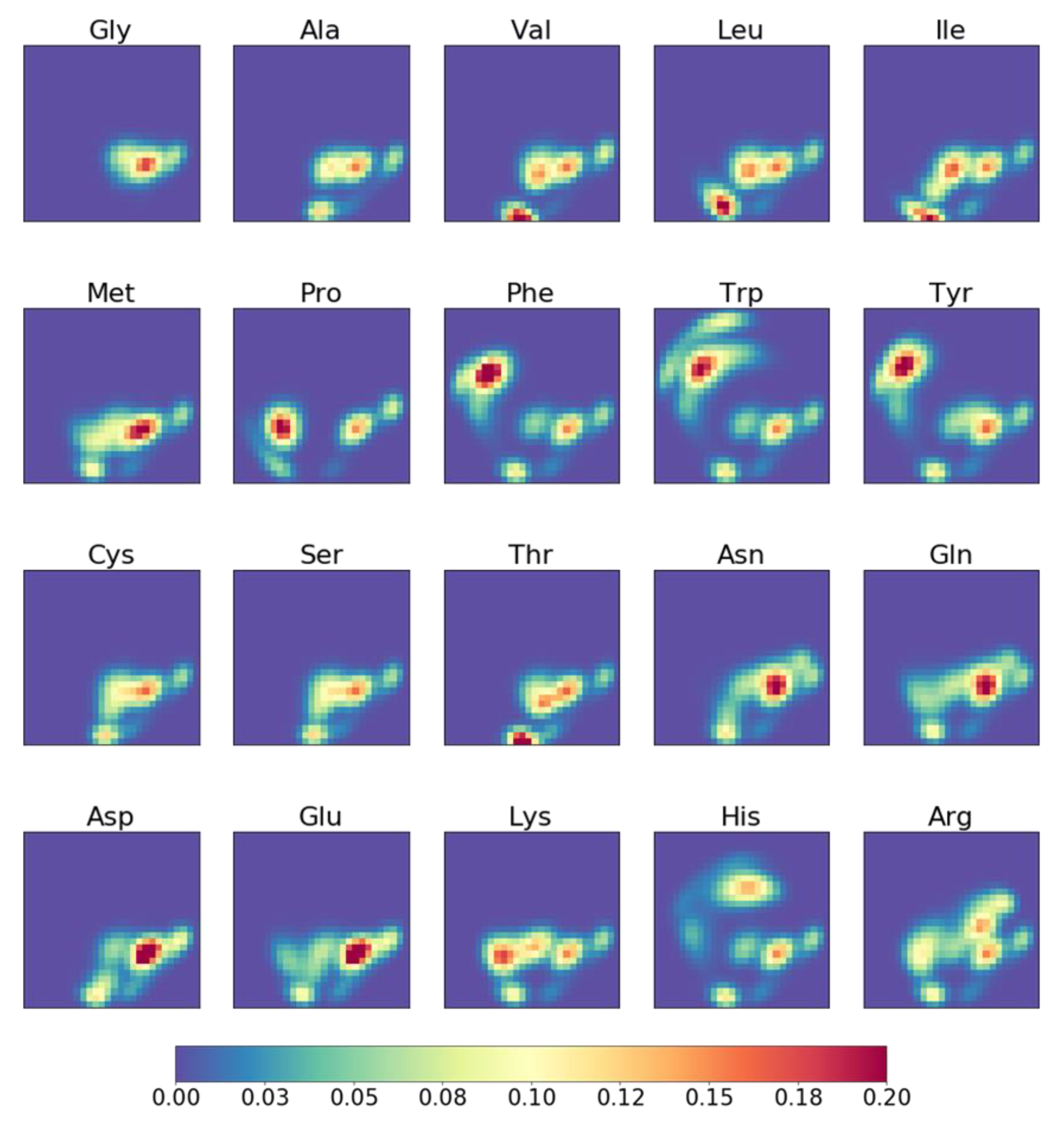
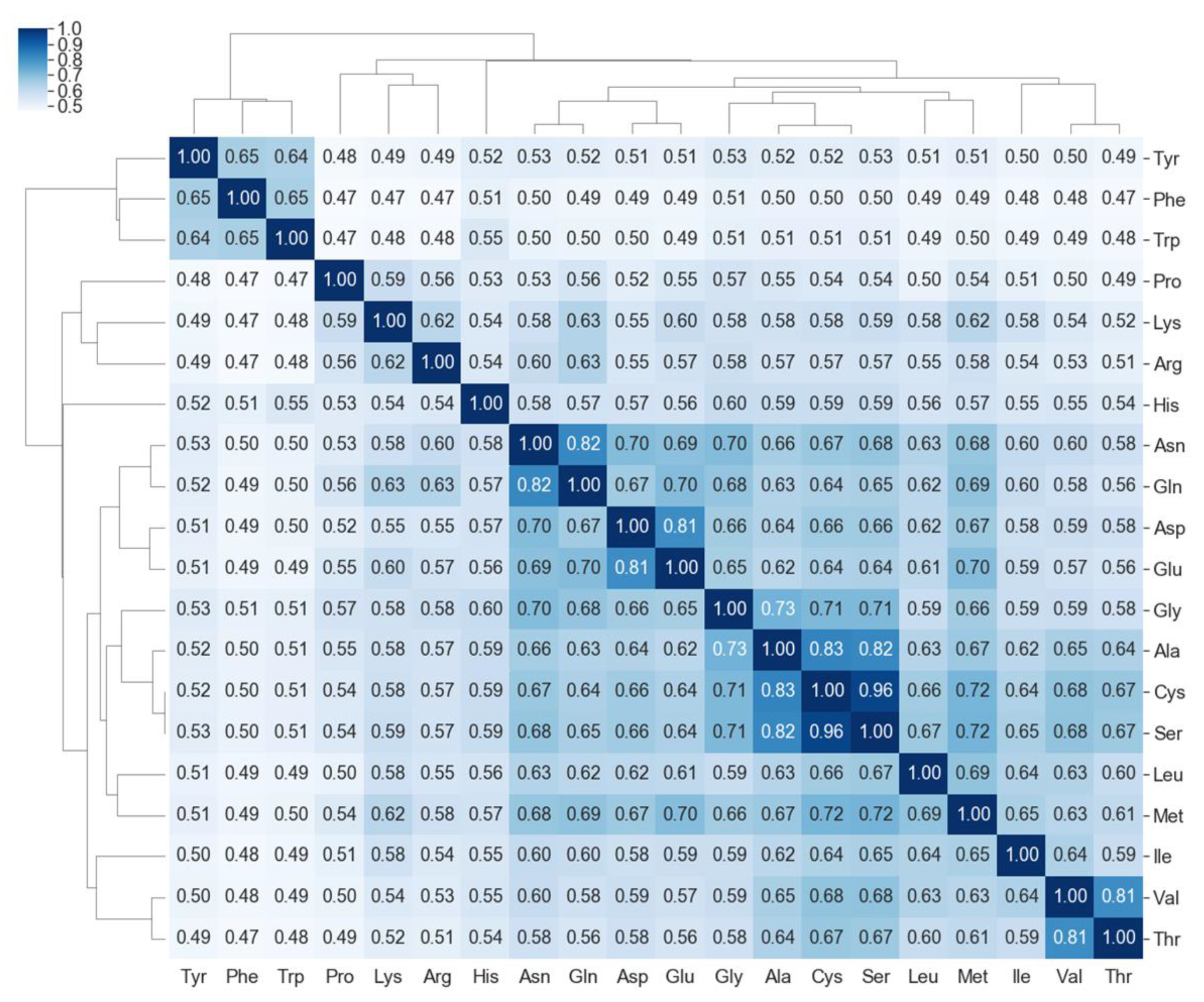
2.3. Molecular Topographic Maps of 20 Amino Acids
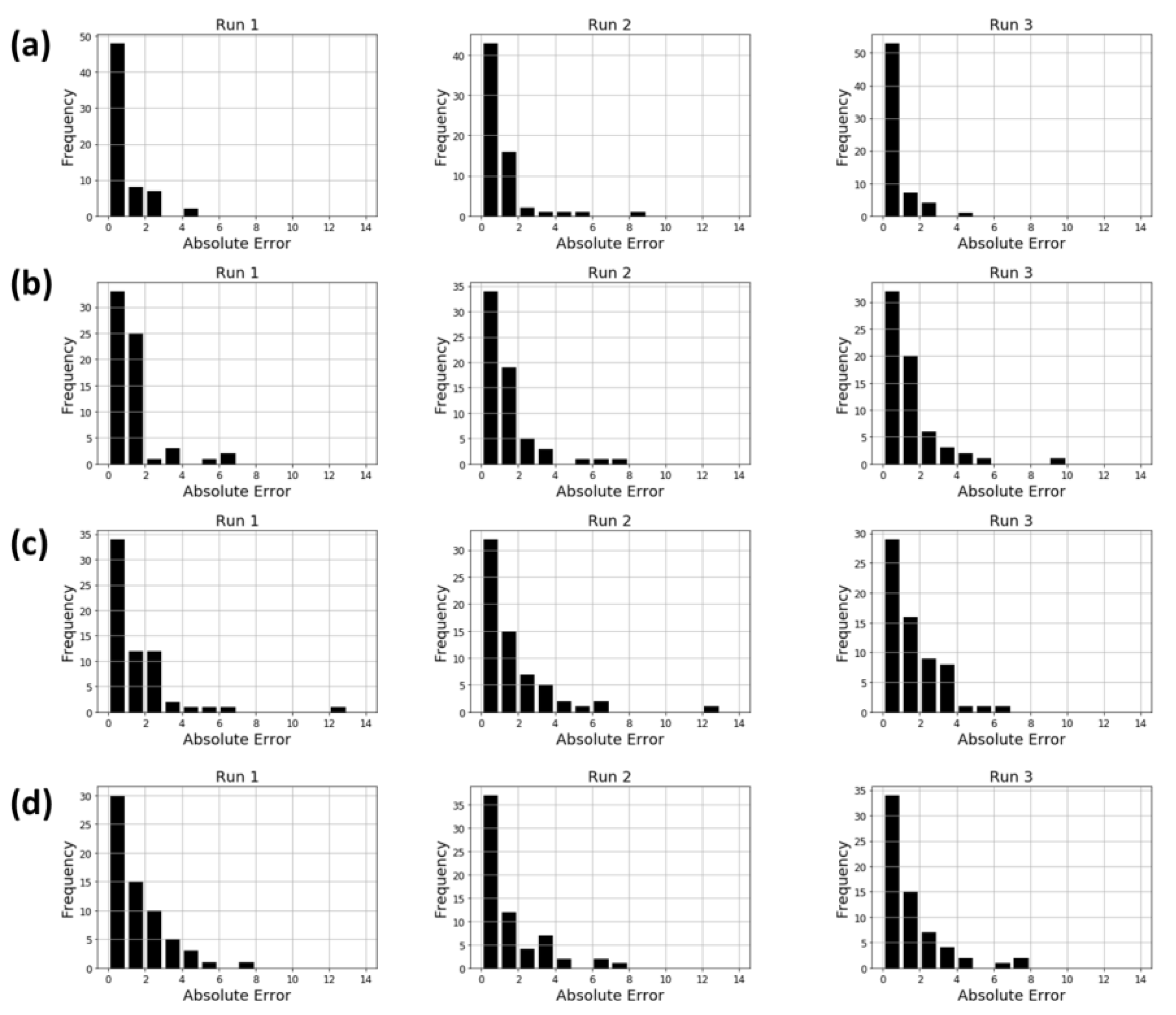
2.4. Property Prediction Using Molecular Topographic Map
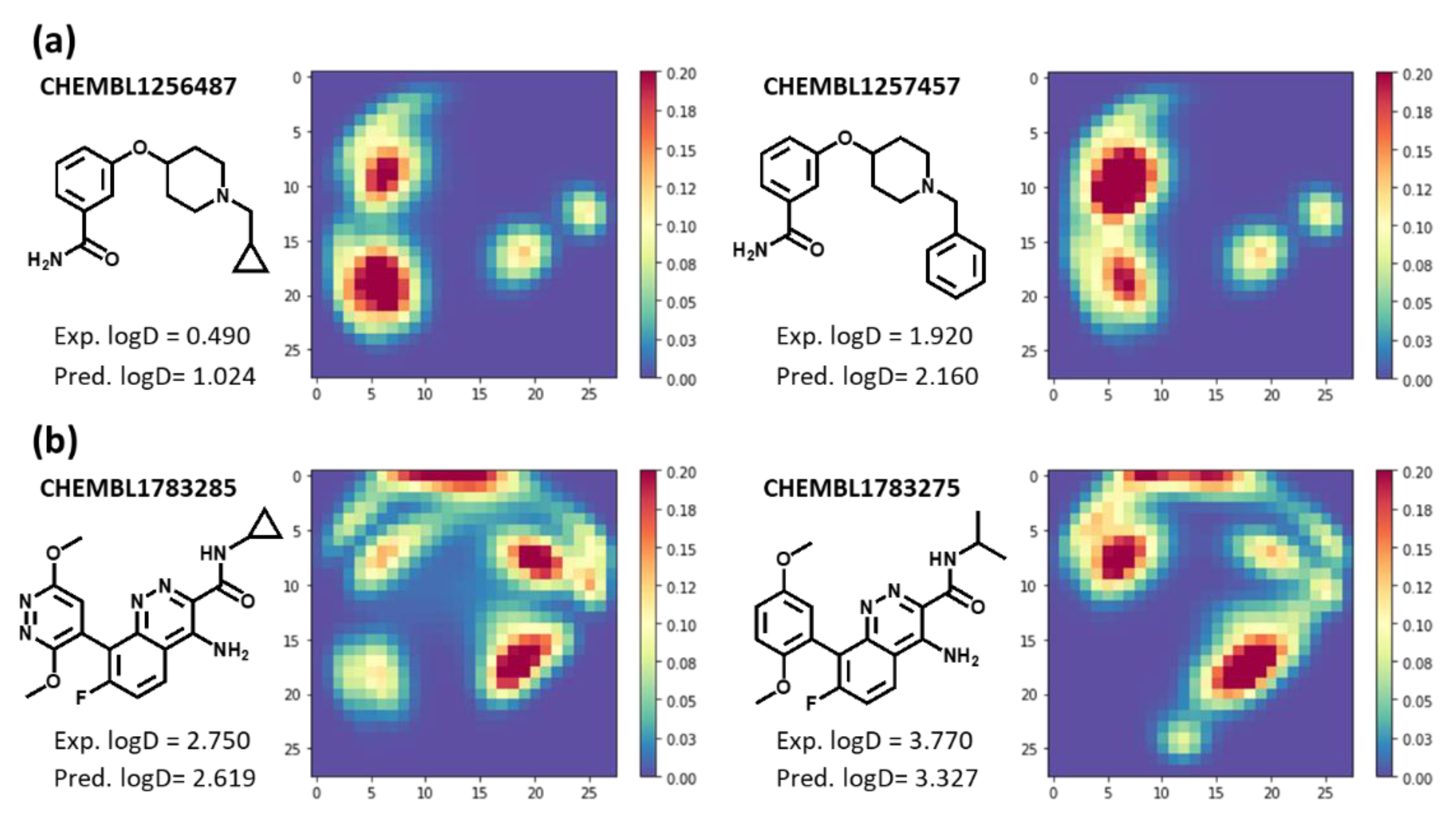
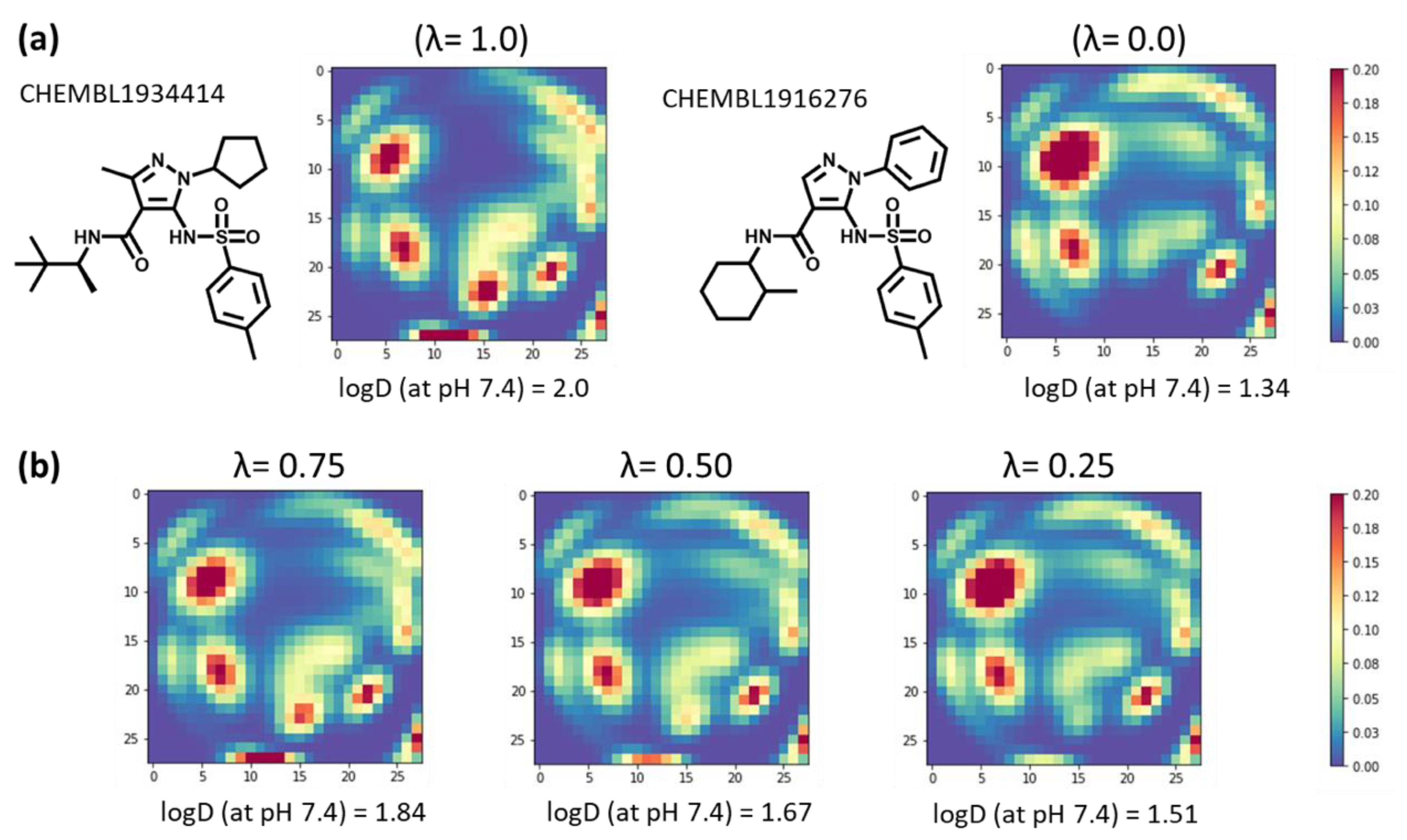
2.5. Examples of Relationship between MTM and Its Predicted Molecular Property
2.6. Data Augmentation of Molecular Topographic Map Using MIXUP
3. Discussion
4. Materials and Methods
4.1. Generation of Atomic Features Set
4.2. Generation of Molecular Topographic Map
4.3. Similarity Matrix Using Molecular Topographic Maps
4.4. Molecular Property Prediction
4.4.1. Property Datasets
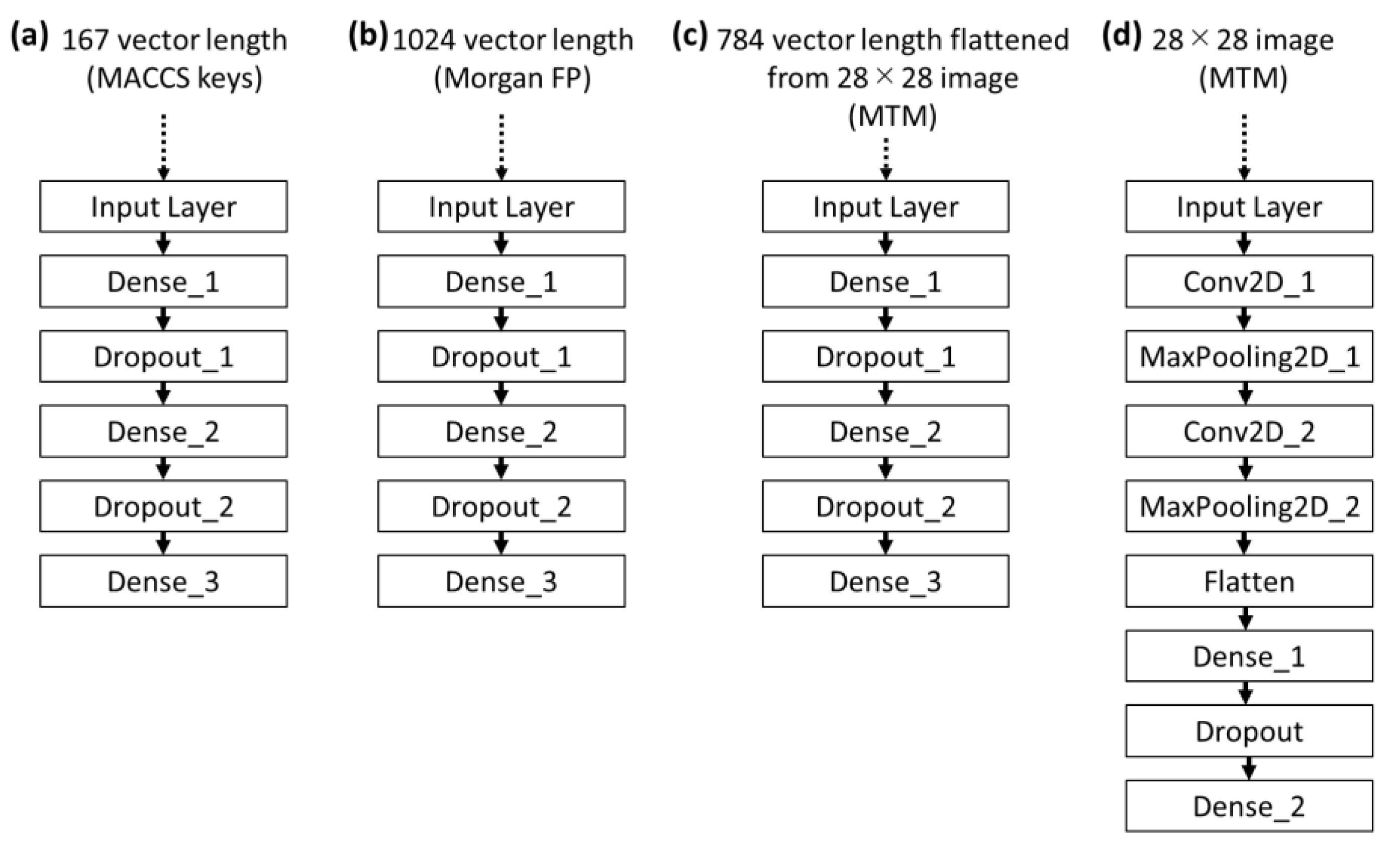
4.4.2. Molecular Representation
4.4.3. Calculation Protocol
4.4.4. Data Augmentation Using Mixup
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Patil, P.S. Drug Discovery and ADMET process: A Review. Int. J. Adv. Res. Biol. Sci. 2016, 3, 181–192. [Google Scholar]
- Shen, J.; Nicolaou, C.A. Molecular property prediction: Recent trends in the era of artificial intelligence. Drug Discov. Today Technol. 2019, 32, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, F.; Desai, P.V.; Arimoto, R.; Desino, K.E.; Fischer, H.; Keefer, C.E.; Petersson, C.; Winiwarter, S.; Broccatelli, F. In Silico Absorption, Distribution, Metabolism, Excretion, and Pharmacokinetics (ADME-PK): Utility and Best Practices. An