
New Trends in Diaziridine Formation and Transformation (a Review)

Abstract
:1. Introduction
2. The Chemistry of Diaziridines
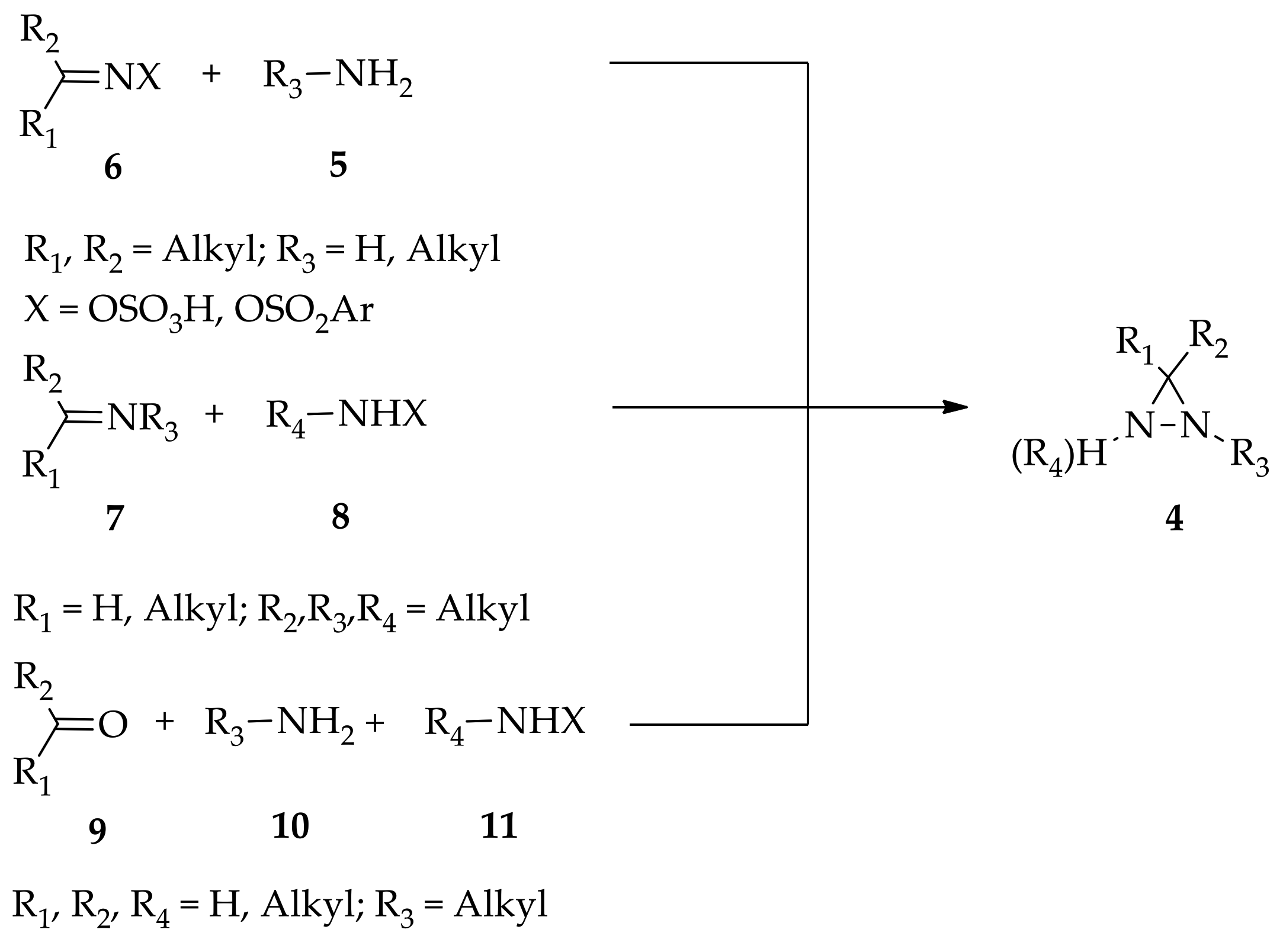
3. Diaziridine Formation and Transformation
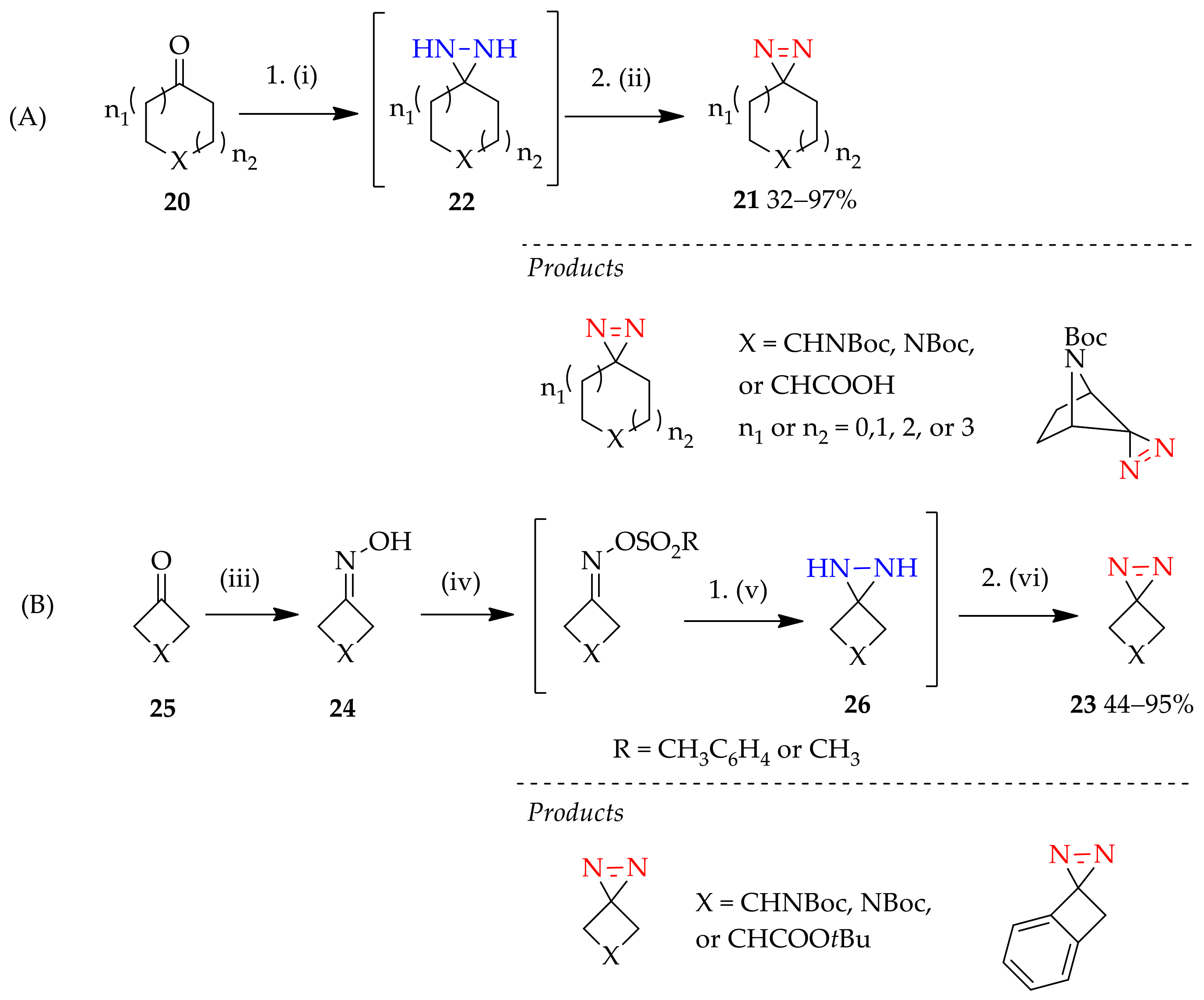
3.1. Non-Substituted Diaziridines
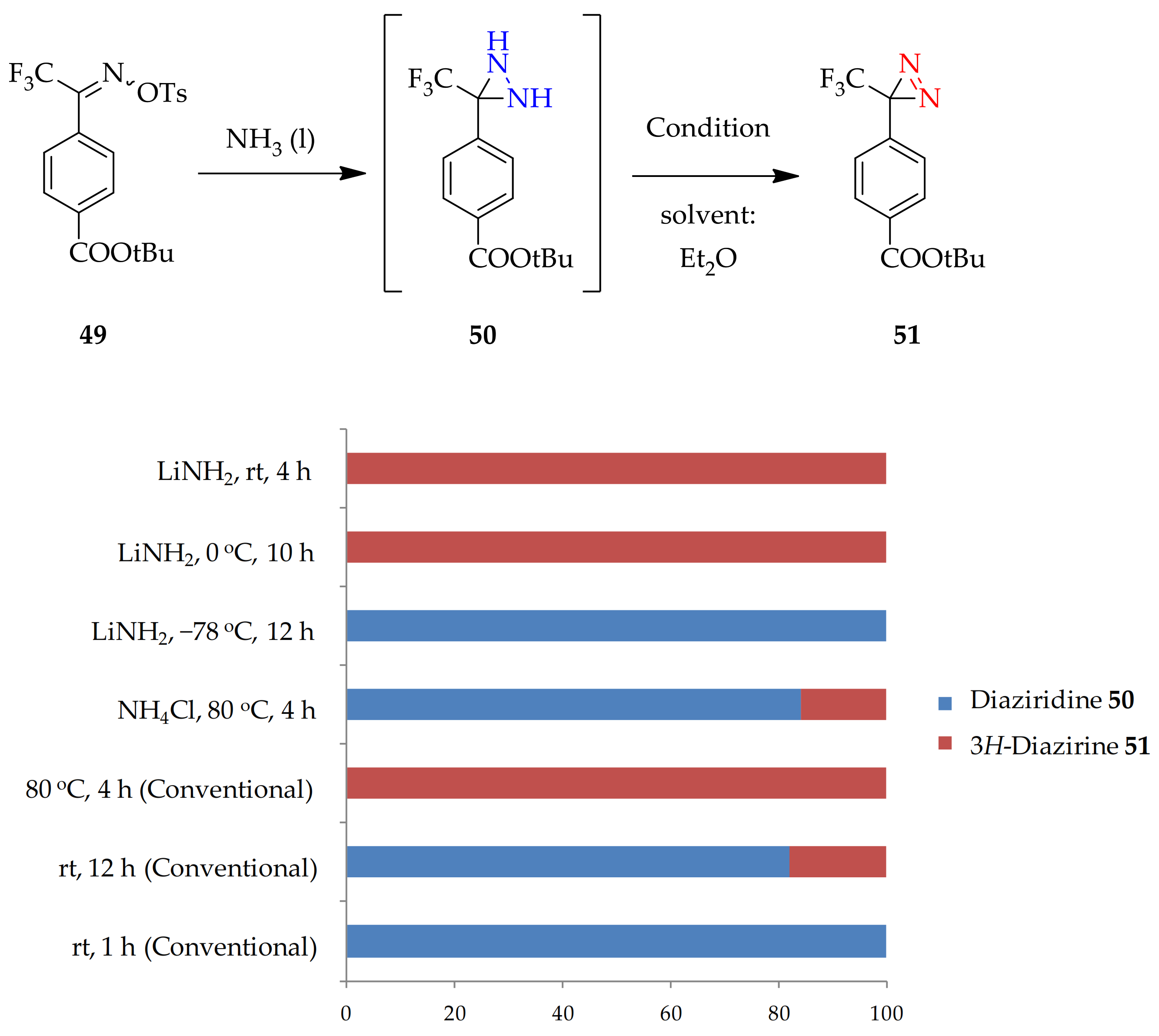
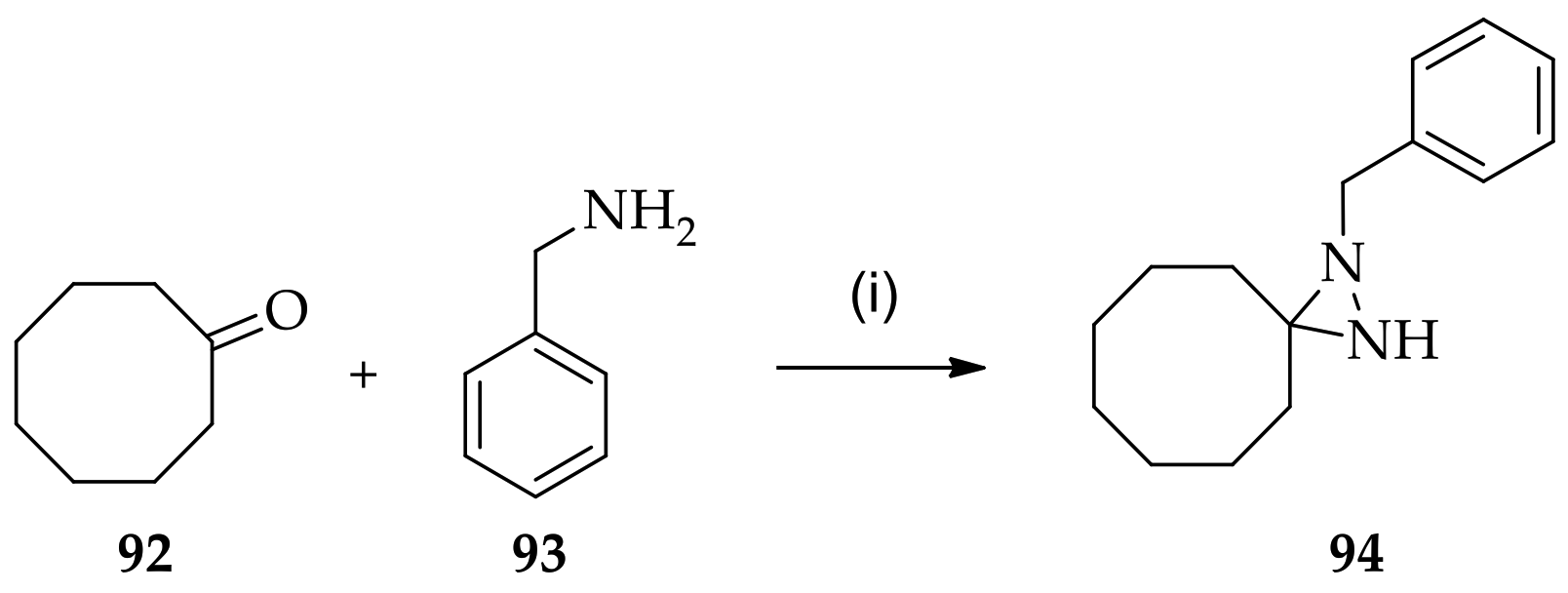
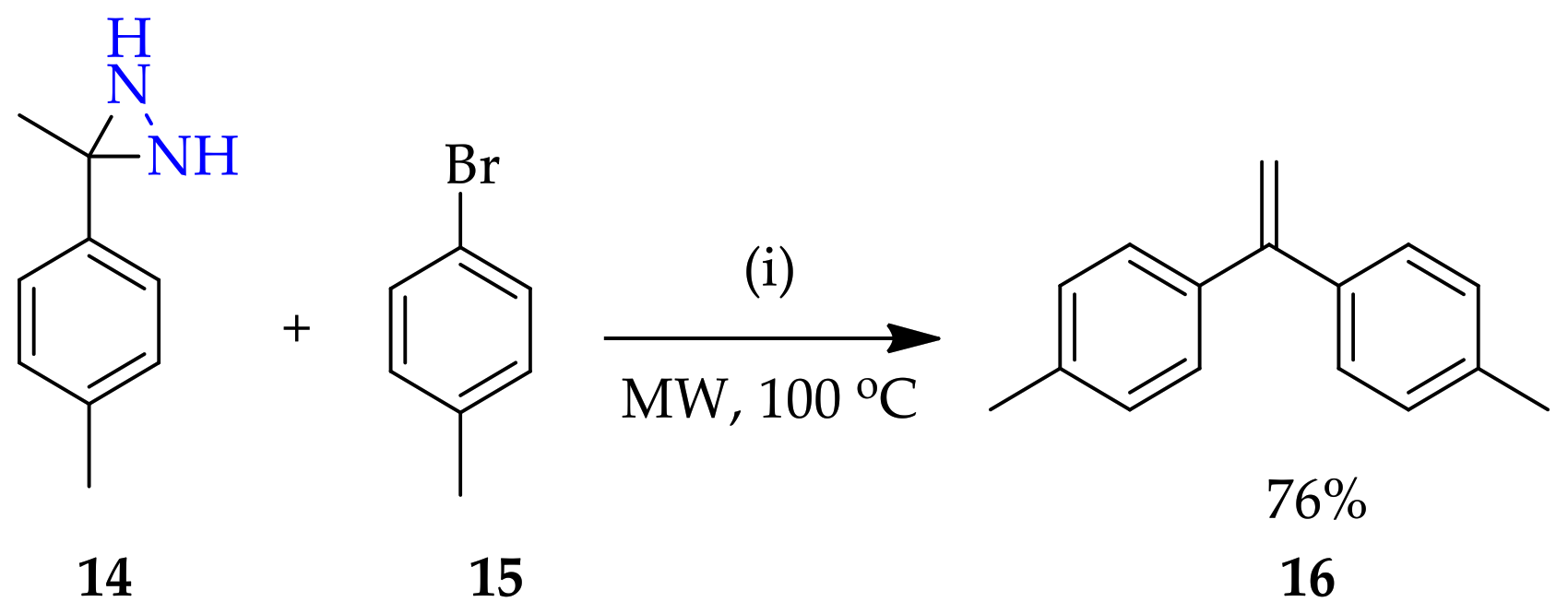
3.1.1. Conventional Method for Recent Uses
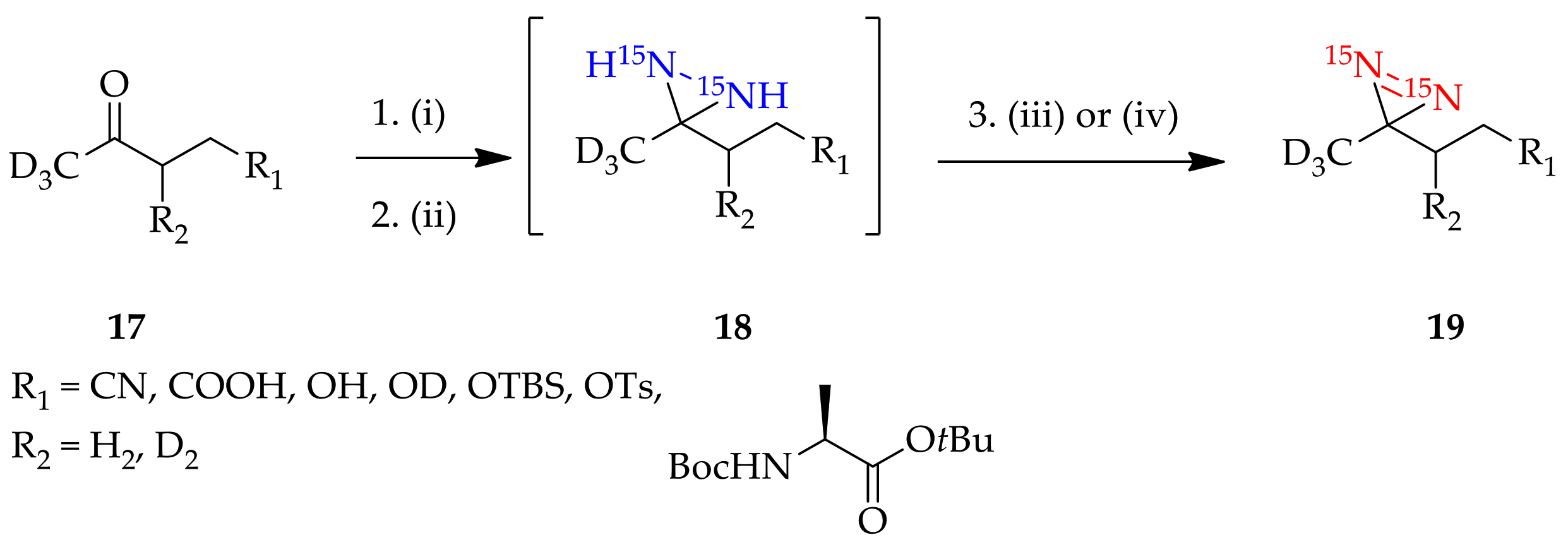
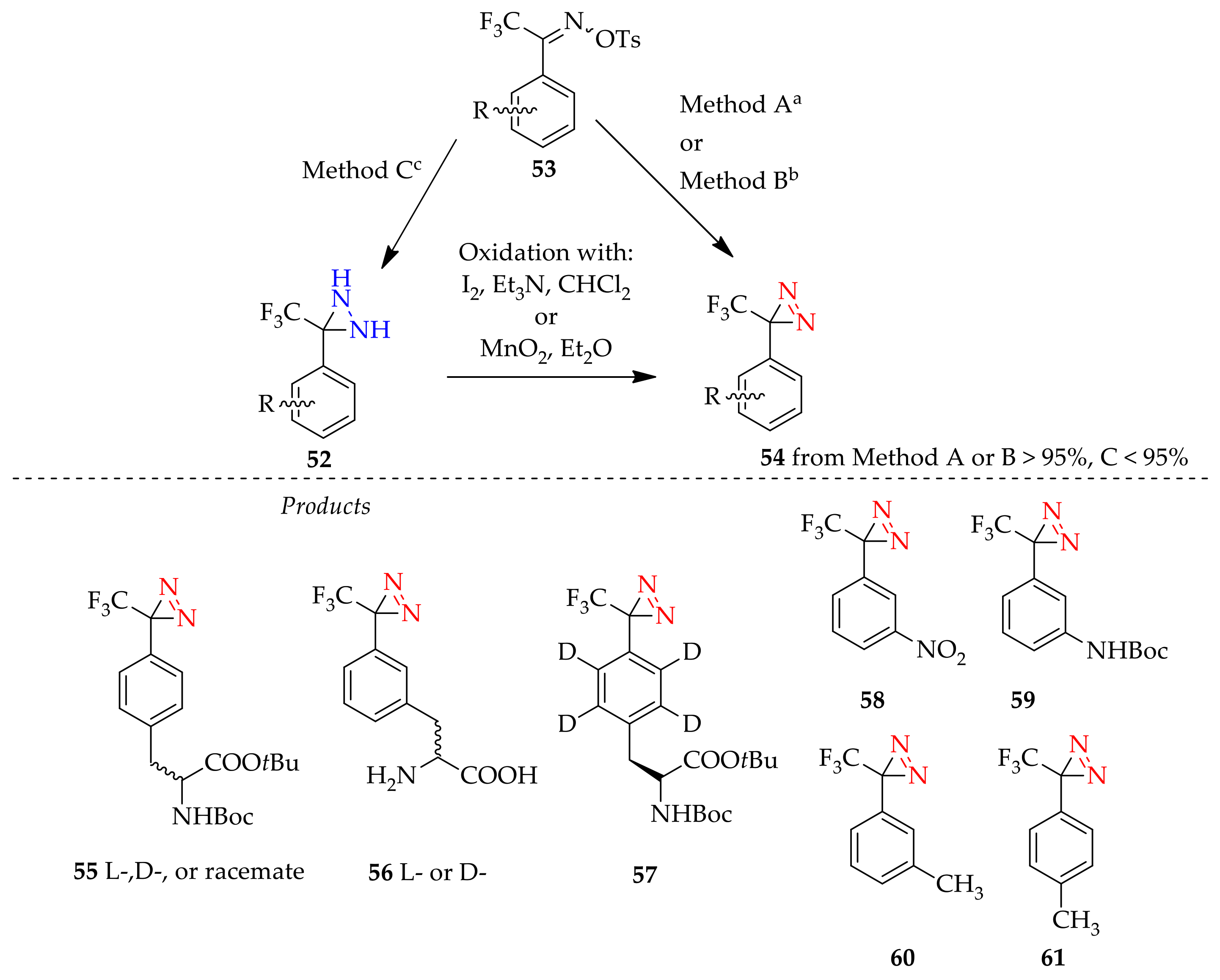
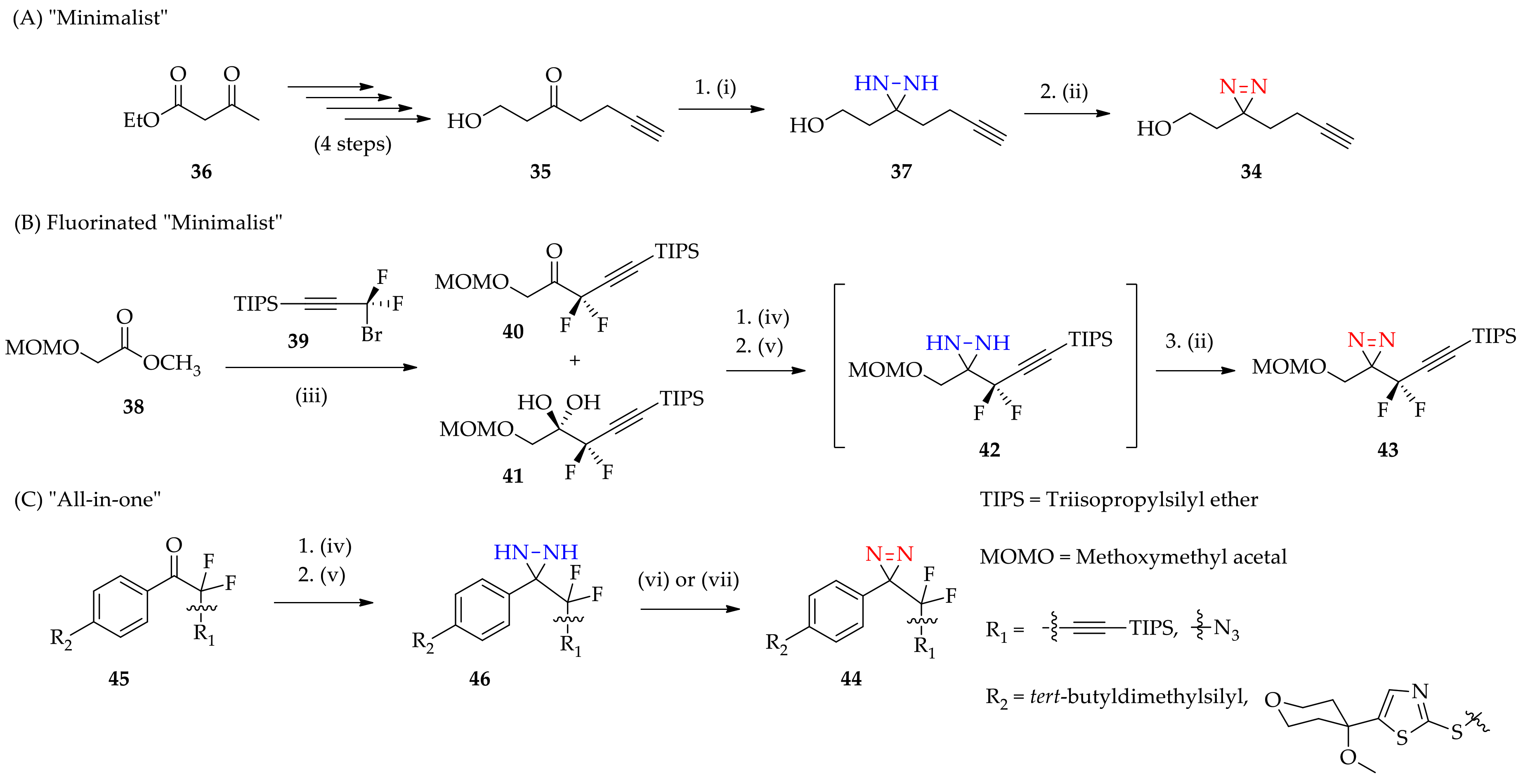
3.1.2. Diaziridine as an Intermediate for “Minimalist” into “All-in-One” 3H-Diazirine

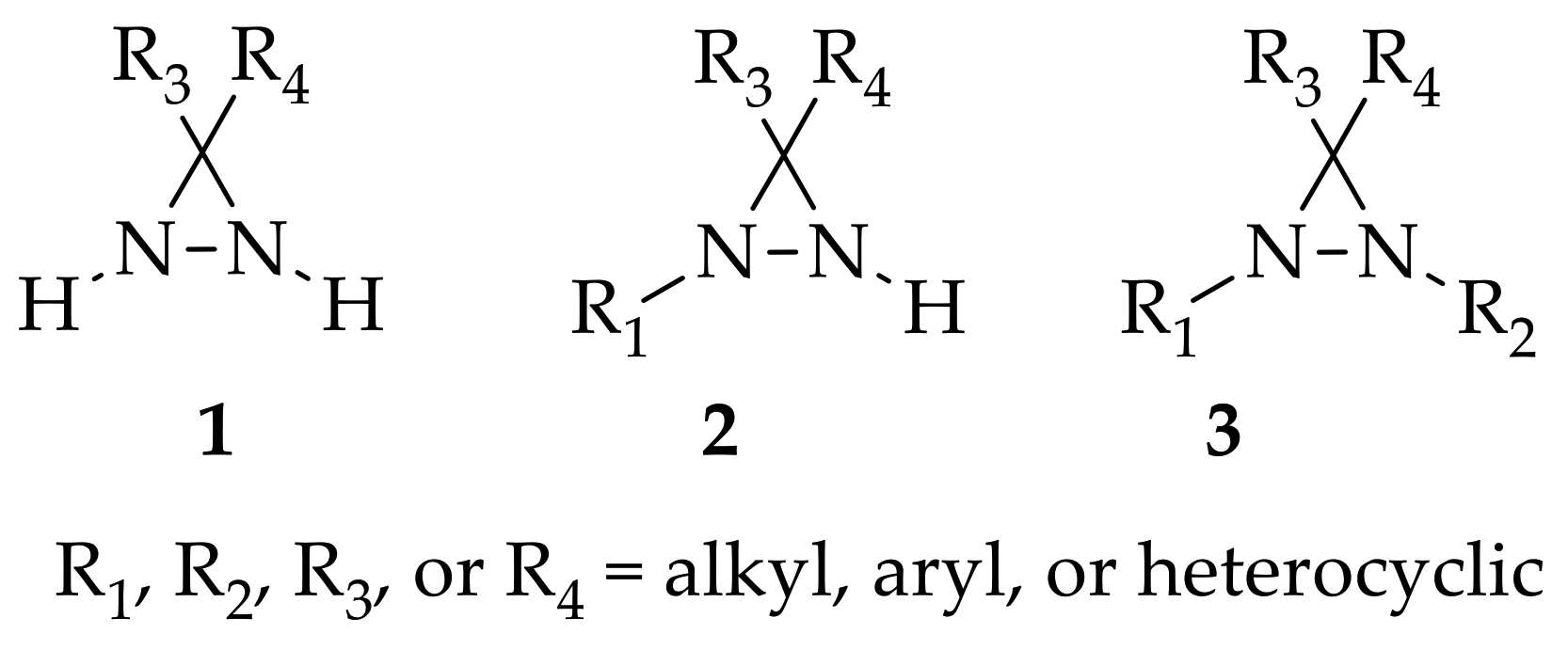{kind=link}
{kind=link}
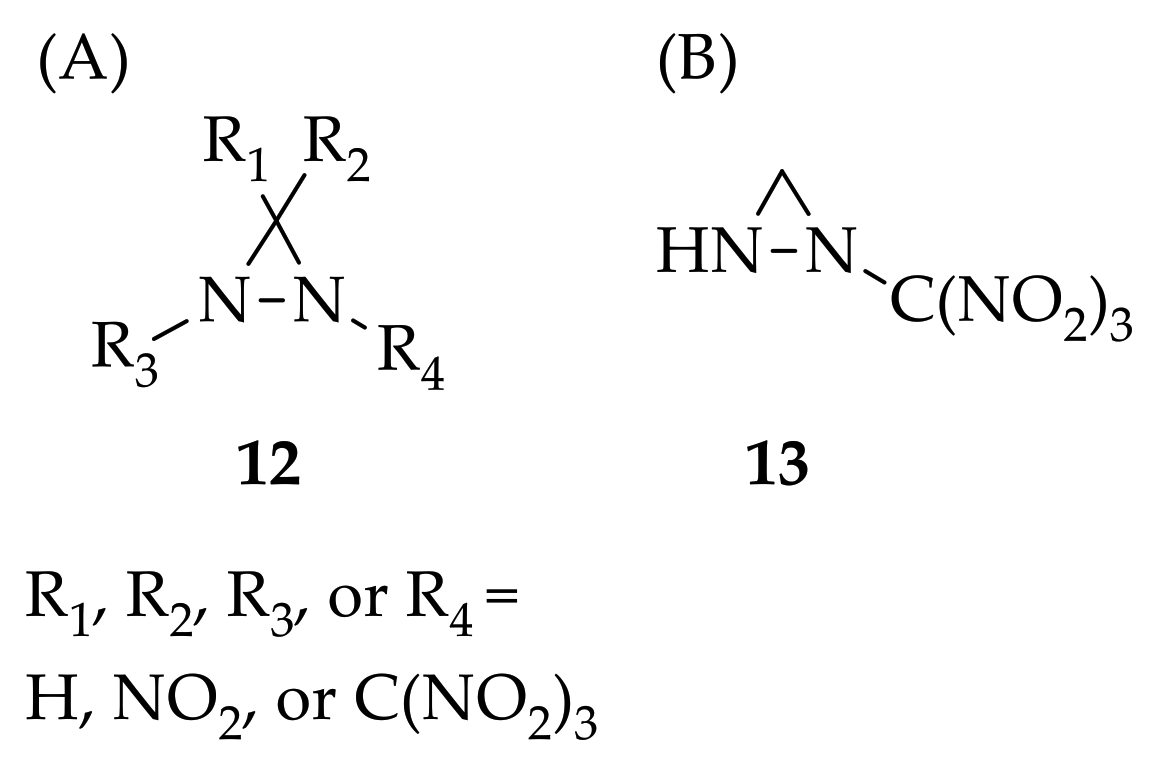{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
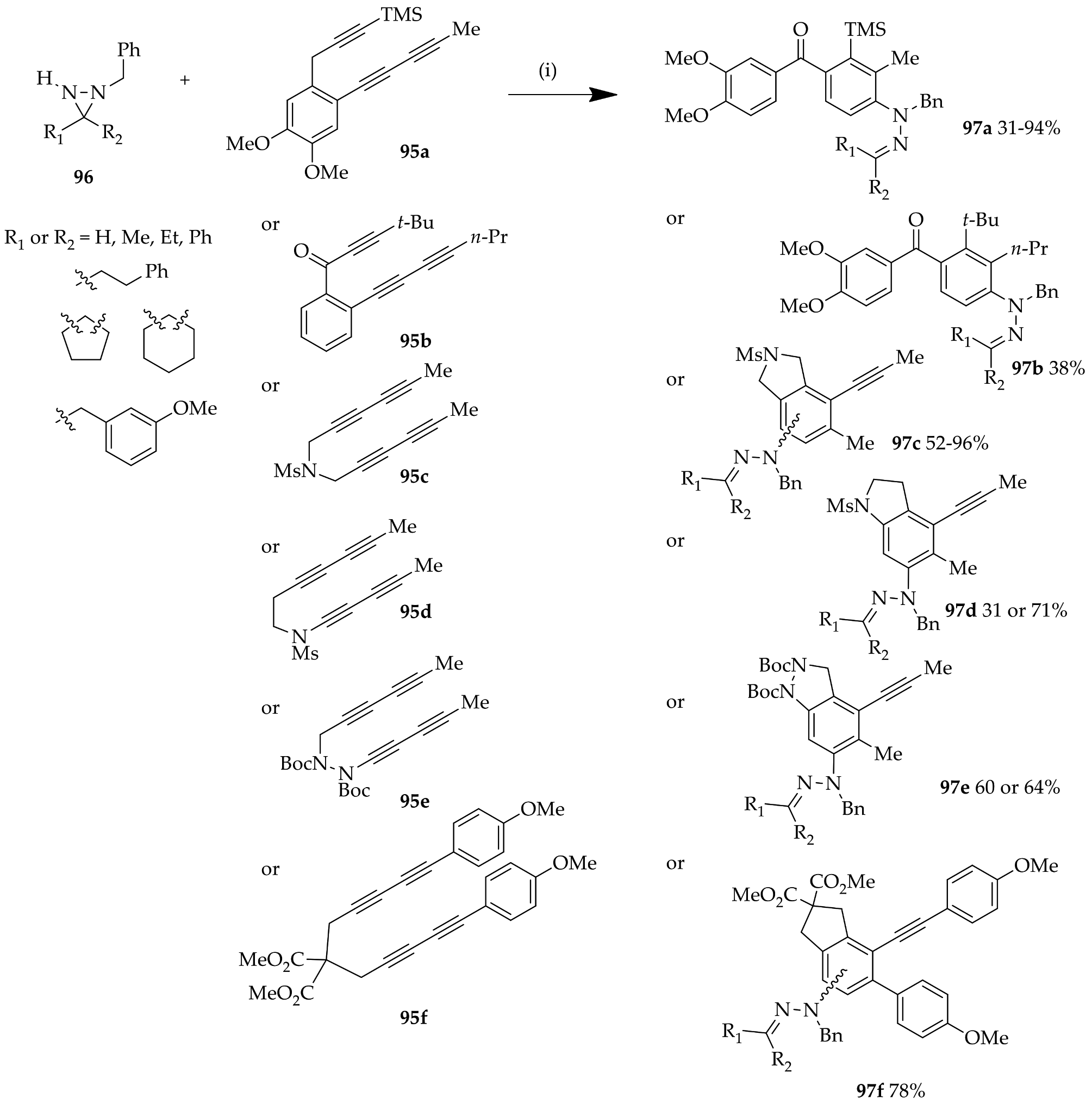{kind=link}
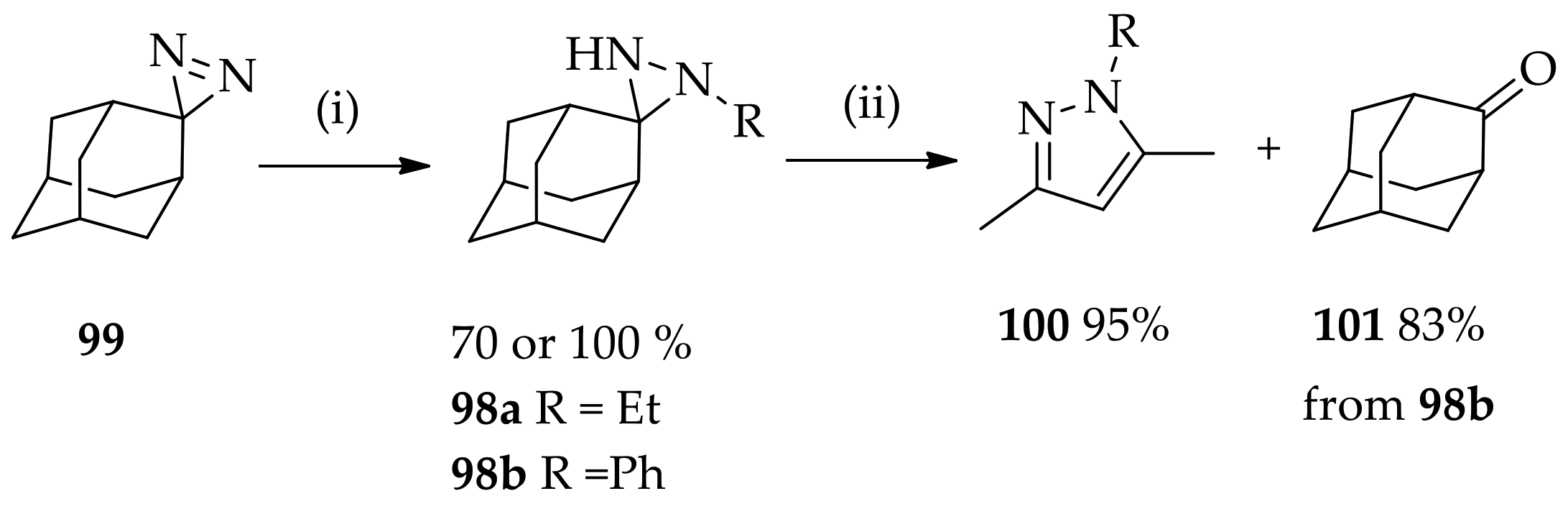{kind=link}
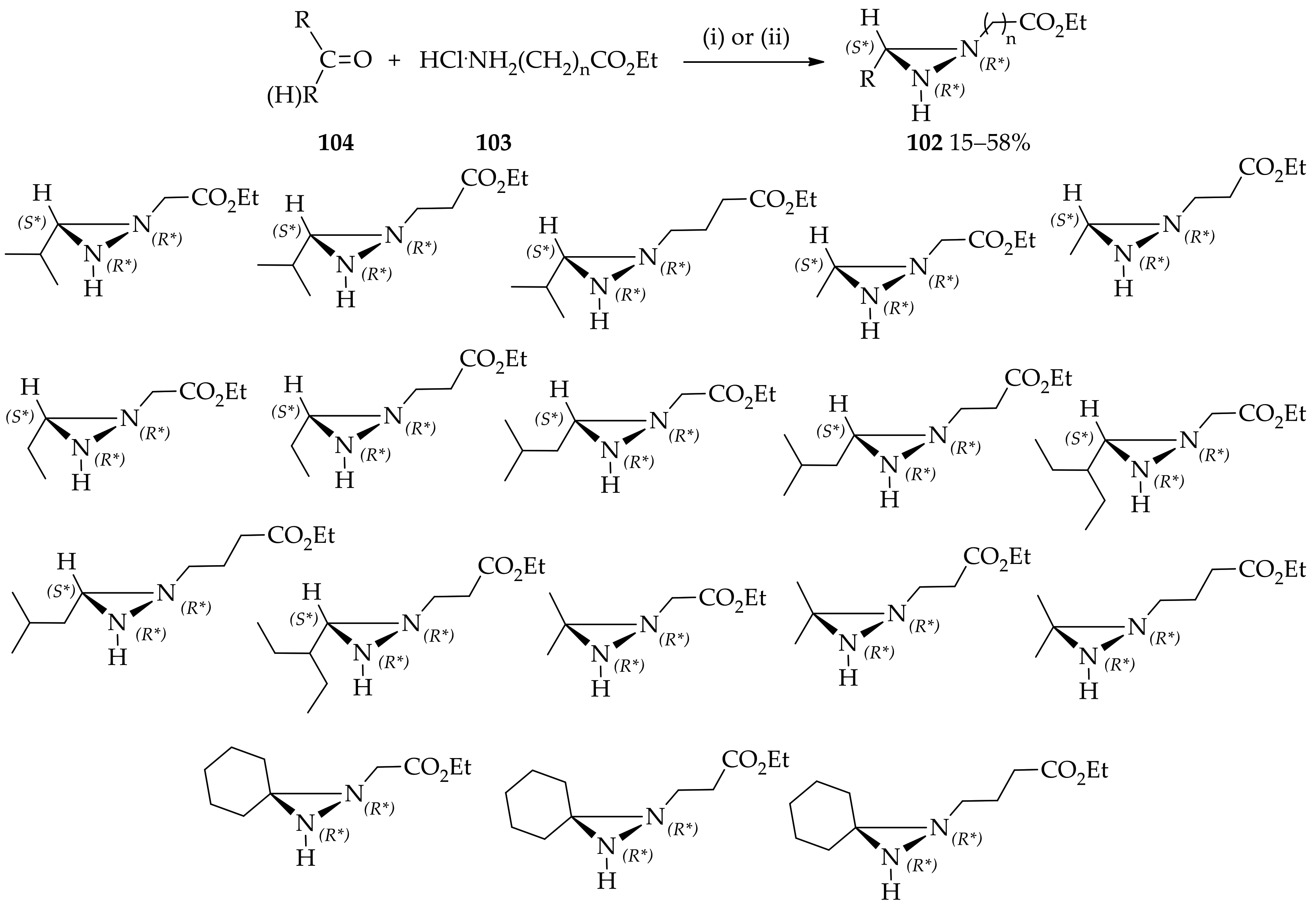{kind=link}
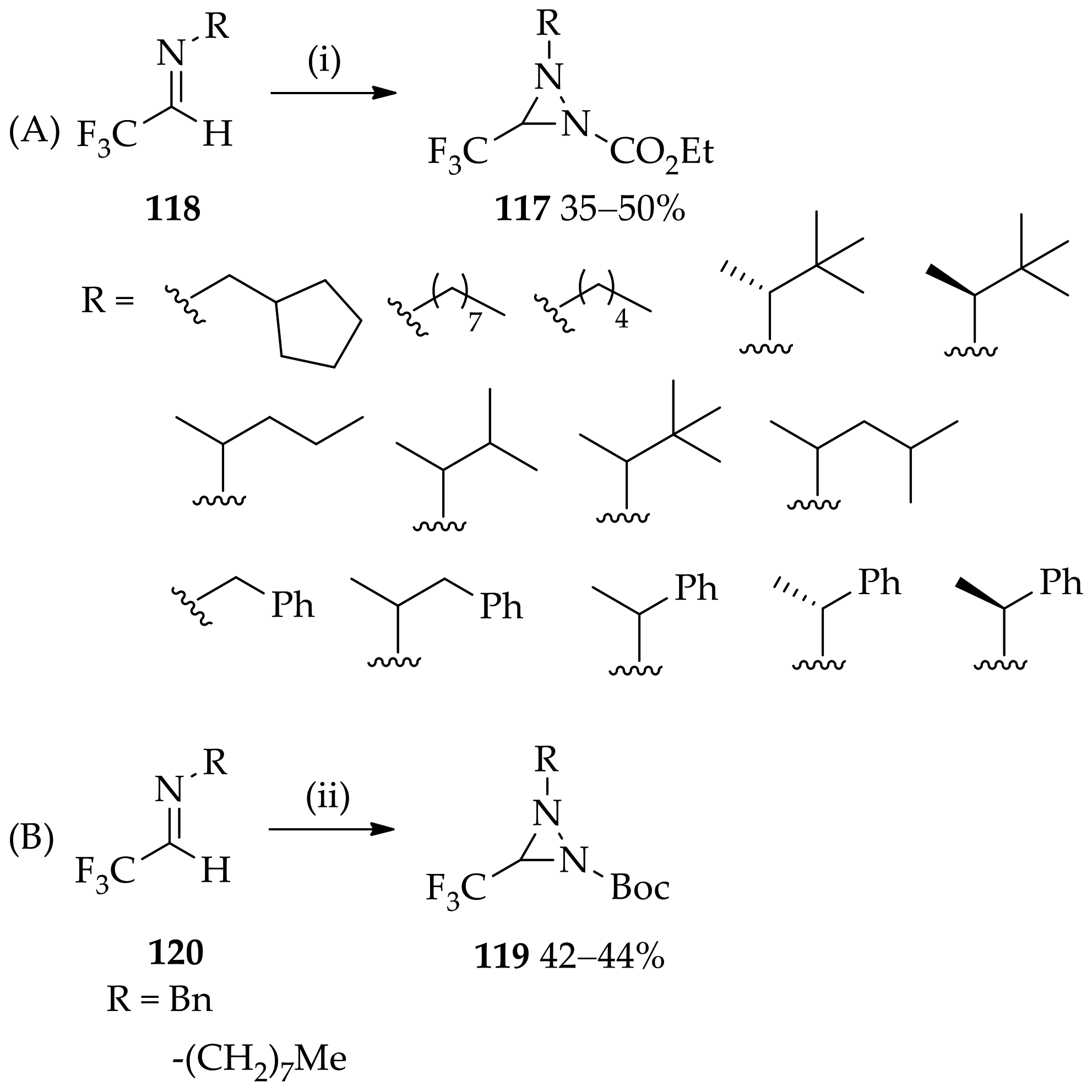{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entries | Possible Diaziridine Formation (In-Situ) | Base (equiv.) | Reference |
|---|---|---|---|
| 1 |  | tBuOK (2.3 equiv.) | [57] |
| 47a | KOH (2.3 equiv.) | [58] | |
| 2 |  | tBuOK (2.3 equiv.) | [57] |
| 47b | KOH (2.3 equiv.) | [58] | |
| 3 |  | tBuOK (3.3 equiv) | [57] |
| 47c | KOH (3.3 equiv.) | [58] | |
| 4 |  | tBuOK (4.3 equiv.) | [57] |
| 47d | KOH (4.3 equiv.) | [58] | |
| 5 |  | KOH (2.3 equiv.) | [58] |
| 47e | |||
| 6 |  | tBuOK (2.3 equiv.) | [57] |
| 47f | KOH (2.3 equiv.) | [58] | |
| 7 |  | tBuOK (2.3 equiv.) | [57] |
| 47g | KOH (2.3 equiv.) | [58] | |
| 8 |  | tBuOK (3.3 equiv.) | [57] |
| 47h | KOH (3.3 equiv.) | [58] |
3.1.3. In Situ Formation of Diaziridines in One-Pot Synthesis of 3H-Diazirine
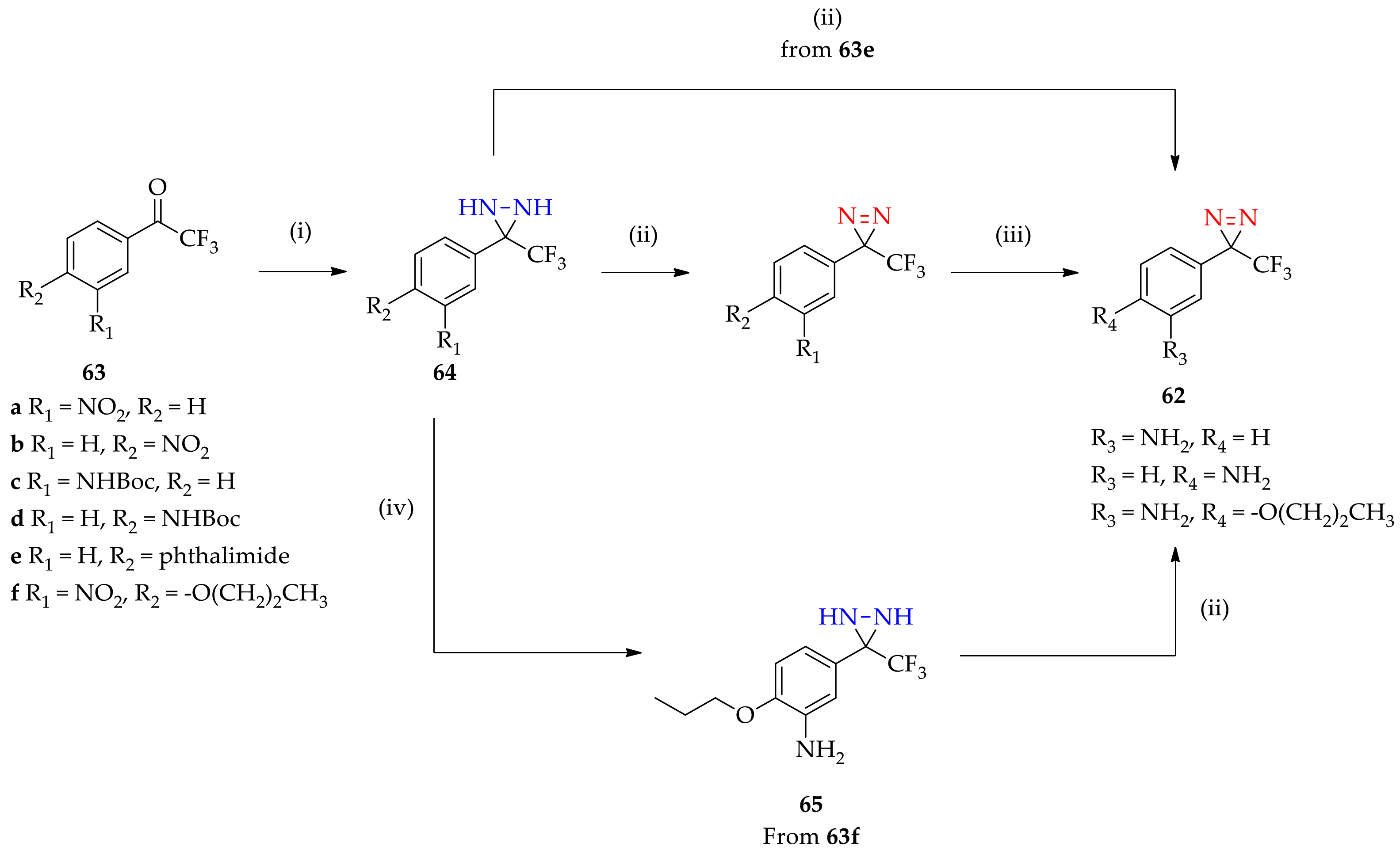
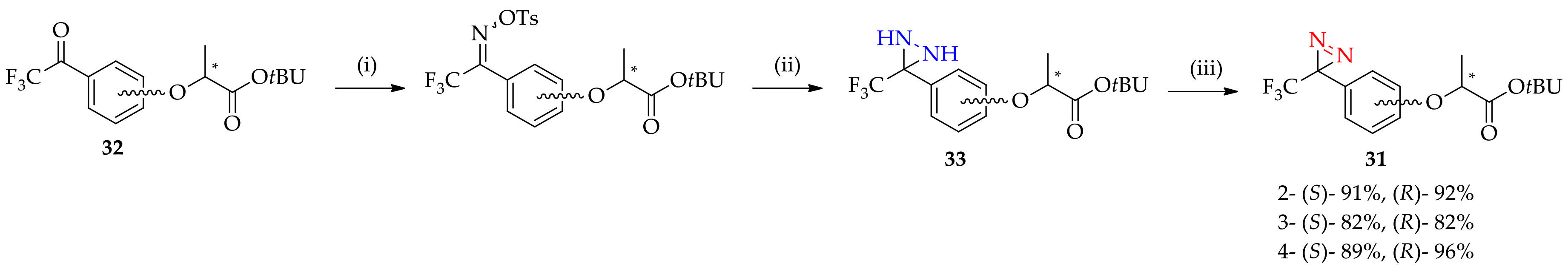
3.1.4. Improved Method for the Synthesis of 3-[3-(Trifluoromethyl)-3H-diazirin-3-yl]aniline Derivatives
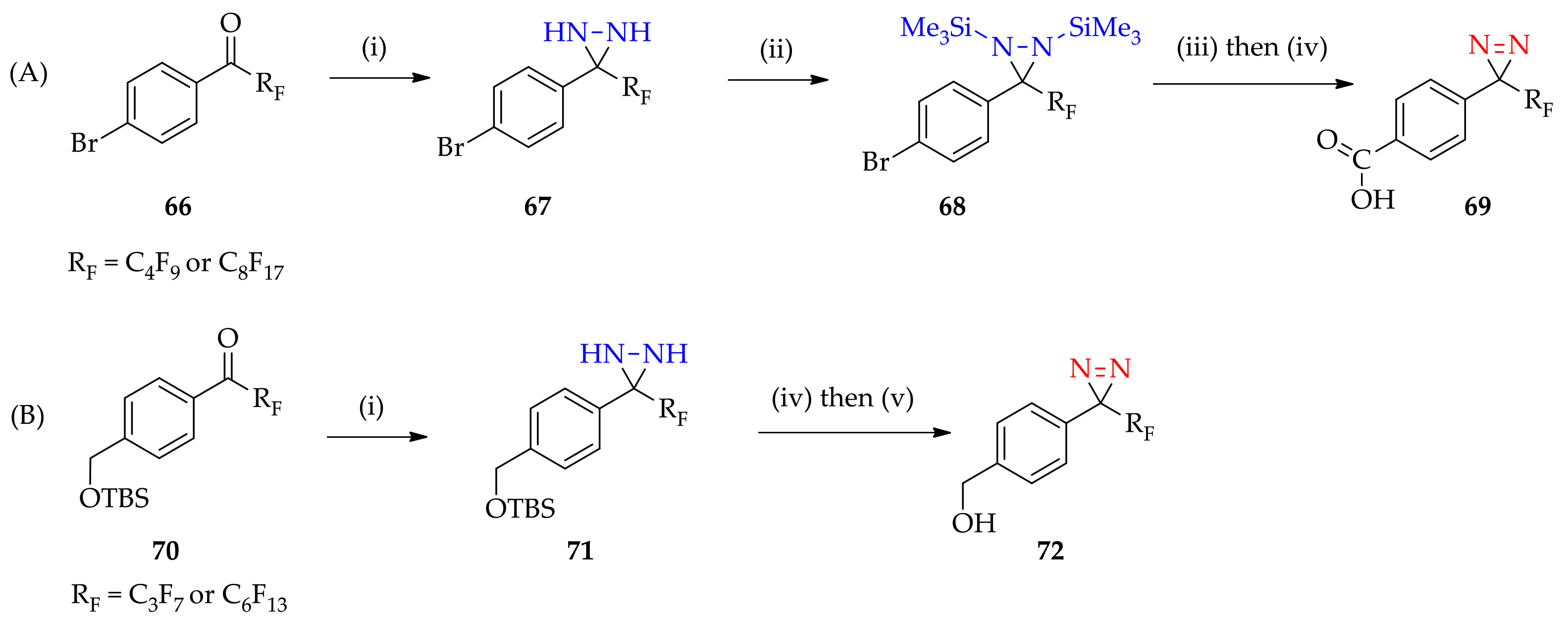
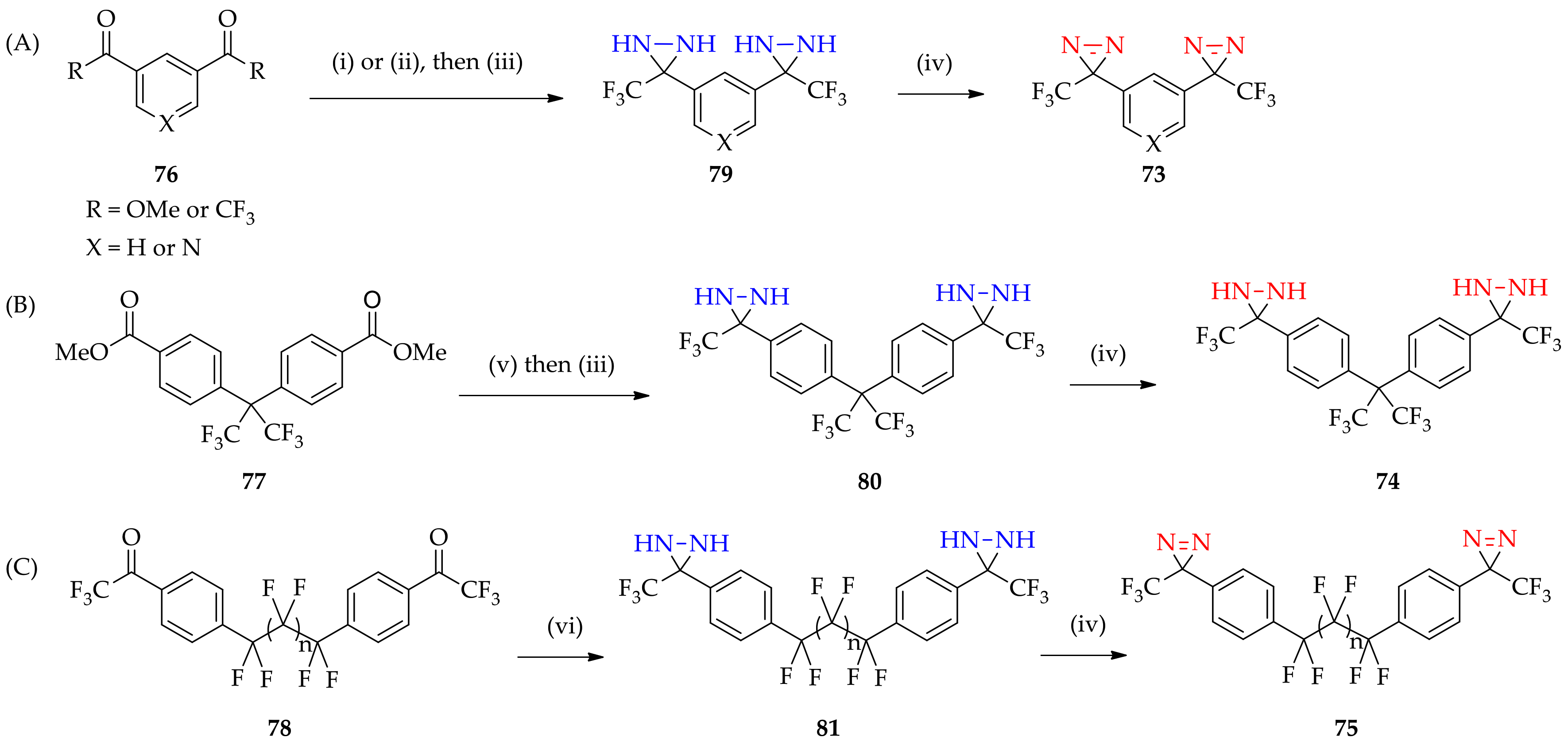
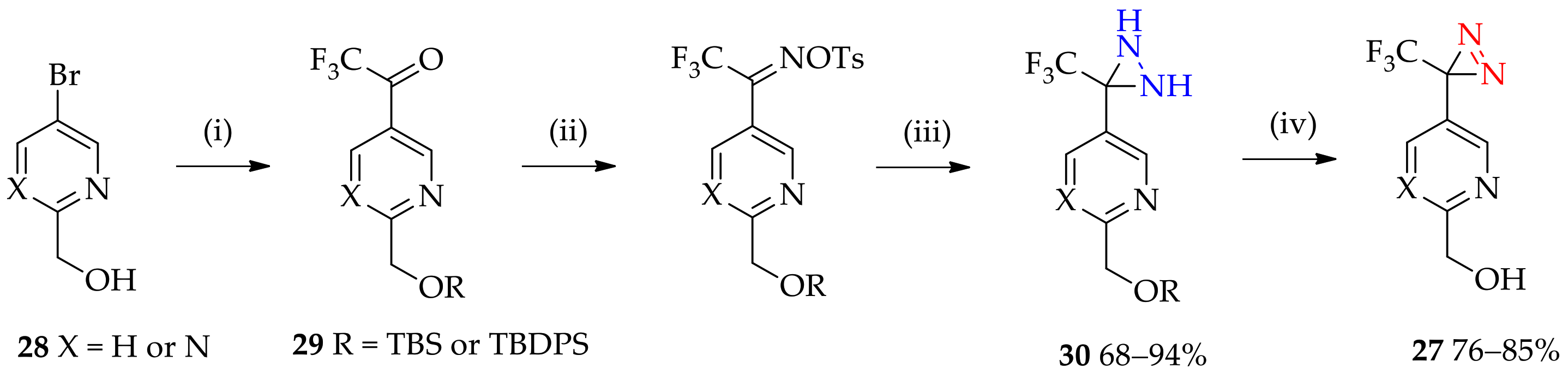
3.1.5. Expansion of Fluorous 3H-Diaziridine as a Basis for 3H-Diazirine Application
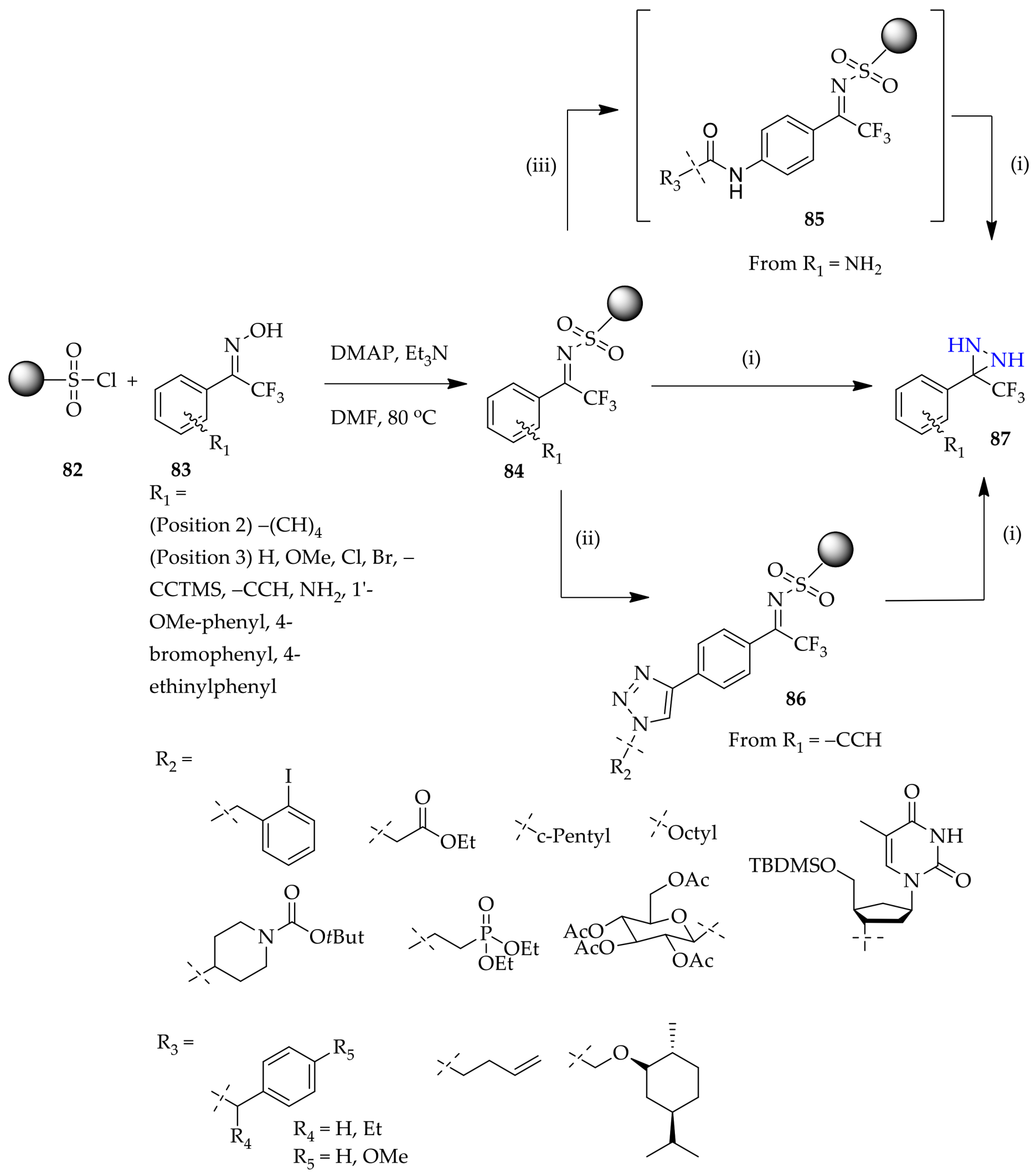
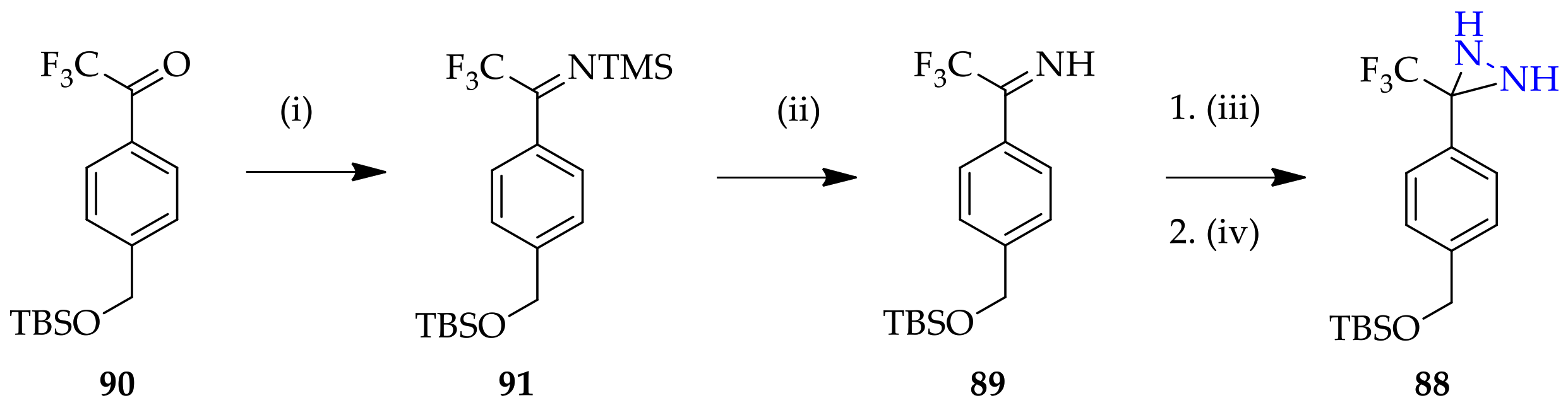
3.1.6. Unconventional Non-Substituted Diaziridine Synthesis Approaches
3.2. N-Monosubstituted Diaziridines
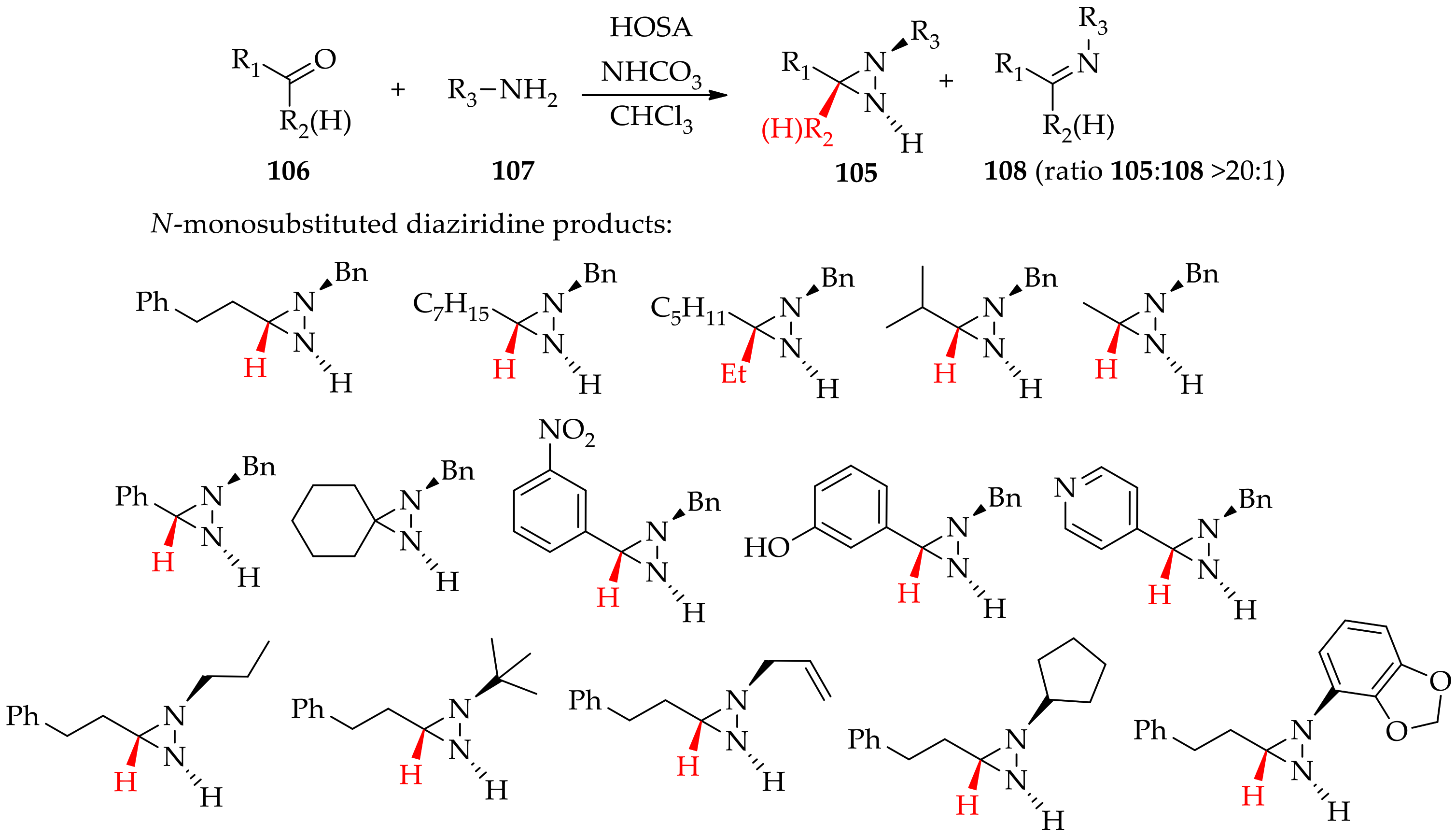
3.2.1. Conventional Method for Recent Uses
3.2.2. Base Addition for Enhancement of N-Monosubstituted Diaziridine Formation
3.2.3. Unconventional N-Monosubstituted Diaziridine Synthesis Approach
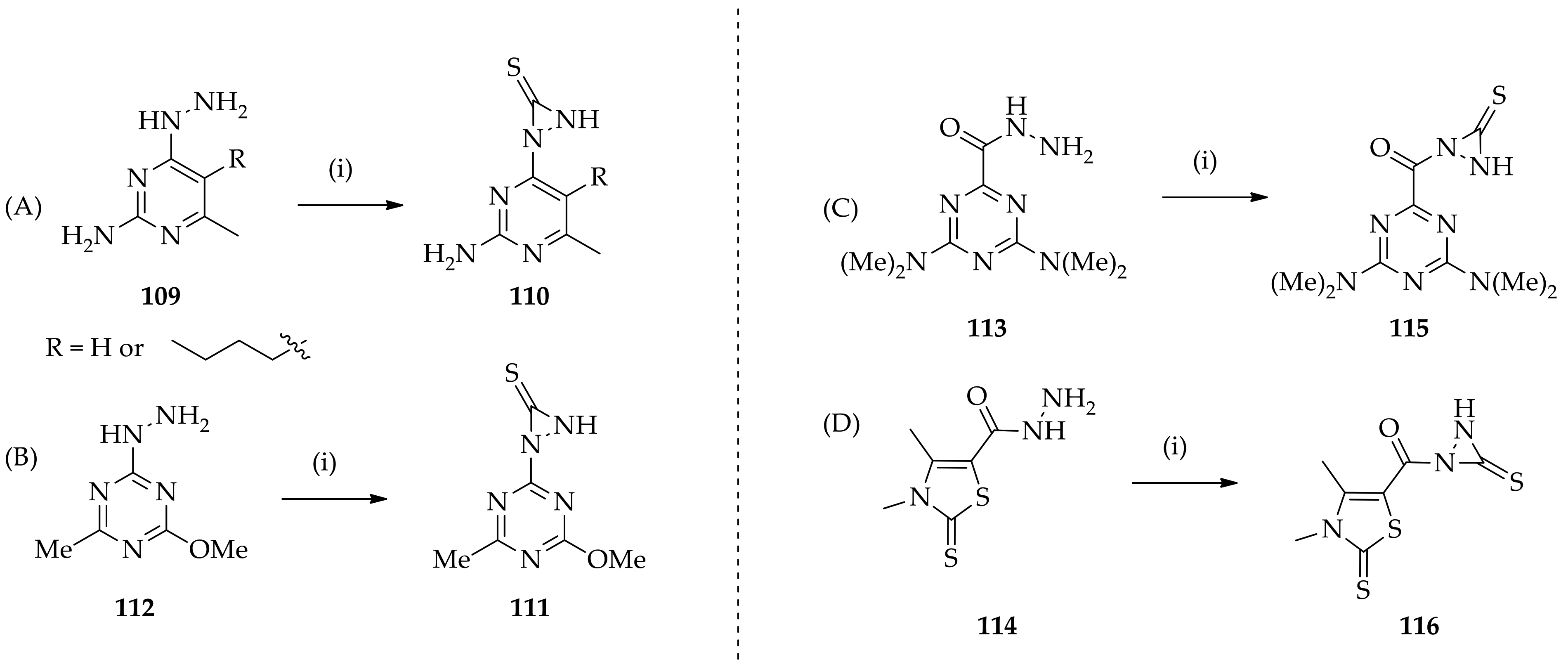
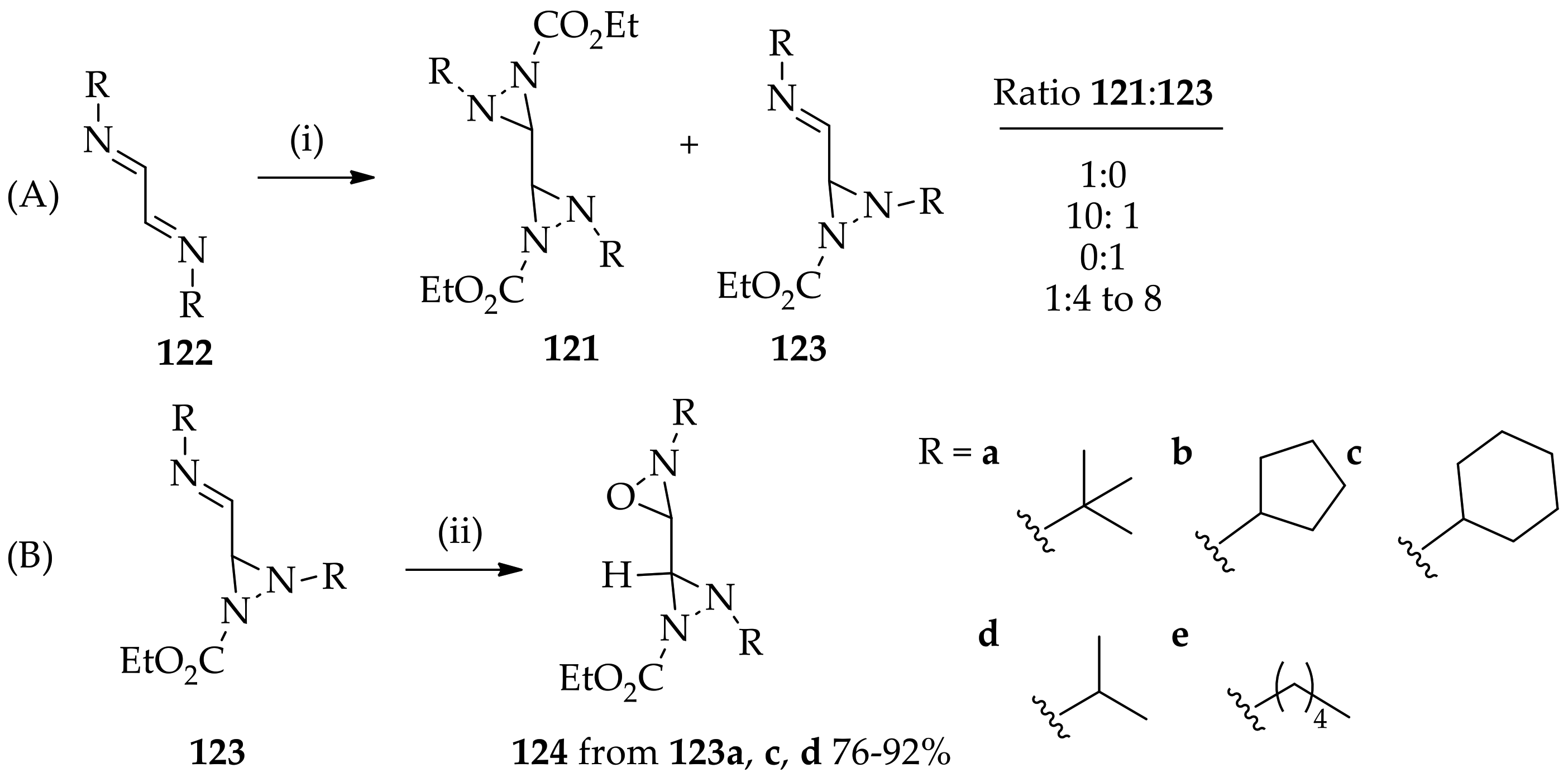
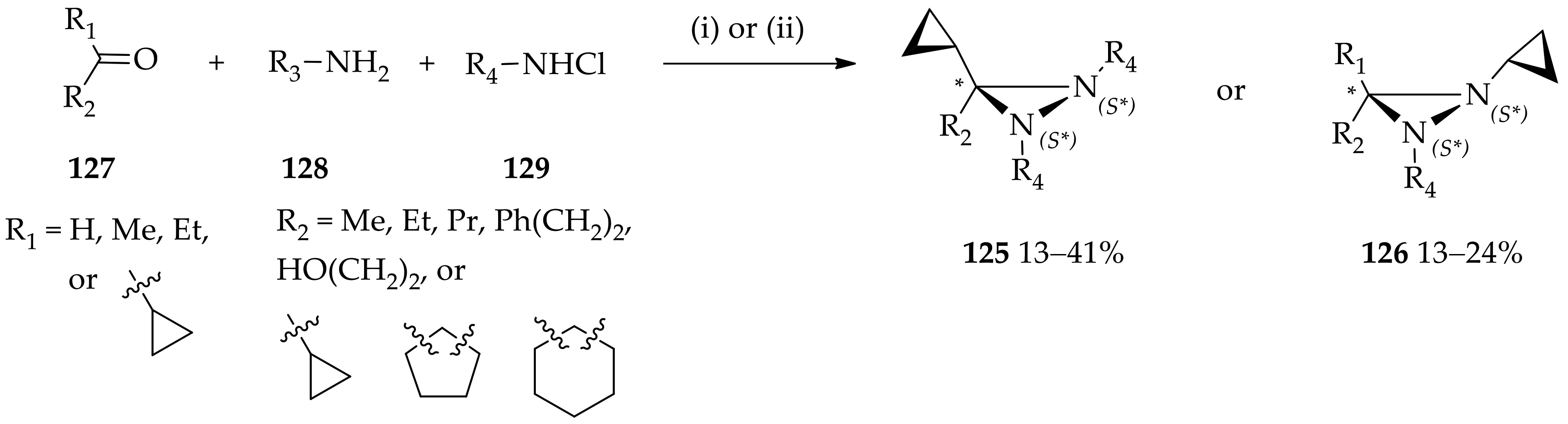
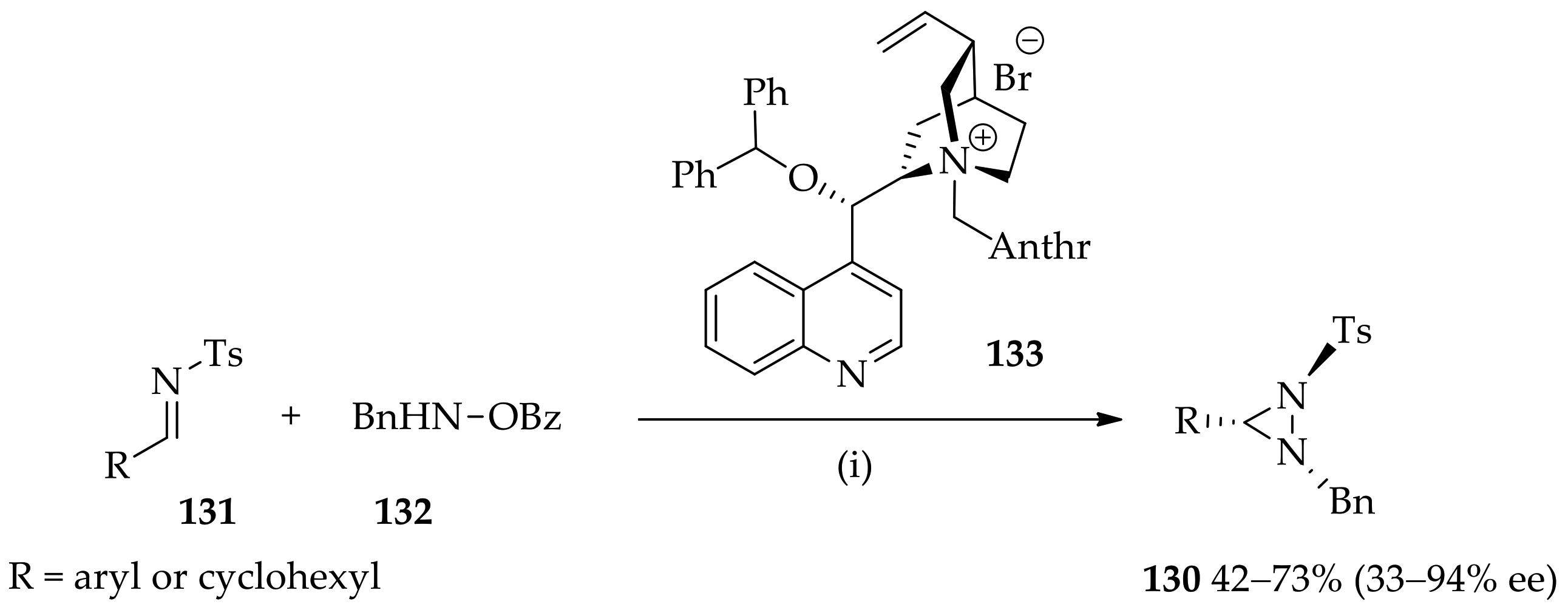
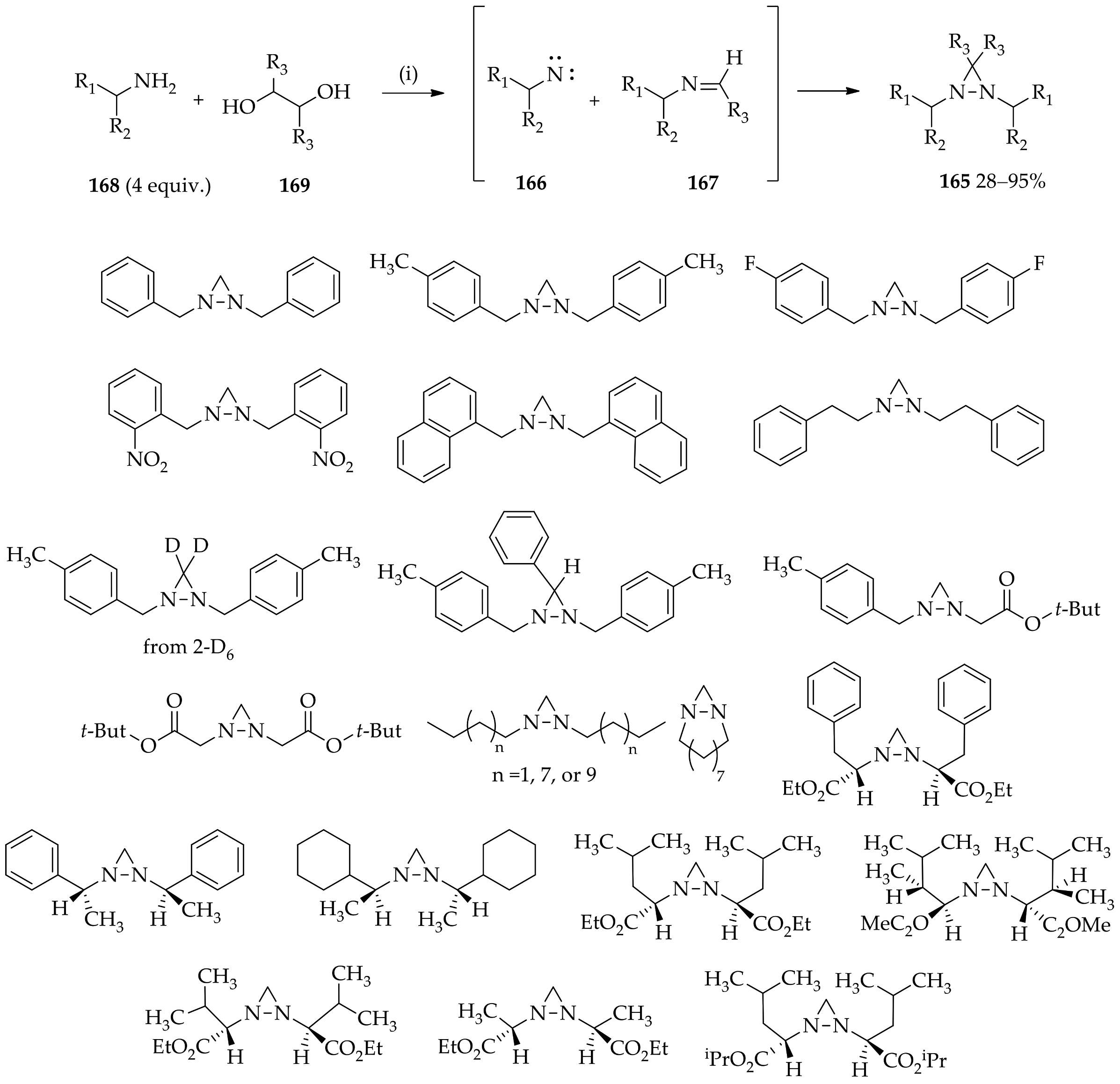
3.3. N,N-Disubstituted Diaziridines
3.3.1. Conventional Method for Recent Uses
3.3.2. The Green Transformation of 6-Aryl-1,5-diazabicyclo[3.1.0]hexanes
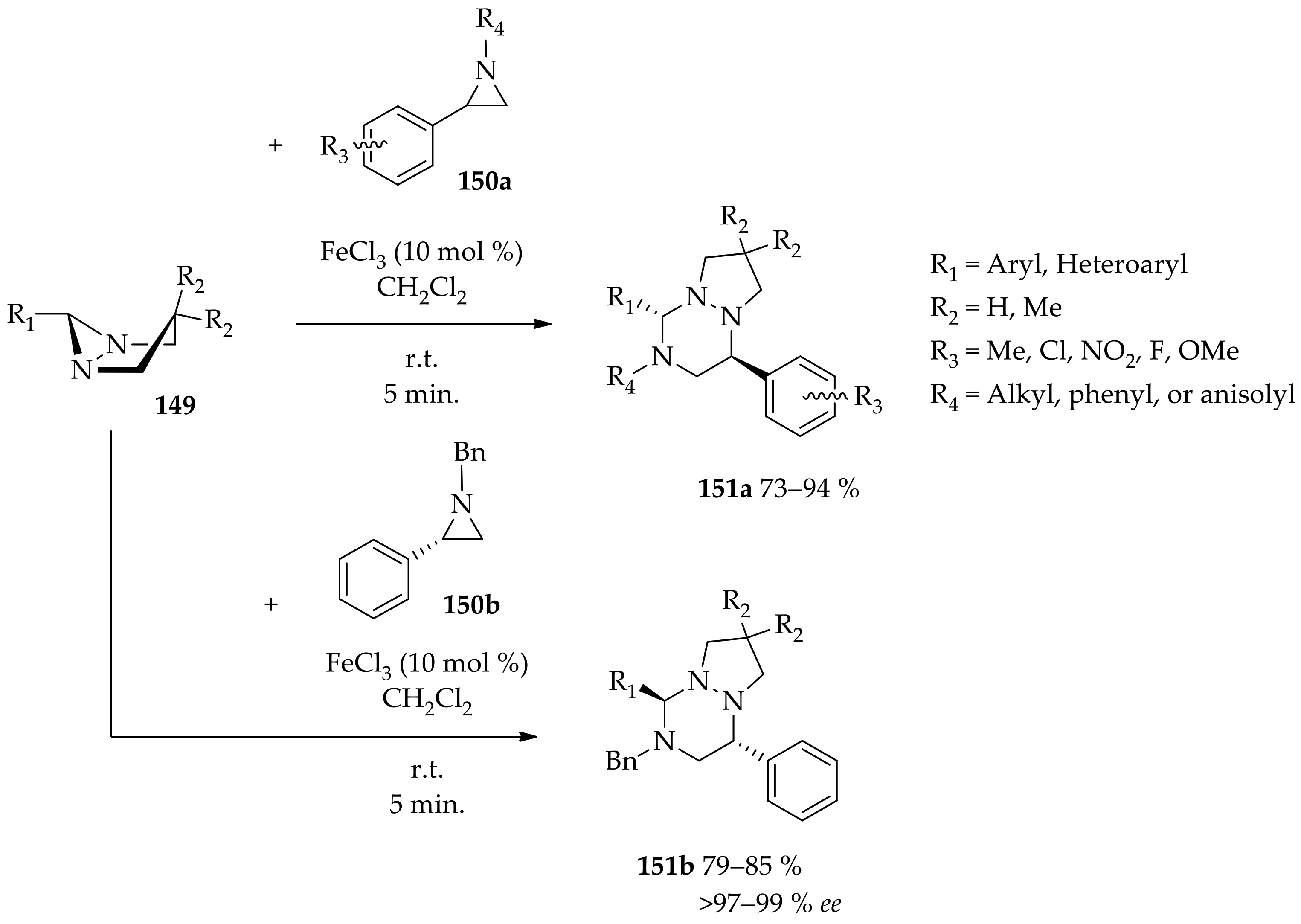
3.3.3. Metal Catalysis of Diaziridines Ring Opening
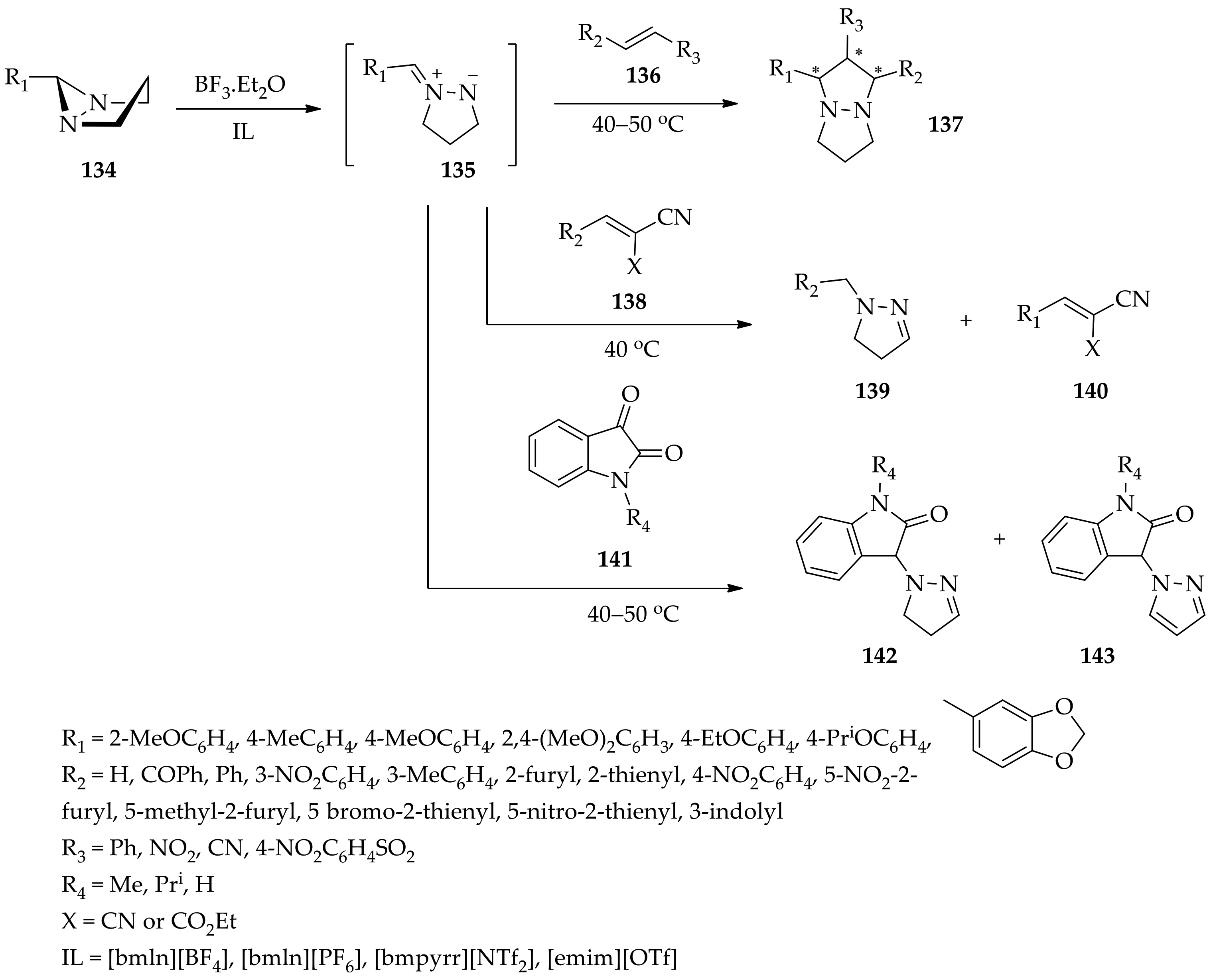
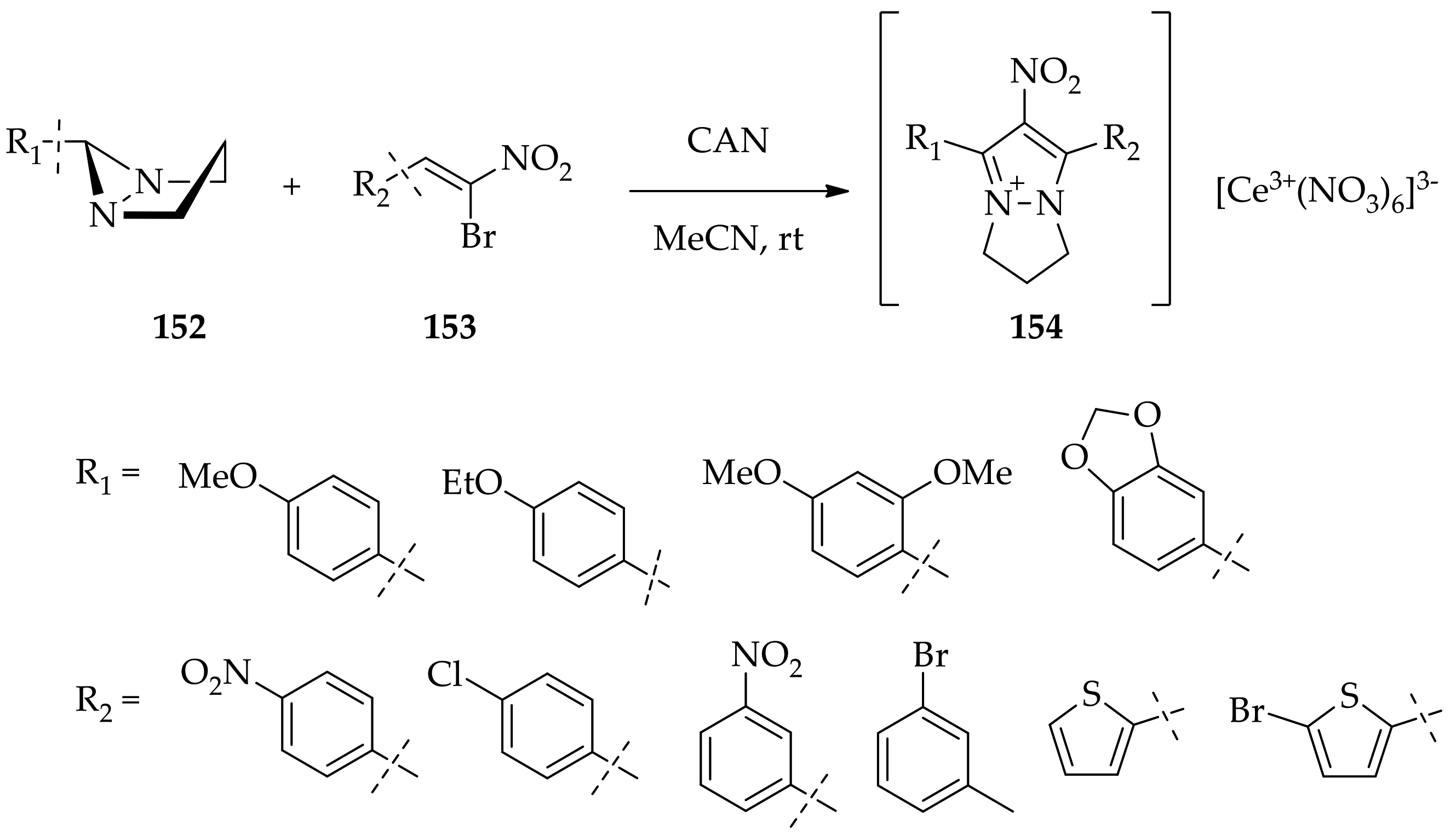
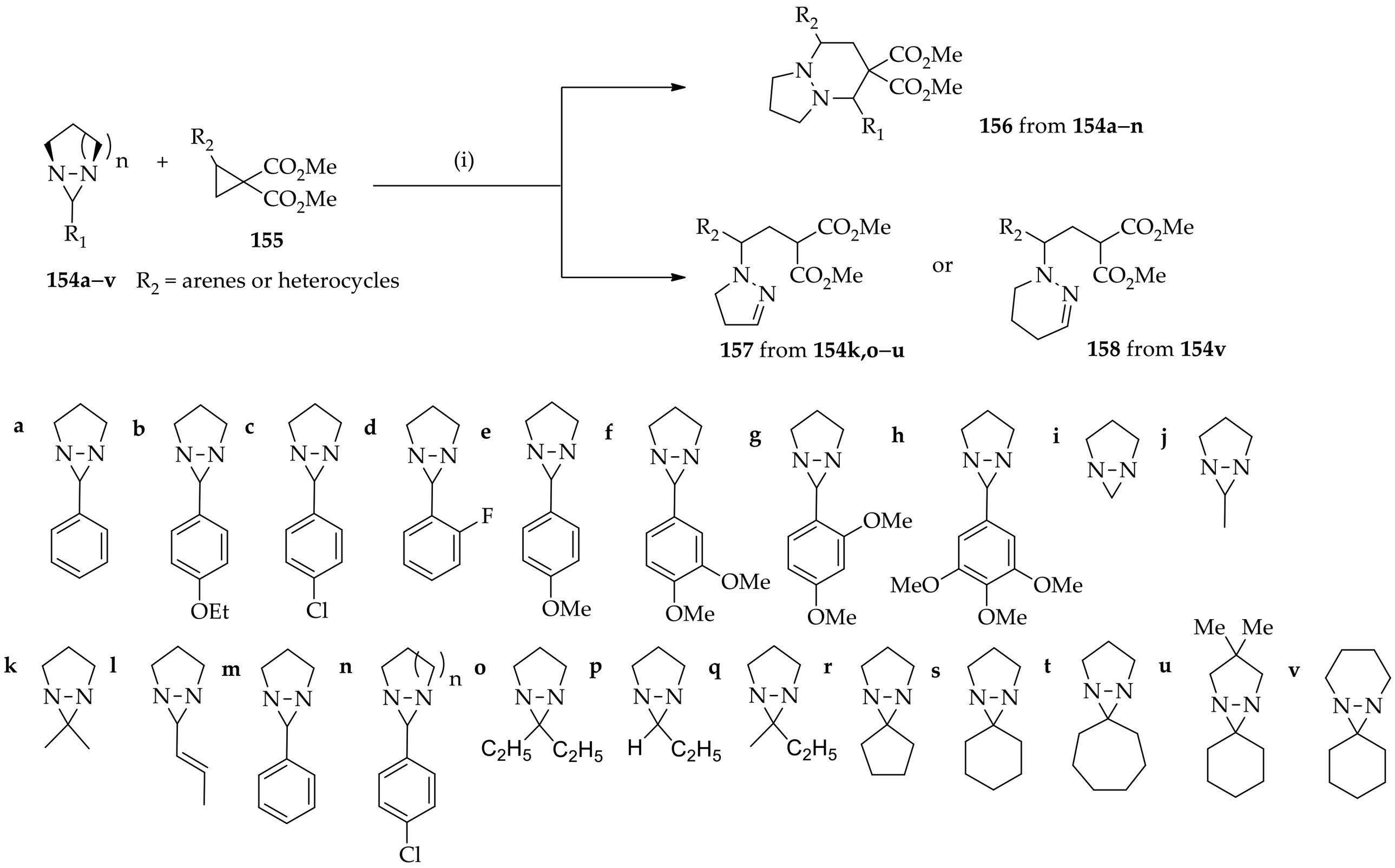
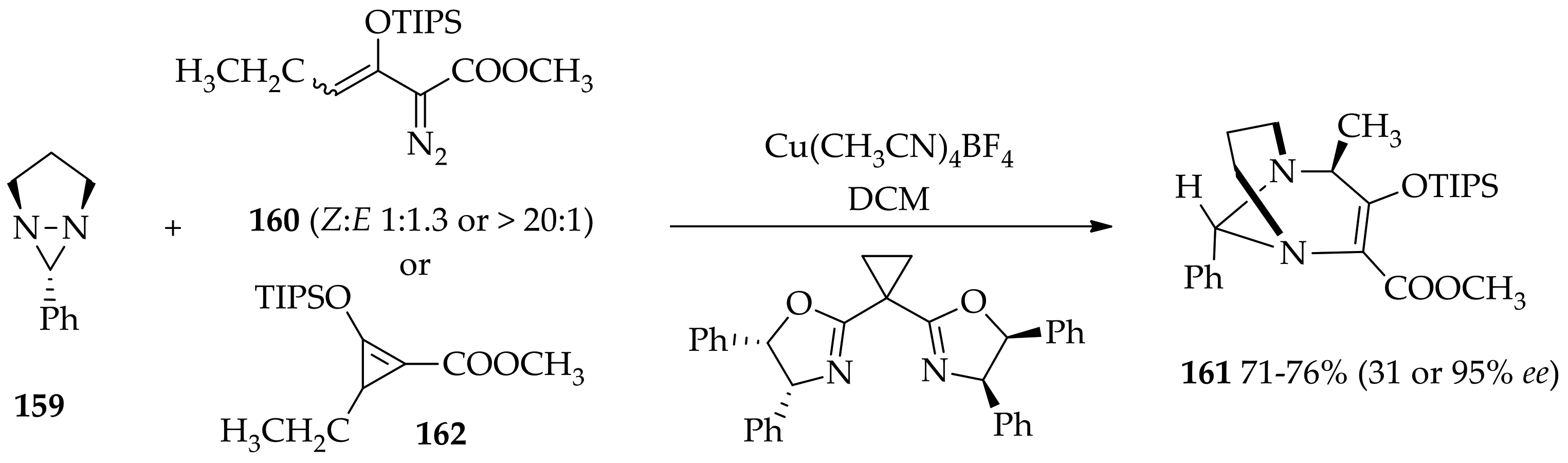
3.3.4. Diaziridine Reaction with Donor–Acceptor Cyclopropanes/Cyclopropenes
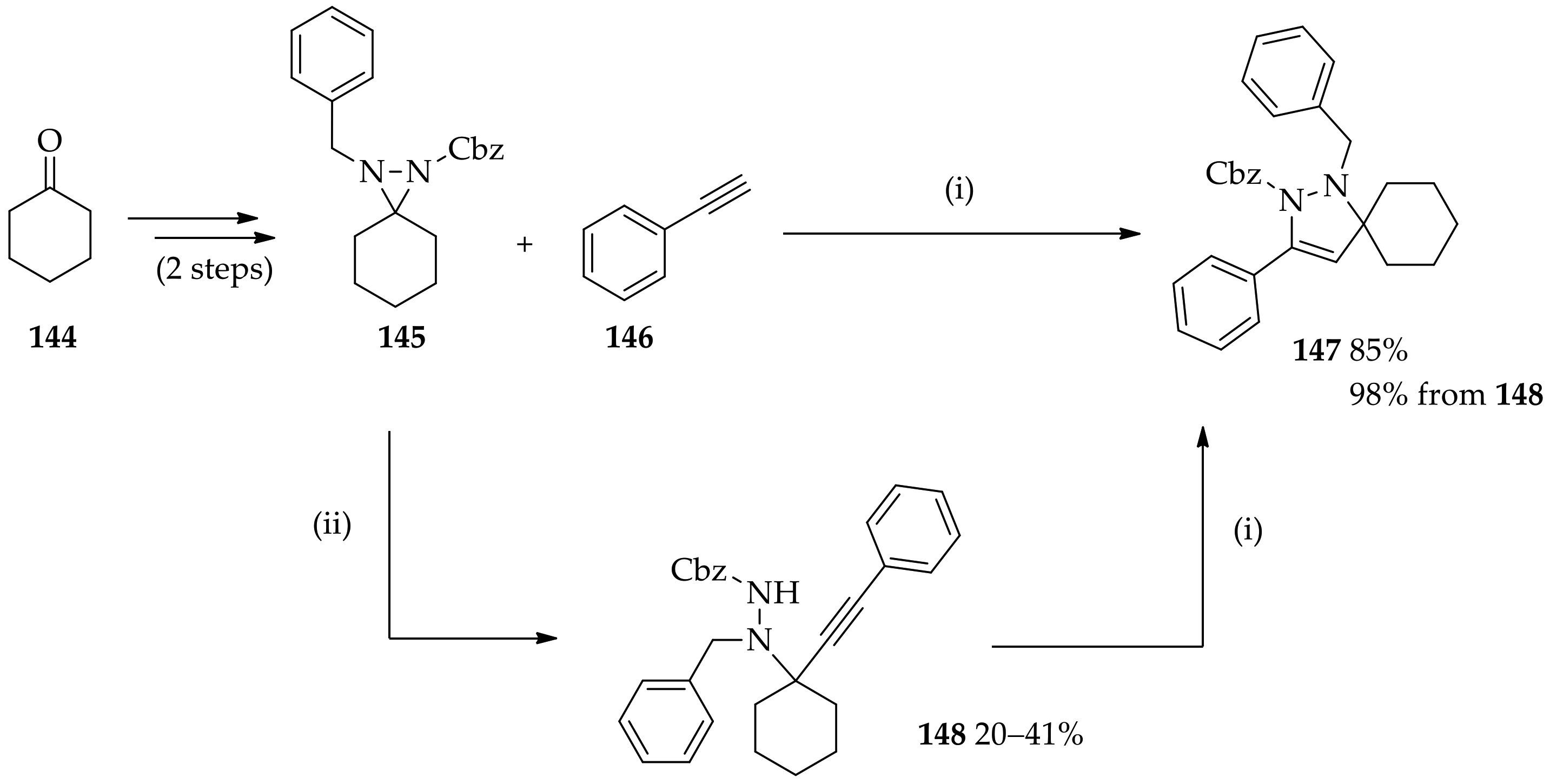
3.3.5. Unconventional N,N-Disubstituted Diaziridine Synthesis Approach
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hashimoto, M. Diaziridines and Diazirines, 4th ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2008; Volume 1, ISBN 9780080449920. [Google Scholar]
