
Strategies and Approaches for Discovery of Small Molecule Disruptors of Biofilm Physiology


Abstract
:1. Introduction
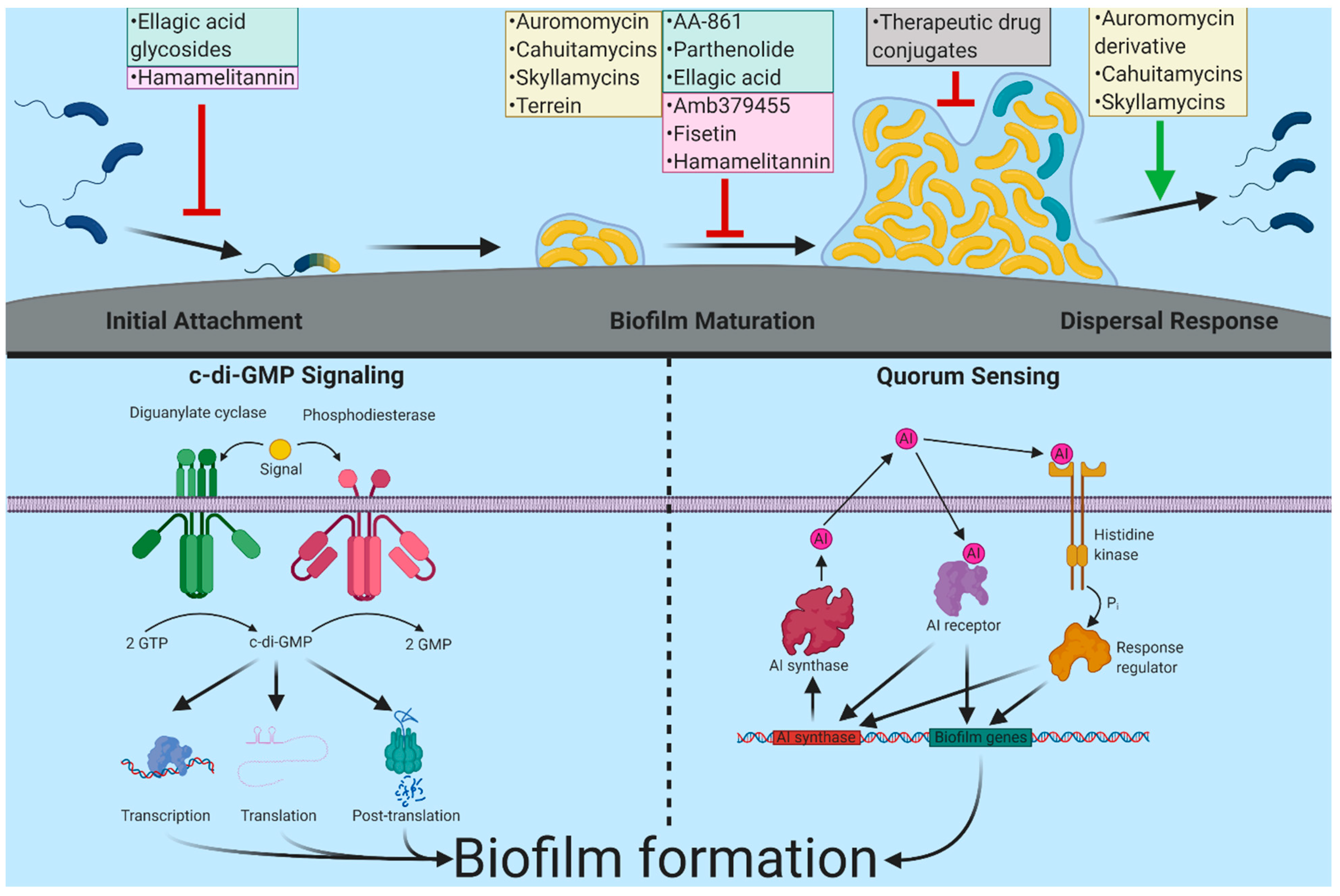
2. Biofilm Formation and Targets for Anti-Biofilm Interventions
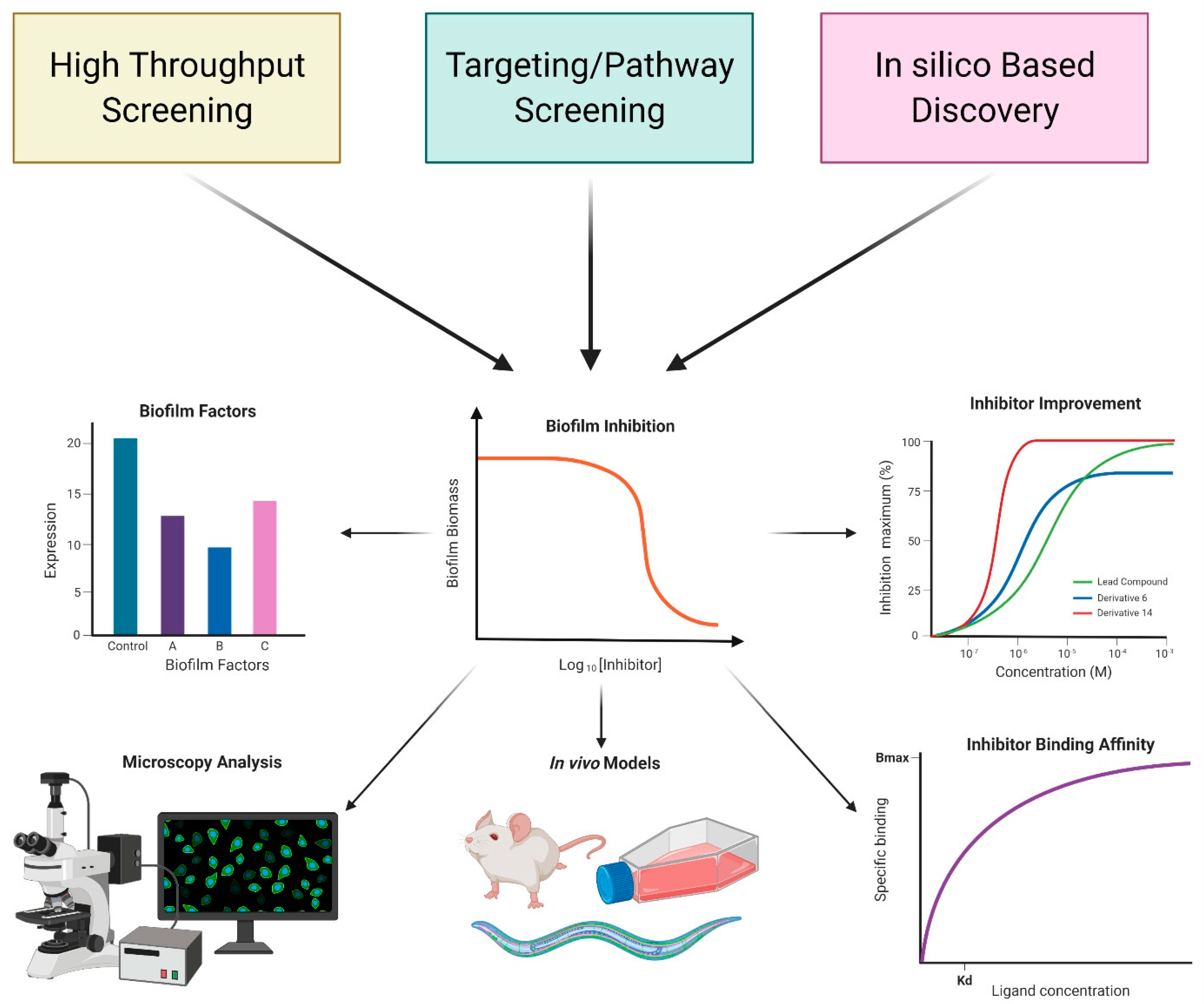
3. Approaches to Discover Anti-Biofilm Compounds
4. Inhibitors Identified via Cell-Based or In Vitro Screens
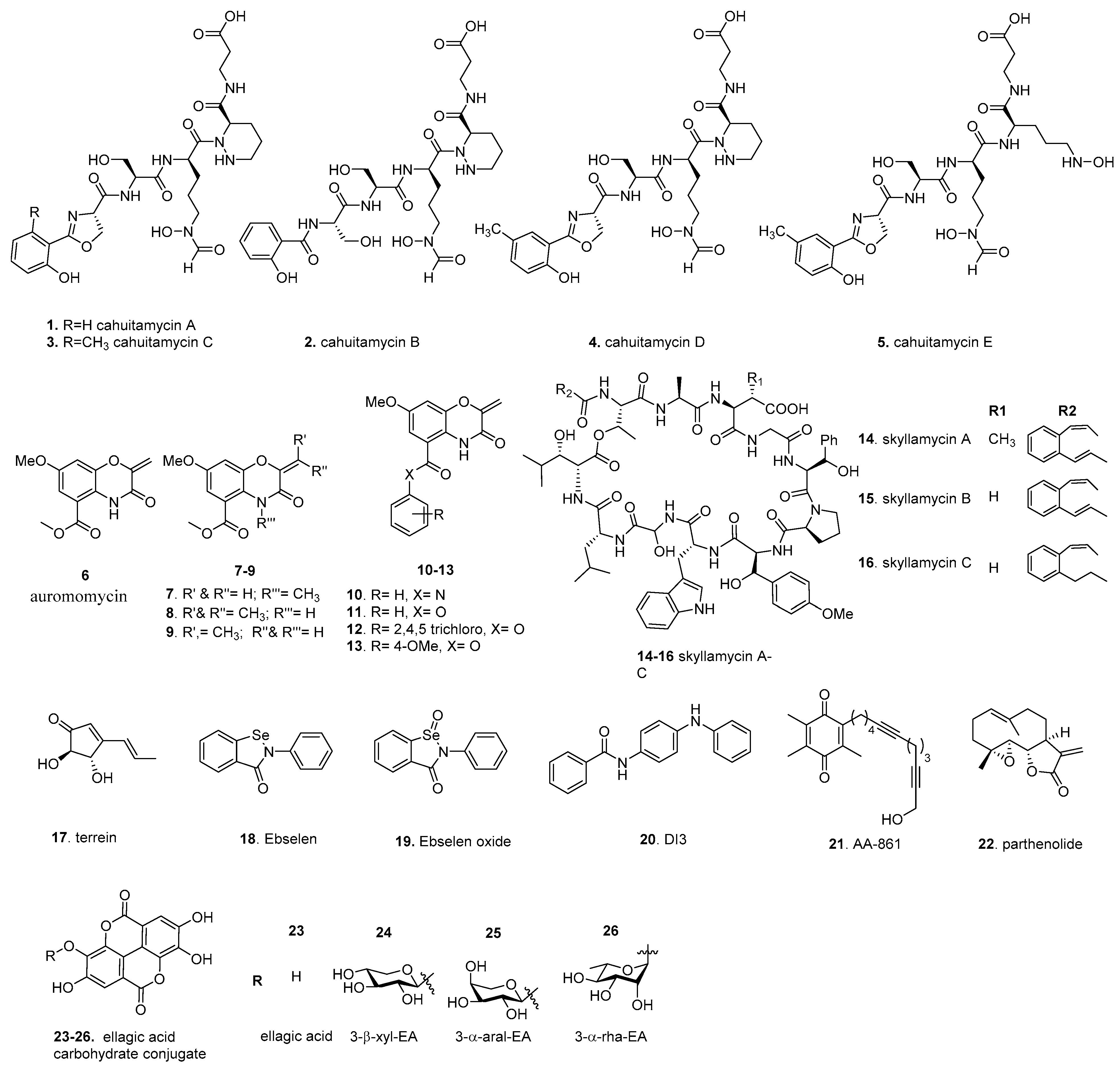
4.1. Cahuitamycins
4.2. Auromomycin
4.3. Skyllamycins
4.4. Terrein
4.5. Ebselen
4.6. DI-3
4.7. AA-861 and Parthenolide
4.8. Ellagic Acid Carbohydrate Conjugates
5. In Silico Discovery
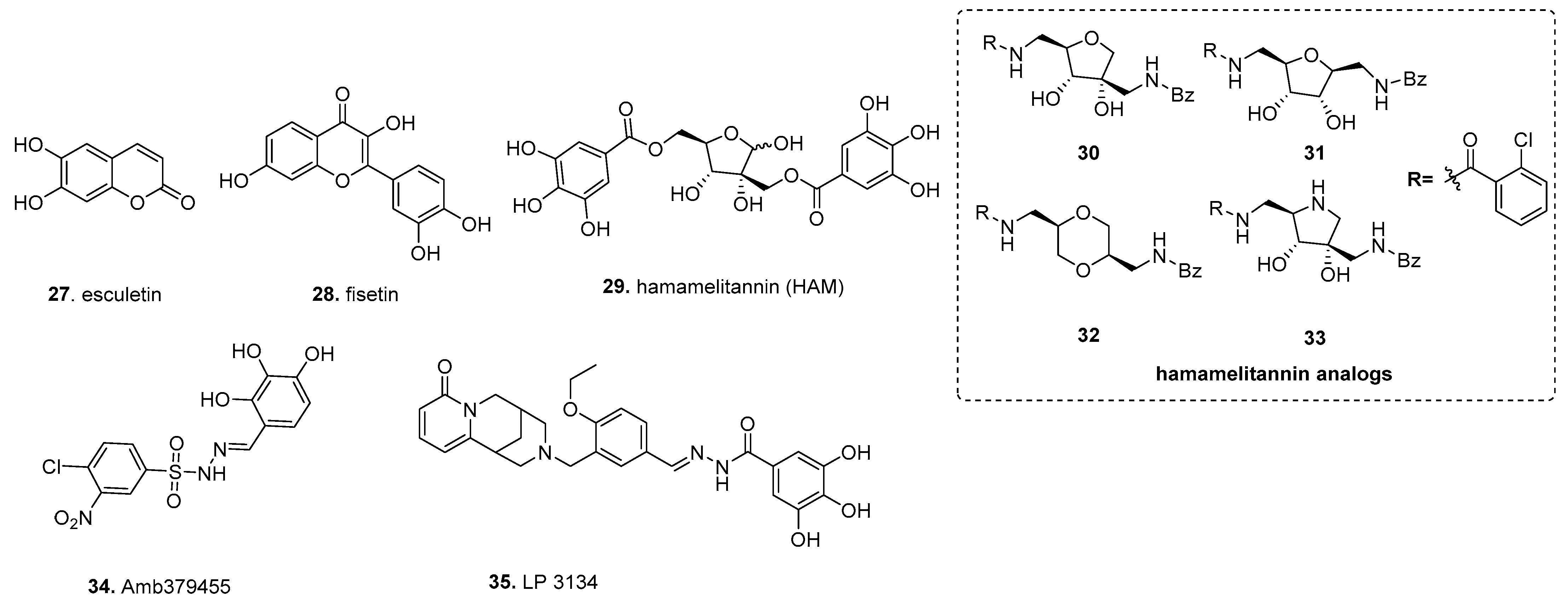
5.1. Fisetin (from Ellagic Acid)
5.2. Hamamelitannin
5.3. Amb379455
5.4. LP 3134
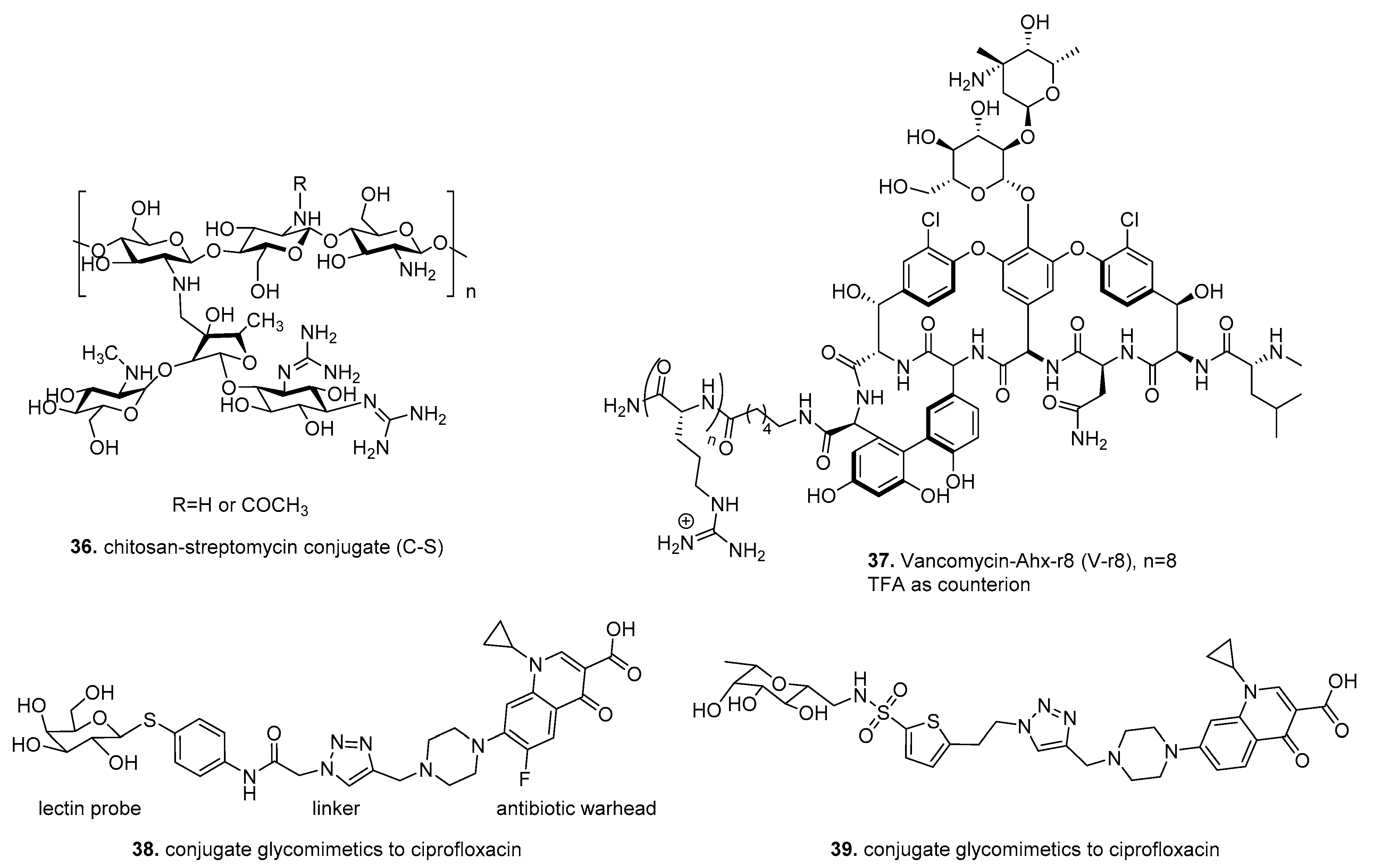
6. Therapeutic Drug Conjugates
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADMET | Absorption, distribution, metabolism, and excretion properties and toxicities which are useful in evaluating pharmecuetical properties of prospective therapeutics. |
| Antibacterial | A compound that kills bacteria or prevents bacterial growth. |
| BDC50 | The concentration at which 50% of the preformed biofilm is dispersed. |
| BIC50 | The concentration at which 50% of biofilm formation is inhibited. |
| Biofilm inhibitor | A compound that inhibits or negatively impacts biofilm formation. This includes the prevention of biofilm formation as well as dispersal and disruption of preformed biofilms. |
| DGC | Diguanylate cyclase |
| EC50 | The concentration at which treatment is 50% effective. |
| EDC condition | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) is used as a carboxyl activating agent to form amide bonds in coupling reactions. |
| IC50 | The concentration at which enzymatic activity is 50% inhibited. |
| MBEC | Minimal biofilm eradication concentration. |
| MIC | Minimal inhibitory concentration. |
| MoTr | Cell associated or cell-penetrating molecular transporter. |
| PDE | Phosphodiesterase |
| QS | Quorum sensing |
References
- Flemming, H.C.; Wuertz, S. Bacteria and Archaea on Earth and Their Abundance in Biofilms. Nat. Rev. Microbiol. 2019, 17, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Bjarnsholt, T. The Role of Bacterial Biofilms in Chronic Infections. APMIS Suppl. 2013, 121, 1–51. [Google Scholar] [CrossRef]
- Stewart, P.S. Mechanisms of Antibiotic Resistance in Bacterial Biofilms. Int. J. Med. Microbiol. 2002, 292, 107–113. [Google Scholar] [CrossRef]
- Tamayo, R.; Patimalla, B.; Camilli, A. Growth in a Biofilm Induces a Hyperinfectious Phenotype in Vibrio cholerae. Infect. Immun. 2010, 78, 3560–3569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallego-Hernandez, A.L.; Depas, W.H.; Park, J.H.; Teschler, J.K.; Hartmann, R.; Jeckel, H.; Drescher, K.; Beyhan, S.; Newman, D.K.; Yildiz, F.H. Upregulation of Virulence Genes Promotes Vibrio cholerae Biofilm Hyperinfectivity. Proc. Natl. Acad. Sci. USA 2020, 117, 11010–11017. [Google Scholar] [CrossRef]
- González, J.F.; Hahn, M.M.; Gunn, J.S. Chronic Biofilm-Based Infections: Skewing of the Immune Response. Pathog. Dis. 2018, 76, 1–7. [Google Scholar] [CrossRef]
- Hall, C.W.; Mah, T.F. Molecular Mechanisms of Biofilm-Based Antibiotic Resistance and Tolerance in Pathogenic Bacteria. FEMS Microbiol. Rev. 2017, 41, 276–301. [Google Scholar] [CrossRef]
- Gunn, J.S.; Bakaletz, L.O.; Wozniak, D.J. What’s on the Outside Matters: The Role of the Extracellular Polymeric Substance of Gram-Negative Biofilms in Evading Host Immunity and as a Target for Therapeutic Intervention. J. Biol. Chem. 2016, 291, 12538–12546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crabbé, A.; Jensen, P.Ø.; Bjarnsholt, T.; Coenye, T. Antimicrobial Tolerance and Metabolic Adaptations in Microbial Biofilms. Trends Microbiol. 2019, 27, 850–863. [Google Scholar] [CrossRef]
- Lewis, K. Riddle of Biofilm Resistance. Antimicrob. Agents Chemother. 2001, 45, 999–1007. [Google Scholar] [CrossRef] [Green Version]
- Kirketerp-Møller, K.; Zulkowski, K.; James, G. Chronic Wound Colonization, Infection, and Biofilms. In Biofilm Infections; Springer: New York, NY, USA, 2011; pp. 11–24. [Google Scholar] [CrossRef]
- Hidron, A.I.; Edwards, J.R.; Patel, J.; Horan, T.C.; Sievert, D.M.; Pollock, D.A.; Fridkin, S.K.; National Healthcare Safety Network Team; Participating National Healthcare Safety Network Facilities. Antimicrobial-Resistant Pathogens Associated With Healthcare-Associated Infections: Annual Summary of Data Reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect. Control. Hosp. Epidemiol. 2008, 29, 996–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, P.S.; Bjarnsholt, T. Risk Factors for Chronic Biofilm-Related Infection Associated with Implanted Medical Devices. Clin. Microbiol. Infect. 2020, 26, 1034–1038. [Google Scholar] [CrossRef]
- Bjarnsholt, T.; Ciofu, O.; Molin, S.; Givskov, M.; Høiby, N. Applying Insights from Biofilm Biology to Drug Development-Can a New Approach Be Developed? Nat. Rev. Drug Discov. 2013, 12, 791–808. [Google Scholar] [CrossRef]
- Ventola, C.L. The Antibiotic Resistance Crisis: Causes and Threats. P T. 2015, 40, 277–283. [Google Scholar] [PubMed]
- Dadgostar, P. Antimicrobial Resistance: Implications and Costs. Infect. Drug Resist. 2019, 12, 3903–3910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, H.; Allan, R.N.; Howlin, R.P.; Stoodley, P.; Hall-Stoodley, L. Targeting Microbial Biofilms: Current and Prospective Therapeutic Strategies. Nat. Rev. Microbiol. 2017, 15, 740–755. [Google Scholar] [CrossRef]
- Ghosh, A.; Jayaraman, N.; Chatterji, D. Small-Molecule Inhibition of Bacterial Biofilm. ACS Omega 2020, 5, 3108–3115. [Google Scholar] [CrossRef]
- Qvortrup, K.; Hultqvist, L.D.; Nilsson, M.; Jakobsen, T.H.; Jansen, C.U.; Uhd, J.; Andersen, J.B.; Nielsen, T.E.; Givskov, M.; Tolker-Nielsen, T. Small Molecule Anti-Biofilm Agents Developed on the Basis of Mechanistic Understanding of Biofilm Formation. Front. Chem. 2019, 7, 742. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.H.; Tryon, R.G.; Kim, J.H. Screening for Diguanylate Cyclase (DGC) Inhibitors Mitigating Bacterial Biofilm Formation. Front. Chem. 2020, 8, 264. [Google Scholar] [CrossRef]