Industry Perspective from the International Consortium for Innovation through Quality in Pharmaceutical Development. J. Med. Chem. 2017, 60, 9097–9113. [Google Scholar] [CrossRef] [PubMed]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random forest: A classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Cheng, F.; Xu, Y.; Li, W.; Tang, Y. Estimation of ADME properties with substructure pattern recognition. J. Chem. Inf. Model. 2010, 50, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, R.P.; Wang, W.M.; Liaw, A.; Ma, J.; Gifford, E.M. Extreme Gradient Boosting as a Method for Quantitative Structure-Activity Relationships. J. Chem. Inf. Model. 2016, 56, 2353–2360. [Google Scholar] [CrossRef]
- Zhang, J.; Mucs, D.; Norinder, U.; Svensson, F. LightGBM: An Effective and Scalable Algorithm for Prediction of Chemical Toxicity-Application to the Tox21 and Mutagenicity Data Sets. J. Chem. Inf. Model. 2019, 59, 4150–4158. [Google Scholar] [CrossRef]
- Xia, X.; Maliski, E.G.; Gallant, P.; Rogers, D. Classification of Kinase Inhibitors Using a Bayesian Model. J. Med. Chem. 2004, 47, 4463–4470. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.Y.; Zhou, L.C.; Yang, L.; Liu, P.Y.; Zhao, B.W.; Liu, H.X. Predicting the aquatic toxicity mode of action using logistic regression and linear discriminant analysis. SAR QSAR Environ. Res. 2016, 27, 721–746. [Google Scholar] [CrossRef]