- Eliazyan, K.A.; Avetisyan, F.V.; Jivanshiryan, T.L.; Pivazyan, V.A.; Ghazaryan, E.A.; Shahbazyan, L.V.; Harutyunyan, S.V.; Yengoyan, A.P. Synthesis and Fungicidal Activity of Novel 1,3-Disubstituted 1H-Diazirine Derivatives. J. Heterocycl. Chem. 2009, 46, 1259–1265. [Google Scholar] [CrossRef]
- Ravindra, S.; Irfana Jesin, C.P.; Shabashini, A.; Nandi, G.C. Recent Advances in the Preparations and Synthetic Applications of Oxaziridines and Diaziridines. Adv. Synth. Catal. 2021, 363, 1756–1781. [Google Scholar] [CrossRef]
- Makhova, N.N.; Belen’kii, L.I.; Gazieva, G.A.; Dalinger, I.L.; Konstantinova, L.S.; Kuznetsov, V.V.; Kravchenko, A.N.; Krayushkin, M.M.; Rakitin, O.A.; Starosotnikov, A.M.; et al. Progress in the chemistry of nitrogen-, oxygen- and sulfur-containing heterocyclic systems. Russ. Chem. Rev. 2020, 89, 55–124. [Google Scholar] [CrossRef]
- Makhova, N.N.; Shevtsov, A.V.; Petukhova, V.Y. Transformations of diaziridines and their fused analogues induced by electrophilic reagents. Russ. Chem. Rev. 2011, 80, 1035–1066. [Google Scholar] [CrossRef]
- Williamson, K.S.; Michaelis, D.J.; Yoon, T.P. Advances in the chemistry of oxaziridines. Chem. Rev. 2014, 114, 8016–8036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubinsky, L.; Krom, B.P.; Meijler, M.M. Diazirine based photoaffinity labeling. Bioorganic Med. Chem. 2012, 20, 554–570. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Hatanaka, Y. Recent progress in diazirine-based photoaffinity labeling. Eur. J. Org. Chem. 2008, 2513–2523. [Google Scholar] [CrossRef]
- Halloran, M.W.; Lumb, J.P. Recent Applications of Diazirines in Chemical Proteomics. Chem. Eur. J. 2019, 25, 4885–4898. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, Y.; Nakayama, H.; Kanaoka, Y. Diazirine-based photoaffinity labeling: Chemical approach to biological interfaces. Rev. Heteroat. Chem. 1996, 14, 213–243. [Google Scholar]
- Hassan, M.M.; Olaoye, O.O. Recent Advances in Chemical Biology Using Benzophenones and Diazirines as Radical Precursors. Molecules 2020, 25, 2285. [Google Scholar] [CrossRef] [PubMed]
- Ichiishi, N.; Moore, K.P.; Wassermann, A.M.; Wolkenberg, S.E.; Krska, S.W. Reducing Limitation in Probe Design: The Development of a Diazirine-Compatible Suzuki-Miyaura Cross Coupling Reaction. ACS Med. Chem. Lett. 2019, 10, 56–61. [Google Scholar] [CrossRef]
- Ge, S.S.; Chen, B.; Wu, Y.Y.; Long, Q.S.; Zhao, Y.L.; Wang, P.Y.; Yang, S. Current advances of carbene-mediated photoaffinity labeling in medicinal chemistry. RSC Adv. 2018, 8, 29428–29454. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.R.; Robertson, A.A.B. Fishing for Drug Targets: A Focus on Diazirine Photoaffinity Probe Synthesis. J. Med. Chem. 2018, 61, 6945–6963. [Google Scholar] [CrossRef]
- Gakh, A.; Shermolovich, Y. Trifluoromethylated Heterocycles. Curr. Top. Med. Chem. 2014, 14, 952–965. [Google Scholar] [CrossRef]
- Ishihara, H.; Hori, K.; Sugihara, H.; Ito, Y.N.; Katsuki, T. Highly diastereo- and enantioselective aziridination of α,β-unsaturated amides with diaziridine and mechanistic consideration on its stereochemistry. Helv. Chim. Acta 2002, 85, 4272–4286. [Google Scholar] [CrossRef]
- Hu, H.; Xu, J.; Wang, F.; Dong, S.; Liu, X.; Feng, X. Chiral ScIII- N, N′-Dioxide-Catalyzed 1,3-Dipolar Cycloaddition of Diaziridines with Chalcones. Org. Lett. 2020, 22, 93–97. [Google Scholar] [CrossRef]
- Pellacani, L.; Fioravanti, S.; Tardella, P.A. Ethyl Nosyloxycarbamate: A Chameleonic Aminating Agent. Curr. Org. Chem. 2011, 15, 1465–1481. [Google Scholar] [CrossRef]
- Huang, C.Y.; Doyle, A.G. The chemistry of transition metals with three-membered ring heterocycles. Chem. Rev. 2014, 114, 8153–8198. [Google Scholar] [CrossRef]