- Verderosa, A.D.; Totsika, M.; Fairfull-Smith, K.E. Bacterial Biofilm Eradication Agents: A Current Review. Front. Chem. 2019, 7, 824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melander, R.; Basak, A.; Melander, C. Natural Products as Inspiration for the Development of Bacterial Antibiofilm Agents. Nat. Prod. Rep. 2020, 37, 1454–1477. [Google Scholar] [CrossRef] [PubMed]
- Hyde, K.D.; Xu, J.; Rapior, S.; Jeewon, R.; Lumyong, S.; Niego, A.G.T.; Abeywickrama, P.D.; Aluthmuhandiram, J.V.S.; Brahamanage, R.S.; Brooks, S.; et al. The Amazing Potential of Fungi: 50 Ways We Can Exploit Fungi Industrially. Fungal Divers. 2019, 97, 1–136. [Google Scholar] [CrossRef] [Green Version]
- Utada, A.S.; Bennett, R.R.; Fong, J.C.N.; Gibiansky, M.L.; Yildiz, F.H.; Golestanian, R.; Wong, G.C.L. Vibrio cholerae Use Pili and Flagella Synergistically to Effect Motility Switching and Conditional Surface Attachment. Nat. Commun. 2014, 5, 4913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibiansky, M.L.; Conrad, J.C.; Jin, F.; Gordon, V.D.; Motto, D.A.; Mathewson, M.A.; Stopka, W.G.; Zelasko, D.C.; Shrout, J.D.; Wong, G.C.L. Bacteria Use Type IV Pili to Walk Upright and Detach from Surfaces. Science 2010, 330, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.C.; Zamorano-Sánchez, D.; Pagliai, F.A.; Park, J.; Id, K.A.F.; Lee, C.K.; Kitts, G.; Rose, C.B.; Bilotta, E.M.; Wong, G.C.L.; et al. Reciprocal C-di-GMP Signaling: Incomplete Flagellum Biogenesis Triggers c-di-GMP Signaling Pathways That Promote Biofilm Formation. PLoS Genet. 2020, 16, e1008703. [Google Scholar] [CrossRef] [Green Version]
- Belas, R. Biofilms, Flagella, and Mechanosensing of Surfaces by Bacteria. Trends Microbiol. 2014, 22, 517–527. [Google Scholar] [CrossRef]
- Guttenplan, S.B.; Kearns, D.B. Regulation of Flagellar Motility during Biofilm Formation. FEMS Microbiol. Rev. 2013, 37, 849–871. [Google Scholar] [CrossRef] [Green Version]
- Laverty, G.; Gorman, S.P.; Gilmore, B.F. Biomolecular Mechanisms of Staphylococcal Biofilm Formation. Future Microbiol. 2013, 8, 509–524. [Google Scholar] [CrossRef] [Green Version]
- Otto, M. Staphylococcal Biofilms. Microbiol. Spectr. 2018, 6, 1–17. [Google Scholar] [CrossRef]
- Le, K.Y.; Park, M.D.; Otto, M. Immune Evasion Mechanisms of Staphylococcus epidermidis Biofilm Infection. Front. Microbiol. 2018, 9, 359. [Google Scholar] [CrossRef]
- Ageorges, V.; Monteiro, R.; Leroy, S.; Burgess, C.M.; Pizza, M.; Chaucheyras-Durand, F.; Desvaux, M. Molecular Determinants of Surface Colonisation in Diarrhoeagenic Escherichia coli (DEC): From Bacterial Adhesion to Biofilm Formation. FEMS Microbiol. Rev. 2020, 44, 314–350. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Conrad, J.C.; Gibiansky, M.L.; Wong, G.C.L. Bacteria Use Type-IV Pili to Slingshot on Surfaces. Proc. Natl. Acad. Sci. USA 2011, 108, 12617–12622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, K.; Tseng, B.S.; Beckerman, B.; Jin, F.; Gibiansky, M.L.; Harrison, J.J.; Luijten, E.; Parsek, M.R.; Wong, G.C.L. Psl Trails Guide Exploration and Microcolony Formation in Pseudomonas aeruginosa Biofilms. Nature 2013, 497, 388–391. [Google Scholar] [CrossRef] [Green Version]
- Maier, B.; Wong, G.C.L. How Bacteria Use Type IV Pili Machinery on Surfaces. Trends Microbiol. 2015, 23, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.D.; Starkey, M.; Kremer, S.; Parsek, M.R.; Wozniak, D.J. Identification of Psl, a Locus Encoding a Potential Exopolysaccharide That Is Essential for Pseudomonas aeruginosa PAO1 Biofilm Formation. J. Bacteriol. 2004, 186, 4466–4475. [Google Scholar] [CrossRef] [Green Version]
- Hobley, L.; Ostrowski, A.; Rao, F.V.; Bromley, K.M.; Porter, M.; Prescott, A.R.; MacPhee, C.E.; Van Aalten, D.M.F.; Stanley-Wall, N.R. BslA Is a Self-Assembling Bacterial Hydrophobin That Coats the Bacillus subtilis Biofilm. Proc. Natl. Acad. Sci. USA 2013, 110, 13600–13605. [Google Scholar] [CrossRef] [Green Version]
- Zogaj, X.; Nimtz, M.; Rohde, M.; Bokranz, W.; Römling, U. The Multicellular Morphotypes of Salmonella Typhimurium and Escherichia coli Produce Cellulose as the Second Component of the Extracellular Matrix. Mol. Microbiol. 2001, 39, 1452–1463. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, F.H.; Schoolnik, G.K. Vibrio cholerae O1 El Tor: Identification of a Gene Cluster Required for the Rugose Colony Type, Exopolysaccharide Production, Chlorine Resistance, and Biofilm Formation. Proc. Natl. Acad. Sci. USA 1999, 96, 4028–4033. [Google Scholar] [CrossRef] [Green Version]
- Hobley, L.; Harkins, C.; Macphee, C.E.; Stanley-Wall, N.R. Giving Structure to the Biofilm Matrix: An Overview of Individual Strategies and Emerging Common Themes. FEMS Microbiol. Rev. 2015, 39, 649–669. [Google Scholar] [CrossRef] [Green Version]
- Whitchurch, C.B.; Tolker-Nielsen, T.; Ragas, P.C.; Mattick, J.S. Extracellular DNA Required for Bacterial Biofilm Formation. Science 2002, 295, 1487. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.J.; Jarrod Smith, T.; Sondermann, H.; O’Toole, G.A. From Input to Output: The Lap/c-di-GMP Biofilm Regulatory Circuit. Annu. Rev. Microbiol. 2020, 74, 607–631. [Google Scholar] [CrossRef] [PubMed]
- Kitts, G.; Giglio, K.M.; Zamorano-Sánchez, D.; Park, J.H.; Townsley, L.; Cooley, R.B.; Wucher, B.R.; Klose, K.E.; Nadell, C.D.; Yildiz, F.H.; et al. A Conserved Regulatory Circuit Controls Large Adhesins in Vibrio cholerae. mBio 2019, 10, e02822-19. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.J.; Wozniaka, D.J. Psl Produced by Mucoid Pseudomonas aeruginosa Contributes to the Establishment of Biofilms and Immune Evasion. mBio 2017, 8, e00864-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, D.P.; Matwichuk, M.L.; Townsend, D.O.; Reichhardt, C.; Lamba, D.; Wozniak, D.J.; Parsek, M.R. The Pseudomonas aeruginosa Lectin LecB Binds to the Exopolysaccharide Psl and Stabilizes the Biofilm Matrix. Nat. Commun. 2019, 10, 2183. [Google Scholar] [CrossRef] [PubMed]
- Reichhardt, C.; Wong, C.; da Silva, D.P.; Wozniak, D.J.; Parsek, M.R. CdrA Interactions within the Pseudomonas aeruginosa Biofilm Matrix Safeguard It from Proteolysis and Promote Cellular Packing. mBio 2018, 9, e01376-18. [Google Scholar] [CrossRef] [Green Version]