- Xue, Y.; Li, Z.R.; Yap, C.W.; Sun, L.Z.; Chen, X.; Chen, Y.Z. Effect of molecular descriptor feature selection in support vector machine classification of pharmacokinetic and toxicological properties of chemical agents. J. Chem. Inf. Comput. Sci. 2004, 44, 1630–1638. [Google Scholar] [CrossRef] [Green Version]
- Stahura, F.L.; Godden, J.W.; Bajorath, J. Differential Shannon Entropy Analysis Identifies Molecular Property Descriptors that Predict Aqueous Solubility of Synthetic Compounds with High Accuracy in Binary QSAR Calculations. J. Chem. Inf. Comput. Sci. 2002, 42, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Awale, M.; Riniker, S.; Kramer, C. Matched Molecular Series Analysis for ADME Property Prediction. J. Chem. Inf. Model. 2020, 60, 2903–2914. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A. Deep learning in drug discovery: Opportunities, challenges and future prospects. Drug Discov. Today 2019, 24, 2017–2032. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Engkvist, O.; Wang, Y.; Olivecrona, M.; Blaschke, T. The rise of deep learning in drug discovery. Drug Discov. Today 2018, 23, 1241–1250. [Google Scholar] [CrossRef]
- Gawehn, E.; Hiss, J.A.; Schneider, G. Deep Learning in Drug Discovery. Mol. Inform. 2016, 35, 3–14. [Google Scholar] [CrossRef]
- Bajorath, J. State-of-the-art of artificial intelligence in medicinal chemistry. Future Sci. OA 2021, FSO702. [Google Scholar] [CrossRef]