- Makhova, N.N.; Petukhova, V.Y.; Kuznetsov, V.V. Synthesis of monocyclic diaziridines and their fused derivatives. Arkivoc 2008, 2008, 128–152. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, V.V.; Marochkin, I.I.; Goloveshkin, A.S.; Makhova, N.N.; Shishkov, I.F. Comparable study of the structure of 1,2-bis(2-acetamidoethyl) diaziridine and 3,3-diethyldiaziridine with structures of related compounds by X-ray diffraction analysis and quantum chemical calculations. Struct. Chem. 2017, 28, 1211–1221. [Google Scholar] [CrossRef]
- Marochkin, I.I.; Kuznetsov, V.V.; Li, Z.; Rykov, A.N.; Makhova, N.N.; Shishkov, I.F. Molecular structure of 1,2-diethyldiaziridine studied by gas electron diffraction supported by quantum chemistry calculations. J. Mol. Struct. 2021, 1225, 129066–129073. [Google Scholar] [CrossRef]
- Marochkin, I.I.; Kuznetsov, V.V.; Rykov, A.N.; Makhova, N.N.; Shishkov, I.F. Molecular structure study of 1,2,3-trimethyldiaziridine by means of gas electron diffraction method. Struct. Chem. 2019, 30, 457–464. [Google Scholar] [CrossRef]
- Altova, E.P.; Kuznetsov, V.V.; Marochkin, I.I.; Rykov, A.N.; Makhova, N.N.; Shishkov, I.F. 3-Cyclopropyl-1,2-dimethyldiaziridine: Synthesis and study of molecular structure by gas electron diffraction method. Struct. Chem. 2018, 29, 815–822. [Google Scholar] [CrossRef]
- Khaikin, L.S.; Kochikov, I.V.; Rykov, A.N.; Grikina, O.E.; Ageev, G.G.; Shishkov, I.F.; Kuznetsov, V.V.; Makhova, N.N. Equilibrium structures of the tetramezine diastereomers and their ratio: Joint analysis of gas phase electron diffraction, quantum chemistry, and spectroscopic data. Phys. Chem. Chem. Phys. 2019, 21, 5598–5613. [Google Scholar] [CrossRef] [PubMed]
- Khaikin, L.S.; Ageev, G.G.; Rykov, A.N.; Grikina, O.E.; Shishkov, I.F.; Kochikov, I.V.; Kuznetsov, V.V.; Makhova, N.N.; Bukalov, S.S.; Leites, L.A. Equilibrium molecular structure and spectra of 6-methyl-1,5-diazabicyclo[3.1.0]hexane: Joint analysis of gas phase electron diffraction, quantum chemistry, and spectroscopic data. Phys. Chem. Chem. Phys. 2020, 22, 22477–22492. [Google Scholar] [CrossRef] [PubMed]
- Kamuf, M.; Trapp, O. Stereodynamics of tetramezine. Chirality 2011, 23, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Kamuf, M.; Trapp, O. Stereodynamics of Small 1,2-Dialkyldiaziridines. Chirality 2013, 25, 224–229. [Google Scholar] [CrossRef]
- Trapp, O.; Sahraoui, L.; Hofstadt, W.; Könen, W. The stereodynamics of 1,2-dipropyldiaziridines. Chirality 2010, 22, 284–291. [Google Scholar] [CrossRef]
- Zawatzky, K.; Kamuf, M.; Trapp, O. Chiral 1,2-Dialkenyl Diaziridines: Synthesis, Enantioselective Separation, and Nitrogen Inversion Barriers. Chirality 2015, 27, 156–162. [Google Scholar] [CrossRef]
- Ameen, R.; Anoop, A. Detonation properties and impact sensitivities of trinitromethane derivatives of three-membered heterocyclic ring compounds. J. Mol. Graph. Model. 2021, 105, 107863. [Google Scholar] [CrossRef]
- Angelone, D.; Hammer, A.J.S.; Rohrbach, S.; Krambeck, S.; Granda, J.M.; Wolf, J.; Zalesskiy, S.; Chisholm, G.; Cronin, L. Convergence of multiple synthetic paradigms in a universally programmable chemical synthesis machine. Nat. Chem. 2021, 13, 63–69. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, G.; Yan, C.; Lu, K.; Li, H.; Zhang, Y.; Wang, J. Microwave-assisted, Pd(0)-catalyzed cross-coupling of diazirines with aryl halides. Org. Lett. 2010, 12, 5580–5583. [Google Scholar] [CrossRef]
- Shen, K.; Logan, A.W.J.; Colell, J.F.P.; Bae, J.; Ortiz, G.X.; Theis, T.; Warren, W.S.; Malcolmson, S.J.; Wang, Q. Diazirines as Potential Molecular Imaging Tags: Probing the Requirements for Efficient and Long-Lived SABRE-Induced Hyperpolarization. Angew. Chem. 2017, 56, 12112–12116. [Google Scholar] [CrossRef]