- Jennings, L.K.; Storek, K.M.; Ledvina, H.E.; Coulon, C.; Marmont, L.S.; Sadovskaya, I.; Secor, P.R.; Tseng, B.S.; Scian, M.; Filloux, A.; et al. Pel Is a Cationic Exopolysaccharide That Cross-Links Extracellular DNA in the Pseudomonas aeruginosa Biofilm Matrix. Proc. Natl. Acad. Sci. USA 2015, 112, 11353–11358. [Google Scholar] [CrossRef] [Green Version]
- Jain, N.; Chapman, M.R. Bacterial Functional Amyloids: Order from Disorder. Science 2019, 295, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Berk, V.; Fong, J.C.N.; Dempsey, G.T.; Develioglu, O.N.; Zhuang, X.; Liphardt, J.; Yildiz, F.H.; Chu, S. Molecular Architecture and Assembly Principles of Vibrio cholerae Biofilms. Science 2012, 337, 236–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colvin, K.M.; Gordon, V.D.; Murakami, K.; Borlee, B.R.; Wozniak, D.J.; Wong, G.C.L.; Parsek, M.R. The Pel Polysaccharide Can Serve a Structural and Protective Role in the Biofilm Matrix of Pseudomonas aeruginosa. PLoS Pathog. 2011, 7, e1001264. [Google Scholar] [CrossRef]
- Earl, C.; Arnaouteli, S.; Bamford, N.C.; Porter, M.; Sukhodub, T.; MacPhee, C.E.; Stanley-Wall, N.R. The Majority of the Matrix Protein TapA Is Dispensable for Bacillus subtilis Colony Biofilm Architecture. Mol. Microbiol. 2020, 114, 920–933. [Google Scholar] [CrossRef]
- Stewart, P.S.; Franklin, M.J. Physiological Heterogeneity in Biofilms. Nat. Rev. Microbiol. 2008, 6, 199–210. [Google Scholar] [CrossRef]
- Walters, M.C.; Roe, F.; Bugnicourt, A.; Franklin, M.J.; Stewart, P.S. Contributions of Antibiotic Penetration, Oxygen Limitation, and Low Metabolic Activity to Tolerance of Pseudomonas aeruginosa Biofilms to Ciprofloxacin and Tobramycin. Antimicrob. Agents Chemother. 2003, 47, 317–323. [Google Scholar] [CrossRef] [Green Version]
- Jenal, U.; Reinders, A.; Lori, C. Cyclic Di-GMP: Second Messenger Extraordinaire. Nat. Rev. Microbiol. 2017, 15, 271–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conner, J.G.; Zamorano-Sánchez, D.; Park, J.H.; Sondermann, H.; Yildiz, F.H. The Ins and Outs of Cyclic Di-GMP Signaling in Vibrio cholerae. Curr. Opin. Microbiol. 2017, 36, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hengge, R. Linking Bacterial Growth, Survival, and Multicellularity—Small Signaling Molecules as Triggers and Drivers. Curr. Opin. Microbiol. 2020, 55, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Valentini, M.; Filloux, A. Multiple Roles of C-di-GMP Signaling in Bacterial Pathogenesis. Annu. Rev. Microbiol. 2019, 8, 387–406. [Google Scholar] [CrossRef]
- Orr, M.W.; Galperin, M.Y.; Lee, V.T. Sustained Sensing as an Emerging Principle in Second Messenger Signaling Systems. Curr. Opin. Microbiol. 2016, 34, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Papenfort, K.; Bassler, B.L. Quorum Sensing Signal-Response Systems in Gram-Negative Bacteria. Nat. Rev. Microbiol. 2016, 14, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Jamal, S.B.; Hassan, S.S.; Carvalho, P.V.S.D.; Almeida, S.; Barh, D.; Ghosh, P.; Silva, A.; Castro, T.L.P.; Azevedo, V. Two-Component Signal Transduction Systems of Pathogenic Bacteria as Targets for Antimicrobial Therapy: An Overview. Front. Microbiol. 2017, 8, 1878. [Google Scholar] [CrossRef]
- Hirakawa, H.; Kurushima, J.; Hashimoto, Y.; Tomita, H. Progress Overview of Bacterial Two-Component Regulatory Systems as Potential Targets for Antimicrobial Chemotherapy. Antibiotics 2020, 9, 635. [Google Scholar] [CrossRef]
- Fong, J.; Yuan, M.; Jakobsen, T.H.; Mortensen, K.T.; Delos Santos, M.M.S.; Chua, S.L.; Yang, L.; Tan, C.H.; Nielsen, T.E.; Givskov, M. Disulfide Bond-Containing Ajoene Analogues As Novel Quorum Sensing Inhibitors of Pseudomonas aeruginosa. J. Med. Chem. 2017, 60, 215–227. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, C.T.; Miller, L.C.; Siryaporn, A.; Drescher, K.; Semmelhack, M.F.; Bassler, B.L. A Quorum-Sensing Inhibitor Blocks Pseudomonas aeruginosa Virulence and Biofilm Formation. Proc. Natl. Acad. Sci. USA 2013, 110, 17981–17986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carle, J.S.; Christophersen, C. Bromo-Substituted Physostigmine Alkaloids from a Marine Bryozoa Flustra foliacea. J. Am. Chem. Soc. 1979, 101, 4012–4013. [Google Scholar] [CrossRef]
- Lee, J.; Bansal, T.; Jayaraman, A.; Bentley, W.E.; Wood, T.K. Enterohemorrhagic Escherichia coli Biofilms Are Inhibited by 7-Hydroxyindole and Stimulated by Isatin. Appl. Environ. Microbiol. 2007, 73, 4100–4109. [Google Scholar] [CrossRef] [Green Version]
- Bunders, C.; Cavanagh, J.; Melander, C. Flustramine Inspired Synthesis and Biological Evaluation of Pyrroloindoline Triazole Amides as Novel Inhibitors of Bacterial Biofilms. Org. Biomol. Chem. 2011, 9, 5476–5481. [Google Scholar] [CrossRef] [PubMed]
- Minvielle, M.J.; Bunders, C.A.; Melander, C. Indole-Triazole Conjugates are Selective Inhibitors and Inducers of Bacterial Biofilms. Med. Chem. Comm. 2013, 4, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Bunders, C.A.; Minvielle, M.J.; Worthington, R.J.; Ortiz, M.; Cavanagh, J.; Melander, C. Intercepting Bacterial Indole Signaling with Flustramine Derivatives. J. Am. Chem. Soc. 2011, 133, 20160–20163. [Google Scholar] [CrossRef] [Green Version]
- Minvielle, M.J.K.; Eguren, K.; Melander, C. Highly Active Modulators of Indole Signaling Alter Pathogenic Behaviors in Gram-Negative and Gram-Positive Bacteria. Chem. Eur. J. 2013, 19, 17595–17602. [Google Scholar] [CrossRef]
- Huggins, W.M.; Barker, W.T.; Baker, J.T.; Hahn, N.A.; Melander, R.J.; Melander, C. Meridianin D Analogues Display Antibiofilm Activity against MRSA and Increase Colistin Efficacy in Gram-Negative Bacteria. ACS Med. Chem. Lett. 2018, 9, 702–707. [Google Scholar] [CrossRef]
- Brackett, S.M.; Cox, K.E.; Barlock, S.L.; Huggins, W.M.; Ackart, D.F.; Bassaraba, R.J.; Melander, R.J.; Melander, C. Meridianin D Analogues Possess Antibiofilm Activity Against Mycobacterium smegmatis. RSC Med. Chem. 2020, 11, 92–97. [Google Scholar] [CrossRef]
- Huigens, R.W.; Richards, J.J.; Parise, G.; Ballard, T.E.; Zeng, W.; Deora, R.; Melander, C. Inhibition of Pseudomonas aeruginosa Biofilm Formation with Bromoageliferin Anlogues. J. Am. Chem. Soc. 2007, 129, 6966–6967. [Google Scholar] [CrossRef]