- Sun, M.; Zhao, S.; Gilvary, C.; Elemento, O.; Zhou, J.; Wang, F. Graph convolutional networks for computational drug development and discovery. Brief. Bioinform. 2020, 21, 919–935. [Google Scholar] [CrossRef]
- Bhhatarai, B.; Walters, W.P.; Hop, C.E.C.A.; Lanza, G.; Ekins, S. Opportunities and challenges using artificial intelligence in ADME/Tox. Nat. Mater. 2019, 18, 418–422. [Google Scholar] [CrossRef]
- Taherkhani, A.; Cosma, G.; McGinnity, T.M. Deep-FS: A feature selection algorithm for Deep Boltzmann Machines. Neurocomputing 2018, 322, 22–37. [Google Scholar] [CrossRef]
- Yang, K.; Swanson, K.; Jin, W.; Coley, C.; Piden, P.; Gao, H.; Guzman-Perez, A.; Hopper, T.; Kelley, B.; Mathea, M.; et al. Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 2019, 59, 3370–3388. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Sheridan, R.P.; Liaw, A.; Dahl, G.E.; Svetnik, V. Deep neural nets as a method for quantitative structure-activity relationships. J. Chem. Inf. Model. 2015, 55, 263–274. [Google Scholar] [CrossRef]
- Kireev, D.B. ChemNet: A Novel Neural Network Based Method for Graph/Property Mapping. J. Chem. Inf. Comput. Sci. 1995, 35, 175–180. [Google Scholar] [CrossRef]
- Duvenaud, D.; Maclaurin, D.; Aguilera-Iparraguirre, J.; Go´mez-Bombarelli, R.; Hirzel, T.; Aspuru-Guzik, A.; Adams, R.P. Convolutional networks on graphs for learning molecular fingerprints. Adv. Neural Inf. Process. Syst. 2015, 2224–2232. [Google Scholar]