- Theis, T.; Ortiz, G.X.; Logan, A.W.J.; Claytor, K.E.; Feng, Y.; Huhn, W.P.; Blum, V.; Malcolmson, S.J.; Chekmenev, E.Y.; Wang, Q.; et al. Direct and cost-efficient hyperpolarization of long-lived nuclear spin states on universal 15N2-diazirine molecular tags. Sci. Adv. 2016, 2, e1501438. [Google Scholar] [CrossRef] [Green Version]
- Martyloga, O.V.; Myronenko, A.; Tkachenko, A.M.; Matvienko, V.O.; Kuchkovska, Y.O.; Grygorenko, O.O. Multigram Synthesis of Functionalized Spirocyclic Diazirines. Eur. J. Org. Chem. 2019, 3744–3750. [Google Scholar] [CrossRef]
- Das, J. Aliphatic diazirines as photoaffinity probes for proteins: Recent developments. Chem. Rev. 2011, 111, 4405–4417. [Google Scholar] [CrossRef]
- Wu, B.; Jayakar, S.S.; Zhou, X.; Titterton, K.; Chiara, D.C.; Szabo, A.L.; Savechenkov, P.Y.; Kent, D.E.; Cohen, J.B.; Forman, S.A.; et al. Inhibitable photolabeling by neurosteroid diazirine analog in the β3-Subunit of human hetereopentameric type A GABA receptors. Eur. J. Med. Chem. 2019, 162, 810–824. [Google Scholar] [CrossRef]
- Savechenkov, P.Y.; Chiara, D.C.; Desai, R.; Stern, A.T.; Zhou, X.; Ziemba, A.M.; Szabo, A.L.; Zhang, Y.; Cohen, J.B.; Forman, S.A.; et al. Synthesis and pharmacological evaluation of neurosteroid photoaffinity ligands. Eur. J. Med. Chem. 2017, 136, 334–347. [Google Scholar] [CrossRef]
- Byrd, K.M.; Arieno, M.D.; Kennelly, M.E.; Estiu, G.; Wiest, O.; Helquist, P. Design and synthesis of a crosslinker for studying intracellular steroid trafficking pathways. Bioorganic Med. Chem. 2015, 23, 3843–3851. [Google Scholar] [CrossRef] [Green Version]
- Van Der Meijden, B.; Robinson, J.A. Synthesis and application of photoproline-a photoactivatable derivative of proline. Arkivoc 2011, 2011, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Wright, M.H.; Fetzer, C.; Sieber, S.A. Chemical Probes Unravel an Antimicrobial Defense Response Triggered by Binding of the Human Opioid Dynorphin to a Bacterial Sensor Kinase. J. Am. Chem. Soc. 2017, 139, 6152–6159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, A.L.; Menzies, S.K.; King, E.F.B.; Tulloch, L.B.; Gould, E.R.; Zacharova, M.K.; Smith, T.K.; Florence, G.J. Design and Synthesis of Broad Spectrum Trypanosomatid Selective Inhibitors. ACS Infect. Dis. 2018, 4, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Correia, B.E.; Niphakis, M.J.; Cravatt, B.F. Mapping the Protein Interaction Landscape for Fully Functionalized Small-Molecule Probes in Human Cells. J. Am. Chem. Soc. 2014, 136, 10777–10782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.B.; Tipton, J.D.; Manetsch, R. 3-Trifluoromethyl-3-aryldiazirine photolabels with enhanced ambient light stability. Chem. Commun. 2016, 52, 2729–2732. [Google Scholar] [CrossRef]
- Nakagita, T.; Ishida, A.; Tachrim, Z.P.; Wang, L.; Misaka, T.; Hashimoto, M. Asymmetric Synthesis of Photophore-Containing Lactisole Derivatives to Elucidate Sweet Taste Receptors. Molecules 2020, 25, 2790. [Google Scholar] [CrossRef]
- Li, Z.; Hao, P.; Li, L.; Tan, C.Y.J.; Cheng, X.; Chen, G.Y.J.; Sze, S.K.; Shen, H.-M.; Yao, S.Q. Design and Synthesis of Minimalist Terminal Alkyne-Containing Diazirine Photo-Crosslinkers and Their Incorporation into Kinase Inhibitors for Cell- and Tissue-Based Proteome Profiling. Angew. Chem. 2013, 125, 8713–8718. [Google Scholar] [CrossRef]
- Pan, S.; Zhang, H.; Wang, C.; Yao, S.C.L.; Yao, S.Q. Target identification of natural products and bioactive compounds using affinity-based probes. Nat. Prod. Rep. 2016, 33, 612–620. [Google Scholar] [CrossRef]