- Huigens, R.W.; Ma, L.; Gambino, C.; Moeller, P.D.R.; Basso, A.; Cavanagh, J.; Wozniak, D.J.; Melander, C. Control of Bacterial Biofilms with Marine Alkaloid Derivatives. Mol. BioSyst. 2008, 4, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, E.A.; Brackett, C.M.; Mullikin, T.; Alcaraz, C.; Melander, C. The Discovery of N-1 Substituted 2-Aminobenzimidazoles as Zinc-Dependent, S. Aureus Biofilm Inhibitors. Med. Chem. Comm. 2012, 3, 1462–1465. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.V.; Peszko, M.T.; Melander, R.J.; Melander, C. Using 2-aminobenzimidazole Derivatives to Inhibit Mycobacterium Smegmatis Biofilm Formation. Med. Chem. Comm. 2019, 10, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Huggins, W.M.; Nguyen, T.V.; Hahn, N.A.; Baker, J.T.; Kuo, L.G.; Kaur, D.; Melander, R.J.; Gunn, J.S.; Melander, C. 2-Aminobenzimidazoles as Antibiofilm Agents Against Salmonella enterica Serovar Typhimurium. Med. Chem. Comm. 2018, 9, 1547–1552. [Google Scholar] [CrossRef]
- Frei, R.; Breitbach, A.S.; Blackwell, H.E. 2-Aminobenzimidazole Derivatives Strongly Inhibit and Disperse Pseudomonas aeruginosa Biofilms. Angew. Chem. Int. Ed. 2012, 51, 5226–5229. [Google Scholar] [CrossRef] [Green Version]
- Richards, J.J.; Huigens, R.W.; Ballard, T.E.; Basso, A.; Cavanagh, J.; Melander, C. Inhibition and Dispersion of Proteobacterial Biofilms. Chem. Commun. 2008, 14, 1698–1700. [Google Scholar] [CrossRef]
- Richards, J.J.; Reed, C.S.; Melander, C. Effects of N-pyrrole Substitution on The Anti-biofilm Activities of Oroidin Derivatives Against Acinetobacter baumannii. Bioorg. Med. Chem. Lett. 2008, 18, 4325–4327. [Google Scholar] [CrossRef]
- Ballard, T.E.; Richards, J.J.; Wolfe, A.L.; Melander, C. Synthesis and Antibiofilm Activity of a Second-Generation Reverse-amide Oroidin Library: A Structure-Activity Relationship Study. Chem. Eur. J. 2008, 14, 10745–10761. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Worthington, R.J.; Melander, C.; Wu, H. A New Small Molecule Specifically Inhibits The Cariogenic Bacterium Streptococcus Mutans in Multispecies Biofilms. Antimicrob. Agents Chemother. 2011, 55, 2679–2687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milton, M.E.; Minrovic, B.M.; Harris, D.L.; Kang, B.; Jung, D.; Lewis, C.P.; Thompson, R.J.; Melander, R.J.; Zeng, D.; Melander, C.; et al. Re-sensitizing Multidrug Resistant Bacteria to Antibiotics by Targeting Bacterial Response Regulators: Characterization and Comparison of Interactions Between 2-Aminoimidazoles and The Response Regulators BfmR from Acinetobacter baumannii and QseB from Francisella spp. Front. Mol. Biosci. 2018, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Milton, M.E.; Allen, C.L.; Feldmann, E.A.; Bobay, B.G.; Jung, D.K.; Stephens, M.D.; Melander, R.J.; Theisen, K.E.; Zeng, D.; Thompson, R.J.; et al. Structure of the Francisella Response Regulator QseB Receiver Domain, and Characterization of QseB Inhibition by Antibiofilm 2-Aminoimidazole-based Compounds. Mol. Microbiol. 2017, 106, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Steenackers, H.P.L.; Ermolat’ev, D.S.; Savaliya, B.; De Weerdt, A.; De Costert, D.; Shah, A.; Van der Eycken, E.V.; De Vos, D.E.; Vanderleyden, J.; De Keersmaecker, S.C.J. Structure Activity Relationship of 4 (5)- aryl-2-amino-1H-imidazoles, N1-Substituted 2-Aminoimidazoles and Imidazo [1,2-a] pyrimidinium Salts as Inhibitors of Biofilm Formation by Salmonella typhimurium and Pseudomonas aeruginosa. J. Med. Chem. 2011, 54, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Ermolat’ev, D.S.; Bariwal, J.B.; Steenackers, H.P.L.; De Keersmaecker, S.C.J.; Van der Eycken, E.V. Concise and Diversity-Oriented Route toward Polysubstituted 2-Aminoimidazole Alkaloids and Their Analogues. Angew. Chem. Int. Ed. 2010, 49, 9465–9468. [Google Scholar] [CrossRef] [PubMed]
- Trang, T.T.T.; Dieltjens, L.; Hooyberghs, G.; Waldrant, K.; Ermolat’ev, D.S.; Van der Eycken, E.V.; Steenackers, H.P.L. Enhancing the Anti-biofilm Activity of 5-aryl-2-aminoimidazoles Through Nature Inspired Dimerisation. Bioorg. Med. Chem. 2018, 26, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, E.A.; Worthington, R.J.; Alcaraz, C.; Melander, C. 2-Aminopyrimidine as a Novel Scaffold for Biofilm Modulation. Org. Biomol. Chem. 2012, 10, 2552–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, K.E.; Melander, C. Anti-biofilm Activity of Quinazoline Derivatives against Mycobacterium smegmatis. Med. Chem. Comm. 2019, 10, 1177–1179. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.J.; Bobay, B.G.; Stowe, S.D.; Olson, A.L.; Peng, L.; Su, Z.; Actis, L.A.; Melander, C.; Cavanagh, J. Identification of BfmR, a Response Regulator Involved in Biofilm Development, as a Target for a 2-aminoimidazole-based Antibiofilm Agent. Biochemistry 2012, 51, 9776–9778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrero, N.V.; Bai, F.; Perez, C.; Duong, B.Q.; Rocca, J.R.; Jin, S.; Huigens, R.W. Phenazine Antibiotic Inspired Discovery of Potent Bromophenazine Antibacterial Agents Against Staphylococcus aureus and Staphylococcus epidermidis. Org. Biomol. Chem. 2014, 12, 881–886. [Google Scholar] [CrossRef]
- Garrison, A.T.; Abouelhassan, Y.; Kallifidas, D.; Bai, F.; Ukhanova, M.; Mai, V.; Jin, S.; Luesch, H.; Huigens, R.W. Halogenated Phenazines that Potently Eradicate Biofilms, MRSA Persister Cells in Non-Biofilm Cultures, and Mycobacterium tuberculosis. Angew. Chem. Int. Ed. 2015, 54, 14819–14823. [Google Scholar] [CrossRef]
- Garrison, A.T.; Bai, F.; Abouelhassan, Y.; Paciaroni, N.G.; Jin, S.; Huigens, R.W. Bromophenazine Derivatives with Potent Inhibition, Dispersion and Eradication Activities against Staphylococcus aureus Biofilms. RSC Adv. 2014, 5, 1120–1124. [Google Scholar] [CrossRef]
- Garrison, A.T.; Abouelhassan, Y.; Norwood, V.M.; Kallifidas, D.; Bai, F.; Nguyen, M.T.; Rolfe, M.; Burch, G.M.; Jin, S.; Luesch, H.; et al. Structure-Activity Relationships of a Diverse Class of Halogenated Phenazines That Targets Persistent, Antibiotic-Tolerant Bacterial Biofilms and Mycobacterium tuberculosis. J. Med. Chem. 2016, 59, 3808–3825. [Google Scholar] [CrossRef]