- Kearnes, S.; McCloskey, K.; Berndl, M.; Pande, V.; Riley, P. Molecular graph convolutions: Moving beyond fingerprints. J. Comput. Aided Mol. Des. 2016, 30, 595–608. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, Z.; Jiang, M.; Wang, S.; Zhang, S.; Wei, Z. Molecule Property Prediction Based on Spatial Graph Embedding. J. Chem. Inf. Model. 2019, 59, 3817–3828. [Google Scholar] [CrossRef]
- Wang, S.; Guo, Y.; Wang, Y.; Sun, H.; Huang, J. SMILES-BERT: Large Scale Unsupervised Pre-training for Molecular Property Prediction. In Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, Niagara Falls, NY, USA, 7–10 September 2019; pp. 429–436. [Google Scholar]
- Chithrananda, S.; Grand, G.; Ramsundar, B. ChemBERTa: Large-Scale SelfSupervised Pretraining for Molecular Property Prediction. arXiv 2020, arXiv:2010.09885. [Google Scholar]
- Weininger, D. SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Devlin, J.; Chang, M.W.; Lee, K.; Toutanova, K. BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. arXiv 2018, arXiv:1810.04805. [Google Scholar]
- Goh, G.B.; Siegel, C.; Vishnu, A.; Hodas, N.O.; Baker, N. Chemception: A deep neural network with minimal chemistry knowledge matches the performance of expert-developed QSAR/QSPR models. arXiv 2017, arXiv:1706.06689. [Google Scholar]
- Uesawa, Y. Quantitative structure—Activity relationship analysis using deep learning based on a novel molecular image input technique. Bioorg. Med. Chem. Lett. 2018, 28, 3400–3403. [Google Scholar] [CrossRef]