- Lang, W.; Yuan, C.; Zhu, B.; Pan, S.; Liu, J.; Luo, J.; Nie, S.; Zhu, Q.; Lee, J.S.; Ge, J. Expanding the “minimalist” small molecule tagging approach to different bioactive compounds. Org. Biomol. Chem. 2019, 17, 3010–3017. [Google Scholar] [CrossRef]
- Su, Y.; Pan, S.; Li, Z.; Li, L.; Wu, X.; Hao, P.; Sze, S.K.; Yao, S.Q. Multiplex Imaging and Cellular Target Identification of Kinase Inhibitors via an Affinity-Based Proteome Profiling Approach. Sci. Rep. 2015, 5, 7724–7734. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wang, D.; Li, L.; Pan, S.; Na, Z.; Tan, C.Y.J.; Yao, S.Q. “Minimalist” cyclopropene-containing photo-cross-linkers suitable for live-cell imaging and affinity-based protein labeling. J. Am. Chem. Soc. 2014, 136, 9990–9998. [Google Scholar] [CrossRef]
- Pan, S.; Jang, S.Y.; Wang, D.; Liew, S.S.; Li, Z.; Lee, J.S.; Yao, S.Q. A Suite of “Minimalist” Photo-Crosslinkers for Live-Cell Imaging and Chemical Proteomics: Case Study with BRD4 Inhibitors. Angew. Chem. 2017, 56, 11816–11821. [Google Scholar] [CrossRef]
- Chang, C.-F.; Mfuh, A.; Gao, J.; Wu, H.-Y.; Woo, C.M. Synthesis of an electronically-tuned minimally interfering alkynyl photo-affinity label to measure small molecule–protein interactions. Tetrahedron 2018, 74, 3273–3277. [Google Scholar] [CrossRef]
- Brunner, J.; Senn, H.; Richards, F.M. 3-Trifluoromethyl-3-phenyldiazirine. A new carbene generating group for photolabeling reagents. J. Biol. Chem. 1980, 255, 3313–3318. [Google Scholar] [CrossRef]
- Kumar, N.S.; Young, R.N. Design and synthesis of an all-in-one 3-(1,1-difluoroprop-2-ynyl)-3H-diazirin-3-yl functional group for photo-affinity labeling. Bioorg. Med. Chem. 2009, 17, 5388–5395. [Google Scholar] [CrossRef]
- Hiramatsu, T.; Guo, Y.; Hosoya, T. 3-Azidodifluoromethyl-3H-diazirin-3-yl group as an all-in-one functional group for radioisotope-free photoaffinity labeling. Org. Biomol. Chem. 2007, 5, 2916–2919. [Google Scholar] [CrossRef]
- Wang, L.; Ishida, A.; Hashidoko, Y.; Hashimoto, M. Dehydrogenation of the NH–NH Bond Triggered by Potassium tert-Butoxide in Liquid Ammonia. Angew. Chem. 2017, 56, 870–873. [Google Scholar] [CrossRef]
- Wang, L.; Tachrim, Z.P.; Kurokawa, N.; Ohashi, F.; Sakihama, Y.; Hashidoko, Y.; Hashimoto, M. Base-mediated one-pot synthesis of aliphatic diazirines for photoaffinity labeling. Molecules 2017, 22, 1389. [Google Scholar] [CrossRef] [Green Version]
- Church, R.F.R.; Kende, A.S.; Weiss, M.J. Diazirines. I. Some Observations on the Scope of the Ammonia-Hydroxylamine-O-sulfonic Acid Diaziridine Synthesis. The Preparation of Certain Steroid Diaziridines and Diazirines. J. Am. Chem. Soc. 1965, 87, 2665–2671. [Google Scholar] [CrossRef]
- Church, R.F.R.; Weiss, M.J. Diazirines. II. Synthesis and Properties of Small Functionalized Diazirine Molecules. Some Observations on the Reaction of a Diaziridine with the Iodine-Iodide Ion System. J. Org. Chem. 1970, 35, 2465–2471. [Google Scholar] [CrossRef]
- Wang, L.; Murai, Y.; Yoshida, T.; Ishida, A.; Masuda, K.; Sakihama, Y.; Hashidoko, Y.; Hatanaka, Y.; Hashimoto, M. Alternative One-Pot Synthesis of (Trifluoromethyl)phenyldiazirines from Tosyloxime Derivatives: Application for New Synthesis of Optically Pure Diazirinylphenylalanines for Photoaffinity Labeling. Org. Lett. 2015, 17, 616–619. [Google Scholar] [CrossRef]
- Smith, R.A.G.; Knowles, J.R. The preparation and photolysis of 3-aryl-3H-diazirines. J. Chem. Soc. Perkin Trans. 2 1975, 686. [Google Scholar] [CrossRef]