- Garrison, A.T.; Abouelhassan, Y.; Kallifidas, D.; Tan, H.; Kim, Y.S.; Jin, S.; Luesch, H.; Huigens, R.W. An Efficient BuchwaldHartwig/Reductive Cyclization for the Scaffold Diversification of Halogenated Phenazines: Potent Antibacterial Targeting, Biofilm Eradication and Prodrug Exploration. J. Med. Chem. 2018, 61, 3962–3983. [Google Scholar] [CrossRef] [PubMed]
- Hifnawy, M.S.; Hassan, H.M.; Mohammed, R.; Fouda, M.M.; Sayed, A.M.; Hamed, A.A.; AbouZid, S.F.; Rateb, M.E.; Alhadrami, H.A.; Abdelmohsen, U.R. Induction of Antibacterial Metabolites by Co-Cultivation of Two Red-Sea-Sponge-Associated Actinomycetes Micromonospora sp. UR56 and Actinokinespora sp. EG49. Mar. Drugs 2020, 18, 243. [Google Scholar] [CrossRef]
- Garrison, A.T.; Abouelhassan, Y.; Yang, H.; Yousaf, H.H.; Nguyen, T.J.; Huigens Iii, R.W. Microwave-Enhanced Friedländer Synthesis for the Rapid Assembly of Halogenated Quinolines with Antibacterial and Biofilm Eradication Activities against Drug Resistant and Tolerant Bacteria. Med. Chem. Comm. 2017, 8, 720–724. [Google Scholar] [CrossRef]
- Basak, A.; Abouelhassan, Y.; Huigens, R.W. Halogenated Quinolines Discovered Through Reductive Amination with Potent eradication Activities Against MRSA, MRSE and VRE Biofilms. Org. Biomol. Chem. 2015, 13, 10290–10294. [Google Scholar] [CrossRef] [PubMed]
- Basak, A.; Abouelhassan, Y.; Norwood, V.M., IV; Bai, F.; Nguyen, M.T.; Jin, S.; Huigens, R.W. Synthetically Tuning the 2-Position of Halogenated Quinolines: Optimizing Antibacterial and Biofilm Eradication Activities via Alkylation and Reductive Amination Pathways. Chem. Eur. J. 2016, 22, 9181–9189. [Google Scholar] [CrossRef]
- Basak, A.; Abouelhassan, Y.; Kim, Y.S.; Norwood, V.M.; Jin, S.; Huigens, R.W. Halogenated Quinolines Bearing Polar Functionality at the 2-Position: Identification of New Antibacterial Agents with Enhanced Activity Against Staphylococcus epidermidis. Eur. J. Med. Chem. 2018, 155, 705–713. [Google Scholar] [CrossRef]
- Kamal, A.; Rahim, A.; Riyaz, S.; Poornachandra, Y.; Balakrishna, M.; Kumar, C.G.; Hussaini, S.M.A.; Sridhar, B.; Machiraju, P.K. Regioselective Synthesis, Antimicrobial Evaluation and Theoretical Studies of 2-styryl Quinolines. Org. Biomol. Chem. 2015, 13, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Leon, B.; Fong, J.C.N.; Peach, K.C.; Wong, W.R.; Yildiz, F.H.; Linington, R.G. Development of Quinoline-Based Disruptors of Biofilm Formation Against Vibrio cholera. Org. Lett. 2013, 15, 1234–1237. [Google Scholar] [CrossRef]
- Abouelhassan, Y.; Garrison, A.T.; Burch, G.M.; Wong, W.; Norwood, V.M.; Huigens, R.W. Discovery of Quinoline Small Molecules with Potent Dispersal Activity Against Methicillin-Resistant Staphylococcus aureus and Staphylococcus epidermidis Biofilms Using a Scaffold Hopping Strategy. Bioorg. Med. Chem. Lett. 2014, 24, 5076–5080. [Google Scholar] [CrossRef]
- Leon, B.; Haeckl, F.P.; Linington, R.G. Optimized Quinoline Amino Alcohols as Disruptors and Dispersal Agents of Vibrio cholerae Biofilms. Org. Biomol. Chem. 2015, 13, 8495–8499. [Google Scholar] [CrossRef]
- Zaheer, Z.; Khan, F.A.; Sangshetti, J.N.; Patil, R.H.; Lohar, K.S. Novel Amalgamation of Phthalazine-quinolines as Biofilm Inhibitors: One-pot Synthesis, Biological Evaluation and in Silico ADME Prediction with Favorable Metabolic Fate. Bioorg. Med. Chem. Lett. 2016, 26, 1696–1703. [Google Scholar] [CrossRef] [PubMed]
- Aleksic, I.; Segan, S.; Andric, F.; Zlatovic, M.; Moric, I.; Opsenica, D.M.; Senerovic, L. Long-Chain 4-Aminoquinolines as Quorum Sensing Inhibitors in Serratia marcescens and Pseudomonas aeruginosa. ACS Chem. Biol. 2017, 12, 1425–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, F.A.K.; Kaduskar, R.N.; Patil, R.; Patil, R.H.; Ansari, S.A.; Alkahtani, H.M.; Almehizia, A.A.; Shinde, D.B.; Sangshetti, J.N. Synthesis, Biological Evaluations and Computational Studies of N-(3-(-2-(7-Chloroquinolin-2-yl)vinyl) benzylidene)anilines as Fungal Biofilm Inhibitors. Bioorg. Med. Chem. Lett. 2019, 29, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.N.; Zimmer, K.R.; Macedo, A.J.; Trentin, D.S. Plant Natural Products Targeting Bacterial Virulence Factors. Chem. Rev. 2016, 116, 9162–9236. [Google Scholar] [CrossRef] [PubMed]
- Huigens, R.W.; Abouelhassan, Y.; Yang, H. Phenazine Antibiotic-Inspired Discovery of Bacterial Biofilm-Eradicating Agents. Chem. Bio. Chem. 2019, 20, 2885–2902. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, L.; Zhang, M.; Liu, H.; Lu, P.; Lin, K. Quorum Sensing Inhibitors: A Patent Review (2014−2018). Expert Opin. Ther. Pat. 2018, 28, 849–865. [Google Scholar] [CrossRef]
- Parrino, B.; Schillaci, D.; Carnevale, I.; Giovannetti, E.; Diana, P.; Cirrincione, G.; Cascioferro, S. Synthetic Small Molecules as AntiBiofilm Agents in the Struggle against Antibiotic Resistance. Eur. J. Med. Chem. 2019, 161, 154–178. [Google Scholar] [CrossRef]
- Gilbert-Girard, S.; Savijoki, K.; Yli-Kauhaluoma, J.; Fallarero, A. Optimization of a High-Throughput 384-Well Plate-Based Screening Platform with Staphylococcus aureus ATCC 25923 and Pseudomonas aeruginosa ATCC 15442 Biofilms. Int. J. Mol. Sci. 2020, 21, 3034. [Google Scholar] [CrossRef]
- Paytubi, S.; de La Cruz, M.; Tormo, J.R.; Martín, J.; González, I.; González-Menendez, V.; Genilloud, O.; Reyes, F.; Vicente, F.; Madrid, C.; et al. A High-Throughput Screening Platform of Microbial Natural Products for the Discovery of Molecules with Antibiofilm Properties against Salmonella. Front. Microbiol. 2017, 8, 326. [Google Scholar] [CrossRef] [Green Version]
- Park, S.R.; Tripathi, A.; Wu, J.; Schultz, P.J.; Yim, I.; McQuade, T.J.; Yu, F.; Arevang, C.J.; Mensah, A.Y.; Tamayo-Castillo, G.; et al. Discovery of Cahuitamycins as Biofilm Inhibitors Derived from a Convergent Biosynthetic Pathway. Nat. Commun. 2016, 7, 10710. [Google Scholar] [CrossRef] [PubMed]
- Ochi, K.; Okamoto, S.; Tozawa, Y.; Inaoka, T.; Hosaka, T.; Xu, J.; Kurosawa, K. Ribosome Engineering and Secondary Metabolite Production. Adv. Appl. Microbiol. 2004, 56, 155–184. [Google Scholar] [CrossRef]
- Tripathi, A.; Park, S.R.; Sikkema, A.P.; Cho, H.J.; Wu, J.; Lee, B.; Xi, C.; Smith, J.L.; Sherman, D.H. A Defined and Flexible Pocket Explains Aryl Substrate Promiscuity of the Cahuitamycin Starter Unit-Activating Enzyme CahJ. Chem. Bio. Chem. 2018, 19, 1595–1600. [Google Scholar] [CrossRef] [PubMed]