- Matsuzaka, Y.; Uesawa, Y. Molecular Image-Based Prediction Models of Nuclear Receptor Agonists and Antagonists Using the DeepSnap-Deep Learning Approach with the Tox21 10K Library. Molecules 2020, 25, 2764. [Google Scholar] [CrossRef]
- Zhong, S.; Hu, J.; Yu, X.; Zhang, H. Molecular image-convolutional neural network (CNN) assisted QSAR models for predicting contaminant reactivity toward OH radicals: Transfer learning, data augmentation and model interpretation. Chem. Eng. J. 2021, 408, 127998. [Google Scholar] [CrossRef]
- Huang, G.; Liu, Z.; Van Der Maaten, L.; Weinberger, K.Q. Densely connected convolutional networks. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition 2017, Honolulu, HI, USA, 21–26 July 2017; pp. 4700–4708. [Google Scholar]
- Bishop, C.M.; Svense’n, M.; Williams, C.K.I. Developments of the generative topographic mapping. Neurocomputing 1998, 21, 203–224. [Google Scholar] [CrossRef]
- Bishop, C.M.; Svense’n, M.; Williams, C.K.I. GTM: The generative topographic mapping. Neural Comput. 1998, 10, 215–234. [Google Scholar] [CrossRef]
- Glem, R.C.; Bender, A.; Arnby, C.H.; Carlsson, L.; Boyer, S.; Smith, J. Circular fingerprints: Flexible molecular descriptors with applications from physical chemistry to ADME. IDrugs 2006, 9, 199–204. [Google Scholar] [PubMed]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- RDKit: Open-source cheminformatics. Available online: https://www.rdkit.org (accessed on 29 May 2021).