- Ishida, A.; Wang, L.; Tachrim, Z.P.; Suzuki, T.; Sakihama, Y.; Hashidoko, Y.; Hashimoto, M. Comprehensive Synthesis of Photoreactive Phenylthiourea Derivatives for the Photoaffinity Labeling. Chemistryselect 2017, 2, 160–164. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Yoshida, T.; Muto, Y.; Murai, Y.; Tachrim, Z.P.; Ishida, A.; Nakagawa, S.; Sakihama, Y.; Hashidoko, Y.; Masuda, K.; et al. Synthesis of Diazirine-Based Photoreactive Saccharin Derivatives for the Photoaffinity Labeling of Gustatory Receptors. Eur. J. Org. Chem. 2015, 3129–3134. [Google Scholar] [CrossRef]
- Darrow, J.W.; Hadac, E.M.; Miller, L.J.; Sugg, E.E. Structurally similar small molecule photoaffinity CCK-A agonists and antagonists as novel tools for directly probing 7TM receptor-ligand interactions. Bioorganic Med. Chem. Lett. 1998, 8, 3127–3132. [Google Scholar] [CrossRef]
- Brunner, J.; Semenza, G. Selective Labeling of the Hydrophobic Core of Membranes with 3-(Trifluoromethyl)-3-(m-[125I]iodophenyl)diazirine, a Carbene-Generating Reagent. Biochemistry 1981, 20, 7174–7182. [Google Scholar] [CrossRef]
- Döring, T.; Mitchell, P.; Osswald, M.; Bochkariov, D.; Brimacombe, R. The decoding region of 16S RNA; A cross-linking study of the ribosomal A, P and E sites using tRNA derivatized at position 32 in the anticodon loop. EMBO J. 1994, 13, 2677–2685. [Google Scholar] [CrossRef]
- Biasotti, B.; Dallavalle, S.; Merlini, L.; Farina, C.; Gagliardi, S.; Parini, C.; Belfiore, P. Synthesis of photoactivable inhibitors of osteoclast vacuolar ATPase. Bioorg. Med. Chem. 2003, 11, 2247–2254. [Google Scholar] [CrossRef]
- Wixe, T.; Almqvist, F. An improved synthesis of 3-[3-(trifluoromethyl)-3H-1,2-diazirin-3-yl]aniline: A key intermediate in the synthesis of photoaffinity probes. Tetrahedron Lett. 2017, 58, 3350–3352. [Google Scholar] [CrossRef]
- Kosemura, S.; Emori, H.; Yamamura, S.; Anai, T.; Tomita, K.; Hasegawa, K. Design of photoaffinity reagents for labeling the auxin receptor in maize. Tetrahedron Lett. 1997, 38, 2125–2128. [Google Scholar] [CrossRef]
- Murai, Y.; Yoshida, T.; Wang, L.; Masuda, K.; Hashidoko, Y.; Monde, K.; Hatanaka, Y.; Hashimoto, M. Efficient Synthesis of Photoreactive 2-Propoxyaniline Derivatives as Artificial Sweeteners. Synlett 2016, 27, 946–950. [Google Scholar] [CrossRef]
- Burkard, N.; Bender, T.; Westmeier, J.; Nardmann, C.; Huss, M.; Wieczorek, H.; Grond, S.; Von Zezschwitz, P. New fluorous photoaffinity labels (F-PAL) and their application in V-ATPase inhibition studies. Eur. J. Org. Chem. 2010, 2176–2181. [Google Scholar] [CrossRef]
- Song, Z.; Zhang, Q. Fluorous aryldiazirine photoaffinity labeling reagents. Org. Lett. 2009, 11, 4882–4885. [Google Scholar] [CrossRef] [PubMed]
- Lepage, M.L.; Simhadri, C.; Liu, C.; Takaffoli, M.; Bi, L.; Crawford, B.; Milani, A.S.; Wulff, J.E. A broadly applicable cross-linker for aliphatic polymers containing C–H bonds. Science 2019, 366, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Simhadri, C.; Bi, L.; Lepage, M.L.; Takaffoli, M.; Pei, Z.; Musolino, S.F.; Milani, A.S.; DiLabio, G.A.; Wulff, J.E. Flexible polyfluorinated bis-diazirines as molecular adhesives. Chem. Sci. 2021, 12, 4147–4153. [Google Scholar] [CrossRef] [PubMed]
- Protasova, I.; Bulat, B.; Jung, N.; Bräse, S. Synthesis of diaziridines and diazirines via resin-bound sulfonyl oximes. Org. Lett. 2017, 19, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.B.; Manetsch, R. Ammonia-free synthesis of 3-trifluoromethyl-3-phenyldiaziridine. Synth. Commun. 2018, 48, 626–631. [Google Scholar] [CrossRef]
- Gosselin, F.; O’Shea, P.D.; Roy, S.; Reamer, R.A.; Chen, C.Y.; Volante, R.P. Unprecedented catalytic asymmetric reduction of N-H imines. Org. Lett. 2005, 7, 355–358. [Google Scholar] [CrossRef]
- Schmitz, E.; Ohme, R. 3,3-Pentamethylenediazirine. In Organic Syntheses; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003; Volume 27, p. 83. ISBN 0471264180. [Google Scholar]