- Peach, K.C.; Cheng, A.T.; Oliver, A.G.; Yildiz, F.H.; Linington, R.G. Discovery and Biological Characterization of the Auromomycin Chromophore as an Inhibitor of Biofilm Formation in Vibrio cholerae. Chem. Bio. Chem. 2013, 14, 2209–2215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Miura, K.; Kumada, Y.; Takeuchi, T.; Tanaka, N. Biological Activities of Non-Protein Chromophores of Antitumor Protein Antibiotics: Auromomycin and Neocarzinostatin. Biochem. Biophys. Res. Commun. 1980, 94, 255–261. [Google Scholar] [CrossRef]
- Shibuya, M.; Sakurai, H.; Maeda, T.; Nishiwaki, E.; Saito, M. Synthesis of the Degradation Product of Auromomycin Chromophore and DNA-Cleaving Activities of Its Derivatives. Tetrahedron Lett. 1986, 27, 1351–1354. [Google Scholar] [CrossRef]
- Kumada, Y.; Miwa, T.; Naoi, N.; Watanabe, K.; Naganawa, H.; Takita, T.; Umezawa, H.; Nnakamura, H.; Iitaka, Y. A Degradation Product of the Chromophore of Auromomycin. J. Antibiot. (Tokyo) 1983, 36, 200–202. [Google Scholar] [CrossRef] [Green Version]
- Peach, K.C.; Bray, W.M.; Shikuma, N.J.; Gassner, N.C.; Lokey, R.S.; Yildiz, F.H.; Linington, R.G. An Image-Based 384-Well High-Throughput Screening Method for the Discovery of Biofilm Inhibitors in Vibrio cholerae. Mol. Biosyst. 2011, 7, 1176. [Google Scholar] [CrossRef] [PubMed]
- Warner, C.J.A.; Cheng, A.T.; Yildiz, F.H.; Linington, R.G. Development of Benzo[1,4]Oxazines as Biofilm Inhibitors and Dispersal Agents against Vibrio cholerae. Chem. Commun. 2015, 51, 1305–1308. [Google Scholar] [CrossRef] [Green Version]
- Navarro, G.; Cheng, A.T.; Peach, K.C.; Bray, W.M.; Bernan, V.S.; Yildiz, F.H.; Linington, R.G. Image-Based 384-Well High-Throughput Screening Method for the Discovery of Skyllamycins A to C as Biofilm Inhibitors and Inducers of Biofilm Detachment in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2014, 58, 1092–1099. [Google Scholar] [CrossRef] [Green Version]
- Giltrap, A.M.; Haeckl, F.P.J.; Kurita, K.L.; Linington, R.G.; Payne, R.J. Total Synthesis of Skyllamycins A-C. J. Org. Chem. 2017, 23, 15046–15049. [Google Scholar] [CrossRef] [PubMed]
- Giltrap, A.M.; Haeckl, F.P.J.; Kurita, K.L.; Linington, R.G.; Payne, R.J. Synthetic Studies Toward the Skyllamycins: Total Synthesis and Generation of Simplified Analogues. J. Org. Chem. 2018, 83, 7250–7270. [Google Scholar] [CrossRef]
- Zhao, T.; Zhang, J.; Tang, M.; Ma, L.Z.; Lei, X. Development of an Effective Fluorescence Probe for Discovery of Aminopeptidase Inhibitors to Suppress Biofilm Formation. J. Antibiot. 2019, 72, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; He, X.; Xie, W.; Xiong, J.; Sheng, H.; Guo, S.; Huang, C.; Zhang, D.; Zhang, K. Elastase LasB of Pseudomonas aeruginosa Promotes Biofilm Formation Partly through Rhamnolipid-Mediated Regulation. Can. J. Microbiol. 2014, 60, 227–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.; Park, J.S.; Choi, H.Y.; Yoon, S.S.; Kim, W.G. Terrein Is an Inhibitor of Quorum Sensing and C-di-GMP in Pseudomonas aeruginosa: A Connection between Quorum Sensing and c-di-GMP. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Raistrick, H.; Smith, G. Studies in the Biochemistry of Micro-Organisms. Biochem. J. 1935, 29, 606–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.H.; Kim, D.S.; Kim, W.G.; Ryoo, I.J.; Lee, D.H.; Huh, C.H.; Youn, S.W.; Yoo, I.D.; Park, K.C. Terrein: A New Melanogenesis Inhibitor and Its Mechanism. Cell. Mol. Life Sci. 2004, 61, 2878–2885. [Google Scholar] [CrossRef]
- Goutam, J.; Sharma, G.; Tiwari, V.K.; Mishra, A.; Kharwar, R.N.; Ramaraj, V.; Koch, B. Isolation and Characterization of “Terrein” an Antimicrobial and Antitumor Compound from Endophytic Fungus Aspergillus Terreus (JAS-2) Associated from Achyranthus Aspera Varanasi, India. Front. Microbiol. 2017, 8, 1334. [Google Scholar] [CrossRef] [Green Version]
- Roelofs, K.G.; Wang, J.; Sintim, H.O.; Lee, V.T. Differential Radial Capillary Action of Ligand Assay for High-Throughput Detection of Protein-Metabolite Interactions. Proc. Natl. Acad. Sci. USA 2011, 108, 15528–15533. [Google Scholar] [CrossRef] [Green Version]
- Lieberman, O.J.; Orr, M.W.; Wang, Y.; Lee, V.T. High-Throughput Screening Using the Differential Radial Capillary Action of Ligand Assay Identifies Ebselen as an Inhibitor of Diguanylate Cyclases. ACS Chem. Biol. 2014, 9, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Lee, V.T.; Matewish, J.M.; Kessler, J.L.; Hyodo, M.; Hayakawa, Y.; Lory, S. A Cyclic-di-GMP Receptor Required for Bacterial Exopolysaccharide Production. Mol. Microbiol. 2007, 65, 1474–1484. [Google Scholar] [CrossRef] [Green Version]
- Schewe, T. Molecular Actions of Ebselen-an Antiinflammatory Antioxidant. Gen. Pharm. 1995, 26, 1153–1169. [Google Scholar] [CrossRef]
- Sambanthamoorthy, K.; Sloup, R.E.; Parashar, V.; Smith, J.M.; Kim, E.E.; Semmelhack, M.F.; Neiditch, M.B.; Waters, C.M. Identification of Small Molecules That Antagonize Diguanylate Cyclase Enzymes To Inhibit Biofilm Formation. Antimicrob. Agents Chemother. 2012, 56, 5202–5211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Römling, U.; Sierralta, W.D.; Eriksson, K.; Normark, S. Multicellular and Aggregative Behaviour of Salmonella typhimurium Strains Is Controlled by Mutations in the AgfD Promoter. Mol. Microbiol. 1998, 28, 249–264. [Google Scholar] [CrossRef]
- Kirisits, M.J.; Prost, L.; Starkey, M.; Parsek, M.R. Characterization of Colony Morphology Variants Isolated from Pseudomonas aeruginosa Biofilms. Appl. Environ. Microbiol. 2005, 71, 4809–4821. [Google Scholar] [CrossRef] [Green Version]
- Romero, D.; Sanabria-Valentin, E.; Vlamakis, H.; Kolter, R. Biofilm Inhibitors That Target Amyloid Proteins. Chem. Biol. 2013, 20, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Caro-Astorga, J.; Pérez-García, A.; de Vicente, A.; Romero, D. A Genomic Region Involved in the Formation of Adhesin Fibers in Bacillus cereus Biofilms. Front. Microbiol. 2014, 5, 745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalia, M.; Yadav, V.K.; Singh, P.K.; Sharma, D.; Narvi, S.S.; Agarwal, V. Exploring the Impact of Parthenolide as Anti-Quorum Sensing and Anti-Biofilm Agent against Pseudomonas aeruginosa. Life Sci. 2018, 199, 96–103. [Google Scholar] [CrossRef]