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Cisse, M.; Dauphin, Y.N.; Lopez-Paz, D. mixup: Beyond empirical risk minimization. In Proceedings of the International Conference on Learning Representations, Vancouver, BC, Canada, 3 May–30 April 2018. [Google Scholar]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The ChEMBL Bioactivity Database: An Update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef] [Green Version]
- Shorten, C.; Khoshgoftaar, T.M. A survey on Image Data Augmentation for Deep Learning. J. Big Data 2019, 6, 60. [Google Scholar] [CrossRef]
- Selvaraju, R.R.; Cogswell, M.; Das, A.; Vedantam, R.; Parikh, D.; Batra, D. Grad-CAM: Visual Explanations from Deep Networks via Gradient-based Localization. arXiv 2016, arXiv:1610.02391. [Google Scholar]
- Setiawan, W.; Utoyo, M.I.; Rulaningtyas, R. Transfer learning with multiple pre-trained network for fundus classification. TELKOMNIKA Telecommunication. Comput. Electron. Control. 2020, 18, 1382–1388. [Google Scholar]
- Hert, J.; Willett, P.; Wilton, D.J.; Acklin, P.; Azzaoui, K.; Jacoby, E.; Schuffenhauer, A. Comparison of topological descriptors for similarity-based virtual screening using multiple bioactive reference structures. Org. Biomol. Chem. 2004, 2, 3256–3266. [Google Scholar] [CrossRef] [Green Version]
- Heikamp, K.; Bajorath, J. Large-Scale Similarity Search Profiling of ChEMBL Compound Data Sets. J. Chem. Inf. Model. 2011, 51, 1831–1839. [Google Scholar] [CrossRef] [PubMed]
- ugtm: Generative Topographic Mapping with Python. Available online: https://ugtm.readthedocs.io (accessed on 29 May 2021).
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A benchmark for molecular machine learning. Chem. Sci. 2017, 9, 513–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seaborn: Statistical Data Visualization. Available online: https://seaborn.pydata.org (accessed on 29 May 2021).
- Wang, N.N.; Dong, J.; Deng, Y.H.; Zhu, M.F.; Wen, M.; Yao, Z.J.; Lu, A.P.; Wang, J.B.; Cao, D.S. ADME Properties Evaluation in Drug Discovery: Prediction of Caco-2 Cell Permeability Using a Combination of NSGA-II and Boosting. J. Chem. Inf. Model. 2016, 56, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Corrado, G.S.; Davis, A.; Dean, J.; Devin, M.; et al. TensorFlow: Large-Scale Machine Learning on Heterogeneous Systems. arXiv 2016, arXiv:1603.04467. [Google Scholar]
- Keras. Deep Learning for Python. Available online: https://github.com/keras-team/keras (accessed on 29 May 2021).
- Optuna. A Hyperparameter Optimization Framework. Available online: https://github.com/optuna/optuna (accessed on 29 May 2021).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Description | ID | Description | ||
|---|---|---|---|---|---|
| 1 | H | Atom type as a one-hot vector | 25 | S | Hybridization of an atom as one-hot vector |
| 2 | C | 26 | SP | ||
| 3 | N | 27 | SP2 | ||
| 4 | O | 28 | SP3 | ||
| 5 | S | 29 | SP3D | ||
| 6 | P | 30 | SP3D2 | ||
| 7 | F | 31 | 0 | Number of hydrogens on an atom as one-hot vector | |
| 8 | Cl | 32 | 1 | ||
| 9 | Br | 33 | 2 | ||
| 10 | I | 34 | 3 | ||
| 11 | 0 | Degree of an atom as one-hot vector, which defined to be its number of directly-bonded neighbors. | 35 | 4 | |
| 12 | 1 | 36 | −1 | Formal charge of an atom as one-hot vector | |
| 13 | 2 | 37 | 0 | ||
| 14 | 3 | 38 | 1 | ||
| 15 | 4 | 39 | Aromatic | Is aromatic | |
| 16 | 5 | 40 | Ring | Is in ring | |
| 17 | 6 | 41 | R | Chirality of an atom as one-hot vector | |
| 18 | 0 | Total valence of an atom as one-hot vector | 42 | S | |
| 19 | 1 | 43 | Non-chiral | ||
| 20 | 2 | ||||
| 21 | 3 | ||||
| 22 | 4 | ||||
| 23 | 5 | ||||
| 24 | 6 | ||||
| Dataset | No. | Molecular Representation | Model | MSE | MAE | R2 | |||