- Bond, M.R.; Zhang, H.; Vu, P.D.; Kohler, J.J. Photocrosslinking of glycoconjugates using metabolically incorporated diazirine-containing sugars. Nat. Protoc. 2009, 4, 1044–1063. [Google Scholar] [CrossRef]
- Bradley, G.F.; Evans, W.B.L.; Stevens, I.D.R. Thermal and photochemical decomposition of cycloalkanespirodiazirines. J. Chem. Soc. Perkin Trans. 1977, 2, 1214–1220. [Google Scholar] [CrossRef]
- Billing, P.; Brinker, U.H. Solvent dependency of the yield of an N-monosubstituted diaziridine: A GC–MS study. Tetrahedron Lett. 2012, 53, 2822–2824. [Google Scholar] [CrossRef]
- Heine, H.W.; Hoye, T.R.; Williard, P.G.; Hoye, R.C. Diaziridines. II. Addition of diaziridines to electrophilic acetylenes. J. Org. Chem. 1973, 38, 2984–2988. [Google Scholar] [CrossRef]
- Arora, S.; Palani, V.; Hoye, T.R. Reactions of Diaziridines with Benzynes Give N-Arylhydrazones. Org. Lett. 2018, 20, 8082–8085. [Google Scholar] [CrossRef]
- Schneider, Y.; Prévost, J.; Gobin, M.; Legault, C.Y. Diazirines as potent electrophilic Nitrogen sources: Application to the synthesis of Pyrazoles. Org. Lett. 2014, 16, 596–599. [Google Scholar] [CrossRef]
- Kuznetsov, V.V.; Shevtsov, A.V.; Pleshchev, M.I.; Strelenko, Y.A.; Makhova, N.N. Diastereoselective synthesis of 1,3-di- and 1,3,3-trisubstituted diaziridines coupled with neurotransmitter amino acids. Mendeleev Commun. 2016, 26, 391–394. [Google Scholar] [CrossRef]
- Beebe, A.W.; Dohmeier, E.F.; Moura-Letts, G. Diastereoselective synthesis of substituted diaziridines from simple ketones and aldehydes. Chem. Commun. 2015, 51, 13511–13514. [Google Scholar] [CrossRef]
- Carroccia, L.; Fioravanti, S.; Pellacani, L.; Sadun, C.; Tardella, P.A. Synthesis of optically active trifluoromethyl substituted diaziridines and oxaziridines. Tetrahedron 2011, 67, 5375–5381. [Google Scholar] [CrossRef]
- Aresu, E.; Carroccia, L.; Fioravanti, S.; Pellacani, L. Desymmetrization of α-diimines: Synthesis of new 3-(diaziridin-3-yl) oxaziridines. Tetrahedron Lett. 2013, 54, 4574–4576. [Google Scholar] [CrossRef]
- Kuznetsov, V.V.; Kachala, V.V.; Makhova, N.N. Synthesis of hybrid structures comprising diaziridine and cyclopropane rings in one molecule. Mendeleev Commun. 2018, 28, 497–500. [Google Scholar] [CrossRef]
- Lykke, L.; Halskov, K.S.; Carlsen, B.D.; Chen, V.X.; Jørgensen, K.A. Catalytic asymmetric diaziridination. J. Am. Chem. Soc. 2013, 135, 4692–4695. [Google Scholar] [CrossRef]
- Syroeshkina, Y.S.; Fershtat, L.L.; Kachala, V.V.; Kuznetsov, V.V.; Makhova, N.N. Diaziridine ring expansion in 6-aryl-1,5-diazabicyclo[3.1.0]hexanes upon reactions with activated olefins in ionic liquids. Russ. Chem. Bull. 2010, 59, 1621–1630. [Google Scholar] [CrossRef]
- Pleshchev, M.I.; Kachala, V.V.; Goloveshkin, A.S.; Bushmarinov, I.S.; Kuznetsov, V.V.; Khakimov, D.V.; Makhova, N.N. Unexpected regioselectivities of [3 + 2] cycloaddition of azomethine imines to acrylonitrile and 4-nitrophenyl vinyl sulfone. Mendeleev Commun. 2013, 23, 271–273. [Google Scholar] [CrossRef]
- Pleshchev, M.I.; Epishina, M.A.; Kachala, V.V.; Kuznetsov, V.V.; Goloveshkin, A.S.; Bushmarinov, I.S.; Makhova, N.N. Ionic liquid-promoted stereoselective [3 + 2] cycloaddition of 1-hetaryl-2-nitroethenes to azomethine imines generated in situ. Mendeleev Commun. 2013, 23, 206–208. [Google Scholar] [CrossRef]
- Pleshchev, M.I.; Petukhova, V.Y.; Kuznetsov, V.V.; Khakimov, D.V.; Pivina, T.S.; Struchkova, M.I.; Nelyubina, Y.V.; Makhova, N.N. Metathesis of Azomethine Imines in Reaction of 6-aryl-1,5-Diazabicyclo[3.1.0]Hexanes with (Het)Arylidenemalononitriles. Mendeleev Commun. 2013, 23, 34–36. [Google Scholar] [CrossRef]