- Huber, B.; Eberl, L.; Feucht, W.; Polster, J. Influence of Polyphenols on Bacterial Biofilm Formation and Quorum-Sensing. Z Naturforsch. C J. Biosci. 2003, 58, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Quave, C.L.; Estévez-Carmona, M.; Compadre, C.M.; Hobby, G.; Hendrickson, H.; Beenken, K.E.; Smeltzer, M.S. Ellagic Acid Derivatives from Rubus ulmifolius Inhibit Staphylococcus aureus Biofilm Formation and Improve Response to Antibiotics. PLoS ONE 2012, 7, e28737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talekar, S.J.; Chochua, S.; Nelson, K.; Klugman, K.P.; Quave, C.L.; Vidal, J.E. 220D-F2 from Rubus Ulmifolius Kills Streptococcus pneumoniae Planktonic Cells and Pneumococcal Biofilms. PLoS ONE 2014, 9, e97314. [Google Scholar] [CrossRef] [Green Version]
- Fontaine, B.M.; Nelson, K.; Lyles, J.T.; Jariwala, P.B.; García-Rodriguez, J.M.; Quave, C.L.; Weinert, E.E. Identification of Ellagic Acid Rhamnoside as a Bioactive Component of a Complex Botanical Extract with Anti-Biofilm Activity. Front. Microbiol. 2017, 8, 496. [Google Scholar] [CrossRef]
- Chambers, S.A.; Gaddy, J.A.; Townsend, S.D. Synthetic Ellagic Acid Glycosides Inhibit Early Stage Adhesion of Streptococcus agalactiae Biofilms as Observed by Scanning Electron Microscopy. Chem. A Eur. J. 2020, 26, 9923–9928. [Google Scholar] [CrossRef]
- Maia, E.H.B.; Assis, L.C.; de Oliveira, T.A.; da Silva, A.M.; Taranto, A.G. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Dürig, A.; Kouskoumvekaki, I.; Vejborg, R.M.; Klemm, P. Chemoinformatics-Assisted Development of New Anti-Biofilm Compounds. Appl. Microbiol. Biotechnol. 2010, 87, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Balaban, I.; Novick, R.P. Autocrine Regulation of Toxin Synthesis by Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 1995, 92, 1619–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiran, M.D.; Adikesavan, N.V.; Cirioni, O.; Giacometti, A.; Silvestri, C.; Scalise, G.; Ghiselli, R.; Saba, V.; Orlando, F.; Shoham, M.; et al. Discovery of a Quorum-Sensing Inhibitor of Drug-Resistant Staphylococcal Infections by Structure-Based Virtual Screening. Mol. Pharmacol. 2008, 73, 1578–1586. [Google Scholar] [CrossRef] [Green Version]
- Brackman, G.; Breyne, K.; De Rycke, R.; Vermote, A.; Van Nieuwerburgh, F.; Meyer, E.; Van Calenbergh, S.; Coenye, T. The Quorum Sensing Inhibitor Hamamelitannin Increases Antibiotic Susceptibility of Staphylococcus aureus Biofilms by Affecting Peptidoglycan Biosynthesis and EDNA Release. Sci. Rep. 2016, 6, 20321. [Google Scholar] [CrossRef]
- Vermote, A.; Brackman, G.; Risseeuw, M.D.P.; Vanhoutte, B.; Cos, P.; Van Hecke, K.; Breyne, K.; Meyer, E.; Coenye, T.; Van Calenbergh, S. Hamamelitannin Analogues That Modulate Quorum Sensing as Potentiators of Antibiotics against Staphylococcus aureus. Angew.Chem. Int. Ed. 2016, 55, 6551–6555. [Google Scholar] [CrossRef]
- Vermote, A.; Brackman, G.; Risseeuw, M.D.P.; Coenye, T.; Van Calenbergh, S. Novel Hamamelitannin Analogues for the Treatment of Biofilm Related MRSA Infections–A Scaffold Hopping Approach. Eur. J. Med. Chem. 2017, 127, 757–770. [Google Scholar] [CrossRef] [Green Version]
- Bouton, J.; Van Hecke, K.; Rasooly, R.; Van Calenbergh, S. Synthesis of Pyrrolidine-Based Hamamelitannin Analogues as Quorum Sensing Inhibitors in Staphylococcus aureus. Beilstein J. Org. Chem. 2018, 14, 2822–2828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soukarieh, F.; Oton, E.V.; Dubern, J.F.; Gomes, J.; Halliday, N.; De Pilar Crespo, M.; Ramírez-Prada, J.; Insuasty, B.; Abonia, R.; Quiroga, J.; et al. In Silico and in Vitro-Guided Identification of Inhibitors of Alkylquinolone-Dependent Quorum Sensing in Pseudomonas aeruginosa. Molecules 2018, 23, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldo, S.; Giardina, G.; Mantoni, F.; Paiardini, A.; Paone, A.; Cutruzzolà, F. Discovering Selective Diguanylate Cyclase Inhibitors: From PleD to Discrimination of the Active Site of Cyclic-di-GMP Phosphodiesterases. Methods Mol. Biol. 2017, 1657, 431–453. [Google Scholar] [CrossRef] [PubMed]
- Fernicola, S.; Paiardini, A.; Giardina, G.; Rampioni, G.; Leoni, L.; Cutruzzolà, F.; Rinaldo, S. In Silico Discovery and In Vitro Validation of Catechol-Containing Sulfonohydrazide Compounds as Potent Inhibitors of the Diguanylate Cyclase PleD. J. Bacteriol. 2016, 198, 147–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, J.R.; Kuwada, N.J.; Huangyutitham, V.; Wiggins, P.A.; Harwood, C.S. Surface Sensing and Lateral Subcellular Localization of WspA, the Receptor in a Chemosensory-like System Leading to c-di-GMP Production. Mol. Microbiol. 2012, 86, 720–729. [Google Scholar] [CrossRef] [Green Version]
- Ueda, A.; Wood, T.K. Connecting Quorum Sensing, c-di-GMP, Pel Polysaccharide, and Biofilm Formation in Pseudomonas aeruginosa through Tyrosine Phosphatase TpbA (PA3885). PLoS Pathog. 2009, 5, e1000483. [Google Scholar] [CrossRef]
- Sambanthamoorthy, K.; Luo, C.; Pattabiraman, N.; Feng, X.; Koestler, B.; Waters, C.M.; Palys, T.J. Identification of Small Molecules Inhibiting Diguanylate Cyclases to Control Bacterial Biofilm Development. Biofouling 2014, 30, 17–28. [Google Scholar] [CrossRef]
- Hafeez, U.; Parakh, S.; Gan, H.K.; Scott, A.M. Antibody–Drug Conjugates for Cancer Therapy. Molecules 2020, 25, 4764. [Google Scholar] [CrossRef]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the Potential of Antibody–Drug Conjugates for Cancer Therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef]
- Liu, Y.; Busscher, H.J.; Zhao, B.; Li, Y.; Zhang, Z.; Van Der Mei, H.C.; Ren, Y.; Shi, L. Surface-Adaptive, Antimicrobially Loaded, Micellar Nanocarriers with Enhanced Penetration and Killing Efficiency in Staphylococcal Biofilms. ACS Nano 2016, 10, 4779–4789. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.F.; Brezden, A.; Mohammad, H.; Chmielewski, J.; Seleem, M.N. Targeting Biofilms and Persisters of ESKAPE Pathogens with P14KanS, a Kanamycin Peptide Conjugate. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 848–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedghizadeh, P.P.; Sun, S.; Junka, A.F.; Richard, E.; Sadrerafi, K.; Mahabady, S.; Bakhshalian, N.; Tjokro, N.; Bartoszewicz, M.; Oleksy, M.; et al. Design, Synthesis, and Antimicrobial Evaluation of a Novel Bone-Targeting Bisphosphonate-Ciprofloxacin Conjugate for the Treatment of Osteomyelitis Biofilms. J. Med. Chem. 2017, 60, 2326–2343. [Google Scholar] [CrossRef]