|---|---|---|---|---|---|---|---|---|---|
| ESOL | 1128 | MACCS keys | DNN | 1.202 | (±0.187) | 0.789 | (±0.048) | 0.781 | (±0.011) |
| Morgan FP | DNN | 1.592 | (±0.108) | 0.942 | (±0.041) | 0.705 | (±0.018) | ||
| flattened MTM | DNN | 0.897 | (±0.178) | 0.681 | (±0.056) | 0.850 | (±0.028) | ||
| MTM | CNN | 0.839 | (±0.166) | 0.621 | (±0.059) | 0.858 | (±0.021) | ||
| FreeSolv | 642 | MACCS keys | DNN | 1.901 | (±0.834) | 0.810 | (±0.149) | 0.902 | (±0.035) |
| Morgan FP | DNN | 5.007 | (±1.495) | 1.402 | (±0.120) | 0.741 | (±0.029) | ||
| flattened MTM | DNN | 7.114 | (±2.293) | 1.701 | (±0.109) | 0.696 | (±0.082) | ||
| MTM | CNN | 4.864 | (±0.180) | 1.531 | (±0.030) | 0.727 | (±0.070) | ||
| Lipophilicity | 4200 | MACCS keys | DNN | 0.685 | (±0.024) | 0.605 | (±0.019) | 0.551 | (±0.012) |
| Morgan FP | DNN | 0.705 | (±0.049) | 0.623 | (±0.025) | 0.539 | (±0.005) | ||
| flattened MTM | DNN | 0.707 | (±0.040) | 0.626 | (±0.016) | 0.537 | (±0.026) | ||
| MTM | CNN | 0.692 | (±0.066) | 0.610 | (±0.029) | 0.554 | (±0.031) | ||
| caco2 | 1272 | MACCS keys | DNN | 0.196 | (±0.040) | 0.351 | (±0.050) | 0.711 | (±0.021) |
| Morgan FP | DNN | 0.210 | (±0.056) | 0.337 | (±0.048) | 0.684 | (±0.059) | ||
| flattened MTM | DNN | 0.196 | (±0.012) | 0.336 | (±0.012) | 0.655 | (±0.051) | ||
| MTM | CNN | 0.179 | (±0.021) | 0.321 | (±0.024) | 0.718 | (±0.024) | ||
| Dataset | No. | α | MSE | MAE | R2 | |||
|---|---|---|---|---|---|---|---|---|
| ESOL | 1128 | - | 0.839 | (±0.166) | 0.621 | (±0.059) | 0.858 | (±0.021) |
| 1128 × 2 | 0.2 | 0.833 | (±0.125) | 0.643 | (±0.067) | 0.848 | (±0.020) | |
| 1128 × 2 | 2.0 | 0.785 | (±0.086) | 0.649 | (±0.034) | 0.863 | (±0.017) | |
| 1128 ×10 | 0.2 | 0.890 | (±0.153) | 0.662 | (±0.069) | 0.839 | (±0.018) | |
| 1128 × 10 | 2.0 | 0.851 | (±0.149) | 0.674 | (±0.049) | 0.851 | (±0.033) | |
| FreeSolv | 642 | - | 4.864 | (±0.18) | 1.531 | (±0.030) | 0.727 | (±0.070) |
| 642 × 2 | 0.2 | 4.215 | (±0.396) | 1.414 | (±0.035) | 0.755 | (±0.065) | |
| 642 × 2 | 2.0 | 4.274 | (±0.467) | 1.469 | (±0.089) | 0.746 | (±0.102) | |
| 642 × 10 | 0.2 | 3.642 | (±0.414) | 1.331 | (±0.161) | 0.770 | (±0.091) | |
| 642 × 10 | 2.0 | 3.469 | (±0.400) | 1.332 | (±0.061) | 0.788 | (±0.081) | |
| Lipophilicity | 4200 | - | 0.692 | (±0.066) | 0.610 | (±0.029) | 0.554 | (±0.031) |
| 4200 × 2 | 0.2 | 0.642 | (±0.050) | 0.588 | (±0.025) | 0.576 | (±0.025) | |
| 4200 × 2 | 2.0 | 0.627 | (±0.075) | 0.588 | (±0.035) | 0.586 | (±0.033) | |
| 4200 × 10 | 0.2 | 0.646 | (±0.076) | 0.595 | (±0.032) | 0.572 | (±0.037) | |
| 4200 × 10 | 2.0 | 0.607 | (±0.054) | 0.576 | (±0.021) | 0.597 | (±0.027) | |
| caco2 | 1272 | - | 0.179 | (±0.021) | 0.321 | (±0.024) | 0.718 | (±0.024) |
| 1271 × 2 | 0.2 | 0.161 | (±0.005) | 0.303 | (±0.007) | 0.722 | (±0.023) | |
| 1272 × 2 | 2.0 | 0.189 | (±0.032) | 0.324 | (±0.022) | 0.696 | (±0.020) | |
| 1272 × 10 | 0.2 | 0.151 | (±0.011) | 0.288 | (±0.011) | 0.736 | (±0.017) | |
| 1272 × 10 | 2.0 | 0.151 | (±0.009) | 0.285 | (±0.007) | 0.732 | (±0.010) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshimori, A. Prediction of Molecular Properties Using Molecular Topographic Map. Molecules 2021, 26, 4475. https://doi.org/10.3390/molecules26154475
Yoshimori A. Prediction of Molecular Properties Using Molecular Topographic Map. Molecules. 2021; 26(15):4475. https://doi.org/10.3390/molecules26154475
Chicago/Turabian StyleYoshimori, Atsushi. 2021. "Prediction of Molecular Properties Using Molecular Topographic Map" Molecules 26, no. 15: 4475. https://doi.org/10.3390/molecules26154475
APA StyleYoshimori, A. (2021). Prediction of Molecular Properties Using Molecular Topographic Map. Molecules, 26(15), 4475. https://doi.org/10.3390/molecules26154475