- Petukhova, V.Y.; Pleshchev, M.I.; Fershtat, L.L.; Kuznetsov, V.V.; Kachala, V.V.; Makhova, N.N. Metathesis of azomethine imines in the reaction of 6-aryl-1,5- diazabicyclo[3.1.0]hexanes with carbonyl compounds. Mendeleev Commun. 2012, 22, 32–34. [Google Scholar] [CrossRef]
- Molchanov, A.P.; Efremova, M.M.; Kryukova, M.A.; Kuznetsov, M.A. Selective and reversible 1,3-dipolar cycloaddition of 6-aryl-1,5-diazabicyclo[3.1.0]hexanes with 1,3-diphenylprop-2-en-1-ones under microwave irradiation. Beilstein J. Org. Chem. 2020, 16, 2679–2686. [Google Scholar] [CrossRef]
- Camp, J.E. Auto-Tandem Catalysis: Activation of Multiple, Mechanistically Distinct Process by a Single Catalyst. Eur. J. Org. Chem. 2017, 425–433. [Google Scholar] [CrossRef] [Green Version]
- Capretto, D.A.; Brouwer, C.; Poor, C.B.; He, C. Gold(I)-catalyzed formation of 3-pyrazolines through cycloaddition of diaziridine to alkynes. Org. Lett. 2011, 13, 5842–5845. [Google Scholar] [CrossRef]
- Sarkar, T.; Talukdar, K.; Roy, S.; Punniyamurthy, T. Expedient iron-catalyzed stereospecific synthesis of triazines: Via cycloaddition of aziridines with diaziridines. Chem. Commun. 2020, 56, 3381–3384. [Google Scholar] [CrossRef]
- Pleshchev, M.I.; Das Gupta, N.V.; Kuznetsov, V.V.; Fedyanin, I.V.; Kachala, V.V.; Makhova, N.N. CAN-mediated new, regioselective one-pot access to bicyclic cationic structures with 2,3-dihydro-1H-pyrazolo[1,2-a]pyrazol-4-ium core. Tetrahedron 2015, 71, 9012–9021. [Google Scholar] [CrossRef]
- Chagarovskiy, A.O.; Vasin, V.S.; Kuznetsov, V.V.; Ivanova, O.A.; Rybakov, V.B.; Shumsky, A.N.; Makhova, N.N.; Trushkov, I.V. (3+3)-Annulation of Donor–Acceptor Cyclopropanes with Diaziridines. Angew. Chem. 2018, 57, 10338–10342. [Google Scholar] [CrossRef]
- Chagarovskiy, A.O.; Kuznetsov, V.V.; Ivanova, O.A.; Goloveshkin, A.S.; Levina, I.I.; Makhova, N.N.; Trushkov, I.V. Synthesis of 1-Substituted Pyrazolines by Reaction of Donor-Acceptor Cyclopropanes with 1,5-Diazabicyclo[3.1.0]hexanes. Eur. J. Org. Chem. 2019, 2019, 5475–5485. [Google Scholar] [CrossRef]
- Dong, K.; Marichev, K.O.; Doyle, M.P. Role of Donor–Acceptor Cyclopropenes in Metal Carbene Reactions. Conversion of E-Substituted Enoldiazoacetates to Z-Substituted Metallo-Enolcarbenes. Organometallics 2019, 38, 4043–4050. [Google Scholar] [CrossRef]
- Mondal, R.R.; Khamarui, S.; Maiti, D.K. Photocatalytic Generation of Nitrenes for Rapid Diaziridination. Org. Lett. 2017, 19, 5964–5967. [Google Scholar] [CrossRef]

































| No. | Analysis * | Possible Conformations/Configurations | ||||
|---|---|---|---|---|---|---|
| GED | XRD | DGC | QC | S | ||
| 1 | [22] | - | [28,29,30] | [22] | [28,29,30] |  |
| 2 | [23] | - | - | [23] | [23] |  |
| 3 | [24] | - | - | [24] | [24] |  |
| 4 | [25] | [27] | [27] | [25] | [25,27] |  |
| 5 | [26] | - | - | [26] | [26] |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tachrim, Z.P.; Wang, L.; Murai, Y.; Hashimoto, M. New Trends in Diaziridine Formation and Transformation (a Review). Molecules 2021, 26, 4496. https://doi.org/10.3390/molecules26154496
Tachrim ZP, Wang L, Murai Y, Hashimoto M. New Trends in Diaziridine Formation and Transformation (a Review). Molecules. 2021; 26(15):4496. https://doi.org/10.3390/molecules26154496
Chicago/Turabian StyleTachrim, Zetryana Puteri, Lei Wang, Yuta Murai, and Makoto Hashimoto. 2021. "New Trends in Diaziridine Formation and Transformation (a Review)" Molecules 26, no. 15: 4496. https://doi.org/10.3390/molecules26154496
APA StyleTachrim, Z. P., Wang, L., Murai, Y., & Hashimoto, M. (2021). New Trends in Diaziridine Formation and Transformation (a Review). Molecules, 26(15), 4496. https://doi.org/10.3390/molecules26154496