- Konai, M.M.; Haldar, J. Fatty Acid Comprising Lysine Conjugates: Anti-MRSA Agents That Display in Vivo Efficacy by Disrupting Biofilms with No Resistance Development. Bioconjug. Chem. 2017, 28, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Thakur, J.; Pal, S.; Gupta, R.; Mishra, D.; Kumar, S.; Yadav, K.; Saini, A.; Yavvari, P.S.; Vedantham, M.; et al. Cholic Acid-Peptide Conjugates as Potent Antimicrobials against Interkingdom Polymicrobial Biofilms. Antimicrob. Agents Chemother. 2019, 63, e00520-19. [Google Scholar] [CrossRef]
- Klahn, P.; Brönstrup, M. Bifunctional Antimicrobial Conjugates and Hybrid Antimicrobials. Nat. Prod. Rep. 2017, 34, 832–885. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Sommer, R.; Hinsberger, S.; Lu, C.; Hartmann, R.W.; Empting, M.; Titz, A. Novel Strategies for the Treatment of Pseudomonas Aeruginosa Infections. J. Med. Chem. 2016, 59, 5929–5969. [Google Scholar] [CrossRef]
- Stanzl, E.G.; Trantow, B.M.; Vargas, J.R.; Wender, P.A. Fifteen Years of Cell-Penetrating, Guanidinium-Rich Molecular Transporters: Basic Science, Research Tools, and Clinical Applications. Acc. Chem. Res. 2013, 46, 2944–2954. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Liu, K.; Jin, S.; Huigens, R.W. Design, Synthesis and Biological Evaluation of a Halogenated Phenazine-Erythromycin Conjugate Prodrug for Antibacterial Applications. Org. Biomol. Chem. 2021, 19, 1483–1487. [Google Scholar] [CrossRef]
- Xiao, T.; Liu, K.; Huigens, R.W. Progress towards a Stable Cephalosporin-Halogenated Phenazine Conjugate for Antibacterial Prodrug Applications. Bioorg. Med. Chem. Lett. 2020, 30, 127515. [Google Scholar] [CrossRef]
- Yilmaz Atay, H. Antibacterial Activity of Chitosan-Based Systems. In Functional Chitosan: Drug Delivery and Biomedical Applications; Springer: Singapore, 2019; pp. 457–489. [Google Scholar] [CrossRef]
- Zhang, A.; Mu, H.; Zhang, W.; Cui, G.; Zhu, J.; Duan, J. Chitosan Coupling Makes Microbial Biofilms Susceptible to Antibiotics. Sci. Rep. 2013, 3, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonoplis, A.; Zang, X.; Huttner, M.A.; Chong, K.K.L.; Lee, Y.B.; Co, J.Y.; Amieva, M.R.; Kline, K.A.; Wender, P.A.; Cegelski, L. A Dual-Function Antibiotic-Transporter Conjugate Exhibits Superior Activity in Sterilizing MRSA Biofilms and Killing Persister Cells. J. Am. Chem. Soc. 2018, 140, 16140–16151. [Google Scholar] [CrossRef] [PubMed]
- Meiers, J.; Zahorska, E.; Röhrig, T.; Hauck, D.; Wagner, S.; Titz, A. Directing Drugs to Bugs: Antibiotic-Carbohydrate Conjugates Targeting Biofilm-Associated Lectins of Pseudomonas Aeruginosa. J. Med. Chem. 2020, 63, 11707–11724. [Google Scholar] [CrossRef] [PubMed]
- Sommer, R.; Wagner, S.; Rox, K.; Varrot, A.; Hauck, D.; Wamhoff, E.C.; Schreiber, J.; Ryckmans, T.; Brunner, T.; Rademacher, C.; et al. Glycomimetic, Orally Bioavailable LecB Inhibitors Block Biofilm Formation of Pseudomonas Aeruginosa. J. Am. Chem. Soc. 2018, 140, 2537–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Target Organism | Discovery Method | BIC50/ IC50 | BDC50/ MBEC50 | Biofilm Reduction | Enzyme Inhibition |
|---|---|---|---|---|---|---|
| Cahuitamycin C (3) | A. baumannii | Cell Based HTS | 14.5 μM | 692 μM | - | - |
| Cahuitamycin D (4) | A. baumannii | Mutasynthetic Studies | 8.4 μM | 535 μM | - | - |
| Cahuitamycin E (5) | A. baumannii | Mutasynthetic Studies | 10.5 μM | - | - | - |
| Auromomycin (6) | V. cholerae | Cell Based HTS | 60.1 μM | - | - | - |
| Derivative 25 (13) | V. cholerae | SAR Studies | 6.0 μM | 13 μM | - | - |
| Skyllamycin A (14) | P. aeruginosa | Cell Based HTS | >250 μM | - | - | - |
| Skyllamycin B (15) | P. aeruginosa | Cell Based HTS | 30 μM | - | - | - |
| Skyllamycin C (16) | P. aeruginosa | Cell Based HTS | 60 μM | - | - | - |
| Terrein (17) | P. aeruginosa | Cell Based HTS | - | - | - | 81.10% |
| Ebselen (18) | P. aeruginosa | In vitro HTS | - | - | - | 80–90% |
| DI-3 (20) | V. cholerae | Cell Based HTS | 1.0 μM | - | - | - |
| AA-861 (21) | E. coli | Phenotypic screen | - | - | Near 40% | - |
| Parthenolide (22) | E. coli | Phenotypic screen | - | - | Near 40% | - |
| Ellagic acid (23) | S. aureus | Targeted screening | 50 μM | - | 50% | - |
| 3-β-xyl-EA (24) | S. aureus | SAR Studies | 512 μg/mL | - | - | - |
| 3-α-ara-EA (25) | S. aureus | SAR Studies | 512 μg/mL | - | - | - |
| Fiscetin (28) | S. aureus | Structure Based In silico Screen | - | - | 90% | - |
| Hamamelitannin (29) | S. aureus | Structure Based In silico Screen | 145.5 μM | - | - | - |
| Derivative 38 (30) | S. aureus | SAR Studies | 0.389 μM | - | - | - |
| Amb379455 (34) | C. crescentus | In silico Docking | 11.1 μM | - | - | - |
| LP3134 (35) | C. crescentus | In silico Docking | 44.9 μM | - | - | - |
| V-r8 (37) | S. aureus | Drug-conjugation | - | 9.5–20 μM | - | - |
| Congujate 7b (39) | P. aeruginosa | Drug-conjugation | - | - | 80–90% | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trebino, M.A.; Shingare, R.D.; MacMillan, J.B.; Yildiz, F.H. Strategies and Approaches for Discovery of Small Molecule Disruptors of Biofilm Physiology. Molecules 2021, 26, 4582. https://doi.org/10.3390/molecules26154582
Trebino MA, Shingare RD, MacMillan JB, Yildiz FH. Strategies and Approaches for Discovery of Small Molecule Disruptors of Biofilm Physiology. Molecules. 2021; 26(15):4582. https://doi.org/10.3390/molecules26154582
Chicago/Turabian StyleTrebino, Michael A., Rahul D. Shingare, John B. MacMillan, and Fitnat H. Yildiz. 2021. "Strategies and Approaches for Discovery of Small Molecule Disruptors of Biofilm Physiology" Molecules 26, no. 15: 4582. https://doi.org/10.3390/molecules26154582
APA StyleTrebino, M. A., Shingare, R. D., MacMillan, J. B., & Yildiz, F. H. (2021). Strategies and Approaches for Discovery of Small Molecule Disruptors of Biofilm Physiology. Molecules, 26(15), 4582. https://doi.org/10.3390/molecules26154582